Prion Disease and the Innate Immune System



Abstract
:
1. Introduction
1.1. Prion Diseases
1.2. The Innate Immune System
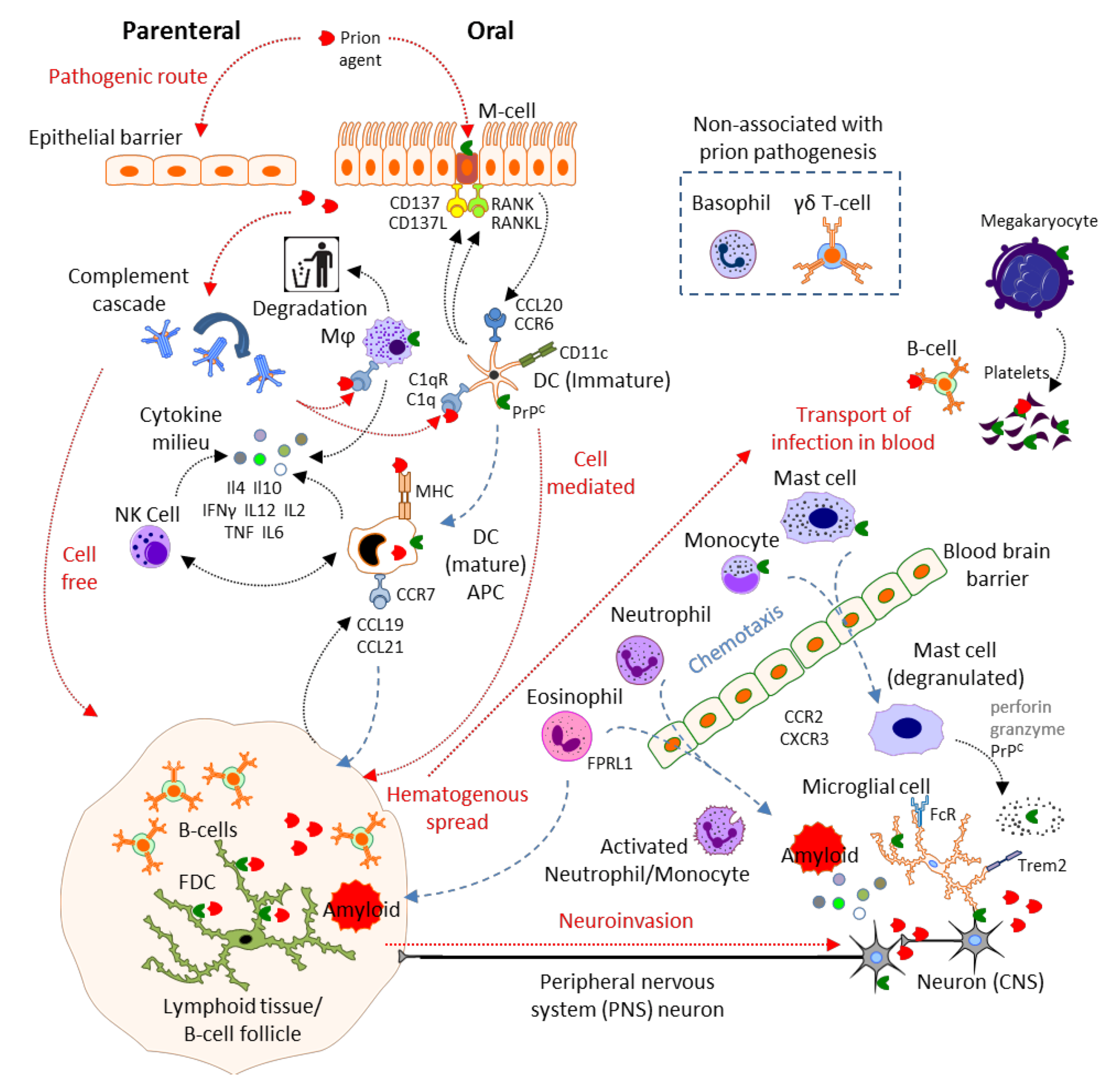
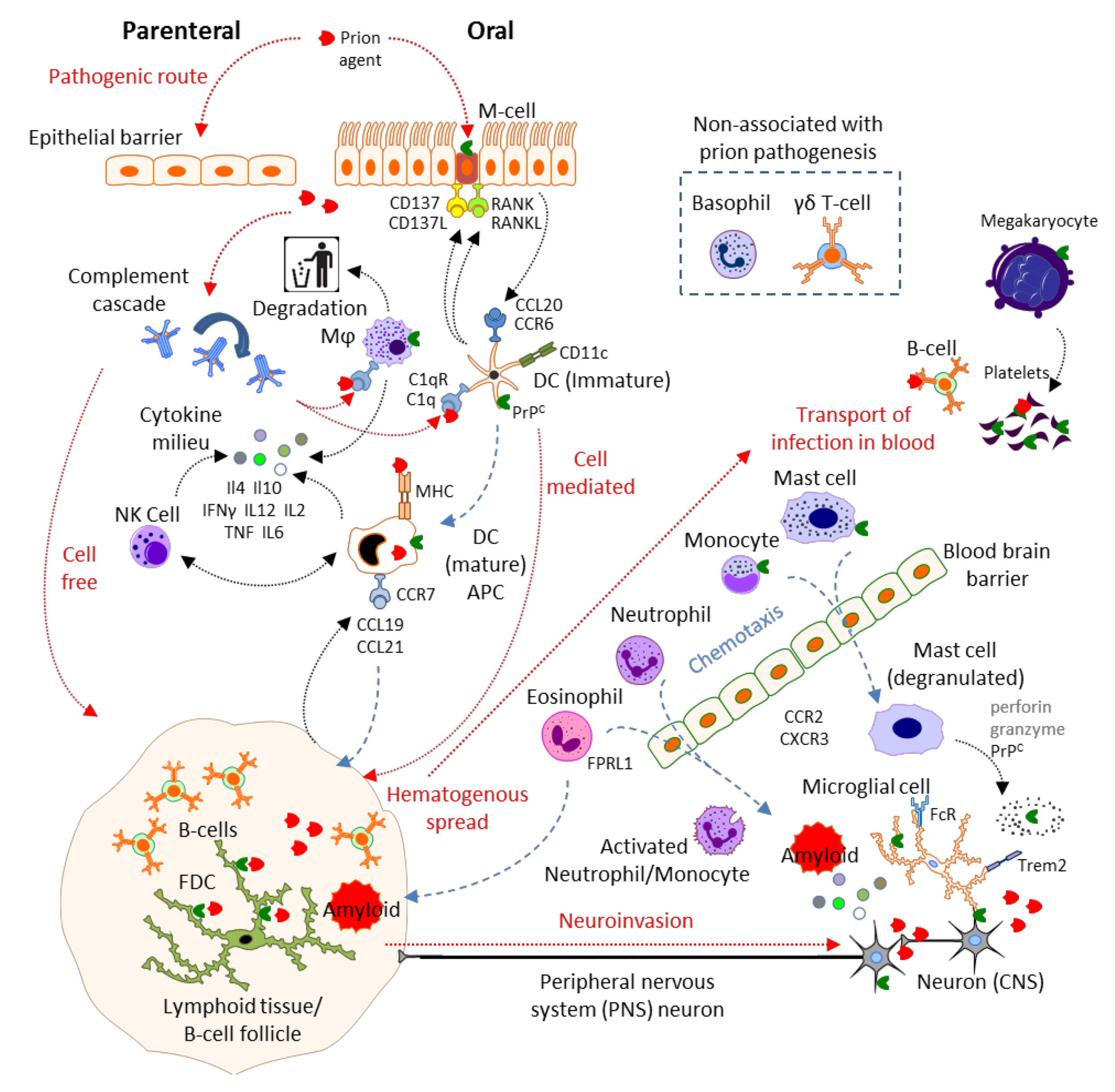
2. Results and Discussion
2.1. Physical Barrier to Infection: Epithelial Cells, Microfold Cells
2.2. Complement
2.3. Mast Cells
2.4. Mononuclear Phagocytes: Microglia, Macrophages, Monocytes, Dendritic Cells and Langerhans Cells
2.4.1. Degradative MNP
2.4.2. Antigen Presenting Cells (APC)
2.4.3. MNP in the CNS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene name | Gene | Uniprot | Phase a | Ref |
|---|---|---|---|---|
| Allograft inflammatory factor 1 | Aif1 / Iba1 | O70200 | M | [99] |
| B-cell differentiation antigen CD72 | Cd72 / Ly32, Lyb-2 | P21855 | M | [98] |
| Beta-2-microglobulin | B2m | P01887 | M | [99,100] |
| Calreticulin | Calr / CRP55 | P14211 | E | [47] |
| C3a anaphylatoxin chemotactic receptor | C3ar1 | O09047 | M | [101] |
| CAMPATH-1 antigen | Cd52 / Mb7 | Q64389 | E | [99,100,102] |
| C–C chemokine receptor type 5 | Ccr5 / MIP-1αR, CD195 | P51682 | E | [96,103] |
| C–C chemokine receptor type 6 | Ccr6 / CD196 | O54689 | E | [104] |
| C–C chemokine receptor type 7 | Ccr7 / MIP-3βR, CD197 | P47774 | E | [82] |
| C–C motif chemokine 2 | Ccl2 / MCP-1 | P10148 | E | [105,106,107] |
| C–C motif chemokine 3 | Ccl3 / MIP-1α | P10855 | E | [100,105,106] |
| C–C motif chemokine 4 | Ccl4 / MIP-1β | P14097 | E | [106] |
| C–C motif chemokine 5 | Ccl5 / RANTES | P30882 | M | [105] |
| C–C motif chemokine 7 | Ccl7 / MCP-3 | Q03366 | M | [103] |
| C–C motif chemokine 9 | Ccl9 / MRP-2 | P51670 | M | [99,102] |
| C–C motif chemokine 12 | Ccl12 / MCP-5 | Q62401 | M | [99,102] |
| C–C motif chemokine 19 | Ccl19 / ELC | O70460 | E | [82] |
| C–C motif chemokine 20 | Ccl20 / MIP-3α, Exodus-1 | O89093 | E | [104] |
| C–C motif chemokine 21a | Ccl21 / Exodus-2 | P84444 | E | [82] |
| C–C motif chemokine 21b | P86792 | |||
| C–C motif chemokine 21c | P86793 | |||
| CD9 antigen | Cd9 | P40240 | M | [99,108] |
| CD40 ligand | Cd40lg / Gp39, CD154 | P27548 | E | [109,110,111] |
| CD48 antigen | Cd48 / BLAST-1 | P18181 | E | [106] |
| CD209 antigen-like protein A b | Cd209a / DC-SIGN | Q91ZX1 | ||
| Cell surface glycoprotein CD200 receptor 4 | Cd200r4 | Q6XJV4 | M | [98] |
| CMRF35-like molecule 8 | Cd300a / MAIR1 | Q6SJQ0 | M | [98] |
| Complement C1q subcomponent subunit A | C1qa | P98086 | E | [41,44,45,47,100,101,106] |
| Complement C1q subcomponent subunit B | C1qb | P14106 | E | |
| Complement C1q subcomponent subunit C | C1qc | Q02105 | E | |
| Complement C2 | C2 | P21180 | E | [44] |
| Complement C3 | C3 / HSE-MSF | P01027 | M | [44,45,100,101] |
| Complement C4-B | C4B | P01029 | E | [41,99,101,102] |
| Complement C5 | C5 | P06684 | [112] | |
| Complement C7 c | C7 | D3YXF5 | E | [113] |
| Complement component C1q receptor | Cd93 / C1qRp, Ly68 | O89103 | ||
| Complement receptor type 1 | CR1 / CD35 | P17927 | E | [44,114] |
| Complement receptor type 2 | CR2 / CD21 | P19070 | E | [44,114,115,116] |
| C-type lectin domain family 4 member K | Cd207 / Langerin | Q8VBX4 | ||
| C-type lectin domain family 7 member A | Clec7a / Dectin1 | Q6QLQ4 | E | [99,101,102] |
| C-type lectin domain family 11 member A | Clec11a / Scgf | O88200 | L | [108] |
| CX3C chemokine receptor 1 | Cx3cr1 | Q9Z0D9 | E | [106,117] |
| C-X-C motif chemokine 9 | Cxcl9 / MIG, Scyb9 | P18340 | M | [118] |
| C-X-C motif chemokine 10 | Cxcl10 / Crg2, Ifi10, Inp10, Scyb10 | P17515 | E | [100,105,109,113] |
| C-X-C motif chemokine 11 | Cxcl11 / I-TAC, Scyb11 | Q9JHH5 | ||
| C-X-C motif chemokine 13 | Cxcl13 / BLC, Scyb13 | O55038 | E | [109] |
| C-X-C chemokine receptor type 3 | Cxcr3 / Cmkar3, CD183 | O88410 | M | [97] |
| C-X-C chemokine receptor type 4 | Cxcr4 / Cmkar4, Lestr, Sdf1r, CD184 | P70658 | M | [103] |
| C-X-C chemokine receptor type 5 | Cxcr5 / Blr1, Gpcr6, CD185 | Q04683 | L | [109] |
| EGF-like module-containing mucin-like hormone receptor-like 1 | Emr1 / GpF4/80 | Q61549 | E | [119] |
| Eotaxin | Ccl11 / Syca11 | P48298 | ||
| Forkhead box protein P3 | Foxp3 | Q99JB6 | [120] | |
| Formyl peptide receptor-related sequence 1 | Fpr-s1 / Fpr2, Fpr3, Fprl1, Lxa4r | O08790 | [121] | |
| Galectin-3 | Lgals3 / MAC-2 | P16110 | E | [106] |
| Galectin-3-binding protein | Lgals3bp / Cycap, Mama | Q07797 | E | [99,101] |
| Granulins | Grn / PCDGF | P28798 | E | [99,117,122] |
| Granulocyte-macrophage colony-stimulating factor | Csf2 / GM-CSF | P01587 | M | [105] |
| Granzyme B | Gzmb / Ctla1 | P04187 | ||
| Growth-regulated alpha protein | Cxcl1 / Gro, Gro1, Mgsa, Scyb1 | P12850 | E | [105] |
| Guanylate-binding protein 4 | Gbp4 / Gbp3 | Q61107 | M | [99] |
| H-2 class I histocompatibility antigen, D-37 alpha chain | H2-T23 | P06339 | M | [99] |
| H-2 class I histocompatibility antigen, D-D alpha chain | H2-D1 | P01900 | M | [99] |
| H-2 class I histocompatibility antigen, K-B alpha chain, | H2-K1 | Q7TN03 | E | [99] |
| Hematopoietic progenitor cell antigen CD34 | Cd34 | Q64314 | [123] | |
| High affinity immunoglobulin epsilon receptor subunit alpha | Fcer1a / FcεR1α | P20489 | ||
| High affinity immunoglobulin epsilon receptor subunit gamma | Fcer1g / FcεR1γ | P20491 | M | [99,101,102,117,122] |
| High affinity immunoglobulin gamma Fc receptor I | Fcgr1 / FcγRI, CD64 | P26151 | E | [44,98] |
| IgG receptor FcRn large subunit p51 | Fcgrt / FcRn | Q61559 | E | [124] |
| Integrin alpha-4 | Itga4 / CD49d | Q00651 | E | [125] |
| Integrin alpha-X | Itgax / CD11c | Q9QXH4 | E | [9,85,90,100,115,126] |
| Integrin alpha-M | Itgam / CD11b | P05555 | E | [96,100,127] |
| Integrin beta-1 | Itgb1 / VLA-4β, CD29 | P09055 | [128] | |
| Integrin beta-2 | Itgb2 / CD18 | P11835 | E | [101,117] |
| Integrin beta-7 | Itgb7 | P26011 | [125] | |
| Interferon alpha-inducible protein 27-like protein 2A | Ifi27 | Q8R412 | E | [99] |
| Interferon-induced protein with tetratricopeptide repeats 1 | Ifit1 | Q64282 | M | [99] |
| Interferon-induced protein with tetratricopeptide repeats 3 | Ifit3 | Q64345 | L | [99,108,122] |
| Interferon-induced transmembrane protein 3 | Ifitm3 | Q9CQW9 | E | [99,108] |
| Interferon gamma | Ifng / IFNγ | P01580 | M | [105,129] |
| Interferon gamma receptor 1 | Ifngr1 / IFNγR1, CD119 | P15261 | ||
| Interleukin-1 alpha | Il1a / Il1α | P01582 | E | [105,106,108,117] |
| Interleukin-1 beta | Il1b / Il1β | P10749 | E | [105,117,118,129] |
| Interleukin-1 receptor type 1 | Il1ra / IL1Rα, CD121a | P13504 | M | [118,130] |
| Interleukin-2 | Il2 | P04351 | E | |
