Immune Responses and Lassa Virus Infection

Abstract
:1. Introduction
2. Pathogenesis of Lassa Fever
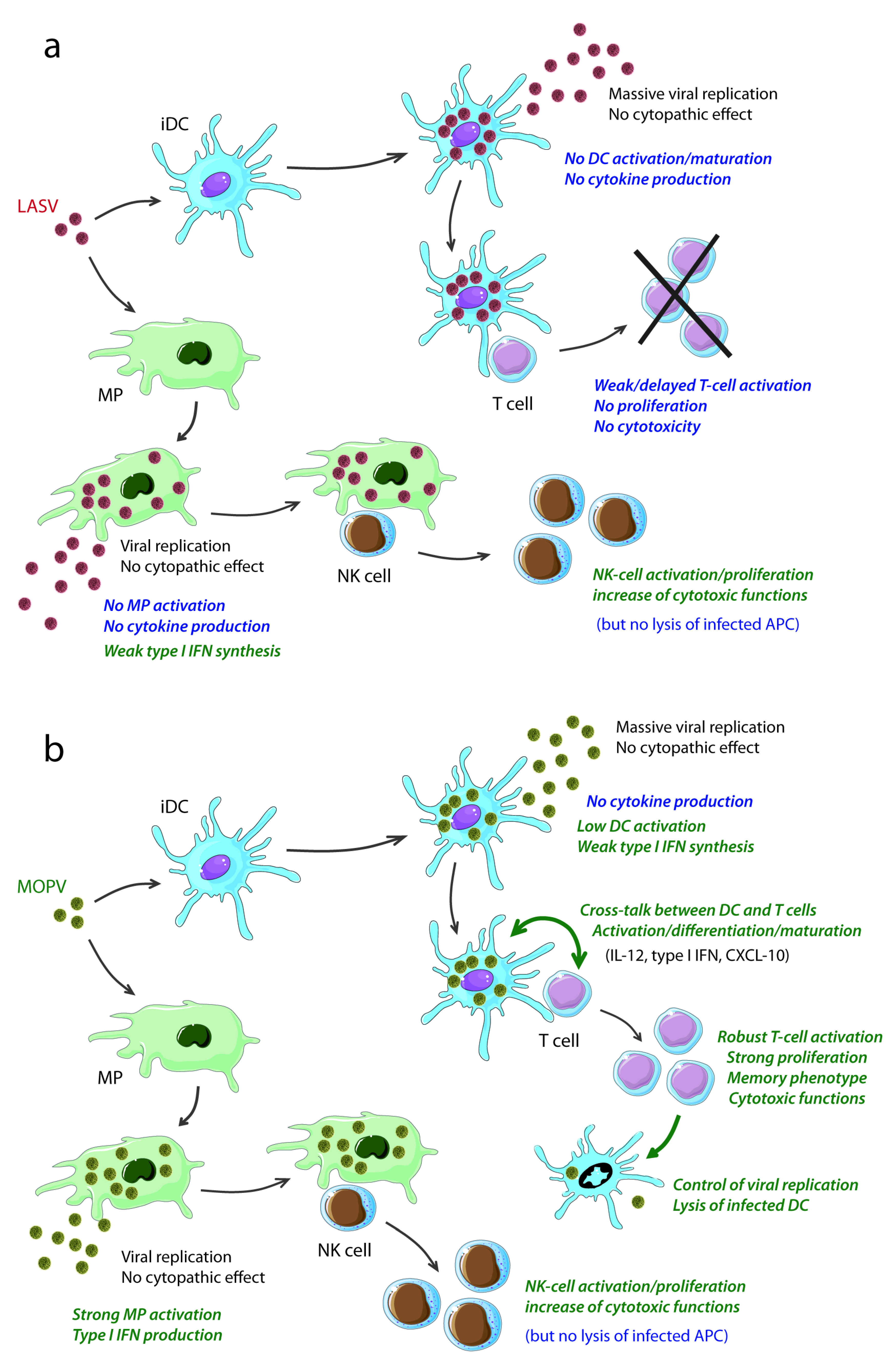
3. Antigen-Presenting Cells Play a Key Role in Lassa Fever
3.1. LASV Infection of APC
3.2. Responses of APC to LASV Infection and Correlation with Pathogenicity
3.3. Inhibition of Innate Immunity by LASV NP
4. Humoral Responses During Lassa Fever
5. NK-Cell Responses During LASV Infection
6. T-Cell Responses and the Control of LASV Infection
6.1. Defective T-Cell Immunity and Fatal Lassa Fever
6.2. The Control of LASV Infection Is Associated with the Induction of T-Cell Responses

| Immunological parameters | Nonfatal LF | Fatal outcome | References |
|---|---|---|---|
| Inflammatory responses | High number of CD80+ circulating monocytes | Low number of CD80+ circulating monocytes | [22,25] |
| Early and transient release of IFNα | Late release of IFNα | ||
| Inflammatory cytokines: not detected (ND) | Inflammatory cytokines: ND, except for IL-6 (late) | ||
| CXCL-10 and 11 mRNA | CXCL-10 and 11 mRNA | ||
| MCP-1 ?, eotaxin ? | MCP-1, eotaxin | ||
| Antibodies | High levels of IgM/IgG | High levels of IgM/IgG | [25,28] |
| No nAb | No nAb | ||
| NK cells | Transient depletion from the circulation | Profound depletion | [25] |
| T-cell responses | T cell-derived cytokines: ND | T cell-derived cytokines: ND | [22,25] |
| Transient lymphopenia followed by lymphocytosis | Marked lymphopenia | ||
| Early and robust activation of CD4+ and CD8+ T cells | Weak and late activation of CD4+ and CD8+ T cells | ||
| In vitro proliferation of T cells in response to LASV | No proliferation in response to LASV |
7. Conclusions

{kind=link}
Conflict of Interest
Acknowledgments
References
- Buchmeier, M.J.; de la Torre, J.-C.; Peters, C.J. Arenaviridae: the viruses and their replication. In Fields virology, 5th; Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Roizman, B., Straus, S.E., Eds.; Lippincott Williams & Wilkins: Philadelphia, Country, 2007; pp. 1791–1827. [Google Scholar]
- Cornu, T.I.; De la Torre, J. Characterization of the arenavirus RING finger Z protein regions required for Z-mediated inhibition of viral RNA synthesis. J. Virol. 2002, 76, 6678–6688. [Google Scholar] [CrossRef]
- Cornu, T.I.; de la Torre, J.C. RING finger Z protein of lymphocytic choriomeningitis virus (LCMV) inhibits transcription and RNA replication of an LCMV S-segment minigenome. J. Virol. 2001, 75, 9415–9426. [Google Scholar] [CrossRef]
- Cornu, T.I.; Feldmann, H.; De la Torre, J. Cells expressing the RING finger Z protein are resistant to Arenavirus infection. J. Virol. 2004, 78, 2979–2983. [Google Scholar] [CrossRef]
- Perez, M.; Craven, R.C.; de la Torre, J.C. The small RING finger protein Z drives arenavirus budding: implications for antiviral strategies. Proc. Natl. Acad. Sci. USA 2003, 100, 12978–12983. [Google Scholar] [CrossRef]
- Perez, M.; Greenwald, D.L.; Carlos de la Torre, J. Myristoylation of the RING finger Z protein is essential for arenavirus budding. J. Virol. 2004, 78, 11443–11448. [Google Scholar] [CrossRef]
- Strecker, T.; Eichler, R.; ter Meulen, J.; Weissenhorn, W.; Klenk, H.-D.; Garten, W.; Lenz, O. Lassa virus Z protein is a matrix protein sufficient for the release of virus-like particles. J. Virol. 2003, 77, 10700–10705. [Google Scholar] [CrossRef]
- Cao, W.; Henry, M.D.; Borrow, P.; Yamada, H.; Elder, J.H.; Ravkov, E.V.; Nichol, S.T.; Compans, R.W.; Campbell, K.P.; Oldstone, M.B.A. Identification of α-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science 1998, 282, 2079–2081. [Google Scholar]
