Forty-Five Years of Marburg Virus Research




Abstract
:
1. Epidemiology
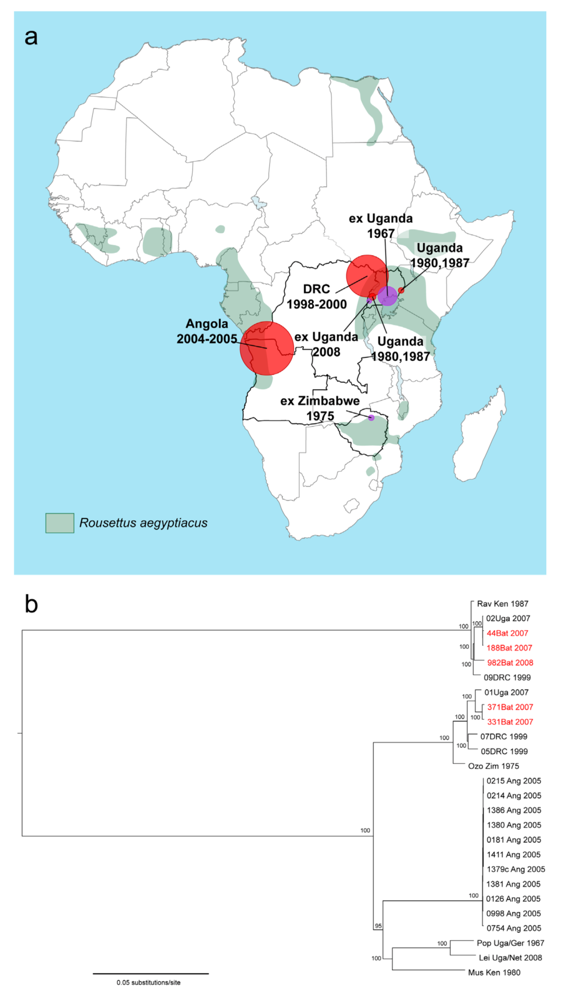

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year(s) | City | Country | Origin (apparent or suspected)/ mode of infection | Fatalities/ number of cases (case fatality rate) | Isolate designation (abbreviation) | Reference |
|---|---|---|---|---|---|---|
| 1967 | Marburg | Germany | Uganda/ | 5/24 (21%) | MARV Ci67, Flak (MARV Flak), Hartz (MARV Hartz), MARV “L”, Porton (MARV Porton), Poppinga (MARV Pop), Ratayczak (MARV Rat), Voege (MARV Voe) | [23] |
| Handling of infected African green monkey tissues | ||||||
| 1967 | Frankfurt | Germany | Uganda/ | 2/6 (33%) | ||
| Handling of infected African green monkey tissues | ||||||
| 1967 | Belgrade | Yugoslavia (now Serbia) | Uganda/ | 0/2 (0%) | ||
| Handling of infected African green monkey tissues | ||||||
| 1975 | Johannes-burg | South Africa | Zimbabwe/ | 1/3 (33%) | Cruikshank (MARV Cru), Hogan (MARV Hogan), Ozolin (MARV Ozo) | [8,24] |
| Unknown- most likely: sleeping in rooms inhabited by bats or visit to Sinoia caves (now Chinhoyi caves) | ||||||
| 1980 | Nairobi | Kenya | Kenya/ | 1/2 (50%) | Musoke (MARV Mus) | [25] |
| Working close to Kitum Cave, Mount Elgon National Park | ||||||
| 1987 | Nairobi | Kenya | Kenya/ | 1/1 (100%) | Ravn (RAVV Ravn), R1 (RAVV R1) | [26] |
| Visit to Kitum Cave/Mount Elgon National Park | ||||||
| 1988 | Koltsovo | Soviet Union (now Russia) | Russia/ | 1/1 (100%) | “Variant U” (MARV “U”) | [16] |
| Laboratory infection:Needlestick injury | ||||||
| 1990 | Koltsovo | Soviet Union (now Russia) | Russia/ | 0/1 (0%) | - | [20] |
| Laboratory infection:Unspecified violation of safety requirements | ||||||
| 1998-2000 | Durba, Watsa (multiple indepen-dent, but simultan-eous or over-lapping outbreaks) | Democratic Republic of the Congo | Durba, Democratic Republic of the Congo/ | 128/154 (83%) | MARV 01DRC99, MARV 02DRC99, MARV 03DRC99, MARV 04DRC99, MARV 05DRC99, MARV 06DRC99, MARV 08DRC, MARV 10DRC99, MARV 11DRC99, MARV 12DRC00, MARV 13DRC00, MARV 14DRC00, MARV15 DRC00, MARV 16DRC00, MARV17 DRC00, MARV 18DRC00, MARV 19DRC00, MARV 20DRC00, MARV 21DRC00, MARV 22DRC00, MARV 22DRC00, MARV 23DRC00, MARV 24DRC00, MARV 25DRC00, MARV 26DRC00, MARV 27DRC00, MARV 28DRC00, MARV 29DRC00, MARV 30DRC00, MARV 31DRC00, MARV 32DRC00, MARV 33DRC00, MARV 34DRC00, MARV DRC 5/99 Aru, MARV DRC 5/99 Dra, RAVV 09DRC99 | [9] |
| Gold mining in Goroumbwa cave | ||||||
| 2004-2005 | Uíge | Angola | Uíge Province, Angola/ | 227/252 (90%) | MARV Angola | [10,27] |
| unknown | ||||||
| 2007 | Kam-wenge | Uganda | Kamwenge District, Uganda/ Gold mining in Kitaka Cave | 1/4 (25%) | MARV-01Uga 2007, RAVV- 02Uga 2007 | [19] |