| Interleukin-2 receptor subunit alpha | Il2ra / Il2Rα, CD25 | P01590 | E | [120] |
| Interleukin-4 | Il4 | P07750 | E | [131] |
| Interleukin-6 | Il6 | P08505 | M | [105,129,132] |
| Interleukin-10 | Il10 | P18893 | E | [130,131] |
| Interleukin-12 subunit alpha | Il12p35 | P43431 | [105,133] | |
| Interleukin-12 subunit beta | IL12p40 | P43432 | E | |
| Interleukin 13 | Il13 | P20109 | M | [105,131] |
| Interleukin 15 | Il15 | P48346 | ||
| Interleukin 18 | Il18 | P70380 | ||
| Interleukin 34 | Il34 | Q8R1R4 | ||
| Killer cell lectin-like receptor subfamily B member 1 | Klrb1 / NK1.1, CD161 | Q0ZUP1 | ||
| Leukocyte-associated immunoglobulin-like receptor 1 | Lair1 / CD305 | Q8BG84 | L | [98] |
| Leukocyte surface antigen CD47 | Cd47 / IAP | Q61735 | L | [108] |
| Leukocyte surface antigen CD53 | Cd53 | Q61451 | M | [99,100] |
| Low affinity immunoglobulin gamma Fc region receptor II | Fcgr2b / FcγRIIB, CD32 | P08101 | M | [44,98,99,101,102,117] |
| Low affinity immunoglobulin gamma Fc region receptor III | Fcgr3 / FcγRIII, CD16 | P08508 | M | [44,98,99,101,102,117] |
| L-selectin | Sell / Ly22, LAM1, LECAM1, CD62L | P18337 | E | [106] |
| Lymphocyte antigen 6C1 | Ly6c1 / Ly-6C | P0CW02 | M | [108,122] |
| Lymphocyte antigen 75 | Ly75 / DEC-205, CD205 | Q60767 | E | [127,134] |
| Lymphocyte antigen 86 | Ly86 / Md1 | O88188 | E | [99,102,106,108,122,135] |
| Lymphotoxin-alpha | Lta / LTα, TNFβ, Tnfsf1 | P09225 | E | [125,136] |
| Lymphotoxin-beta | Ltb / LTβ, Tnfc, Tnfsf3 | P41155 | E | [136] |
| Lysozyme C-2 | Lyz2 / Lyzs | P08905 | M | [99,102,137] |
| Macrophage colony-stimulating factor 1 | Csf1 / MCSF | P07141 | M | |
| Macrophage colony-stimulating factor 1 receptor | Csf1r / c-fms, CD115 | P09581 | M | [117] |
| Macrophage receptor MARCO | Marco | Q60754 | E | [138] |
| Macrophage scavenger receptor types I and II | Msr1 / Scvr, SRA, CD204 | P30204 | E | [138] |
| Macrosialin | Cd68 | P31996 | E | [99,100,101,106,115,127] |
| Mannose-binding protein A | Mbl1 / MBP-A | P39039 | ||
| Mannose-binding protein C | Mbl2 / MBP-C | P41317 | ||
| Mast cell surface glycoprotein Gp49A | Gp49a | Q61450 | M | [98] |
| Mast/stem cell growth factor receptor Kit | Kit / SCFR, c-Kit, CD117 | P05532 | ||
| MicroRNA 146a | miR-146a | E | [139] | |
| Monocyte differentiation antigen CD14 | Cd14 | P10810 | E | [101,106,115] |
| Myeloid cell surface antigen CD33 | Cd33 / Siglec3 | Q63994 | M | [98] |
| Myeloid differentiation primary response protein MyD88 | Myd88 | P22366 | [69] | |
| Perforin | Prf1 / Pfp | P10820 | ||
| Probable C-C chemokine receptor type 3 | Ccr3/ Cmkbr1l2, Cmkbr3, MIP-1αRl2, CD193 | P51678 | M | [103] |
| Prion protein | Prnp / PrP, CD230 | P04925 | E | [140,141] |
| Receptor-transporting protein 4 | Rtp4 / Ifrg28 | Q9ER80 | E | [99] |
| Scavenger receptor cysteine-rich type 1 protein M130 | Cd163 / M130 | Q2VLH6 | E | [113] |
| Sialic acid-binding Ig-like lectin 5 | Siglec5 / SiglecF, CD170 | Q920G3 | M | [98] |
| Sialoadhesin | Siglec1 / Sa, Sn, CD169 | Q62230 | E | [142] |
| SLAM family member 5 | Cd84 / Slamf5 | Q18PI6 | L | [101] |
| Sphingosine 1-phosphate receptor 1 | S1pr1 / Edg1, CD363 | O08530 | E | [143] |
| Stromal cell-derived factor 1 | Cxcl12 / SDF-1 | P40224 | E | [113] |
| T-cell surface glycoprotein CD3 gamma chain | Cd3g / TCRγ | P11942 | M | [116] |
| T-cell surface glycoprotein CD3 delta chain | Cd3d / TCRδ | P04235 | M | [116] |
| T-cell surface glycoprotein CD4 | Cd4 | P06332 | M | [116,120,144] |
| T-cell surface glycoprotein CD8 alpha chain | Cd8a / CD8α | P01731 | E | [9,116,144,145] |
| TGF-beta receptor type-2 | Tgfbr2 / TGFβR2 | Q62312 | M | [130] |
| TNF receptor-associated factor 5 | Traf5 | P70191 | L | [108] |
| Toll-like receptor 2 | Tlr2 / CD282 | Q9QUN7 | M | [101] |
| Transforming growth factor beta-1 | Tgfb1 / TGFβ1 | P04202 | M | [108,130,146,147] |
| Triggering receptor expressed on myeloid cells 2 | Trem2 | Q99NH8 | M | [98,99] |
| Tumor necrosis factor | Tnfa / TNFα | P06804 | E | [106,108,130,131,132] |
| Tumor necrosis factor ligand superfamily member 9 | Tnfsf9 / Cd137l, Cd157l, Ly63l | P41274 | E | |
| Tumor necrosis factor receptor superfamily member 1A | Tnfr1 / CD120a | P25118 | E | [130,148,149] |
| Tumor necrosis factor receptor superfamily member 1B | Tnfr2 / CD120b | P25119 | E | [130] |
| Tumor necrosis factor receptor superfamily member 9 | Tnfrsf9 / Ly63, CD137 | P20334 | E | |
| Tumor necrosis factor ligand superfamily member 11 | Tnfsf11 / RANKL, CD254 | O35235 | E | [29] |
| TYRO protein tyrosine kinase-binding protein | Tyrobp / DAP12, CD300d | O54885 | E | [98,99,101] |
| Tyrosine-protein phosphatase non-receptor type substrate 1 | Sirpa / Sirpα1, CD172a | P97797 | E | [115] |
2.5. Granulocytes: Neutrophils, Basophils and Eosinophils
2.6. Natural Killer Cells and γδ T Cells
2.7. Megakaryocytes and Platelets
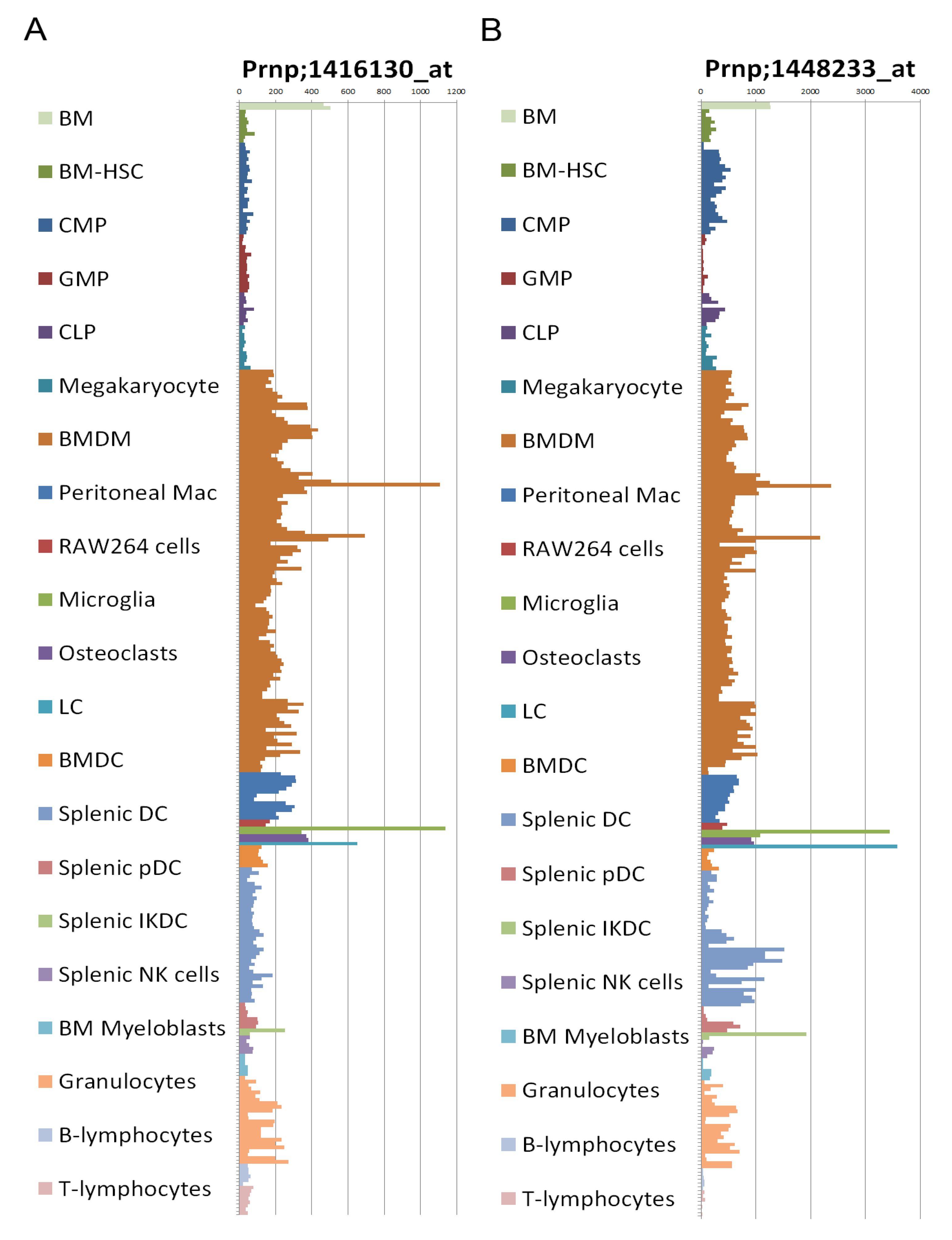
2.8. Prion Protein Expression
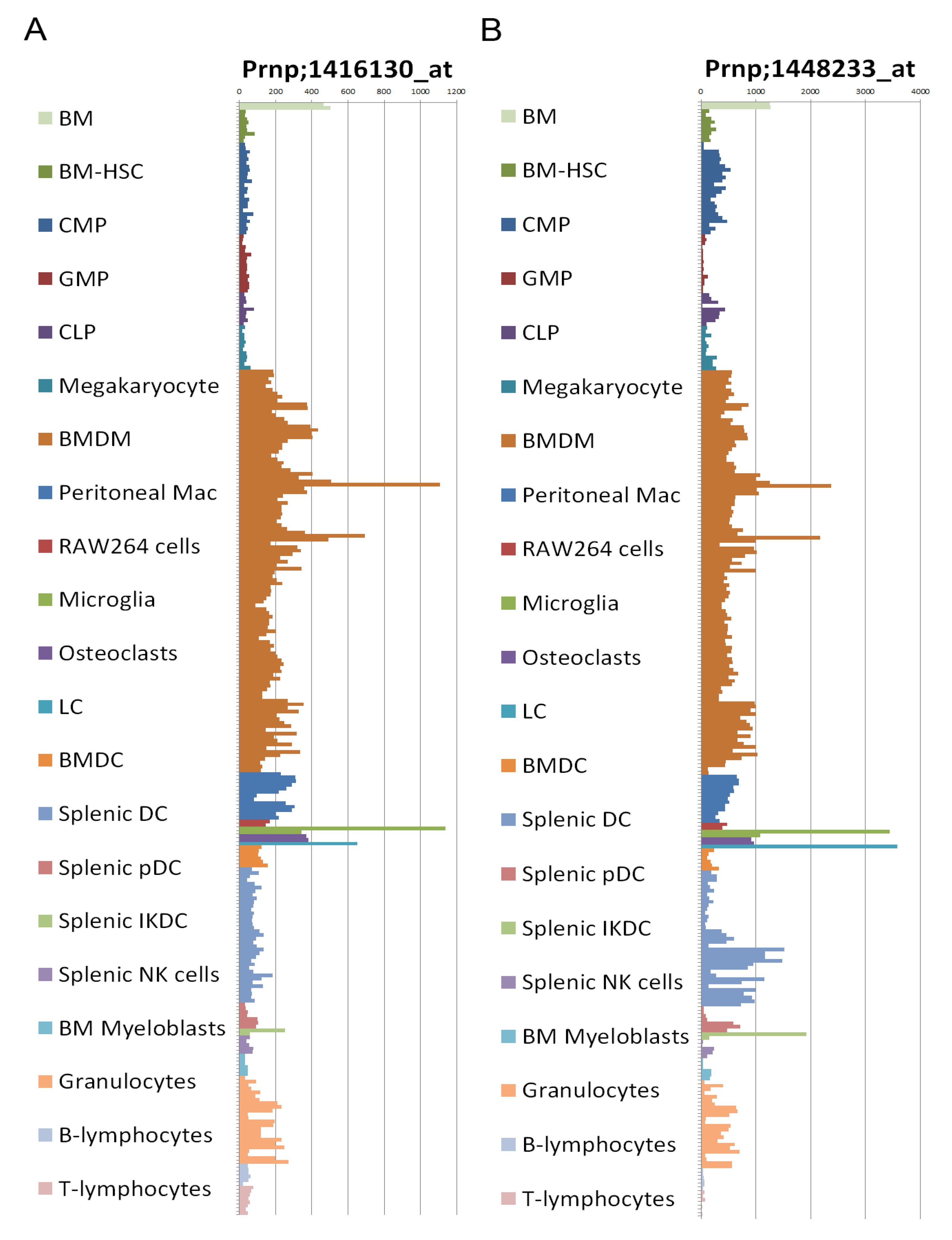
2.9. Transgenic Mice Models
| Upstream regulator | Acronym | P-value of overlap | # of genes |
|---|---|---|---|
| Interleukin-4 | Il-4 | 5.85E-68 | 73 |
| Interleukin-12 | IL-12 | 4.89E-64 | 49 |
| Interferon gamma | IFNγ | 1.32E-63 | 80 |
| Interleukin-10 | IL-10 | 3.85E-63 | 56 |
| Interleukin-2 | IL-2 | 1.05E-56 | 58 |
| Tumor necrosis factor | TNF | 2.70E-53 | 80 |
| Interleukin-6 | IL-6 | 9.31E-49 | 57 |
| Colony stimulating factor 2 | CSF2 | 2.85E-46 | 43 |

3. Summary and Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar]
- Fraser, H.; Dickinson, A.G. Pathogenesis of Scrapie in the Mouse: The Role of the Spleen. Nature 1970, 226, 462–463. [Google Scholar]
- Kimberlin, R.H.; Walker, C.A. The role of the spleen in the neuroinvasion of scrapie in mice. Virus Res. 1989, 12, 201–211. [Google Scholar] [CrossRef]
- Glaysher, B.R.; Mabbott, N.A. Role of the GALT in Scrapie Agent Neuroinvasion from the Intestine. J. Immunol. 2007, 178, 3757–3766. [Google Scholar]
- Glaysher, B.R.; Mabbott, N.A. Role of the draining lymph node in scrapie agent transmission from the skin. Immunol. Lett. 2007, 109, 64–71. [Google Scholar] [CrossRef]
- McCulloch, L.; Brown, K.L.; Bradford, B.M.; Hopkins, J.; Bailey, M.; Rajewsky, K.; Manson, J.C.; Mabbott, N.A. Follicular Dendritic Cell-Specific Prion Protein (PrPc) Expression Alone Is Sufficient to Sustain Prion Infection in the Spleen. PLoS Pathog. 2011, 7, e1002402. [Google Scholar]
- Brown, K.L.; Stewart, K.; Ritchie, D.L.; Mabbott, N.A.; Williams, A.; Fraser, H.; Morrison, W.I.; Bruce, M.E. Scrapie replication in lymphoid tissues depends on prion protein-expressing follicular dendritic cells. Nat. Med. 1999, 5, 1308–1312. [Google Scholar] [CrossRef]