- Kunz, S.; Rojek, J.M.; Perez, M.; Spiropoulou, C.F.; Oldstone, M.B.A. Characterization of the interaction of Lassa fever virus with its cellular receptor α-dystroglycan. J. Virol. 2005, 79, 5979–5987. [Google Scholar] [CrossRef]
- Safronetz, D.; Lopez, J.; Sogoba, N.; Traoré, S.; Raffel, S.; Fischer, E.; Ebihara, H.; Branco, L.; Garry, R.; Schwan, T.; Feldmann, H. Detection of Lassa virus, Mali. Emerg. Infect. Dis. 2010, 16, 1123–1126. [Google Scholar] [CrossRef]
- Günther, S.; Emmerich, P.; Laue, T.; Kühle, O.; Asper, M.; Jung, A.; Grewing, T.; Ter Meulen, J.; Schmitz, H. Imported Lassa fever in Germany: Molecular characterization of a new Lassa virus strain. Emerg. Infect. Dis. 2000, 6, 466–476. [Google Scholar] [CrossRef]
- Ogbu, O.; Ajuluchukwu, E.; Uneke, C.J. Lassa fever in West African sub-region: an overview. J Vector Borne Dis 2007, 44, 1–11. [Google Scholar]
- McCormick, J.B.; Webb, P.A.; Krebs, J.W.; Johnson, K.M.; Smith, E.S. A prospective study of the epidemiology and ecology of Lassa fever. J. Infect. Dis. 1987, 155, 437–444. [Google Scholar] [CrossRef]
- Fisher-Hoch, S.P.; Tomori, O.; Nasidi, A.; Perez-Oronoz, G.; Fakile, Y.; Hutwagner, L.; McCormick, J.B. Review of cases of nosocomial Lassa fever in Nigeria: the high price of poor medical practice. Br. Med. J. 1995, 311, 857–859. [Google Scholar]
- Edington, G.M.; White, H.A. The pathology of Lassa fever. Trans. Roy. Soc. Trop. Med. Hyg. 1972, 66, 381–389. [Google Scholar] [CrossRef]
- Cummins, D.; McCormick, J.B.; Bennett, D.; Samba, J.A.; Farrar, B.; Machin, S.J.; Fisher-Hoch, S.P. Acute sensorineural deafness in Lassa fever. JAMA 1990, 264, 2093–2096. [Google Scholar] [CrossRef]
- McCormick, J.B.; King, I.J.; Webb, P.A.; Scribner, C.L.; Craven, R.B.; Johnson, K.M.; Elliott, L.H.; Belmont-Williams, R. Lassa Fever. Effective therapy with ribavirin. New Eng. J. Med. 1986, 314, 20–26. [Google Scholar]
- Fisher-Hoch, S.P.; McCormick, J.B.; Sasso, D.; Craven, R.B. Hematologic dysfunction in Lassa fever. J. Med. Virol. 1988, 26, 127–135. [Google Scholar] [CrossRef]
- Jahrling, P.B.; Smith, S.; Hesse, R.A.; Rhoderick, J.B. Pathogenesis of Lassa virus infection in guinea pigs. Infect. Immun. 1982, 37, 771–778. [Google Scholar]
- Baize, S.; Kaplon, J.; Faure, C.; Pannetier, D.; Georges-Courbot, M.C.; Deubel, V. Lassa virus infection of dendritic cells and macrophages is productive but fails to activate cells. J. Immunol. 2004, 172, 2861–2869. [Google Scholar]
- Mahanty, S.; Hutchinson, K.; Agarwal, S.; Mcrae, M.; Rollin, P.E.; Pulendran, B. Cutting Edge: Impairment of dendritic cells and adaptive immunity by Ebola and Lassa viruses. J. Immunol. 2003, 170, 2797–2801. [Google Scholar]
- Hensley, L.; Smith, M.; Geisbert, J.; Fritz, E.; Daddario-DiCaprio, K.; Larsen, T.; Geisbert, T. Pathogenesis of lassa fever in cynomolgus macaques. Virol. J. 2011, 8, 205. [Google Scholar] [CrossRef]
- Walker, D.H.; Johnson, K.M.; Lange, J.V.; Gardner, J.J.; Kiley, M.P.; McCormick, J.B. Experimental infection of Rhesus monkeys with Lassa virus and a closely related Arenavirus, Mozambique virus. J. Infect. Dis. 1982, 146, 360–368. [Google Scholar] [CrossRef]
- Walker, D.H.; McCormick, J.B.; Johnson, K.M.; Webb, P.A.; Komba-Kono, G.; Elliott, L.H.; Gardner, J.J. Pathologic and virologic study of fatal Lassa fever in man. Am. J. Pathol. 1982, 107, 349–356. [Google Scholar]