| 2008 | Colorado, City unreported | USA | Uganda/ | 0/1 (0%) | - | [17] |
| Visit of Python Cave in Maramagambo Forest | ||||||
| 2008 | Leiden | Nether-lands | Uganda/ | 1/1 (100%) | MARV Leiden | [18] |
| Visit of Python Cave in Maramagambo Forest | ||||||
| Total: | 368/452 (81%) |

2. Ecology
3. Taxonomy
4. Transmission
5. Clinical manifestations
5.1. Incubation Period
5.2. Generalization Phase (Day One-to Four)
5.3. Early Organ Phase (Day Five to Thirteen)
5.4. Late Organ/Convalescence Phase (Day Thirteen+)
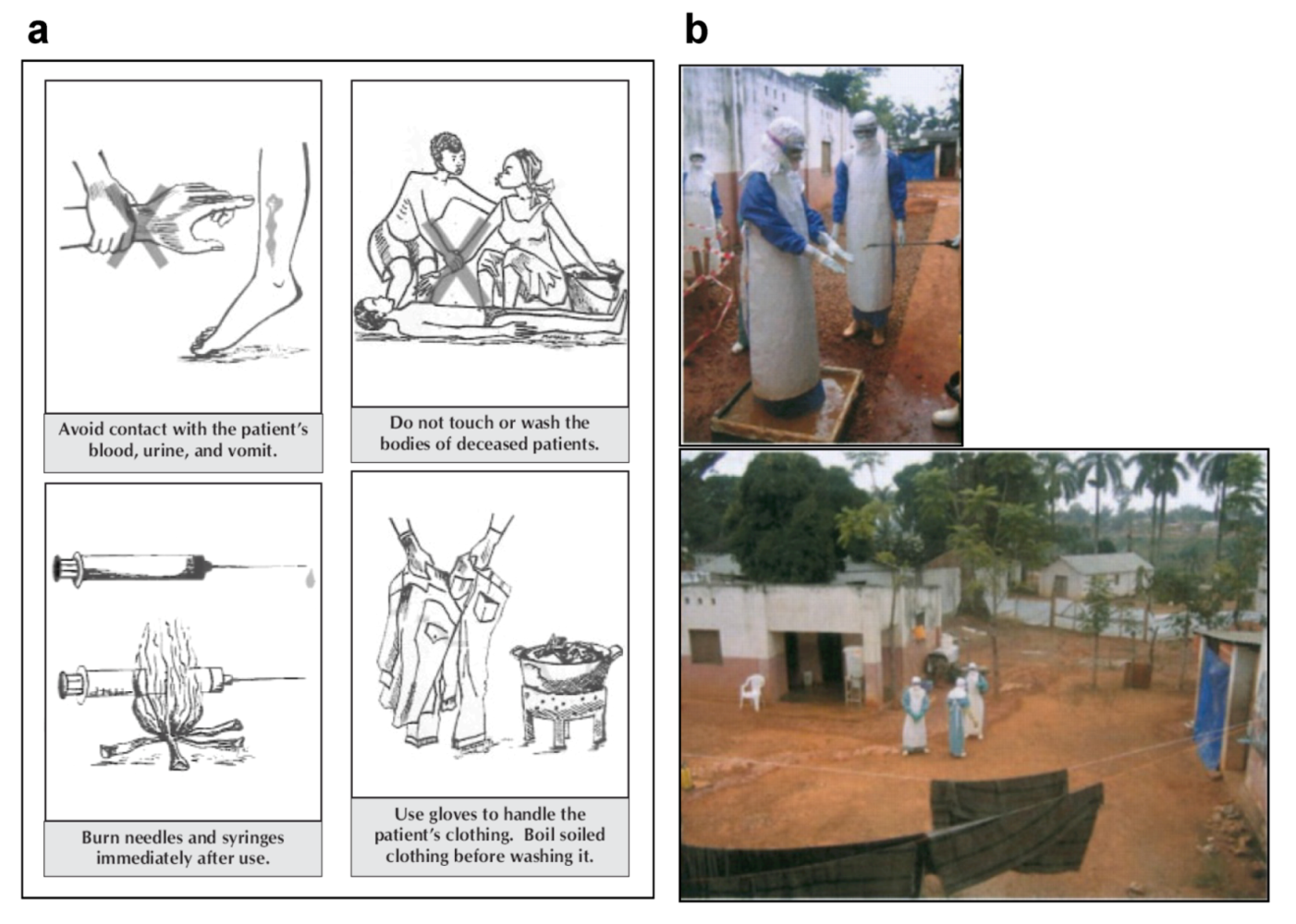
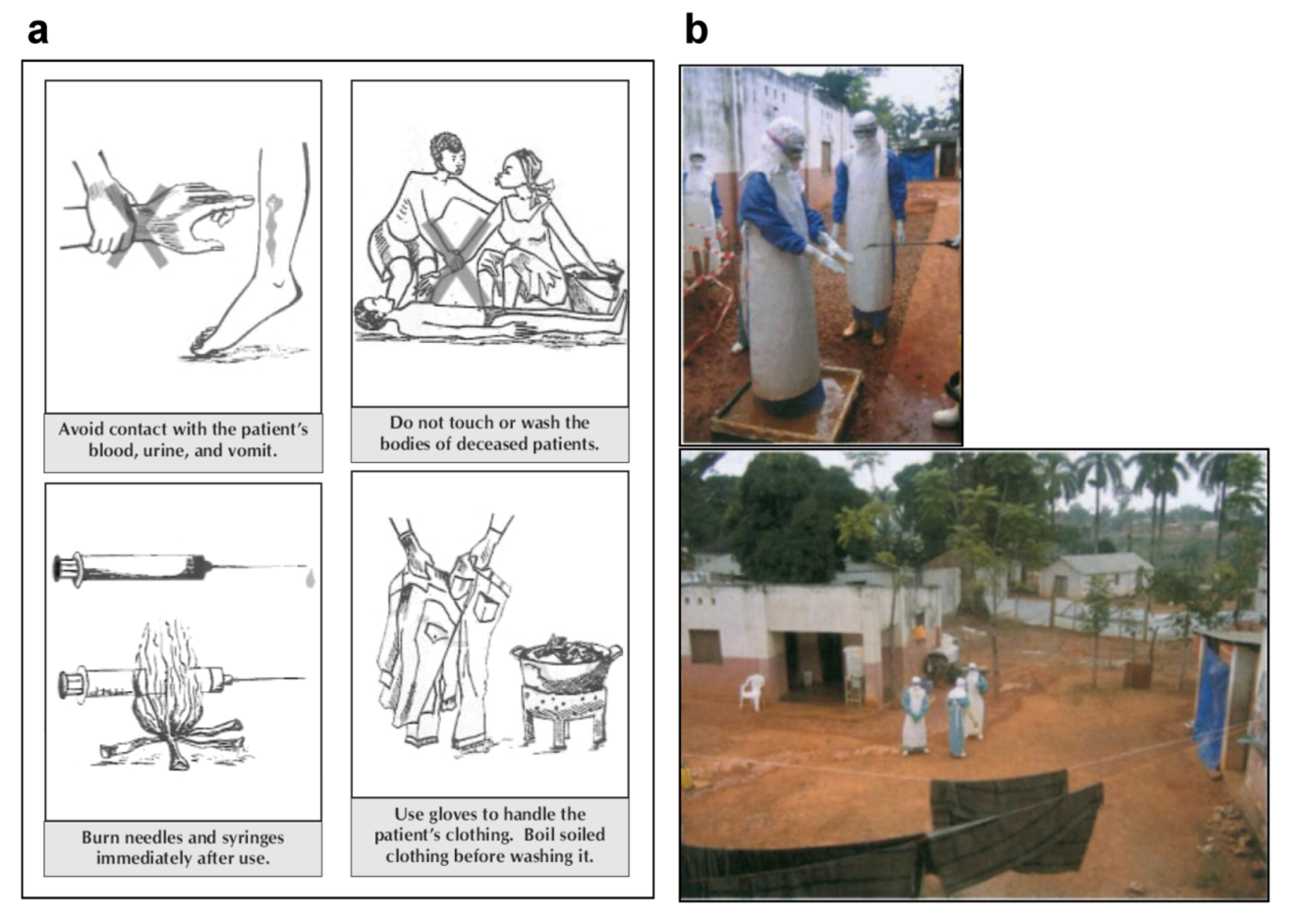
6. Prevention
7. Virus Structure
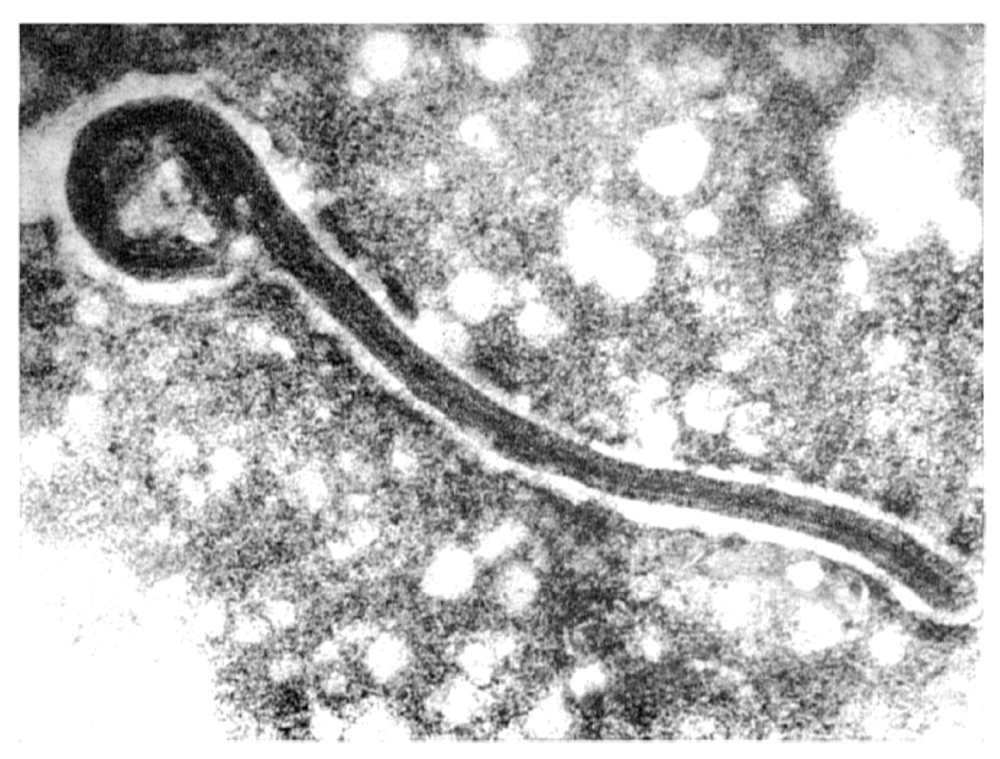
7.1. Virion Structure
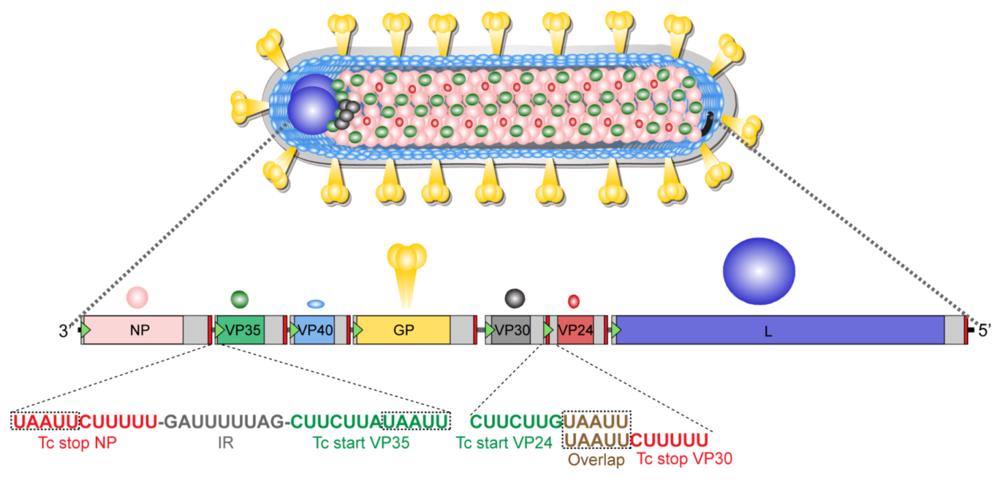
7.2. Genome Organization
7.3. Viral Proteins
| Protein | Amino Acids | Apparent Molecular Mass | Function |
|---|---|---|---|
| NP | 695 | 94 kDa | encapsidation of RNA genome, nucleocapsid formation, budding, essential for transcription and replication |
| VP35 | 329 | 32 kDa | polymerase cofactor, nucleocapsid formation, IFN antagonist |
| VP40 | 303 | 38 kDa | budding, antagonist of IFN signaling |
| GP | 681 | 170-200 kDa | attachment, receptor binding, fusion, tetherin antagonist |
| VP30 | 281 | 28 kDa | nucleocapsid formation |
| VP24 | 253 | 24 kDa | maturation of nucleocapsids, budding |
| L | 2331 | ~220 kDa | catalytic domain of RNA-dependent RNA polymerase |
7.3.1. Glycoprotein (GP)
7.3.2. Viral Protein 40 (VP40)
7.3.3. Viral protein 24 (VP24)
7.3.4. The Nucleocapsid Proteins NP, VP35, VP30 and L
7.3.5. Nucleoprotein (NP)
7.3.6. Viral Protein 35 (VP35)
7.3.7. Viral Protein 30 (VP30)
7.3.8. Large Protein (L)
8. Replication Cycle
8.1. Entry
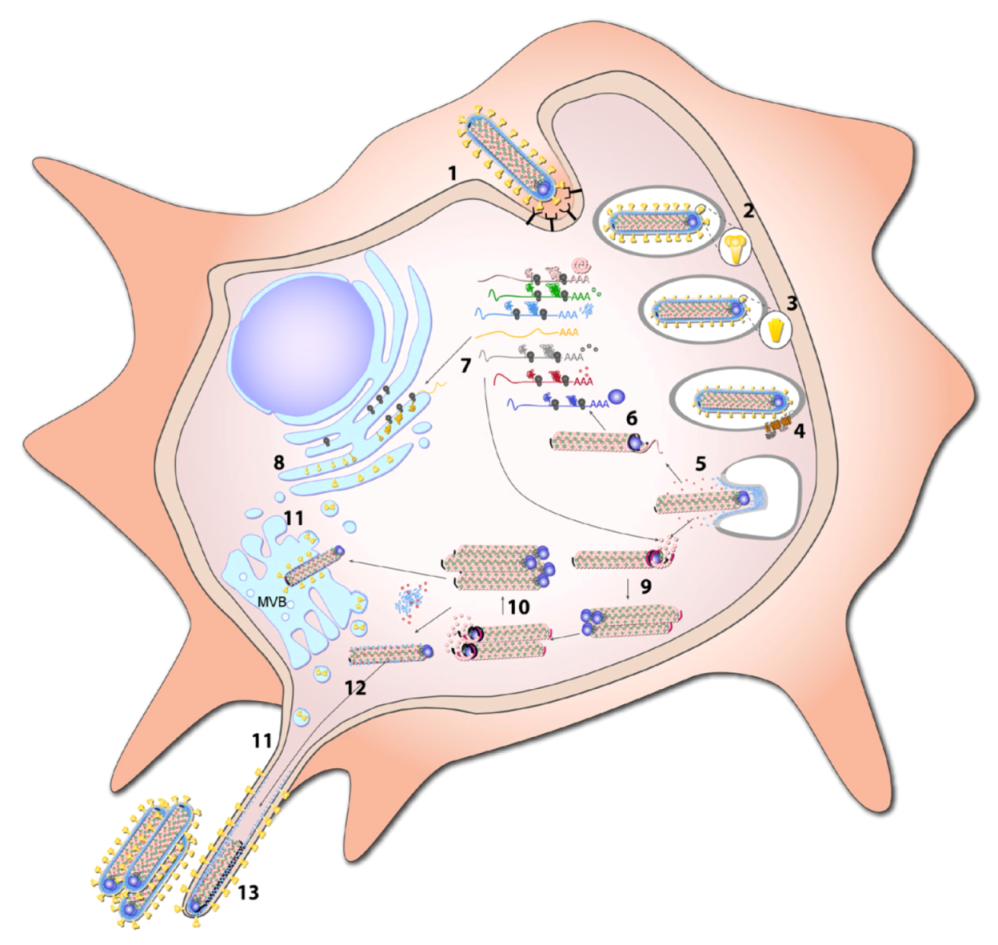
8.1.1. Attachment
8.1.2. Endocytosis
8.1.3. Fusion
8.2. Transcription and Replication
8.3. Budding
9. Pathogenesis
10. Animal Models
11. Diagnosis
12. Vaccine Development
13. Treatment
Acknowledgments
Conflict of Interest
References
- Slenczka, W.; Klenk, H.D. Forty years of marburg virus. J. Infect. Dis. 2007, 196, S131–S135. [Google Scholar] [CrossRef]
- Siegert, R.; Shu, H.L.; Slenczka, W.; Peters, D.; Müller, G. The aetiology of an unknown human infection transmitted by monkeys [premilinary communication]. Ger. Med. Mon. 1968, 13, 1–2. [Google Scholar]
- Kunz, C.; Hofmann, H.; Kovac, W.; Stockinger, L. Biologische und morphologische charakteristika des virus des in Deutschland aufgetretenen "Haemmorhagischen Fiebers". Wien. Klin. Wochenschr. 1968, 80, 161–162. [Google Scholar]
- Kissling, R.E.; Robinson, R.Q.; Murphy, F.A.; Whitfield, S.G. Agent of disease contracted from green monkeys. Science 1968, 160, 888–890. [Google Scholar]