- Kimberlin, R.H.; Walker, C.A. Pathogenesis of scrapie in mice after intragastric infection. Virus Res. 1989, 12, 213–220. [Google Scholar]
- Sethi, S.; Kerksiek, K.M.; Brocker, T.; Kretzschmar, H. Role of the CD8+ Dendritic Cell Subset in Transmission of Prions. J. Virol. 2007, 81, 4877–4880. [Google Scholar]
- Bartz, J.C.; Kincaid, A.E.; Bessen, R.A. Rapid prion neuroinvasion following tongue infection. J. Virol. 2003, 77, 583–591. [Google Scholar]
- Haybaeck, J.; Heikenwalder, M.; Klevenz, B.; Schwarz, P.; Margalith, I.; Bridel, C.; Mertz, K.; Zirdum, E.; Petsch, B.; Fuchs, T.J.; et al. Aerosols Transmit Prions to Immunocompetent and Immunodeficient Mice. PLoS Pathog. 2011, 7, e1001257. [Google Scholar] [CrossRef] [Green Version]
- Kimberlin, R.H.; Hall, S.M.; Walker, C.A. Pathogenesis of mouse scrapie: Evidence for direct neural spread of infection to the CNS after injection of sciatic nerve. J. Neurol. Sci. 1983, 61, 315–325. [Google Scholar] [CrossRef]
- Kimberlin, R.H.; Cole, S.; Walker, C.A. Pathogenesis of scrapie is faster when infection is intraspinal instead of intracerebral. Microb. Pathog. 1987, 2, 405–415. [Google Scholar]
- González, L.; Chianini, F.; Martin, S.; Sisó, S.; Gibbard, L.; Reid, H.W.; Jeffrey, M. Comparative titration of experimental ovine BSE infectivity in sheep and mice. J. Gen. Virol. 2007, 88, 714–717. [Google Scholar] [CrossRef]
- Fraser, H.; Brown, K.L.; Stewart, K.; McConnell, I.; McBride, P.; Williams, A. Replication of scrapie in spleens of SCID mice follows reconstitution with wild-type mouse bone marrow. J. Gen. Virol. 1996, 77, 1935–1940. [Google Scholar]
- Friedman-Levi, Y.; Hoftberger, R.; Budka, H.; Mayer-Sonnenfeld, T.; Abramsky, O.; Ovadia, H.; Gabizon, R. Targeting of prion-infected lymphoid cells to the central nervous system accelerates prion infection. J. Neuroinflammation 2012, 9. [Google Scholar] [CrossRef] [Green Version]
- Mabbott, N.A.; Kenneth Baillie, J.; Kobayashi, A.; Donaldson, D.S.; Ohmori, H.; Yoon, S.-O.; Freedman, A.S.; Freeman, T.C.; Summers, K.M. Expression of mesenchyme-specific gene signatures by follicular dendritic cells: Insights from the meta-analysis of microarray data from multiple mouse cell populations. Immunology 2011, 133, 482–498. [Google Scholar] [CrossRef]
- Fu, Y.-X.; Molina, H.; Matsumoto, M.; Huang, G.; Min, J.; Chaplin, D.D. Lymphotoxin-α (LTα) Supports Development of Splenic Follicular Structure That Is Required for IgG Responses. J. Exp. Med. 1997, 185, 2111–2120. [Google Scholar]
- Ansel, K.M.; Ngo, V.N.; Hyman, P.L.; Luther, S.A.; Forster, R.; Sedgwick, J.D.; Browning, J.L.; Lipp, M.; Cyster, J.G. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature 2000, 406, 309–314. [Google Scholar]
- Glatzel, M.; Heppner, F.L.; Albers, K.M.; Aguzzi, A. Sympathetic innervation of lymphoreticular organs is rate limiting for prion neuroinvasion. Neuron 2001, 31, 25–34. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar]
- Geijtenbeek, T.B.H.; Gringhuis, S.I. Signalling through C-type lectin receptors: Shaping immune responses. Nat. Rev. Immunol. 2009, 9, 465–479. [Google Scholar] [CrossRef]
- Fritz, J.H.; Ferrero, R.L.; Philpott, D.J.; Girardin, S.E. Nod-like proteins in immunity, inflammation and disease. Nat. Immunol. 2006, 7, 1250–1257. [Google Scholar]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef]
- Taylor, D.M.; McConnell, I.; Fraser, H. Scrapie infection can be established readily through skin scarification in immunocompetent but not immunodeficient mice. J. Gen. Virol. 1996, 77, 1595–1599. [Google Scholar] [CrossRef]
- Sigurdson, C.J.; Heikenwalder, M.; Manco, G.; Barthel, M.; Schwarz, P.; Stecher, B.; Krautler, N.J.; Hardt, W.-D.; Seifert, B.; MacPherson, A.J.S.; et al. Bacterial Colitis Increases Susceptibility to Oral Prion Disease. J. Infect. Dis. 2009, 199, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Bessen, R.A.; Wilham, J.M.; Lowe, D.; Watschke, C.P.; Shearin, H.; Martinka, S.; Caughey, B.; Wiley, J.A. Accelerated shedding of prions following damage to the olfactory epithelium. J. Virol 2012, 86, 1777–1788. [Google Scholar]
- Foster, N.; Macpherson, G.G. Murine Cecal Patch M Cells Transport Infectious Prions in Vivo. J. Infect. Dis. 2010, 202, 1916–1919. [Google Scholar] [CrossRef]
- Donaldson, D.S.; Kobayashi, A.; Ohno, H.; Yagita, H.; Williams, I.R.; Mabbott, N.A. M cell-depletion blocks oral prion disease pathogenesis. Mucosal Immunol. 2012, 5, 216–225. [Google Scholar] [CrossRef]
- Hsieh, E.H.; Fernandez, X.; Wang, J.; Hamer, M.; Calvillo, S.; Croft, M.; Kwon, B.S.; Lo, D.D. CD137 is Required for M Cell Functional Maturation but not Lineage Commitment. Am. J. Pathol. 2010, 177, 666–676. [Google Scholar] [CrossRef]
- Lugering, A.; Floer, M.; Westphal, S.; Maaser, C.; Spahn, T.W.; Schmidt, M.A.; Domschke, W.; Williams, I.R.; Kucharzik, T. Absence of CCR6 Inhibits CD4+ Regulatory T-Cell Development and M-Cell Formation inside Peyer’s Patches. Am. J. Pathol. 2005, 166, 1647–1654. [Google Scholar] [CrossRef]
- Westphal, S.; Lügering, A.; von Wedel, J.; von Eiff, C.; Maaser, C.; Spahn, T.; Heusipp, G.; Schmidt, M.A.; Herbst, H.; Williams, I.R.; et al. Resistance of Chemokine Receptor 6-Deficient Mice to Yersinia Enterocolitica Infection: Evidence of Defective M-Cell Formation in Vivo. Am. J. Pathol. 2008, 172, 671–680. [Google Scholar] [CrossRef]
- Nakato, G.; Hase, K.; Suzuki, M.; Kimura, M.; Ato, M.; Hanazato, M.; Tobiume, M.; Horiuchi, M.; Atarashi, R.; Nishida, N.; et al. Cutting Edge: Brucella abortus Exploits a Cellular Prion Protein on Intestinal M Cells as an Invasive Receptor. J. Immunol. 2012, 189, 1540–1544. [Google Scholar] [CrossRef]
- Kujala, P.; Raymond, C.R.; Romeijn, M.; Godsave, S.F.; van Kasteren, S.I.; Wille, H.; Prusiner, S.B.; Mabbott, N.A.; Peters, P.J. Prion Uptake in the Gut: Identification of the First Uptake and Replication Sites. PLoS Pathog. 2011, 7, e1002449. [Google Scholar]
- Rescigno, M.; Urbano, M.; Valzasina, B.; Francolini, M.; Rotta, G.; Bonasio, R.; Granucci, F.; Kraehenbuhl, J.-P.; Ricciardi-Castagnoli, P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2001, 2, 361–367. [Google Scholar]
- Chieppa, M.; Rescigno, M.; Huang, A.Y.C.; Germain, R.N. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J. Exp. Med. 2006, 203, 2841–2852. [Google Scholar] [CrossRef]