- Baize, S.; Marianneau, P.; Loth, P.; Reynard, S.; Journeaux, A.; Chevallier, M.; Tordo, N.; Deubel, V.; Contamin, H. Early and strong immune responses are associated with control of viral replication and recovery in Lassa virus-infected cynomolgus monkeys. J. Virol. 2009, 83, 5890–5903. [Google Scholar] [CrossRef]
- Carrion, R., Jr.; Brasky, K.; Mansfield, K.; Johnson, C.; Gonzales, M.; Ticer, A.; Lukashevich, I.; Tardif, S.; Patterson, J. Lassa virus infection in experimentally infected marmosets: Liver pathology and immunophenotypic alterations in target tissues. J. Virol. 2007, 81, 6482–6490. [Google Scholar] [CrossRef]
- Winn, W.C.J.; Walker, D.H. The pathology of human Lassa fever. Bull. WHO 1975, 52, 535–545. [Google Scholar]
- Fisher-Hoch, S.P.; Mitchell, S.W.; Sasso, D.R.; Lange, J.V.; Ramsey, R.; McCormick, J.B. Physiological and immunologic disturbances associated with shock in a primate model of Lassa fever. J. Infect. Dis. 1987, 155, 465–474. [Google Scholar] [CrossRef]
- Lange, J.V.; Mitchell, S.W.; McCormick, J.B.; Walker, D.H.; Evatt, B.L.; Ramsey, R.R. Kinetic study of platelets and fibrinogen in Lassa virus-infected monkeys and early pathologic events in Mopeia virus-infected monkeys. Am. J. Trop. Med. Hyg. 1985, 34, 999–1007. [Google Scholar]
- Callis, R.T.; Jahrling, P.B.; DePaoli, A. Pathology of Lassa virus infection in the Rhesus monkey. Am. J. Trop. Med. Hyg. 1982, 31, 1038–1045. [Google Scholar]
- Jahrling, P.B.; Hesse, R.A.; Eddy, G.A.; Johnson, K.M.; callis, R.T.; Stephen, E.L. Lassa virus infection of rhesus monkeys: pathogenesis and treatment with ribavirin. J. Infect. Dis. 1980, 141, 580–589. [Google Scholar] [CrossRef]
- Schmitz, H.; Köhler, B.; Laue, T.; Drosten, C.; Veldkamp, P.J.; Günther, S.; Emmerich, P.; Geisen, H.P.; Fleischer, K.; Beersma, M.F.C.; Hoerauf, A. Monitoring of clinical and laboratory data in two cases of imported Lassa fever. Microb. Infect. 2002, 4, 43–50. [Google Scholar] [CrossRef]
- Johnson, K.M.; McCormick, J.B.; Webb, P.A.; Smith, E.S.; Elliott, L.H.; King, I.J. Clinical virology of Lassa fever in hospitalized patients. J. Infect. Dis. 1987, 155, 456–463. [Google Scholar] [CrossRef]
- McCormick, J.B.; Walker, D.H.; King, I.J.; Webb, P.A.; Elliott, L.H.; Whitfield, S.G.; Johnson, K.M. Lassa virus hepatitis: a study of fatal Lassa fever in humans. Am. J. Trop. Med. Hyg. 1986, 35, 401–407. [Google Scholar]
- Romani, L.; Mencacci, A.; Cenci, E.; Spaccapelo, R.; Toniatti, C.; Puccetti, P.; Bistoni, F.; Poli, V. Impaired neutrophil response and CD4+ T helper cell 1 development in interleukin 6-deficient mice infected with Candida albicans. J. Exp. Med. 1996, 183, 1345–1355. [Google Scholar] [CrossRef]
- Ulich, T.R.; del Castillo, J.; Guo, K.Z. In vivo hematologic effects of recombinant interleukin-6 on hematopoiesis and circulating numbers of RBCs and WBCs. Blood 1989, 73, 108–110. [Google Scholar]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.-J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Ann. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar]
- Lukashevich, I.S.; Maryankova, R.; Vladyko, A.S.; Nashkevic, N.; Koleda, S.; Djavani, M.; Horejsh, D.; Voitenok, N.N.; Salvato, M.S. Lassa and Mopeia virus replication in human monocytes/macrophages and in endothelial cells: Different effects on IL-8 and TNF-α gene expression. J. Med. Virol. 1999, 59, 552–560. [Google Scholar] [CrossRef]