- Smith, C.E.; Simpson, D.I.; Bowen, E.T.; Zlotnik, I. Fatal human disease from vervet monkeys. Lancet 1967, 7526, 1119–1121. [Google Scholar]
- WHO. Ebola haemorrhagic fever in Sudan, 1976. Bull. World Health Organ. 1978, 56, 247–270.
- WHO. Ebola haemorrhagic fever in Zaire, 1976. Bull World Health Organ 1978, 56, 271–293.
- Gear, J.S.; Cassel, G.A.; Gear, A.J.; Trappler, B.; Clausen, L.; Meyers, A.M.; Kew, M.C.; Bothwell, T.H.; Sher, R.; Miller, G.B.; et al. Outbreak of Marburg virus disease in Johannesburg. Br. Med. J. 1975, 489–493. [Google Scholar]
- Bausch, D.G.; Nichol, S.T.; Muyembe-Tamfum, J.J.; Borchert, M.; Rollin, P.E.; Sleurs, H.; Campbell, P.; Tshioko, F.K.; Roth, C.; Colebunders, R.; et al. Marburg hemorrhagic fever associated with multiple genetic lineages of virus. N. Engl. J. Med. 2006, 355, 909–919. [Google Scholar] [CrossRef]
- Towner, J. S.; Khristova, M. L.; Sealy, T. K.; Vincent, M. J.; Erickson, B. R.; Bawiec, D. A.; Hartman, A. L.; Comer, J. A.; Zaki, S. R.; Ströher, U.; et al. Marburgvirus genomics and association with a large hemorrhagic fever outbreak in Angola. J. Virol. 2006, 80, 6497–6516. [Google Scholar]
- Feldmann, H. Marburg hemorrhagic fever-the forgotten cousin strikes. N. Engl. J. Med. 2006, 355, 866–869. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Daddario-DiCaprio, K.M.; Geisbert, J.B.; Young, H.A.; Formenty, P.; Fritz, E.A.; Larsen, T.; Hensley, L.E. Marburg virus Angola infection of rhesus macaques: Pathogenesis and treatment with recombinant nematode anticoagulant protein c2. J. Infect. Dis. 2007, 196, S372–S381. [Google Scholar] [CrossRef]
- Daddario-DiCaprio, K.M.; Geisbert, T.W.; Geisbert, J.B.; Stroher, U.; Hensley, L.E.; Grolla, A.; Fritz, E.A.; Feldmann, F.; Feldmann, H.; Jones, S.M. Cross-protection against Marburg virus strains by using a live, attenuated recombinant vaccine. J. Virol. 2006, 80, 9659–9666. [Google Scholar]
- Alves, D.A.; Glynn, A.R.; Steele, K.E.; Lackemeyer, M.G.; Garza, N.L.; Buck, J.G.; Mech, C.; Reed, D.S. Aerosol exposure to the angola strain of marburg virus causes lethal viral hemorrhagic Fever in cynomolgus macaques. Vet. Pathol. 2010, 47, 831–851. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Dodd, L.E.; Wahl-Jensen, V.; Radoshitzky, S.R.; Bavari, S.; Jahrling, P.B. Evaluation of perceived threat differences posed by filovirus variants. Biosecur. Bioterror. 2011, 9, 361–371. [Google Scholar]
- Kuhn, J.H. Filoviruses; A Compendium of 40 years of Epidemiological, Clinical, and Laboratory Studies; Springer Verlag: Vienna, Austria, 2008. [Google Scholar]
- Centers for Disease Control and Prevention. Imported case of Marburg hemorrhagic fever - Colorado, 2008. MMWR Morb. Mortal Wkly. Rep. 2009, 58, 1377–1381.