- Prinz, M.; Heikenwalder, M.; Junt, T.; Schwarz, P.; Glatzel, M.; Heppner, F.L.; Fu, Y.-X.; Lipp, M.; Aguzzi, A. Positioning of follicular dendritic cells within the spleen controls prion neuroinvasion. Nature 2003, 425, 957–962. [Google Scholar]
- Colten, H.R.; Ooi, Y.M.; Edelson, P.J. Synthesis and Secretion of Complement Proteins by Macrophages. Ann. NY Acad. Sci. 1979, 332, 482–490. [Google Scholar]
- Colten, H.R.; Perlmutter, R.C.; Schlessinger, D.H.; Cole, F.S. Regulation of Complement Protein Biosynthesis in Mononuclear Phagocytes; John Wiley & Sons, Ltd.: Chichester, UK, 1986; pp. 141–154. [Google Scholar]
- Schwaeble, W.; Schäfer, M.K.; Petry, F.; Fink, T.; Knebel, D.; Weihe, E.; Loos, M. Follicular dendritic cells, interdigitating cells, and cells of the monocyte-macrophage lineage are the C1q-producing sources in the spleen. Identification of specific cell types by in situ hybridization and immunohistochemical analysis. J. Immunol. 1995, 155, 4971–4978. [Google Scholar]
- Mitchell, D.A.; Kirby, L.; Paulin, S.M.; Villiers, C.L.; Sim, R.B. Prion protein activates and fixes complement directly via the classical pathway: Implications for the mechanism of scrapie agent propagation in lymphoid tissue. Mol. Immunol. 2007, 44, 2997–3004. [Google Scholar]
- Blanquet-Grossard, F.; Thielens, N.M.; Vendrely, C.; Jamin, M.; Arlaud, G.J. Complement Protein C1q Recognizes a Conformationally Modified Form of the Prion Protein. Biochemistry 2005, 44, 4349–4356. [Google Scholar]
- Mabbott, N.A. The complement system in prion diseases. Curr. Opin. Immunol. 2004, 16, 587–593. [Google Scholar]
- Klein, M.A.; Kaeser, P.S.; Schwarz, P.; Weyd, H.; Xenarios, I.; Zinkernagel, R.M.; Carroll, M.C.; Verbeek, J.S.; Botto, M.; Walport, M.J.; et al. Complement facilitates early prion pathogenesis. Nat. Med. 2001, 7, 488–492. [Google Scholar]
- Mabbott, N.A.; Bruce, M.E.; Botto, M.; Walport, M.J.; Pepys, M.B. Temporary depletion of complement component C3 or genetic deficiency of C1q significantly delays onset of scrapie. Nat. Med. 2001, 7, 485–487. [Google Scholar]
- Hartung, H.P.; Hadding, U. Synthesis of complement by macrophages and modulation of their functions through complement activation. Springer Semin. Immunopathol. 1983, 6, 283–326. [Google Scholar] [CrossRef]
- Flores-Langarica, A.; Sebti, Y.; Mitchell, D.A.; Sim, R.B.; MacPherson, G.G. Scrapie Pathogenesis: The Role of Complement C1q in Scrapie Agent Uptake by Conventional Dendritic Cells. J. Immunol. 2009, 182, 1305–1313. [Google Scholar]
- Bergtold, A.; Desai, D.D.; Gavhane, A.; Clynes, R. Cell Surface Recycling of Internalized Antigen Permits Dendritic Cell Priming of B Cells. Immunity 2005, 23, 503–514. [Google Scholar]
- Lewis, V.; Hill, A.F.; Haigh, C.L.; Klug, G.M.; Masters, C.L.; Lawson, V.A.; Collins, S.J. Increased proportions of C1 truncated prion protein protect against cellular M1000 prion infection. J. Neuropathol. Exp. Neurol. 2009, 68, 1125–1135. [Google Scholar] [CrossRef]
- Yadavalli, R.; Guttmann, R.P.; Seward, T.; Centers, A.P.; Williamson, R.A.; Telling, G.C. Calpain-dependent Endoproteolytic Cleavage of PrPSc Modulates Scrapie Prion Propagation. J. Biol. Chem. 2004, 279, 21948–21956. [Google Scholar]
- Hosszu, K.K.; Santiago-Schwarz, F.; Peerschke, E.I.B.; Ghebrehiwet, B. Evidence that a C1q/C1qR system regulates monocyte-derived dendritic cell differentiation at the interface of innate and acquired immunity. Innate Immun. 2010, 16, 115–127. [Google Scholar]
- Hosszu, K.K.; Valentino, A.; Vinayagasundaram, U.; Vinayagasundaram, R.; Joyce, M.G.; Ji, Y.; Peerschke, E.I.B.; Ghebrehiwet, B. DC-SIGN, C1q, and gC1qR form a trimolecular receptor complex on the surface of monocyte-derived immature dendritic cells. Blood 2012, 120, 1228–1236. [Google Scholar] [CrossRef]
- Teh, B.K.; Yeo, J.G.; Chern, L.M.; Lu, J. C1q regulation of dendritic cell development from monocytes with distinct cytokine production and T cell stimulation. Mol. Immunol. 2011, 48, 1128–1138. [Google Scholar]
- vajger, U.; Obermajer, N.; Anderluh, M.; Kos, J.; Jeras, M. DC-SIGN ligation greatly affects dendritic cell differentiation from monocytes compromising their normal function. J. Leukoc. Biol. 2011, 89, 893–905. [Google Scholar] [CrossRef]
- Hasebe, R.; Raymond, G.J.; Horiuchi, M.; Caughey, B. Reaction of complement factors varies with prion strains in vitro and in vivo. Virology 2012, 423, 205–213. [Google Scholar] [CrossRef]
- Michel, B.; Meyerett-Reid, C.; Johnson, T.; Ferguson, A.; Wyckoff, C.; Pulford, B.; Bender, H.; Avery, A.; Telling, G.; Dow, S.; et al. Incunabular Immunological Events in Prion Trafficking. Sci. Rep. 2012, 2. [Google Scholar] [CrossRef]
- Yanamadala, V.; Friedlander, R.M. Complement in neuroprotection and neurodegeneration. Trends Mol. Med. 2010, 16, 69–76. [Google Scholar]
- Veerhuis, R.; Nielsen, H.M.; Tenner, A.J. Complement in the brain. Mol. Immunol. 2011, 48, 1592–1603. [Google Scholar]
- Haddon, D.J.; Hughes, M.R.; Antignano, F.; Westaway, D.; Cashman, N.R.; McNagny, K.M. Prion Protein Expression and Release by Mast Cells After Activation. J. Infect. Dis. 2009, 200, 827–831. [Google Scholar] [CrossRef]
- Shanas, U.; Bhasin, R.; Sutherland, A.K.; Silverman, A.-J.; Silver, R. Brain mast cells lack the c-kit receptor: Immunocytochemical evidence. J. Neuroimmunol. 1998, 90, 207–211. [Google Scholar]
- Gilfillan, A.M.; Tkaczyk, C. Integrated signalling pathways for mast-cell activation. Nat. Rev. Immunol. 2006, 6, 218–230. [Google Scholar] [CrossRef]
- Silver, R.; Silverman, A.-J.; Vitković, L.; Lederhendler, I.I. Mast cells in the brain: Evidence and functional significance. Trends Neurosci. 1996, 19, 25–31. [Google Scholar]
- Parkin, E.T.; Watt, N.T.; Turner, A.J.; Hooper, N.M. Dual Mechanisms for Shedding of the Cellular Prion Protein. J. Biol. Chem. 2004, 279, 11170–11178. [Google Scholar]
- Chesebro, B.; Trifilo, M.; Race, R.; Meade-White, K.; Teng, C.; LaCasse, R.; Raymond, L.; Favara, C.; Baron, G.; Priola, S.; et al. Anchorless Prion Protein Results in Infectious Amyloid Disease without Clinical Scrapie. Science 2005, 308, 1435–1439. [Google Scholar]
- Raymond, G.J.; Race, B.; Hollister, J.R.; Offerdahl, D.K.; Moore, R.A.; Kodali, R.; Raymond, L.D.; Hughson, A.G.; Rosenke, R.; Long, D.; et al. Isolation of Novel Synthetic Prion Strains by Amplification in Transgenic Mice Coexpressing Wild-Type and Anchorless Prion Proteins. J. Virol. 2012, 86, 11763–11778. [Google Scholar]
- Bruce, M.E. Scrapie strain variation and mutation. Br. Med. Bull. 1993, 49, 822–838. [Google Scholar]
- Weissmann, C. Mutation and Selection of Prions. PLoS Pathog. 2012, 8, e1002582. [Google Scholar] [CrossRef]