- Macal, M.; Lewis, Gavin, M.; Kunz, S.; Flavell, R.; Harker, James, A.; Zúñiga, E.I. Plasmacytoid dendritic cells are productively infected and activated through TLR-7 early after Arenavirus infection. Cell. Host. Microbe. 2012, 11, 617–630. [Google Scholar] [CrossRef]
- Kiley, M.P.; Lange, J.V.; Johnson, K.M. Protection of rhesus monkeys from Lassa virus by immunisation with closely related arenavirus. Lancet 1979, 2, 738–745. [Google Scholar]
- Ruo, S.L.; Mitchell, S.W.; Kiley, M.P.; Roumillat, L.F.; Fisher-Hoch, S.P.; McCormick, J.B. Antigenic relatedness between arenaviruses defined at the epitope level by monoclonal antibodies. J. Gen. Virol. 1991, 72, 549–555. [Google Scholar]
- Wulff, H.; McIntosh, B.; Hamner, D.; Johnson, K. Isolation of an arenavirus closely related to Lassa virus from Mastomys natalensis in south-east Africa. Bull. World Health Organ. 1977, 55, 441–444. [Google Scholar]
- Pannetier, D.; Faure, C.; Georges-Courbot, M.C.; Deubel, V.; Baize, S. Human macrophages, but not dendritic cells, are activated and produce type I interferons in response to Mopeia virus infection. J. Virol. 2004, 78, 10516–10524. [Google Scholar] [CrossRef]
- Baize, S.; Pannetier, D.; Faure, C.; Marianneau, P.; Marendat, I.; Georges-Courbot, M.C.; Deubel, V. Role of interferons in the control of Lassa virus replication in human dendritic cells and macrophages. Microb. Infect. 2006, 8, 1193–1422. [Google Scholar] [CrossRef]
- Dhodapkar, M.V.; Steinman, R.M.; Krasovsky, J.; Munz, C.; Bhardwaj, N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J. Exp. Med. 2001, 193, 233–238. [Google Scholar] [CrossRef]
- Hawiger, D.; Inaba, K.; Dorsett, Y.; Guo, M.; Mahnke, K.; Rivera, M.; Ravetch, J.V.; Steinman, R.M.; Nussenzweig, M.C. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 2001, 194, 769–779. [Google Scholar] [CrossRef]
- Curtsinger, J.M.; Schmidt, C.S.; Mondino, A.; Lins, D.C.; Kedl, R.M.; Jenkins, M.K.; Mescher, M.F. Inflammatory cytokines provides a third signal for activation of naive CD4+ and CD8+ T cells. J. Immunol. 1999, 162, 3256–3262. [Google Scholar]
- Oswald, I.P.; Wynn, T.A.; Sher, A.; James, S.L. Interleukin 10 inhibits macrophage microbicidal activity by blocking the endogenous production of tumor necrosis factor α required as a costimulatory factor for interferon γ-induced activation. Proc. Natl. Acad. Sci. USA 1992, 89, 8676–8680. [Google Scholar] [CrossRef]
- Mahanty, S.; Bausch, D.G.; Thomas, R.L.; Goba, A.; Bah, A.; Peters, C.J.; Rollin, P.E. Low levels of interleukin-8 and interferon-inducible protein-10 in serum are associated with fatal infections in acute Lassa fever. J. Infect. Dis. 2001, 183, 1713–1721. [Google Scholar]
- Fennewald, S.M.; Aronson, J.F.; Zhang, L.; Herzog, N.K. Alterations in NF-κB and RBP-Jκ by arenavirus infection of macrophages in vitro and in vivo. J. Virol. 2002, 76, 1154–1162. [Google Scholar] [CrossRef]
- Asper, M.; Sternsdorf, T.; Hass, M.; Drosten, C.; Rhode, A.; Schmitz, H.; Günther, S. Inhibition of different Lassa virus strains by alpha and gamma interferons and comparison with a less pathogenic Arenavirus. J. Virol. 2004, 78, 3162–3169. [Google Scholar]
- Le Bon, A.; Tough, D.F. Links between innate and adaptive immunity via type I interferon. Curr. Opin. Immunol. 2002, 14, 432–436. [Google Scholar] [CrossRef]
- Binder, D.; Fehr, J.; Hengartner, H.; Zinkernagel, R.M. Virus-induced transient bone marrow aplasia: major role of interferon-α/β during acute infection with the noncytopathic lymphocytic choriomeningitis virus. J. Exp. Med. 1997, 185, 517–530. [Google Scholar] [CrossRef]