- Timen, A.; Koopmans, M.P.; Vossen, A.C.; van Doornum, G.J.; Gunther, S.; van den Berkmortel, F.; Verduin, K.M.; Dittrich, S.; Emmerich, P.; Osterhaus, A.D.; van Dissel, J.T.; Coutinho, R.A. Response to imported case of Marburg hemorrhagic fever, the Netherlands. Emerg. Infect. Dis. 2009, 15, 1171–1175. [Google Scholar]
- Adjemian, J.; Farnon, E.C.; Tschioko, F.; Wamala, J.F.; Byaruhanga, E.; Bwire, G.S.; Kansiime, E.; Kagirita, A.; et al. Outbreak of Marburg hemorrhagic fever among miners in Kamwenge and Ibanda Districts, Uganda, 2007. J. Infect. Dis. 2011, 204, S796–S799. [Google Scholar] [CrossRef]
- Nikiforov, V.V.; Turovskii Iu, I.; Kalinin, P.P.; Akinfeeva, L.A.; Katkova, L.R.; Barmin, V.S.; Riabchikova, E.I.; Popkova, N.I.; Shestopalov, A.M.; Nazarov, V.P.; et al. [A case of a laboratory infection with Marburg fever]. Zh. Mikrobiol. Epidemiol. Immunobiol. 1994, 3, 104–106. [Google Scholar]
- U.S. Department of Health and Human Services, Biosafety in Microbiological and Biomedical Laboratories, 5th edU.S. Department of Health and Human Services: Washington, DC, USA, 2009.
- Towner, J.S.; Amman, B.R.; Sealy, T.K.; Carroll, S.A.; Comer, J.A.; Kemp, A.; Swanepoel, R.; Paddock, C.D.; Balinandi, S.; Khristova, M.L.; et al. Isolation of genetically diverse Marburg viruses from Egyptian fruit bats. PLoS Pathog. 2009, 5, e1000536. [Google Scholar]
- Martini, G.A.; Siegert, R. Marburg virus disease; Springer: New York, NY, USA, 1971. [Google Scholar]
- Conrad, J.L.; Isaacson, M.; Smith, E.B.; Wulff, H.; Crees, M.; Geldenhuys, P.; Johnston, J. Epidemiologic investigation of Marburg virus disease, Southern Africa, 1975. Am. J. Trop. Med. Hyg. 1978, 27, 1210–1215. [Google Scholar]
- Smith, D.H.; Johnson, B.K.; Isaacson, M.; Swanapoel, R.; Johnson, K.M.; Killey, M.; Bagshawe, A.; Siongok, T.; Keruga, W.K. Marburg-virus disease in Kenya. Lancet 1982, 1, 816–820. [Google Scholar]
- Johnson, E.D.; Johnson, B.K.; Silverstein, D.; Tukei, P.; Geisbert, T.W.; Sanchez, A.N.; Jahrling, P.B. Characterization of a new Marburg virus isolated from a 1987 fatal case in Kenya. Arch. Virol. Suppl. 1996, 11, 101–114. [Google Scholar]
- Ligon, B.L. Outbreak of Marburg hemorrhagic fever in Angola: A review of the history of the disease and its biological aspects. Semin. Pediatr. Infect. Dis. 2005, 16, 219–224. [Google Scholar] [CrossRef]
- Breman, J.G.; Johnson, K.M.; van der Groen, G.; Robbins, C.B.; Szczeniowski, M.V.; Ruti, K.; Webb, P.A.; Meier, F.; Heymann, D.L. A search for Ebola virus in animals in the Democratic Republic of the Congo and Cameroon: ecologic, virologic, and serologic surveys, 1979-1980. Ebola Virus Study Teams. J. Infect. Dis. 1999, 179, S139–S147. [Google Scholar]
- Leirs, H.; Mills, J.N.; Krebs, J.W.; Childs, J.E.; Akaibe, D.; Woollen, N.; Ludwig, G.; Peters, C.J.; Ksiazek, T.G. Search for the Ebola virus reservoir in Kikwit, Democratic Republic of the Congo: Reflections on a vertebrate collection. J. Infect. Dis. 1999, 179, S155–S163. [Google Scholar]
- Reiter, P.; Turell, M.; Coleman, R.; Miller, B.; Maupin, G.; Liz, J.; Kuehne, A.; Barth, J.; Geisbert, J.; Dohm, D.; et al. Field investigations of an outbreak of Ebola hemorrhagic fever, Kikwit, Democratic Republic of the Congo, 1995: Arthropod studies. J. Infect. Dis. 1999, 179, S148–S154. [Google Scholar]
- Monath, T.P. Ecology of Marburg and Ebola viruses: Speculations and directions for future research. J. Infect. Dis. 1999, 179, S127–S138. [Google Scholar]
- Peterson, A.T.; Carroll, D.S.; Mills, J.N.; Johnson, K.M. Potential mammalian filovirus reservoirs. Emerg. Infect. Dis. 2004, 10, 2073–2081. [Google Scholar]
- Peterson, A.T.; Lash, R.R.; Carroll, D.S.; Johnson, K.M. Geographic potential for outbreaks of Marburg hemorrhagic fever. Am. J. Trop. Med. Hyg. 2006, 75, 9–15. [Google Scholar]
- Bausch, D.G.; Borchert, M.; Grein, T.; Roth, C.; Swanepoel, R.; Libande, M.L.; Talarmin, A.; Bertherat, E.; Muyembe-Tamfum, J.J.; Tugume, B.; et al. Risk factors for Marburg hemorrhagic fever, Democratic Republic of the Congo. Emerg. Infect. Dis. 2003, 9, 1531–1537. [Google Scholar] [CrossRef]
- Towner, J.S.; Pourrut, X.; Albarino, C.G.; Nkogue, C.N.; Bird, B.H.; Grard, G.; Ksiazek, T.G.; Gonzalez, J.P.; Nichol, S.T.; Leroy, E.M. Marburg virus infection detected in a common African bat. PLoS ONE 2007, 2, e764. [Google Scholar]
- Swanepoel, R.; Smit, S.B.; Rollin, P.E.; Formenty, P.; Leman, P.A.; Kemp, A.; Burt, F.J.; Grobbelaar, A.A.; Croft, J.; Bausch, D.G.; Zeller, H.; Leirs, H.; et al. Studies of reservoir hosts for Marburg virus. Emerg. Infect. Dis. 2007, 13, 1847–1851. [Google Scholar] [CrossRef]
- Maganga, G.D.; Bourgarel, M.; Ella, G.E.; Drexler, J.F.; Gonzalez, J P.; Drosten, C.; Leroy, E.M. Is Marburg virus enzootic in Gabon? J. Infect. Dis. 2011, 204, S800–S803. [Google Scholar] [CrossRef]
- Pourrut, X.; Souris, M.; Towner, J.S.; Rollin, P.E.; Nichol, S.T.; Gonzalez, J.P.; Leroy, E. Large serological survey showing cocirculation of Ebola and Marburg viruses in Gabonese bat populations, and a high seroprevalence of both viruses in Rousettus aegyptiacus. BMC Infect. Dis. 2009, 9, 159. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Becker, S.; Ebihara, H.; Geisbert, T.W.; Jahrling, P.B.; Kawaoka, Y.; Netesov, S. V.; Nichol, S.T.; Peters, C.J.; Volchkov, V.E.; Ksiazek, T.G. Filoviridae. In Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Academic Press: Amsterdam, Netherlands, 2011; Volume 9, pp. 665–671. [Google Scholar]
- Adams, M.J.; Carstens, E.B. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2012). Arch. Virol. 2012, 157, 1411–1422. [Google Scholar]
- Martini, G.A. Marburg virus disease. Postgrad. Med. J. 1973, 49, 542–546. [Google Scholar]
- Martini, G.A. Marburg agent disease: In man. Trans. R. Soc. Trop. Med. Hyg. 1969, 63, 295–302. [Google Scholar]
- Slenczka, W.G. The Marburg virus outbreak of 1967 and subsequent episodes. Curr. Top. Microbiol. Immunol. 1999, 235, 49–75. [Google Scholar]
- Gear, J.S.; Cassel, G.A.; Gear, A.J.; Trappler, B.; Clausen, L.; Meyers, A.M.; Kew, M.C.; Bothwell, T.H.; Sher, R.; Miller, G.B.; et al. Outbreake of Marburg virus disease in Johannesburg. Br. Med. J. 1975, 4, 489–493. [Google Scholar]
- Stille, W.; Böhle, E. Clinical course and prognosis of Marburg virus (green monkey) disease. In Marburg virus disease; Martini, G.A., Siegert, R., Eds.; Springer: Berlin, Germay, 1971; pp. 10–18. [Google Scholar]
- Hartman, A.L.; Towner, J.S.; Nichol, S.T. Ebola and marburg hemorrhagic fever. Clin. Lab. Med. 2010, 30, 161–177. [Google Scholar]
- Mehedi, M.; Groseth, A.; Feldmann, H.; Ebihara, H. Clinical aspects of Marburg hemorrhagic fever. Future Virol. 2011, 6, 1091–1106. [Google Scholar]
- Kortepeter, M.G.; Bausch, D.G.; Bray, M. Basic clinical and laboratory features of filoviral hemorrhagic fever. J. Infect. Dis. 2011, 204, S810–6. [Google Scholar]
- Bray, M. Epidemiology, pathogenesis, and clinical manifestations of Ebola and Marburg hemorrhagic fever. Available online: http://www.uptodate.com/contents/epidemiology-pathogenesis-and-clinical-manifestations-of-ebola-and-marburg-hemorrhagic-fever (accessed on 26 September 2012).