- Wathne, G.J.; Mabbott, N.A. The diverse roles of mononuclear phagocytes in prion disease pathogenesis. Prion 2012, 6, 124–133. [Google Scholar] [CrossRef]
- Prinz, M.; Heikenwalder, M.; Schwarz, P.; Takeda, K.; Akira, S.; Aguzzi, A. Prion pathogenesis in the absence of Toll-like receptor signalling. EMBO Rep. 2003, 4, 195–199. [Google Scholar] [Green Version]
- de Almeida, C.J.G.; Chiarini, L.B.; da Silva, J.P.; e Silva, P.M.R.; Martins, M.A.; Linden, R. The cellular prion protein modulates phagocytosis and inflammatory response. J. Leukoc. Biol. 2005, 77, 238–246. [Google Scholar]
- Uraki, R.; Sakudo, A.; Ando, S.; Kitani, H.; Onodera, T. Enhancement of phagocytotic activity by prion protein in PrP-deficient macrophage cells. Int. J. Mol. Med. 2010, 26, 527–532. [Google Scholar]
- Nitta, K.; Sakudo, A.; Masuyama, J.; Xue, G.; Sugiura, K.; Onodera, T. Role of Cellular Prion Proteins in the Function of Macrophages and Dendritic Cells. Protein Pept. Lett. 2009, 16, 239–246. [Google Scholar] [CrossRef]
- Sassa, Y.; Inoshima, Y.; Ishiguro, N. Bovine macrophage degradation of scrapie and BSE PrPSc. Vet. Immunol. Immunopathol. 2010, 133, 33–39. [Google Scholar]
- Gilch, S.; Schmitz, F.; Aguib, Y.; Kehler, C.; Bülow, S.; Bauer, S.; Kremmer, E.; Schätzl, H.M. CpG and LPS can interfere negatively with prion clearance in macrophage and microglial cells. FEBS J. 2007, 274, 5834–5844. [Google Scholar]
- Moore, R.A.; Vorberg, I.; Priola, S.A. Species barriers in prion diseases—Brief review. In Infectious Diseases from Nature: Mechanisms of Viral Emergence and Persistence; Peters, C.J., Calisher, C.H., Eds.; Springer: Vienna, Austria, 2005; pp. 187–202. [Google Scholar]
- Neale, M.H.; Mountjoy, S.J.; Edwards, J.C.; Vilette, D.; Laude, H.; Windl, O.; Saunders, G.C. Infection of Cell Lines with Experimental and Natural Ovine Scrapie Agents. J. Virol. 2010, 84, 2444–2452. [Google Scholar] [CrossRef]
- Luhr, K.M.; Nordstrom, E.K.; Low, P.; Ljunggren, H.-G.; Taraboulos, A.; Kristensson, K. Scrapie Protein Degradation by Cysteine Proteases in CD11c+ Dendritic Cells and GT1-1 Neuronal Cells. J. Virol. 2004, 78, 4776–4782. [Google Scholar]
- Peters, P.J.; Mironov, A.; Peretz, D.; van Donselaar, E.; Leclerc, E.; Erpel, S.; DeArmond, S.J.; Burton, D.R.; Williamson, R.A.; Vey, M.; et al. Trafficking of prion proteins through a caveolae-mediated endosomal pathway. J. Cell Biol. 2003, 162, 703–717. [Google Scholar] [CrossRef]
- Marijanovic, Z.; Caputo, A.; Campana, V.; Zurzolo, C. Identification of an Intracellular Site of Prion Conversion. PLoS Pathog. 2009, 5, e1000426. [Google Scholar]
- Rybner-Barnier, C.; Jacquemot, C.; Cuche, C.; Doré, G.; Majlessi, L.; Gabellec, M.-M.; Moris, A.; Schwartz, O.; Di Santo, J.; Cumano, A.; et al. Processing of the Bovine Spongiform Encephalopathy-Specific Prion Protein by Dendritic Cells. J. Virol. 2006, 80, 4656–4663. [Google Scholar]
- Paquet, S.; Daude, N.; Courageot, M.-P.; Chapuis, J.; Laude, H.; Vilette, D. PrPc Does Not Mediate Internalization of PrPSc but is Required at an Early Stage for De Novo Prion Infection of Rov Cells. J. Virol. 2007, 81, 10786–10791. [Google Scholar]
- Levavasseur, E.; Metharom, P.; Dorban, G.; Nakano, H.; Kakiuchi, T.; Carnaud, C.; Sarradin, P.; Aucouturier, P. Experimental scrapie in ‘plt’ mice: An assessment of the role of dendritic-cell migration in the pathogenesis of prion diseases. J. Gen. Virol. 2007, 88, 2353–2360. [Google Scholar]
- Dieu, M.-C.; Vanbervliet, B.; Vicari, A.; Bridon, J.-M.; Oldham, E.; Ait-Yahia, S.; Briere, F.; Zlotnik, A.; Lebecque, S.; Caux, C. Selective Recruitment of Immature and Mature Dendritic Cells by Distinct Chemokines Expressed in Different Anatomic Sites. J. Exp. Med. 1998, 188, 373–386. [Google Scholar]
- Dorban, G.; Defaweux, V.; Levavasseur, E.; Demonceau, C.; Thellin, O.; Flandroy, S.; Piret, J.; Falisse, N.; Heinen, E.; Antoine, N. Oral scrapie infection modifies the homeostasis of Peyer’s patches’ dendritic cells. Histochem. Cell Biol. 2007, 128, 243–251. [Google Scholar] [CrossRef]
- Raymond, C.R.; Aucouturier, P.; Mabbott, N.A. In vivo Depletion of CD11c+ Cells Impairs Scrapie Agent Neuroinvasion from the Intestine. J. Immunol. 2007, 179, 7758–7766. [Google Scholar]
- Cordier-Dirikoc, S.; Chabry, J. Temporary Depletion of CD11c+ Dendritic Cells Delays Lymphoinvasion after Intraperitonal Scrapie Infection. J. Virol. 2008, 82, 8933–8936. [Google Scholar]
- Bradford, B.M.; Sester, D.P.; Hume, D.A.; Mabbott, N.A. Defining the anatomical localisation of subsets of the murine mononuclear phagocyte system using integrin alpha X (Itgax, CD11c) and colony stimulating factor 1 receptor (Csf1r, CD115) expression fails to discriminate dendritic cells from macrophages. Immunobiology 2011, 216, 1228–1237. [Google Scholar] [CrossRef]
- Klein, M.A.; Frigg, R.; Flechsig, E.; Raeber, A.J.; Kalinke, U.; Bluethmann, H.; Bootz, F.; Suter, M.; Zinkernagel, R.M.; Aguzzi, A. A crucial role for B cells in neuroinvasive scrapie. Nature 1997, 390, 687–690. [Google Scholar]
- Wathne, G.J.; Kissenpfennig, A.; Malissen, B.; Zurzolo, C.; Mabbott, N.A. Determining the role of mononuclear phagocytes in prion neuroinvasion from the skin. J. Leukoc. Biol. 2012, 91, 817–828. [Google Scholar]
- Aucouturier, P.; Geissmann, F.; Damotte, D.; Saborio, G.P.; Meeker, H.C.; Kascsak, R.; Kascsak, R.; Carp, R.I.; Wisniewski, T. Infected splenic dendritic cells are sufficient for prion transmission to the CNS in mouse scrapie. J. Clin. Invest. 2001, 108, 703–708. [Google Scholar]
- Castro-Seoane, R.; Hummerich, H.; Sweeting, T.; Tattum, M.H.; Linehan, J.M.; Fernandez de Marco, M.; Brandner, S.; Collinge, J.; Klöhn, P.-C. Plasmacytoid Dendritic Cells Sequester High Prion Titres at Early Stages of Prion Infection. PLoS Pathog. 2012, 8, e1002538. [Google Scholar] [CrossRef]
- Erblich, B.; Zhu, L.; Etgen, A.M.; Dobrenis, K.; Pollard, J.W. Absence of Colony Stimulation Factor-1 Receptor Results in Loss of Microglia, Disrupted Brain Development and Olfactory Deficits. PLoS One 2011, 6, e26317. [Google Scholar]
- Wang, Y.; Szretter, K.J.; Vermi, W.; Gilfillan, S.; Rossini, C.; Cella, M.; Barrow, A.D.; Diamond, M.S.; Colonna, M. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 2012, 13, 753–760. [Google Scholar]
- Kondo, Y.; Duncan, I.D. Selective reduction in microglia density and function in the white matter of colony-stimulating factor-1–deficient mice. J. Neurosci. Res. 2009, 87, 2686–2695. [Google Scholar] [CrossRef]