- McNally, J.M.; Zarozinski, C.C.; Lin, M.-Y.; Brehm, M.A.; Chen, H.D.; Welsh, R.M. Attrition of bystander CD8 T cells during virus-induced T-cell and interferon responses. J. Virol. 2001, 75, 5965–5976. [Google Scholar]
- Iannacone, M.; Sitia, G.; Isogawa, M.; Whitmire, J.K.; Marchese, P.; Chisari, F.V.; Ruggeri, Z.M.; Guidotti, L.G. Platelets prevent IFN-α/β -induced lethal hemorrhage promoting CTL-dependent clearance of lymphocytic choriomeningitis virus. Proc Natl Acad Sci USA 2008, 105, 629–634. [Google Scholar]
- Martinez-Sobrido, L.; Giannakas, P.; Cubitt, B.; Garcia-Sastre, A.; de La Torre, J.C. Differential inhibition of type I interferon induction by arenavirus nucleoproteins. J. Virol. 2007, 81, 12696–12703. [Google Scholar] [CrossRef]
- Martinez-Sobrido, L.; Zuniga, E.I.; Rosario, D.; Garcia-Sastre, A.; de la Torre, J.C. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2006, 80, 9192–9199. [Google Scholar]
- Hastie, K.M.; Kimberlin, C.R.; Zandonatti, M.A.; MacRae, I.J.; Saphire, E.O. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3′ to 5′ exonuclease activity essential for immune suppression. Proc Natl Acad Sci USA 2011, 108, 2396–2401. [Google Scholar]
- Qi, X.; Lan, S.; Wang, W.; Schelde, L.M.; Dong, H.; Wallat, G.D.; Ly, H.; Liang, Y.; Dong, C. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 2010, 468, 779–785. [Google Scholar]
- Habjan, M.; Andersson, I.; Klingström, J.; Schümann, M.; Martin, A.; Zimmermann, P.; Wagner, V.; Pichlmair, A.; Schneider, U.; Mühlberger, E.; Mirazimi, A.; Weber, F. Processing of Genome 5' Termini as a Strategy of Negative-Strand RNA Viruses to Avoid RIG-I-Dependent Interferon Induction. PLoS ONE 2008, 3, e2032. [Google Scholar]
- Zhou, S.; Cerny, A.M.; Zacharia, A.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Finberg, R.W. Induction and inhibition of Type I interferon responses by distinct components of Lymphocytic Choriomeningitis virus. J. Virol. 2010, 84, 9452–9462. [Google Scholar]
- Yoneyama, M.; Fujita, T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 2009, 227, 54–65. [Google Scholar] [CrossRef]
- Martinez-Sobrido, L.; Emonet, S.; Giannakas, P.; Cubitt, B.; Garcia-Sastre, A.; de la Torre, J.C. Identification of amino acid residues critical for the anti-interferon activity of the nucleoprotein of the prototypic Arenavirus Lymphocytic Choriomeningitis virus. J. Virol. 2009, 83, 11330–11340. [Google Scholar]
- Carnec, X.; Baize, S.; Reynard, S.; Diancourt, L.; Caro, V.; Tordo, N.; Bouloy, M. Lassa virus nucleoprotein mutants generated by reverse genetics induce robust type I IFN response in human dendritic cells and macrophages. J. Virol. 2011, 85, 12093–12097. [Google Scholar]
- Pythoud, C.; Rodrigo, W.W.S.I.; Pasqual, G.; Rothenberger, S.; Martínez-Sobrido, L.; de la Torre, J.C.; Kunz, S. Arenavirus nucleoprotein targets interferon regulatory fact or-activating kinase IKKε. J. Virol. 2012, 86, 7728–7738. [Google Scholar] [CrossRef]
- Rodrigo, W.W.S.I.; Ortiz-Riaño, E.; Pythoud, C.; Kunz, S.; de la Torre, J.C.; Martínez-Sobrido, L. Arenavirus nucleoproteins prevent activation of nuclear factor kappa B. J. Virol. 2012, 86, 8185–8197. [Google Scholar]
- Günther, S.; Kühle, O.; Rehder, D.; Odaibo, G.N.; Olaleye, D.O.; Emmerich, P.; Ter Meulen, J.; Schmitz, H. Antibodies to Lassa virus Z protein and nucleoprotein co-occur in human sera from Lassa fever endemic regions. Med. Microbiol. Immunol. 2001, 189, 225–229. [Google Scholar] [CrossRef]