- Miranda, M.E.; Miranda, N.L. Reston ebolavirus in humans and animals in the Philippines: A review. J. Infect. Dis. 2011, 204, S757–S760. [Google Scholar]
- Roberts, J.A.; Andrews, K. Nonhuman primate quarantine: its evolution and practice. ILAR J. 2008, 49, 145–156. [Google Scholar]
- Schou, S.; Hansen, A.K. Marburg and Ebola virus infections in laboratory non-human primates: A literature review. Comp. Med. 2000, 50, 108–123. [Google Scholar]
- Roddy, P.; Weatherill, D.; Jeffs, B.; Abaakouk, Z.; Dorion, C.; Rodriguez-Martinez, J.; Palma, P.P.; de la Rosa, O.; Villa, L.; Grovas, I.; et al. The Medecins Sans Frontieres intervention in the Marburg hemorrhagic fever epidemic, Uige, Angola, 2005. II. Lessons learned in the community. J. Infect. Dis. 2007, 196, S162–S167. [Google Scholar] [CrossRef] [Green Version]
- Jeffs, B.; Roddy, P.; Weatherill, D.; de la Rosa, O.; Dorion, C.; Iscla, M.; Grovas, I.; Palma, P. P.; Villa, L.; Bernal, O.; et al. The Medecins Sans Frontieres intervention in the Marburg hemorrhagic fever epidemic, Uige, Angola, 2005. I. Lessons learned in the hospital. J. Infect. Dis. 2007, 196, S154–S161. [Google Scholar] [CrossRef] [Green Version]
- Leroy, E.M.; Kumulungui, B.; Pourrut, X.; Rouquet, P.; Hassanin, A.; Yaba, P.; Delicat, A.; Paweska, J.T.; Gonzalez, J.P.; Swanepoel, R. Fruit bats as reservoirs of Ebola virus. Nature 2005, 438, 575–576. [Google Scholar]
- Kuzmin, I.V.; Niezgoda, M.; Franka, R.; Agwanda, B.; Markotter, W.; Breiman, R.F.; Shieh, W.J.; Zaki, S.R.; Rupprecht, C.E. Marburg virus in fruit bat, Kenya. Emerg. Infect. Dis. 2010, 16, 352–354. [Google Scholar]
- Centers for Disease Control and PreventionWorld Health OrganizationWorld Health OrganizationInfection Control for Viral Haemorrhagic Fevers in the African Health Care Setting. Centers for Disease Control and Prevention: Atlanta, GA, USA, 1998.
- Peters, D.; Muller, G. The Marburg agent and structures associated with leptospira. Lancet 1969, 1, 923–925. [Google Scholar]
- Siegert, R.; Shu, H.-L.; Slenczka, W.; Peters, D.; Müller, G. Zur Aetiologie einer unbekannten, von Affen ausgegangenen menschlichen Infektioskrankheit. Dtsch. Med. Wschr. 1967, 51, 2341–2343. [Google Scholar]
- Bharat, T.A.; Riches, J.D.; Kolesnikova, L.; Welsch, S.; Krahling, V.; Davey, N.; Parsy, M.L.; Becker, S.; Briggs, J.A. Cryo-Electron tomography of Marburg Virus particles and their Morphogenesis within infected cells. PLoS Biol. 2011, 9, e1001196. [Google Scholar]
- Peters, D.; Müller, G.; Slenczka, W. Morphology, development, and classification of the Marburg virus. In Marburg virus disease; Martini, G.A., Siegert, R., Eds.; Springer: Berlin, Germany, 1971; pp. 68–83. [Google Scholar]
- Geisbert, T.W.; Jahrling, P.B. Differentiation of filoviruses by electron microscopy. Virus Res. 1995, 39, 129–150. [Google Scholar]
- Ryabchikova, E.; Price, B.B.S. Ebola and Marburg Viruses: A View of Infection Using Electron Microscopy; Battelle Press: Columbus, Ohio, USA, 2004. [Google Scholar]
- Schnittler, H.J.; Mahner, F.; Drenckhahn, D.; Klenk, H.D.; Feldmann, H. Replication of Marburg virus in human endothelial cells. A possible mechanism for the development of viral hemorrhagic disease. J. Clin. Invest. 1993, 91, 1301–1309. [Google Scholar] [CrossRef]
- Feldmann, H.; Will, C.; Schikore, M.; Slenczka, W.; Klenk, H.D. Glycosylation and oligomerization of the spike protein of Marburg virus. Virology 1991, 182, 353–356. [Google Scholar]
- Feldmann, H.; Mühlberger, E.; Randolf, A.; Will, C.; Kiley, M. P.; Sanchez, A.; Klenk, H. D. Marburg virus, a filovirus: Messenger RNAs, gene order, and regulatory elements of the replication cycle. Virus Res. 1992, 24, 1–19. [Google Scholar] [CrossRef]
- Mühlberger, E.; Sanchez, A.; Randolf, A.; Will, C.; Kiley, M.P.; Klenk, H.D.; Feldmann, H. The nucleotide sequence of the L gene of Marburg virus, a filovirus: Homologies with paramyxoviruses and rhabdoviruses. Virology 1992, 187, 534–547. [Google Scholar]
- Mühlberger, E. Filovirus replication and transcription. Future Virology 2007, 2, 205–215. [Google Scholar]
- Kolakofsky, D.; Roux, L.; Garcin, D.; Ruigrok, R.W. Paramyxovirus mRNA editing, the "rule of six" and error catastrophe: a hypothesis. J. Gen. Virol. 2005, 86, 1869–1877. [Google Scholar]
- Tapparel, C.; Maurice, D.; Roux, L. The activity of Sendai virus genomic and antigenomic promoters requires a second element past the leader template regions: a motif (GNNNNN)3 is essential for replication. J. Virol. 1998, 72, 3117–3128. [Google Scholar]
- Enterlein, S.; Schmidt, K.M.; Schümann, M.; Conrad, D.; Krahling, V.; Olejnik, J.; Mühlberger, E. The marburg virus 3' noncoding region structurally and functionally differs from that of ebola virus. J. Virol. 2009, 83, 4508–4519. [Google Scholar]
- Whelan, S.P.; Barr, J.N.; Wertz, G.W. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr. Top. Microbiol. Immunol. 2004, 283, 61–119. [Google Scholar]
- Crary, S.M.; Towner, J.S.; Honig, J.E.; Shoemaker, T.R.; Nichol, S.T. Analysis of the role of predicted RNA secondary structures in Ebola virus replication. Virology 2003, 306, 210–218. [Google Scholar]
- Volchkov, V.E.; Volchkova, V.A.; Chepurnov, A.A.; Blinov, V.M.; Dolnik, O.; Netesov, S.V.; Feldmann, H. Characterization of the L gene and 5' trailer region of Ebola virus. J. Gen. Virol. 1999, 80, 355–362. [Google Scholar]
- Weik, M.; Enterlein, S.; Schlenz, K.; Mühlberger, E. The Ebola virus genomic replication promoter is bipartite and follows the rule of six. J Virol 2005, 79, 10660–10671. [Google Scholar]
- Will, C.; Mühlberger, E.; Linder, D.; Slenczka, W.; Klenk, H.D.; Feldmann, H. Marburg virus gene 4 encodes the virion membrane protein, a type I transmembrane glycoprotein. J. Virol. 1993, 67, 1203–1210. [Google Scholar]
- Volchkov, V.E.; Becker, S.; Volchkova, V.A.; Ternovoj, V.A.; Kotov, A.N.; Netesov, S.V.; Klenk, H D. GP mRNA of Ebola virus is edited by the Ebola virus polymerase and by T7 and vaccinia virus polymerases. Virology 1995, 214, 421–430. [Google Scholar] [CrossRef]
- Sanchez, A.; Trappier, S.G.; Mahy, B.W.; Peters, C.J.; Nichol, S.T. The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 3602–3607. [Google Scholar]
- Volchkova, V.A.; Feldmann, H.; Klenk, H.D.; Volchkov, V.E. The nonstructural small glycoprotein sGP of Ebola virus is secreted as an antiparallel-orientated homodimer. Virology 1998, 250, 408–414. [Google Scholar]
- Mehedi, M.; Falzarano, D.; Seebach, J.; Hu, X.; Carpenter, M.S.; Schnittler, H.J.; Feldmann, H. A new Ebola Virus nonstructural glycoprotein expressed through RNA editing. J. Virol. 2011, pages. [Google Scholar]
- Becker, S.; Klenk, H.-D.; Mühlberger, E. Intracellular transport and processing of the Marburg virus surface protein in vertebrate and insect cells. Virology 1996, 225, 145–155. [Google Scholar]
- Funke, C.; Becker, S.; Dartsch, H.; Klenk, H.D.; Mühlberger, E. Acylation of the Marburg virus glycoprotein. Virology 1995, 208, 289–297. [Google Scholar]
- Sänger, C.; Mühlberger, E.; Lötfering, B.; Klenk, H.D.; Becker, S. The Marburg virus surface protein GP is phosphorylated at its ectodomain. Virology 2002, 295, 20–29. [Google Scholar]
- Geyer, H.; Will, C.; Feldmann, H.; Klenk, H.D.; Geyer, R. Carbohydrate structure of Marburg virus glycoprotein. Glycobiology 1992, 2, 299–312. [Google Scholar]