- Hamilton, J.A.; Whitty, G.; White, A.R.; Jobling, M.F.; Thompson, A.; Barrow, C.J.; Cappai, R.; Beyreuther, K.; Masters, C.L. Alzheimer’s disease amyloid beta and prion protein amyloidogenic peptides promote macrophage survival, DNA synthesis and enhanced proliferative response to CSF-1 (M-CSF). Brain Res. 2002, 940, 49–54. [Google Scholar]
- Marella, M.; Chabry, J. Neurons and Astrocytes Respond to Prion Infection by Inducing Microglia Recruitment. J. Neurosci. 2004, 24, 620–627. [Google Scholar]
- Riemer, C.; Schultz, J.; Burwinkel, M.; Schwarz, A.; Mok, S.W.F.; Gultner, S.; Bamme, T.; Norley, S.; van Landeghem, F.; Lu, B.; et al. Accelerated Prion Replication in, but Prolonged Survival Times of, Prion-Infected CXCR3−/− Mice. J. Virol. 2008, 82, 12464–12471. [Google Scholar]
- Lunnon, K.; Teeling, J.L.; Tutt, A.L.; Cragg, M.S.; Glennie, M.J.; Perry, V.H. Systemic Inflammation Modulates Fc Receptor Expression on Microglia during Chronic Neurodegeneration. J. Immunol. 2011, 186, 7215–7224. [Google Scholar]
- Kim, H.O.; Snyder, G.P.; Blazey, T.M.; Race, R.E.; Chesebro, B.; Skinner, P.J. Prion disease induced alterations in gene expression in spleen and brain prior to clinical symptoms. Adv. Appl. Bioinform. Chem. 2008, 1, 29–50. [Google Scholar]
- Riemer, C.; Neidhold, S.; Burwinkel, M.; Schwarz, A.; Schultz, J.; Krätzschmar, J.; Mönning, U.; Baier, M. Gene expression profiling of scrapie-infected brain tissue. Biochem. Biophys. Res. Commun. 2004, 323, 556–564. [Google Scholar]
- Hwang, D.; Lee, I.Y.; Yoo, H.; Gehlenborg, N.; Cho, J.-H.; Petritis, B.; Baxter, D.; Pitstick, R.; Young, R.; Spicer, D.; et al. A systems approach to prion disease. Mol. Syst. Biol. 2009, 5. [Google Scholar] [CrossRef]
- Xiang, W.; Windl, O.; Wunsch, G.; Dugas, M.; Kohlmann, A.; Dierkes, N.; Westner, I.M.; Kretzschmar, H.A. Identification of Differentially Expressed Genes in Scrapie-Infected Mouse Brains by Using Global Gene Expression Technology. J. Virol. 2004, 78, 11051–11060. [Google Scholar]
- Song, C.-H.; Honmou, O.; Furuoka, H.; Horiuchi, M. Identification of chemoattractive factors involved in the migration of bone marrow-derived mesenchymal stem cells to brain lesions caused by prion. J. Virol. 2011, 85, 11069–11078. [Google Scholar] [CrossRef]
- Heppner, F.L.; Christ, A.D.; Klein, M.A.; Prinz, M.; Fried, M.; Kraehenbuhl, J.-P.; Aguzzi, A. Transepithelial prion transport by M cells. Nat. Med. 2001, 7, 976–977. [Google Scholar]
- Tribouillard-Tanvier, D.; Striebel, J.F.; Peterson, K.E.; Chesebro, B. Analysis of Protein Levels of 24 Cytokines in Scrapie Agent-Infected Brain and Glial Cell Cultures from Mice Differing in Prion Protein Expression Levels. J. Virol. 2009, 83, 11244–11253. [Google Scholar] [CrossRef]
- Lu, Z.Y.; Baker, C.A.; Manuelidis, L. New molecular markers of early and progressive CJD brain infection. J. Cell. Biochem. 2004, 93, 644–652. [Google Scholar]
- Felton, L.M.; Cunningham, C.; Rankine, E.L.; Waters, S.; Boche, D.; Perry, V.H. MCP-1 and murine prion disease: Separation of early behavioural dysfunction from overt clinical disease. Neurobiol. Dis. 2005, 20, 283–295. [Google Scholar] [CrossRef]
- Sorensen, G.; Medina, S.; Parchaliuk, D.; Phillipson, C.; Robertson, C.; Booth, S. Comprehensive transcriptional profiling of prion infection in mouse models reveals networks of responsive genes. BMC Genomics 2008, 9. [Google Scholar] [CrossRef]
- Riemer, C.; Queck, I.; Simon, D.; Kurth, R.; Baier, M. Identification of Upregulated Genes in Scrapie-Infected Brain Tissue. J. Virol. 2000, 74, 10245–10248. [Google Scholar]
- Burwinkel, M.; Schwarz, A.; Riemer, C.; Schultz, J.; van Landeghem, F.; Baier, M. Rapid disease development in scrapie-infected mice deficient for CD40 ligand. EMBO Rep. 2004, 5, 527–531. [Google Scholar]
- Mohan, J.; Bruce, M.E.; Mabbott, N.A. Neuroinvasion by Scrapie following Inoculation via the Skin Is Independent of Migratory Langerhans Cells. J. Virol. 2005, 79, 1888–1897. [Google Scholar]
- Mabbott, N.A.; Bruce, M.E. Complement component C5 is not involved in scrapie pathogenesis. Immunobiology 2004, 209, 545–549. [Google Scholar] [CrossRef]
- Gossner, A.; Roupaka, S.; Foster, J.; Hunter, N.; Hopkins, J. Transcriptional profiling of peripheral lymphoid tissue reveals genes and networks linked to SSBP/1 scrapie pathology in sheep. Vet. Microbiol. 2011, 153, 218–228. [Google Scholar] [CrossRef]
- Zabel, M.D.; Heikenwalder, M.; Prinz, M.; Arrighi, I.; Schwarz, P.; Kranich, J.; von Teichman, A.; Haas, K.M.; Zeller, N.; Tedder, T.F.; et al. Stromal complement receptor CD21/35 facilitates lymphoid prion colonization and pathogenesis. J. Immunol. 2007, 179, 6144–6152. [Google Scholar]
- Sein, L.; Yasuo, I.; Yasuro, A.; Hiroshi, U.; Naotaka, I. Immune cell types involved in early uptake and transport of recombinant mouse prion protein in Peyer’s patches of calves. Cell Tissue Res. 2009, 338, 343–354. [Google Scholar]
- Eaton, S.L.; Rocchi, M.; Gonzalez, L.; Hamilton, S.; Finlayson, J.; Sales, J.; Jeffrey, M.; Steele, P.J.; Dagleish, M.P.; Rodger, S.M.; et al. Immunological differences between susceptible and resistant sheep during the preclinical phase of scrapie infection. J. Gen. Virol. 2007, 88, 1384–1391. [Google Scholar] [CrossRef]
- Baker, C.A.; Manuelidis, L. Unique inflammatory RNA profiles of microglia in Creutzfeldt-Jakob disease. Proc. Natl. Acad. Sci. USA 2003, 100, 675–679. [Google Scholar]
- Schultz, J.; Schwarz, A.; Neidhold, S.; Burwinkel, M.; Riemer, C.; Simon, D.; Kopf, M.; Otto, M.; Baier, M. Role of Interleukin-1 in Prion Disease-Associated Astrocyte Activation. Am. J. Pathol. 2004, 165, 671–678. [Google Scholar] [CrossRef]
- Williams, A.E.; Lawson, L.J.; Perry, V.H.; Fraser, H. Characterization of the microglial response in murine scrapie. Neuropathol. Appl. Neurobiol. 1994, 20, 47–55. [Google Scholar]
- Isaacs, J.D.; Garden, O.A.; Kaur, G.; Collinge, J.; Jackson, G.S.; Altmann, D.M. The cellular prion protein is preferentially expressed by CD4+ CD25+ Foxp3+ regulatory T cells. Immunology 2008, 125, 313–319. [Google Scholar] [CrossRef]
- Le, Y.; Yazawa, H.; Gong, W.; Yu, Z.; Ferrans, V.J.; Murphy, P.M.; Wang, J.M. Cutting Edge: The Neurotoxic Prion Peptide Fragment PrP106–126 is a Chemotactic Agonist for the G Protein-Coupled Receptor Formyl Peptide Receptor-Like 1. J. Immunol. 2001, 166, 1448–1451. [Google Scholar]
- Booth, S.; Bowman, C.; Baumgartner, R.; Sorensen, G.; Robertson, C.; Coulthart, M.; Phillipson, C.; Somorjai, R.L. Identification of central nervous system genes involved in the host response to the scrapie agent during preclinical and clinical infection. J. Gen. Virol. 2004, 85, 3459–3471. [Google Scholar] [CrossRef]