- Jahrling, P.B.; Frame, J.D.; Rhoderick, J.B.; Monson, M.H. Endemic Lassa fever in Liberia. IV. Selection of optimally effective plasma for treatment by passive immunization. Trans. R. Soc. Trop. Med. Hyg. 1985, 79, 380–384. [Google Scholar] [CrossRef]
- Enria, D.A.; Briggiler, A.M.; Sanchez, Z. Treatment of Argentine hemorrhagic fever. Antiviral Res. 2008, 78, 132–139. [Google Scholar] [CrossRef]
- Pinschewer, D.D.; Perez, M.; Jeetendra, E.; Bächi, T.; Horvath, E.; Hengartner, H.; Whitt, M.A.; De la Torre, J.C.; Zinkernagel, R.M. Kinetics of protective antibodies are determined by the viral surface antigen. J. Clin. Invest. 2004, 114, 988–993. [Google Scholar]
- Frame, J.D.; Verbrugge, G.P.; Gill, R.G.; Pinneo, L. The use of Lassa fever convalescent plasma in Nigeria. Trans. R. Soc. Trop. Med. Hyg. 1984, 78, 319–324. [Google Scholar] [CrossRef]
- Jahrling, P.B.; Peters, C.J. Passive antibody therapy of Lassa fever in cynomolgus monkeys: importance of neutralizing antibody and Lassa virus strain. Infect. Immun. 1984, 44, 528–533. [Google Scholar]
- Jahrling, P.B.; Stephen, E.L.; Peters, C.J. Enhanced treatment of Lassa fever by immune plasma combined with ribavirin in cynomolgus monkeys. J. Infect. Dis. 1984, 149, 420–427. [Google Scholar] [CrossRef]
- Fisher-Hoch, S.P.; Hutwagner, L.; Brown, B.; McCormick, J.B. Effective vaccine for Lassa fever. J. Virol. 2000, 74, 6777–6783. [Google Scholar] [CrossRef]
- Fisher-Hoch, S.P.; McCormick, J.B. Lassa fever vaccine. Expert Rev. Vaccines 2004, 3, 189–197. [Google Scholar] [CrossRef]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar]
- Biron, C.A.; Nguyen, K.B.; Pien, G.C. Innate immune responses to LCMV infections: natural killer cells and cytokines. In Arenaviruses; Oldstone, M.B.A., Ed.; Springer: Berlin, Country, 2002; Volume 262, pp. 7–27. [Google Scholar]
- Biron, C.A.; Nguyen, K.B.; Pien, G.C.; Cousens, L.P.; Salazar-Mather, T.P. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu. Rev. Immunol. 1999, 17, 189–220. [Google Scholar] [CrossRef]
- Russier, M.; Reynard, S.; Tordo, N.; Baize, S. NK cells are early activated by Lassa and Mopeia virus-infected human macrophages, but do not mediate virus suppression. Eur J Immunol 2012, 42, 1822–1832. [Google Scholar] [CrossRef]
- Bukowski, J.F.; Woda, B.A.; Habu, S.; Okumura, K.; Welsh, R.M. Natural killer cell depletion enhances virus synthesis and virus-induced hepatitis in vivo. J. Immun. 1983, 131, 1531–1538. [Google Scholar]
- Welsh, R.; Dundon, P.; Eynon, E.; Brubaker, J.; Koo, G.; O'Donnell, C. Demonstration of the antiviral role of natural killer cells in vivo with a natural killer cell-specific monoclonal antibody (NK 1.1). Nat. Immun. Cell Growth Regul. 1990, 9, 112–120. [Google Scholar]
- Orange, J.S.; Biron, C.A. An absolute and restricted requirement for IL-12 in natural killer cell IFN-γ production and antiviral defense. Studies of natural killer and T cell responses in contrasting viral infections. J. Immunol. 1996, 156, 1138–1142. [Google Scholar]
- Nguyen, K.B.; Cousens, L.P.; Doughty, L.A.; Pien, G.C.; Durbin, J.E.; Biron, C.A. Interferon α/β-mediated inhibition and promotion of interferon γ: STAT1 resolves a paradox. Nat. Immunol. 2000, 1, 70–76. [Google Scholar]
- Cousens, L.P.; Orange, J.S.; Su, H.C.; Biron, C. Interferon-α/β inhibition of interleukin 12 and interferon-γ production in vitro and endogenously during viral infection. Proc. Natl. Acad. Sci. USA 1997, 94, 634–639. [Google Scholar] [CrossRef]