- Feldmann, H.; Nichol, S.T.; Klenk, H.D.; Peters, C.J.; Sanchez, A. Characterization of filoviruses based on differences in structure and antigenicity of the virion glycoprotein. Virology 1994, 199, 469–473. [Google Scholar]
- Volchkov, V.E.; Volchkova, V.A.; Stroher, U.; Becker, S.; Dolnik, O.; Cieplik, M.; Garten, W.; Klenk, H.D.; Feldmann, H. Proteolytic processing of Marburg virus glycoprotein. Virology 2000, 268, 1–6. [Google Scholar]
- Becker, S.; Spiess, M.; Klenk, H.D. The asialoglycoprotein receptor is a potential liver-specific receptor for Marburg virus. J. Gen. Virol. 1995, 76, 393–399. [Google Scholar]
- Chan, S.Y.; Empig, C.J.; Welte, F.J.; Speck, R.F.; Schmaljohn, A.; Kreisberg, J.F.; Goldsmith, M.A. Folate receptor-alpha is a cofactor for cellular entry by Marburg and Ebola viruses. Cell 2001, 106, 117–126. [Google Scholar]
- Marzi, A.; Gramberg, T.; Simmons, G.; Möller, P.; Rennekamp, A.J.; Krumbiegel, M.; Geier, M.; Eisemann, J.; Turza, N.; Saunier, B.; et al. DC-SIGN and DC-SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J. Virol. 2004, 78, 12090–12095. [Google Scholar] [CrossRef]
- Shimojima, M.; Takada, A.; Ebihara, H.; Neumann, G.; Fujioka, K.; Irimura, T.; Jones, S.; Feldmann, H.; Kawaoka, Y. Tyro3 family-mediated cell entry of Ebola and Marburg viruses. J. Virol. 2006, 80, 10109–10116. [Google Scholar]
- Takada, A.; Fujioka, K.; Tsuiji, M.; Morikawa, A.; Higashi, N.; Ebihara, H.; Kobasa, D.; Feldmann, H.; Irimura, T.; Kawaoka, Y. Human macrophage C–type lectin specific for galactose and N-acetylgalactosamine promotes filovirus entry. J. Viro.l 2004, 78, 2943–2947. [Google Scholar]
- Matsuno, K.; Kishida, N.; Usami, K.; Igarashi, M.; Yoshida, R.; Nakayama, E.; Shimojima, M.; Feldmann, H.; Irimura, T.; Kawaoka, Y.; Takada, A. Different potential of C-type lectin-mediated entry between Marburg virus strains. J. Virol. 2010, 84, 5140–5147. [Google Scholar]
- Kondratowicz, A.S.; Lennemann, N.J.; Sinn, P.L.; Davey, R.A.; Hunt, C.L.; Moller-Tank, S.; Meyerholz, D.K.; Rennert, P.; Mullins, R.F.; Brindley, M.; Sandersfeld, L.M.; Quinn, K.; et al. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 8426–8431. [Google Scholar]
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 2011, 477, 340–343. [Google Scholar] [CrossRef]
- Cote, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.M.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; Cunningham, J. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 2011, 477, 344–348. [Google Scholar]
- Weissenhorn, W.; Carfi, A.; Lee, K.H.; Skehel, J.J.; Wiley, D.C. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol. Cell. 1998, 2, 605–616. [Google Scholar] [CrossRef]
- Koellhoffer, J.F.; Malashkevich, V.N.; Harrison, J.S.; Toro, R.; Bhosle, R.C.; Chandran, K.; Almo, S.C.; Lai, J.R. Crystal Structure of the Marburg Virus GP2 core domain in its post-fusion conformation. Biochemistry 2012. [Google Scholar]
- Mittler, E.; Kolesnikova, L.; Strecker, T.; Garten, W.; Becker, S. Role of the transmembrane domain of marburg virus surface protein GP in assembly of the viral envelope. J. Virol. 2007, 81, 3942–3948. [Google Scholar]
- Mittler, E.; Kolesnikova, L.; Hartlieb, B.; Davey, R.; Becker, S. The cytoplasmic domain of Marburg virus GP modulates early steps of viral infection. J. Virol. 2011, 85, 8188–8196. [Google Scholar]
- Kuhn, J.H.; Radoshitzky, S.R.; Guth, A.C.; Warfield, K.L.; Li, W.; Vincent, M.J.; Towner, J.S.; Nichol, S.T.; Bavari, S.; Choe, H.; et al. Conserved receptor-binding domains of Lake Victoria marburgvirus and Zaire ebolavirus bind a common receptor. J. Biol. Chem. 2006, 281, 15951–15958. [Google Scholar]
- Manicassamy, B.; Wang, J.; Rumschlag, E.; Tymen, S.; Volchkova, V.; Volchkov, V.; Rong, L. Characterization of Marburg virus glycoprotein in viral entry. Virology 2007, 358, 79–88. [Google Scholar]
- Neil, S.J.; Sandrin, V.; Sundquist, W.I.; Bieniasz, P.D. An interferon-alpha-induced tethering mechanism inhibits HIV-1 and Ebola virus particle release but is counteracted by the HIV-1 Vpu protein. Cell Host Microbe. 2007, 2, 193–203. [Google Scholar]
- Jouvenet, N.; Neil, S.J.; Zhadina, M.; Zang, T.; Kratovac, Z.; Lee, Y.; McNatt, M.; Hatziioannou, T.; Bieniasz, P.D. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 2009, 83, 1837–1844. [Google Scholar]
- Kaletsky, R.L.; Francica, J.R.; Agrawal-Gamse, C.; Bates, P. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 2886–2891. [Google Scholar]
- Kuhl, A.; Banning, C.; Marzi, A.; Votteler, J.; Steffen, I.; Bertram, S.; Glowacka, I.; Konrad, A.; Sturzl, M.; Guo, J.T.; et al. The Ebola virus glycoprotein and HIV-1 Vpu employ different strategies to counteract the antiviral factor tetherin. J. Infect. Dis. 2011, 204, S850–S860. [Google Scholar]
- Bukreyev, A.; Volchkov, V.E.; Blinov, V.M.; Netesov, S.V. The GP-protein of Marburg virus contains the region similar to the 'immunosuppressive domain' of oncogenic retrovirus P15E proteins. FEBS Lett. 1993, 323, 183–187. [Google Scholar]
- Yaddanapudi, K.; Palacios, G.; Towner, J.S.; Chen, I.; Sariol, C.A.; Nichol, S.T.; Lipkin, W.I. Implication of a retrovirus-like glycoprotein peptide in the immunopathogenesis of Ebola and Marburg viruses. Faseb. J. 2006, 20, 2519–2530. [Google Scholar]
- Dolnik, O.; Volchkova, V.; Garten, W.; Carbonnelle, C.; Becker, S.; Kahnt, J.; Stroher, U.; Klenk, H.D.; Volchkov, V. Ectodomain shedding of the glycoprotein GP of Ebola virus. Embo. J. 2004, 23, 2175–2184. [Google Scholar]
- Sänger, C.; Mühlberger, E.; Ryabchikova, E.; Kolesnikova, L.; Klenk, H.D.; Becker, S. Sorting of Marburg virus surface protein and virus release take place at opposite surfaces of infected polarized epithelial cells. J. Virol. 2001, 75, 1274–1283. [Google Scholar]
- Dolnik, O.; Kolesnikova, L.; Becker, S. Filoviruses: Interactions with the host cell. Cell. Mol. Life Sci. 2008, 65, 756–776. [Google Scholar]
- Dolnik, O.; Kolesnikova, L.; Stevermann, L.; Becker, S. Tsg101 is recruited by a late domain of the nucleocapsid protein to support budding of Marburg virus-like particles. J. Virol. 2010, 84, 7847–7856. [Google Scholar]
- Kolesnikova, L.; Mittler, E.; Schudt, G.; Shams-Eldin, H.; Becker, S. Phosphorylation of Marburg virus matrix protein VP40 triggers assembly of nucleocapsids with the viral envelope at the plasma membrane. Cell Microbiol. 2012, 14, 182–197. [Google Scholar]
- Wenigenrath, J.; Kolesnikova, L.; Hoenen, T.; Mittler, E.; Becker, S. Establishment and application of an infectious virus-like particle system for Marburg virus. J. Gen. Virol. 2010, 91, 1325–1334. [Google Scholar]
- Kolesnikova, L.; Bugany, H.; Klenk, H.D.; Becker, S. VP40, the matrix protein of Marburg virus, is associated with membranes of the late endosomal compartment. J. Virol. 2002, 76, 1825–1838. [Google Scholar] [CrossRef]
- Becker, S.; Rinne, C.; Hofsäss, U.; Klenk, H.-D.; Mühlberger, E. Interactions of Marburg virus nucleocapsid proteins. Virology 1998, 249, 406–417. [Google Scholar]
- Kolesnikova, L.; Bamberg, S.; Berghofer, B.; Becker, S. The matrix protein of Marburg virus is transported to the plasma membrane along cellular membranes: exploiting the retrograde late endosomal pathway. J. Virol. 2004, 78, 2382–2393. [Google Scholar]