- Starke, R.; Harrison, P.; Mackie, I.; Wang, G.; Erusalimsky, J.D.; Gale, R.; MassÉ, J.M.; Cramer, E.; Pizzey, A.; Biggerstaff, J.; et al. The expression of prion protein (PrPC) in the megakaryocyte lineage. J. Thromb. Haemost. 2005, 3, 1266–1273. [Google Scholar] [CrossRef]
- Uraki, R.; Sakudo, A.; Michibata, K.; Ano, Y.; Kono, J.; Yukawa, M.; Onodera, T. Blocking of FcR Suppresses the Intestinal Invasion of Scrapie Agents. PLoS One 2011, 6, e17928. [Google Scholar]
- Prinz, M.; Huber, G.; Macpherson, A.J.S.; Heppner, F.L.; Glatzel, M.; Eugster, H.-P.; Wagner, N.; Aguzzi, A. Oral Prion Infection Requires Normal Numbers of Peyer’s Patches but Not of Enteric Lymphocytes. Am. J. Pathol. 2003, 162, 1103–1111. [Google Scholar]
- Luhr, K.M.; Wallin, R.P.A.; Ljunggren, H.-G.; Low, P.; Taraboulos, A.; Kristensson, K. Processing and Degradation of Exogenous Prion Protein by CD11c+ Myeloid Dendritic Cells in vitro. J. Virol. 2002, 76, 12259–12264. [Google Scholar]
- Piercey Åkesson, C.; Press, C.M.; Tranulis, M.A.; Jeffrey, M.; Aleksandersen, M.; Landsverk, T.; Espenes, A. Phenotypic characterization of cells participating in transport of prion protein aggregates across the intestinal mucosa of sheep. Prion 2012, 6, 261–275. [Google Scholar]
- Li, C.; Yu, S.; Nakamura, F.; Pentikäinen, O.T.; Singh, N.; Yin, S.; Xin, W.; Sy, M.-S. Pro-prion Binds Filamin A, Facilitating its Interaction with Integrin β1, and Contributes to Melanomagenesis. J. Biol. Chem. 2010, 285, 30328–30339. [Google Scholar]
- Walsh, D.T.; Betmouni, S.; Perry, V.H. Absence of detectable IL-1 beta production in murine prion disease: A model of chronic neurodegeneration. J. Neuropathol. Exp. Neurol. 2001, 60, 173–182. [Google Scholar]
- Tamguney, G.; Giles, K.; Glidden, D.V.; Lessard, P.; Wille, H.; Tremblay, P.; Groth, D.F.; Yehiely, F.; Korth, C.; Moore, R.C.; et al. Genes contributing to prion pathogenesis. J. Gen. Virol. 2008, 89, 1777–1788. [Google Scholar] [CrossRef]
- Thackray, A.M.; McKenzie, A.N.; Klein, M.A.; Lauder, A.; Bujdoso, R. Accelerated Prion Disease in the Absence of Interleukin-10. J. Virol. 2004, 78, 13697–13707. [Google Scholar]
- Mabbott, N.A.; Williams, A.; Farquhar, C.F.; Pasparakis, M.; Kollias, G.; Bruce, M.E. Tumor Necrosis Factor Alpha-Deficient, but Not Interleukin-6-Deficient, Mice Resist Peripheral Infection with Scrapie. J. Virol. 2000, 74, 3338–3344. [Google Scholar] [CrossRef]
- Tribouillard-Tanvier, D.; Race, B.; Striebel, J.F.; Carroll, J.A.; Phillips, K.; Chesebro, B. Early cytokine elevation, PrPres deposition and gliosis in mouse scrapie: No effect on disease by deletion of cytokine genes, IL-12p40 and IL-12p35. J. Virol. 2012, 86, 10377–10383. [Google Scholar] [CrossRef]
- Rosicarelli, B.; Serafini, B.; Sbriccoli, M.; Lu, M.; Cardone, F.; Pocchiari, M.; Aloisi, F. Migration of dendritic cells into the brain in a mouse model of prion disease. J. Neuroimmunol. 2005, 165, 114–120. [Google Scholar]
- Baker, C.A.; Lu, Z.Y.; Manuelidis, L. Early induction of interferon-responsive mRNAs in Creutzfeldt-Jakob disease. J. Neurovirol. 2004, 10, 29–40. [Google Scholar]
- Oldstone, M.B.A.; Race, R.; Thomas, D.; Lewicki, H.; Homann, D.; Smelt, S.; Holz, A.; Koni, P.; Lo, D.; Chesebro, B.; et al. Lymphotoxin-α- and Lymphotoxin-β- Deficient Mice Differ in Susceptibility to Scrapie: Evidence against Dendritic Cell Involvement in Neuroinvasion. J. Virol. 2002, 76, 4357–4363. [Google Scholar]
- Kopacek, J.; Sakaguchi, S.; Shigematsu, K.; Nishida, N.; Atarashi, R.; Nakaoke, R.; Moriuchi, R.; Niwa, M.; Katamine, S. Upregulation of the Genes Encoding Lysosomal Hydrolases, a Perforin-Like Protein, and Peroxidases in the Brains of Mice Affected with an Experimental Prion Disease. J. Virol. 2000, 74, 411–417. [Google Scholar]
- Cunningham, C.; Wilcockson, D.C.; Boche, D.; Perry, V.H. Comparison of Inflammatory and Acute-Phase Responses in the Brain and Peripheral Organs of the ME7 Model of Prion Disease. J. Virol. 2005, 79, 5174–5184. [Google Scholar]
- Saba, R.; Gushue, S.; Huzarewich, R.L.C.H.; Manguiat, K.; Medina, S.; Robertson, C.; Booth, S.A. MicroRNA 146a (miR-146a) is Over-Expressed during Prion Disease and Modulates the Innate Immune Response and the Microglial Activation State. PLoS One 2012, 7, e30832. [Google Scholar]
- Bueler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar]
- Manson, J.C.; Clarke, A.R.; McBride, P.A.; McConnell, I.; Hope, J. PrP gene dosage determines the timing but not the final intensity or distribution of lesions in scrapie pathology. Neurodegeneration 1994, 3, 331–340. [Google Scholar]
- Betmouni, S.; Perry, V.H. The acute inflammatory response in CNS following injection of prion brain homogenate or normal brain homogenate. Neuropathol. Appl. Neurobiol. 1999, 25, 19–27. [Google Scholar] [CrossRef]
- Mok, S.W.F.; Proia, R.L.; Brinkmann, V.; Mabbott, N.A. B Cell-Specific S1PR1 Deficiency Blocks Prion Dissemination between Secondary Lymphoid Organs. J. Immunol. 2012, 188, 5032–5040. [Google Scholar]
- Segundo, F.D.S.; Sevilla, N.; Gutierrez, J.P.; Brun, A. Altered lymphocyte homeostasis after oral prion infection in mouse. Vet. Immunol. Immunopathol. 2008, 122, 204–215. [Google Scholar] [CrossRef]
- Martinez del Hoyo, G.; Lopez-Bravo, M.; Metharom, P.; Ardavin, C.; Aucouturier, P. Prion Protein Expression by Mouse Dendritic Cells is Restricted to the Nonplasmacytoid Subsets and Correlates with the Maturation State. J. Immunol. 2006, 177, 6137–6142. [Google Scholar]
- Cunningham, C.; Boche, D.; Perry, V.H. Transforming growth factor β1, the dominant cytokine in murine prion disease: influence on inflammatory cytokine synthesis and alteration of vascular extracellular matrix. Neuropathol. Appl. Neurobiol. 2002, 28, 107–119. [Google Scholar] [CrossRef]
- Baker, C.A.; Lu, Z.Y.; Zaitsev, I.; Manuelidis, L. Microglial Activation Varies in Different Models of Creutzfeldt-Jakob Disease. J. Virol. 1999, 73, 5089–5097. [Google Scholar]
- Raymond, C.R.; Mabbott, N.A. Assessing the involvement of migratory dendritic cells in the transfer of the scrapie agent from the immune to peripheral nervous systems. J. Neuroimmunol. 2007, 187, 114–125. [Google Scholar]
- Mabbott, N.A.; McGovern, G.; Jeffrey, M.; Bruce, M.E. Temporary Blockade of the Tumor Necrosis Factor Receptor Signaling Pathway Impedes the Spread of Scrapie to the Brain. J. Virol. 2002, 76, 5131–5139. [Google Scholar] [CrossRef]