- Rodas, J.D.; Cairo, C.; Djavani, M.; Zapata, J.C.; Ruckwardt, T.; Bryant, J.; Pauza, C.D.; Lukashevich, I.S.; Salvato, M.S. Circulating natural killer and γδ T cells decrease soon after infection of rhesus macaques with lymphocytic choriomeningitis virus. Memórias do Instituto Oswaldo Cruz 2009, 104, 583–591. [Google Scholar] [CrossRef]
- McIntyre, K.W.; Welsh, R.M. Accumulation of natural killer and cytotoxic T large granular lymphocytes in the liver during virus infection. J. Exp. Med. 1986, 164, 1667–1681. [Google Scholar]
- Pien, G.C.; Biron, C.A. Compartmental differences in NK Cell responsiveness to IL-12 during Lymphocytic Choriomeningitis virus infection. J. Immunol. 2000, 164, 994–1001. [Google Scholar]
- Sun, J.C.; Beilke, J.N.; Lanier, L.L. Adaptive immune features of natural killer cells. Nature 2009, 457, 557–561. [Google Scholar]
- Lang, P.A.; Lang, K.S.; Xu, H.C.; Grusdat, M.; Parish, I.A.; Recher, M.; Elford, A.R.; Dhanji, S.; Shaabani, N.; Tran, C.W.; Dissanayake, D.; Rahbar, R.; Ghazarian, M.; Brüstle, A.; Fine, J.; Chen, P.; Weaver, C. T.; Klose, C.; Diefenbach, A.; Häussinger, D.; Carlyle, J. R.; Kaech, S. M.; Mak, T. W.; Ohashi, P. S. Natural killer cell activation enhances immune pathology and promotes chronic infection by limiting CD8+ T-cell immunity. Proc. Natl. Acad. Sci. USA 2012, 109, 1210–1215. [Google Scholar]
- Waggoner, S.N.; Cornberg, M.; Selin, L.K.; Welsh, R.M. Natural killer cells act as rheostats modulating antiviral T cells. Nature 2011. [Google Scholar]
- Geisbert, T.W.; Jones, S.; Fritz, E.A.; Shurtleff, A.C.; Geisbert, J.B.; Liebscher, R.; Grolla, A.; Ströher, U.; Fernando, L.; Daddario, K.M.; Guttieri, M.C.; Mothé, B.R.; Larsen, T.; Hensley, L.E.; Jahrling, P.B.; Feldmann, H. Development of a new vaccine for the prevention of Lassa fever. PLOS Med. 2005, 2, 537–545. [Google Scholar]
- Pannetier, D.; Reynard, S.; Russier, M.; Journeaux, A.; Tordo, N.; Deubel, V.; Baize, S. Human dendritic cells infected with the non-pathogenic Mopeia virus induce stronger T-cell responses than with Lassa virus. J. Virol. 2011, 85, 8293–8306. [Google Scholar] [CrossRef]
- Flatz, L.; Rieger, T.; Merkler, D.; Bergthaler, A.; Regen, T.; Schedensack, M.; Bestmann, L.; Verschoor, A.; Kreutzfeldt, M.; Brück, W.; Hanisch, U.-K.; Günther, S.; Pinschewer, D.D. T Cell-dependence of Lassa fever pathogenesis. PLoS Pathog. 2010, 6, e1000836. [Google Scholar]
- Kim, J.V.; Kang, S.S.; Dustin, M.L.; McGavern, D.B. Myelomonocytic cell recruitment causes fatal CNS vascular injury during acute viral meningitis. Nature 2009, 457, 191–195. [Google Scholar]
- Oldstone, M.B.A. Biology and Pathogenesis of Lymphocytic Choriomeningitis Virus Infection. In Arenaviruses II. The Molecular Pathogenesis of Arenavirus Infections; Oldstone, M.B.A., Ed.; Springer: Berlin, Country, 2002; Volume 263, pp. 83–117. [Google Scholar]
- ter Meulen, J.; Badusche, M.; Kuhnt, K.; Doetze, A.; Satoguina, J.; Marti, T.; Loeliger, C.; Koulemou, K.; Koivogui, L.; Schmitz, H.; Fleischer, B.; Hoerauf, A. Characterization of human CD4+ T cell clones recognizing conserved and variable epitopes of the Lassa virus nucleoprotein. J. Virol. 2000, 74, 2186–2192. [Google Scholar] [CrossRef]
- ter Meulen, J.; Badusche, M.; Satoguina, J.; Strecker, T.; Lenz, O.; Loeliger, C.; Sakho, M.; Koulemou, K.; Koivogui, L.; Hoerauf, A. Old and New world arenaviruses share a highly conserved epitope in the fusion domain of the glycoprotein 2, which is recognized by Lassa virus-specific human CD4+ T-cell clones. Virology 2004, 321, 134–143. [Google Scholar] [CrossRef]