- Swenson, D.L.; Warfield, K.L.; Kuehl, K.; Larsen, T.; Hevey, M. C.; Schmaljohn, A.; Bavari, S.; Aman, M.J. Generation of Marburg virus-like particles by co-expression of glycoprotein and matrix protein. FEMS Immunol. Med. Microbiol. 2004, 40, 27–31. [Google Scholar]
- Kolesnikova, L.; Berghofer, B.; Bamberg, S.; Becker, S. Multivesicular bodies as a platform for formation of the Marburg virus envelope. J. Virol. 2004, 78, 12277–12287. [Google Scholar]
- Kolesnikova, L.; Strecker, T.; Morita, E.; Zielecki, F.; Mittler, E.; Crump, C.; Becker, S. Vacuolar protein sorting pathway contributes to the release of Marburg virus. J. Virol. 2009, 83, 2327–2337. [Google Scholar] [CrossRef]
- Timmins, J.; Schoehn, G.; Kohlhaas, C.; Klenk, H.D.; Ruigrok, R.W.; Weissenhorn, W. Oligomerization and polymerization of the filovirus matrix protein VP40. Virology 2003, 312, 359–368. [Google Scholar] [CrossRef]
- Urata, S.; Noda, T.; Kawaoka, Y.; Morikawa, S.; Yokosawa, H.; Yasuda, J. Interaction of Tsg101 with Marburg virus VP40 depends on the PPPY motif, but not the PT/SAP motif as in the case of Ebola virus, and Tsg101 plays a critical role in the budding of Marburg virus-like particles induced by VP40, NP, and GP. J. Virol. 2007, 81, 4895–4899. [Google Scholar] [CrossRef]
- Urata, S.; Yasuda, J. Regulation of Marburg virus (MARV) budding by Nedd4.1: a different WW domain of Nedd4.1 is critical for binding to MARV and Ebola virus VP40. J. Gen. Virol. 2010, 91, 228–234. [Google Scholar] [CrossRef]
- Liu, Y.; Cocka, L.; Okumura, A.; Zhang, Y. A.; Sunyer, J. O.; Harty, R. N. Conserved motifs within Ebola and Marburg virus VP40 proteins are important for stability, localization, and subsequent budding of virus-like particles. J. Virol. 2010, 84, 2294–2303. [Google Scholar] [CrossRef]
- Makino, A.; Yamayoshi, S.; Shinya, K.; Noda, T.; Kawaoka, Y. Identification of amino acids in Marburg virus VP40 that are important for virus-like particle budding. J. Infect. Dis. 2011, 204, S871–S877. [Google Scholar]
- Valmas, C.; Basler, C.F. Marburg Virus VP40 Antagonizes Interferon Signaling in a Species-Specific Manner. J. Virol. 2011, 85, 4309–4317. [Google Scholar]
- Valmas, C.; Grosch, M.N.; Schümann, M.; Olejnik, J.; Martinez, O.; Best, S.M.; Krähling, V.; Basler, C.F.; Mühlberger, E. Marburg virus evades interferon responses by a mechanism distinct from ebola virus. PLoS Pathog. 2010, 6, e1000721. [Google Scholar]
- Kash, J.C.; Mühlberger, E.; Carter, V.; Grosch, M.; Perwitasari, O.; Proll, S.C.; Thomas, M.J.; Weber, F.; Klenk, H.D.; Katze, M.G. Global suppression of the host antiviral response by Ebola- and Marburgviruses: increased antagonism of the type I interferon response is associated with enhanced virulence. J. Virol. 2006, 80, 3009–3020. [Google Scholar]
- Zhang, A.P.; Bornholdt, Z.A.; Liu, T.; Abelson, D.M.; Lee, D.E.; Li, S.; Woods, V.L., Jr.; Saphire, E.O. The ebola virus interferon antagonist VP24 directly binds STAT1 and has a novel, pyramidal fold. PLoS Pathog. 2012, 8, e1002550. [Google Scholar] [CrossRef]
- Ramanan, P.; Shabman, R.S.; Brown, C.S.; Amarasinghe, G.K.; Basler, C.F.; Leung, D.W. Filoviral immune evasion mechanisms. Viruses 2011, 3, 1634–1649. [Google Scholar]
- Warfield, K.L.; Bradfute, S.B.; Wells, J.; Lofts, L.; Cooper, M.T.; Alves, D.A.; Reed, D.K.; VanTongeren, S.A.; Mech, C. A.; Bavari, S. Development and characterization of a mouse model for Marburg hemorrhagic fever. J. Viro.l 2009, 83, 6404–6415. [Google Scholar]
- Lofts, L.L.; Wells, J.B.; Bavari, S.; Warfield, K.L. Key genomic changes necessary for an in vivo lethal mouse marburgvirus variant selection process. J. Virol. 2011, 85, 3905–3917. [Google Scholar] [CrossRef]
- Bamberg, S.; Kolesnikova, L.; Möller, P.; Klenk, H.D.; Becker, S. VP24 of Marburg virus influences formation of infectious particles. J. Virol. 2005, 79, 13421–13433. [Google Scholar]
- Lee, M.S.; Lebeda, F.J.; Olson, M.A. Fold prediction of VP24 protein of Ebola and Marburg viruses using de novo fragment assembly. J. Struct. Biol. 2009, 167, 136–144. [Google Scholar]
- Mühlberger, E.; Lötfering, B.; Klenk, H.-D.; Becker, S. Three of the four nucleocapsid proteins of Marburg virus, NP, VP35, and L, are sufficient to mediate replication and transcription of Marburg virus-specific monocistronic minigenomes. J. Virol. 1998, 72, 8756–8764. [Google Scholar]
- Kolesnikova, L.; Mühlberger, E.; Ryabchikova, E.; Becker, S. Ultrastructural organization of recombinant Marburg virus nucleoprotein: comparison with Marburg virus inclusions. J. Virol. 2000, 74, 3899–3904. [Google Scholar]
- Mavrakis, M.; Kolesnikova, L.; Schoehn, G.; Becker, S.; Ruigrok, R.W. Morphology of Marburg virus NP-RNA. Virology 2002, 296, 300–307. [Google Scholar] [CrossRef]
- DiCarlo, A.; Möller, P.; Lander, A.; Kolesnikova, L.; Becker, S. Nucleocapsid formation and RNA synthesis of Marburg virus is dependent on two coiled coil motifs in the nucleoprotein. Virol. J. 2007, 4, 105. [Google Scholar] [CrossRef]
- Liu, Y.; Stone, S.; Harty, R.N. Characterization of filovirus protein-protein interactions in mammalian cells using bimolecular complementation. J. Infect. Dis. 2011, 204, S817–S824. [Google Scholar]
- Spiegelberg, L.; Wahl-Jensen, V.; Kolesnikova, L.; Feldmann, H.; Becker, S.; Hoenen, T. Genus-specific recruitment of filovirus ribonucleoprotein complexes into budding particles. J. Gen. Virol. 2011, 92, 2900–2905. [Google Scholar]
- Becker, S.; Huppertz, S.; Klenk, H.D.; Feldmann, H. The nucleoprotein of Marburg virus is phosphorylated. J. Gen. Virol. 1994, 75, 809–818. [Google Scholar] [CrossRef]
- Lötfering, B.; Mühlberger, E.; Tamura, T.; Klenk, H.D.; Becker, S. The nucleoprotein of Marburg virus is target for multiple cellular kinases. Virology 1999, 255, 50–62. [Google Scholar]
- DiCarlo, A.; Biedenkopf, N.; Hartlieb, B.; Klussmeier, A.; Becker, S. Phosphorylation of Marburg virus NP region II modulates viral RNA synthesis. J. Infect. Dis. 2011, 204, S927–S933. [Google Scholar]
- Mühlberger, E.; Weik, M.; Volchkov, V.E.; Klenk, H.-D.; Becker, S. Comparison of the transcription and replication strategies of marburg virus and Ebola virus by using artificial replication systems. J. Viro.l 1999, 73, 2333–2342. [Google Scholar]
- Möller, P.; Pariente, N.; Klenk, H.D.; Becker, S. Homo-oligomerization of Marburgvirus VP35 is essential for its function in replication and transcription. J. Virol. 2005, 79, 14876–14886. [Google Scholar]
- Becker, S.; Mühlberger, E. Co- and posttranslational modifications and functions of Marburg virus proteins. Curr. Top Microbiol. Immunol. 1999, 235, 23–34. [Google Scholar]
- Bosio, C.M.; Aman, M.J.; Grogan, C.; Hogan, R.; Ruthel, G.; Negley, D.; Mohamadzadeh, M.; Bavari, S.; Schmaljohn, A. Ebola and Marburg viruses replicate in monocyte-derived dendritic cells without inducing the production of cytokines and full maturation. J. Infect. Dis. 2003, 188, 1630–1638. [Google Scholar]
- Hartman, A.L.; Towner, J.S.; Nichol, S.T. A C-terminal basic amino acid motif of Zaire ebolavirus VP35 is essential for type I interferon antagonism and displays high identity with the RNA-binding domain of another interferon antagonist, the NS1 protein of influenza A virus. Virology 2004, 328, 177–184. [Google Scholar]
- Modrof, J.; Mühlberger, E.; Klenk, H. D.; Becker, S. Phosphorylation of VP30 Impairs Ebola Virus transcription. J. Biol. Chem. 2002, 277, 33099–33104. [Google Scholar]