- Gehlenborg, N.; Hwang, D.; Lee, I.Y.; Yoo, H.; Baxter, D.; Petritis, B.; Pitstick, R.; Marzolf, B.; DeArmond, S.J.; Carlson, G.A.; et al. The Prion Disease Database: A comprehensive transcriptome resource for systems biology research in prion diseases. Database 2009, 2009. [Google Scholar] [CrossRef]
- Rybner, C.; Hillion, J.; Sahraoui, T.; Lanotte, M.; Botti, J. All-trans retinoic acid down-regulates prion protein expression independently of granulocyte maturation. Leukemia 2002, 16, 940–948. [Google Scholar]
- Cordier-Dirikoc, S.; Zsürger, N.; Cazareth, J.; Ménard, B.; Chabry, J. Expression profiles of prion and doppel proteins and of their receptors in mouse splenocytes. Eur. J. Immunol. 2008, 38, 2131–2141. [Google Scholar]
- Miragliotta, G.; Fumarulo, R.; Fumarola, D. Inhibition of Neutrophil Functions by Scrapie Prion Protein—Description of Some Inhibitory Properties. Acta Virol. 1990, 34, 517–522. [Google Scholar]
- Le, Y.; Zhou, Y.; Tao, H.; Wang, J.M. Formylpeptide receptors and their potential roles in inflammatory airway diseases. Clin. Exp. Allergy Rev. 2004, 4, 155–161. [Google Scholar]
- Orange, J.S.; Fassett, M.S.; Koopman, L.A.; Boyson, J.E.; Strominger, J.L. Viral evasion of natural killer cells. Nat. Immunol. 2002, 3, 1006–1012. [Google Scholar] [CrossRef]
- Jonjić, S.; Babić, M.; Polić, B.; Krmpotić, A. Immune evasion of natural killer cells by viruses. Curr. Opin. Immunol. 2008, 20, 30–38. [Google Scholar]
- Lanier, L.L. Evolutionary struggles between NK cells and viruses. Nat. Rev. Immunol. 2008, 8, 259–268. [Google Scholar] [CrossRef]
- Raslova, H.; Kauffmann, A.; Sekkaï, D.; Ripoche, H.; Larbret, F.; Robert, T.; Le Roux, D.T.; Kroemer, G.; Debili, N.; Dessen, P.; et al. Interrelation between polyploidization and megakaryocyte differentiation: A gene profiling approach. Blood 2007, 109, 3225–3234. [Google Scholar] [CrossRef]
- Opalinska, J.B.; Bersenev, A.; Zhang, Z.; Schmaier, A.A.; Choi, J.; Yao, Y.; D'Souza, J.; Tong, W.; Weiss, M.J. MicroRNA expression in maturing murine megakaryocytes. Blood 2010, 116, e128–e138. [Google Scholar]
- Robertson, C.; Booth, S.A.; Beniac, D.R.; Coulthart, M.B.; Booth, T.F.; McNicol, A. Cellular prion protein is released on exosomes from activated platelets. Blood 2006, 107, 3907–3911. [Google Scholar]
- Wang, G.; Zhou, X.; Bai, Y.; Zhang, Z.; Zhao, D. Cellular prion protein released on exosomes from macrophages binds to Hsp70. Acta Biochim. Biophys. Sin. 2010, 42, 345–350. [Google Scholar] [CrossRef]
- Beaulieu, L.M.; Lin, E.; Morin, K.M.; Tanriverdi, K.; Freedman, J.E. Regulatory effects of TLR2 on megakaryocytic cell function. Blood 2011, 117, 5963–5974. [Google Scholar]
- McCutcheon, S.; Alejo Blanco, A.R.; Houston, E.F.; de Wolf, C.; Tan, B.C.; Smith, A.; Groschup, M.H.; Hunter, N.; Hornsey, V.S.; MacGregor, I.R.; et al. All Clinically-Relevant Blood Components Transmit Prion Disease following a Single Blood Transfusion: A Sheep Model of vCJD. PLoS One 2011, 6, e23169. [Google Scholar]
- Houston, F.; McCutcheon, S.; Goldmann, W.; Chong, A.; Foster, J.; Siso, S.; Gonzalez, L.; Jeffrey, M.; Hunter, N. Prion diseases are efficiently transmitted by blood transfusion in sheep. Blood 2008, 112, 4739–4745. [Google Scholar]
- Hunter, N.; Foster, J.; Chong, A.; McCutcheon, S.; Parnham, D.; Eaton, S.; MacKenzie, C.; Houston, F. Transmission of prion diseases by blood transfusion. J. Gen. Virol. 2002, 83, 2897–2905. [Google Scholar]
- Mathiason, C.K.; Hayes-Klug, J.; Hays, S.A.; Powers, J.; Osborn, D.A.; Dahmes, S.J.; Miller, K.V.; Warren, R.J.; Mason, G.L.; Telling, G.C.; et al. B Cells and Platelets Harbor Prion Infectivity in the Blood of Deer Infected with Chronic Wasting Disease. J. Virol. 2010, 84, 5097–5107. [Google Scholar]
- McBride, P.A.; Schulz-Schaeffer, W.J.; Donaldson, M.; Bruce, M.; Diringer, H.; Kretzschmar, H.A.; Beekes, M. Early Spread of Scrapie from the Gastrointestinal Tract to the Central Nervous System Involves Autonomic Fibers of the Splanchnic and Vagus Nerves. J. Virol. 2001, 75, 9320–9327. [Google Scholar] [CrossRef]
- Beekes, M.; McBride, P.A. The spread of prions through the body in naturally acquired transmissible spongiform encephalopathies. FEBS J. 2007, 274, 588–605. [Google Scholar]
- Sisó, S.; Jeffrey, M.; González, L. Neuroinvasion in sheep transmissible spongiform encephalopathies: The role of the haematogenous route. Neuropathol. Appl. Neurobiol. 2009, 35, 232–246. [Google Scholar] [CrossRef]
- Loeuillet, C.; Lemaire-Vieille, C.; Naquet, P.; Cesbron-Delauw, M.-F.; Gagnon, J.; Cesbron, J.-Y. Prion Replication in the Hematopoietic Compartment Is Not Required for Neuroinvasion in Scrapie Mouse Model. PLoS One 2010, 5, e13166. [Google Scholar]
- Magalhães, A.C.; Baron, G.S.; Lee, K.S.; Steele-Mortimer, O.; Dorward, D.; Prado, M.A.M.; Caughey, B. Uptake and Neuritic Transport of Scrapie Prion Protein Coincident with Infection of Neuronal Cells. J. Neurosci. 2005, 25, 5207–5216. [Google Scholar] [CrossRef]
- Liu, T.; Li, R.; Wong, B.-S.; Liu, D.; Pan, T.; Petersen, R.B.; Gambetti, P.; Sy, M.-S. Normal Cellular Prior Protein is Preferentially Expressed on Subpopulations of Murine Hemopoietic Cells. J. Immunol. 2001, 166, 3733–3742. [Google Scholar]
- Kubosaki, A.; Yusa, S.; Nasu, Y.; Nishimura, T.; Nakamura, Y.; Saeki, K.; Matsumoto, Y.; Itohara, S.; Onodera, T. Distribution of Cellular Isoform of Prion Protein in T Lymphocytes and Bone Marrow, Analyzed by Wild-Type and Prion Protein Gene-Deficient Mice. Biochem. Biophys. Res. Commun. 2001, 282, 103–107. [Google Scholar] [CrossRef]
- Ford, M.J.; Burton, L.J.; Li, H.; Graham, C.H.; Frobert, Y.; Grassi, J.; Hall, S.M.; Morris, R.J. A marked disparity between the expression of prion protein and its message by neurones of the CNS. Neuroscience 2002, 111, 533–551. [Google Scholar] [CrossRef]
- Ford, M.J.; Burton, L.J.; Morris, R.J.; Hall, S.M. Selective expression of prion protein in peripheral tissues of the adult mouse. Neuroscience 2002, 113, 177–192. [Google Scholar]
- Mabbott, N.A.; Kenneth Baillie, J.; Hume, D.A.; Freeman, T.C. Meta-analysis of lineage-specific gene expression signatures in mouse leukocyte populations. Immunobiology 2010, 215, 724–736. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bradford, B.M.; Mabbott, N.A. Prion Disease and the Innate Immune System. Viruses 2012, 4, 3389-3419. https://doi.org/10.3390/v4123389
Bradford BM, Mabbott NA. Prion Disease and the Innate Immune System. Viruses. 2012; 4(12):3389-3419. https://doi.org/10.3390/v4123389
Chicago/Turabian StyleBradford, Barry M., and Neil A. Mabbott. 2012. "Prion Disease and the Innate Immune System" Viruses 4, no. 12: 3389-3419. https://doi.org/10.3390/v4123389