- Christensen, J.E.; de Lemos, C.; Moos, T.; Christensen, J.P.; Thomsen, A.R. CXCL10 is the key ligand for CXCR3 on CD8+ effector T cells involved in immune surveillance of the lymphocytic choriomeningitis virus-infected central nervous system. J. Immunol. 2006, 176, 4235–4243. [Google Scholar]
- Dufour, J.H.; Dziejman, M.; Liu, M.T.; Leung, J.H.; Lane, T.E.; Luster, A.D. IFN-γ-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J. Immunol. 2002, 168, 3195–3204. [Google Scholar]
- Curtsinger, J.M.; Valenzuela, J.O.; Agarwal, P.; Lins, D.; Mescher, M.F. Cutting edge: Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 2005, 174, 4465–4469. [Google Scholar]
- Seder, R.A.; Gazzinelli, R.; Sher, A.; Paul, W.E. Interleukin 12 acts directly on CD4+ T cells to enhance priming for interferon γ production and diminishes interleukin 4 inhibition of such priming. Proc. Natl. Acad. Sci. USA 1993, 90, 10188–10192. [Google Scholar] [CrossRef]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nature Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef]
- Fisher-Hoch, S.P.; McCormick, J.B. Towards a human Lassa fever vaccine. Rev. Med. Virol. 2001, 11, 331–341. [Google Scholar] [CrossRef]
- Boesen, A.; Sundar, K.; Coico, R. Lassa fever virus peptides predicted by computational analysis induce epitope-specific cytotoxic-T-lymphocyte responses in HLA-A2.1 transgenic mice. Clin. Diag. Lab. Immunol. 2005, 12, 1223–1230. [Google Scholar]
- Botten, J.; Alexander, J.; Pasquetto, V.; Sidney, J.; Barrowman, P.; Ting, J.; Peters, B.; Southwood, S.; Stewart, B.; Rodriguez-Carreno, M.P.; Mothe, B.; Whitton, J.L.; Sette, A.; Buchmeier, M.J. Identification of protective Lassa virus epitopes that are restricted by HLA-A2. J. Virol. 2006, 80, 8351–8361. [Google Scholar]
- Oldstone, M.B.A.; Lewicki, H.; Homann, D.; Nguyen, C.; Julien, S.; Gairin, J.E. Common antiviral cytotoxic T-lymphocyte epitope for diverse arenaviruses. J. Virol. 2001, 75, 6273–6278. [Google Scholar] [CrossRef]
- Lukashevich, I.S.; Djavani, M.; rodas, J.D.; Zapata, J.C.; Usborne, A.; Emerson, C.; Mitchen, J.; Jahrling, P.B.; Salvato, M.S. Hemorrhagic fever occurs after intravenous, but not after intragastric, inoculation of Rhesus macaques with lymphocytic choriomeningitis virus. J. Med. Virol. 2002, 67, 171–186. [Google Scholar] [CrossRef]
- Sevilla, N.; Kunz, S.; Holz, A.; Lewicki, H.; Homann, D.; Yamada, H.; Campbell, K. P.; de la Torre, J.C.; Oldstone, M.B.A. Immunosuppression and resultant viral persistence by specific viral targeting of dendritic cells. J. Exp. Med. 2000, 192, 1249–1260. [Google Scholar] [CrossRef]
- Merkler, D.; Horvath, E.; Bruck, W.; Zinkernagel, R.M.; de la Torre, J.C.; Pinschewer, D.D. Viral déja vu elicits organ-specific immune disease independent of reactivity to self. J. Clin. Invest. 2006, 116, 1254–1263. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Russier, M.; Pannetier, D.; Baize, S. Immune Responses and Lassa Virus Infection. Viruses 2012, 4, 2766-2785. https://doi.org/10.3390/v4112766
Russier M, Pannetier D, Baize S. Immune Responses and Lassa Virus Infection. Viruses. 2012; 4(11):2766-2785. https://doi.org/10.3390/v4112766
Chicago/Turabian StyleRussier, Marion, Delphine Pannetier, and Sylvain Baize. 2012. "Immune Responses and Lassa Virus Infection" Viruses 4, no. 11: 2766-2785. https://doi.org/10.3390/v4112766
APA StyleRussier, M., Pannetier, D., & Baize, S. (2012). Immune Responses and Lassa Virus Infection. Viruses, 4(11), 2766-2785. https://doi.org/10.3390/v4112766