- Modrof, J.; Moritz, C.; Kolesnikova, L.; Konakova, T.; Hartlieb, B.; Randolf, A.; Mühlberger, E.; Becker, S. Phosphorylation of Marburg virus VP30 at serines 40 and 42 is critical for its interaction with NP inclusions. Virology 2001, 287, 171–182. [Google Scholar]
- Modrof, J.; Becker, S.; Mühlberger, E. Ebola virus transcription activator VP30 is a zinc-binding protein. J Virol 2003, 77, 3334–3338. [Google Scholar]
- Hartlieb, B.; Modrof, J.; Mühlberger, E.; Klenk, H.D.; Becker, S. Oligomerization of Ebola virus VP30 is essential for viral transcription and can be inhibited by a synthetic peptide. J. Biol. Chem. 2003, 278, 41830–41836. [Google Scholar]
- Hartlieb, B.; Muziol, T.; Weissenhorn, W.; Becker, S. Crystal structure of the C-terminal domain of Ebola virus VP30 reveals a role in transcription and nucleocapsid association. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 624–629. [Google Scholar]
- John, S.P.; Wang, T.; Steffan, S.; Longhi, S.; Schmaljohn, C.S.; Jonsson, C.B. The Ebola Virus VP30 is an RNA Binding Protein. J. Virol. 2007, 81, 8967–8976. [Google Scholar]
- Groseth, A.; Charton, J.E.; Sauerborn, M.; Feldmann, F.; Jones, S.M.; Hoenen, T.; Feldmann, H. The Ebola virus ribonucleoprotein complex: a novel VP30-L interaction identified. Virus Res. 2009, 140, 8–14. [Google Scholar]
- Weik, M.; Modrof, J.; Klenk, H.D.; Becker, S.; Mühlberger, E. Ebola Virus VP30-Mediated Transcription Is Regulated by RNA Secondary Structure Formation. J. Virol. 2002, 76, 8532–8539. [Google Scholar]
- Martinez, M.J.; Biedenkopf, N.; Volchkova, V.; Hartlieb, B.; Alazard-Dany, N.; Reynard, O.; Becker, S.; Volchkov, V. Role of Ebola virus VP30 in transcription reinitiation. J. Virol. 2008, 82, 12569–12573. [Google Scholar] [CrossRef]
- Martinez, M.J.; Volchkova, V.A.; Raoul, H.; Alazard-Dany, N.; Reynard, O.; Volchkov, V.E. Role of VP30 phosphorylation in the Ebola virus replication cycle. J. Infect. Dis. 2011, 204, S934–S940. [Google Scholar]
- Enterlein, S.; Volchkov, V.; Weik, M.; Kolesnikova, L.; Volchkova, V.; Klenk, H.D.; Mühlberger, E. Rescue of recombinant Marburg virus from cDNA is dependent on nucleocapsid protein VP30. J. Virol. 2006, 80, 1038–1043. [Google Scholar]
- Fowler, T.; Bamberg, S.; Möller, P.; Klenk, H.D.; Meyer, T.F.; Becker, S.; Rudel, T. Inhibition of Marburg virus protein expression and viral release by RNA interference. J. Gen. Virol. 2005, 86, 1181–1188. [Google Scholar]
- Collins, P.L.; Hill, M.G.; Cristina, J.; Grosfeld, H. Transcription elongation factor of respiratory syncytial virus, a nonsegmented negative-strand RNA virus. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 81–85. [Google Scholar] [CrossRef]
- Poch, O.; Blumberg, B.M.; Bougueleret, L.; Tordo, N. Sequence comparison of five polymerases (L proteins) of unsegmented negative-strand RNA viruses: theoretical assignment of functional domains. J. Gen. Virol. 1990, 71, 1153–1162. [Google Scholar]
- Wang, J.; Manicassamy, B.; Caffrey, M.; Rong, L. Characterization of the receptor-binding domain of Ebola glycoprotein in viral entry. Virol. Sin. 2011, 26, 156–170. [Google Scholar]
- Manicassamy, B.; Wang, J.; Jiang, H.; Rong, L. Comprehensive analysis of ebola virus GP1 in viral entry. J. Virol. 2005, 79, 4793–4805. [Google Scholar]
- Weissenhorn, W.; Dessen, A.; Calder, L.J.; Harrison, S.C.; Skehel, J.J.; Wiley, D.C. Structural basis for membrane fusion by enveloped viruses. Mol. Membr. Biol. 1999, 16, 3–9. [Google Scholar]
- Jeffers, S.A.; Sanders, D.A.; Sanchez, A. Covalent modifications of the ebola virus glycoprotein. J. Virol. 2002, 76, 12463–12472. [Google Scholar]
- Gramberg, T.; Hofmann, H.; Möller, P.; Lalor, P.F.; Marzi, A.; Geier, M.; Krumbiegel, M.; Winkler, T.; Kirchhoff, F.; Adams, D.H.; Becker, S.; Munch, J.; Pohlmann, S. LSECtin interacts with filovirus glycoproteins and the spike protein of SARS coronavirus. Virology 2005, 340, 224–236. [Google Scholar] [CrossRef]
- Dominguez-Soto, A.; Aragoneses-Fenoll, L.; Martin-Gayo, E.; Martinez-Prats, L.; Colmenares, M.; Naranjo-Gomez, M.; Borras, F. E.; Munoz, P.; Zubiaur, M.; Toribio, M. L.; et al. The DC-SIGN-related lectin LSECtin mediates antigen capture and pathogen binding by human myeloid cells. Blood 2007, 109, 5337–5345. [Google Scholar] [Green Version]
- Sinn, P.L.; Hickey, M.A.; Staber, P.D.; Dylla, D.E.; Jeffers, S.A.; Davidson, B.L.; Sanders, D. A.; McCray, P.B., Jr. Lentivirus vectors pseudotyped with filoviral envelope glycoproteins transduce airway epithelia from the apical surface independently of folate receptor alpha. J. Virol. 2003, 77, 5902–5910. [Google Scholar]
- Empig, C.J.; Goldsmith, M.A. Association of the caveola vesicular system with cellular entry by filoviruses. J. Virol. 2002, 76, 5266–5270. [Google Scholar]
- Simmons, G.; Rennekamp, A.J.; Chai, N.; Vandenberghe, L.H.; Riley, J.L.; Bates, P. Folate receptor alpha and caveolae are not required for Ebola virus glycoprotein-mediated viral infection. J. Virol. 2003, 77, 13433–13438. [Google Scholar]
- Bhattacharyya, S.; Hope, T.J.; Young, J.A. Differential requirements for clathrin endocytic pathway components in cellular entry by Ebola and Marburg glycoprotein pseudovirions. Virology 2011, 419, 1–9. [Google Scholar]
- Sanchez, A. Analysis of filovirus entry into vero e6 cells, using inhibitors of endocytosis, endosomal acidification, structural integrity, and cathepsin (B and L) activity. J. Infect. Dis. 2007, 196, S251–S258. [Google Scholar] [CrossRef]
- Aleksandrowicz, P.; Marzi, A.; Biedenkopf, N.; Beimforde, N.; Becker, S.; Hoenen, T.; Feldmann, H.; Schnittler, H.J. Ebola virus enters host cells by macropinocytosis and clathrin-mediated endocytosis. J. Infect. Dis. 2011, 204, S957–S967. [Google Scholar]
- Hunt, C.L.; Kolokoltsov, A.A.; Davey, R.A.; Maury, W. The Tyro3 receptor kinase Axl enhances macropinocytosis of Zaire ebolavirus. J. Virol. 2011, 85, 334–347. [Google Scholar]
- Mulherkar, N.; Raaben, M.; de la Torre, J.C.; Whelan, S.P.; Chandran, K. The Ebola virus glycoprotein mediates entry via a non-classical dynamin-dependent macropinocytic pathway. Virology 2011, 419, 72–83. [Google Scholar]
- Nanbo, A.; Imai, M.; Watanabe, S.; Noda, T.; Takahashi, K.; Neumann, G.; Halfmann, P.; Kawaoka, Y. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog. 2010, 6, e1001121. [Google Scholar]
- Saeed, M.F.; Kolokoltsov, A.A.; Albrecht, T.; Davey, R.A. Cellular entry of Ebola Virus involves uptake by a Macropinocytosis-Like mechanism and Subsequent trafficking through early and aate endosomes. PLoS Pathog. 2010, 6, e1001110. [Google Scholar]
- Mercer, J.; Helenius, A. Virus entry by macropinocytosis. Nat. Cell. Biol. 2009, 11, 510–20. [Google Scholar]
- Misasi, J.; Chandran, K.; Yang, J.Y.; Considine, B.; Filone, C.M.; Cote, M.; Sullivan, N.; Fabozzi, G.; Hensley, L.; Cunningham, J. Filoviruses require endosomal cysteine proteases for entry but exhibit distinct protease preferences. J. Virol. 2012, 86, 3284–3292. [Google Scholar]
- Gnirss, K.; Kuhl, A.; Karsten, C.; Glowacka, I.; Bertram, S.; Kaup, F.; Hofmann, H.; Pohlmann, S. Cathepsins B and L activate Ebola but not Marburg virus glycoproteins for efficient entry into cell lines and macrophages independent of TMPRSS2 expression. Virology 2012, 424, 3–10. [Google Scholar]
- Chandran, K.; Sullivan, N. J.; Felbor, U.; Whelan, S. P.; Cunningham, J. M. Endosomal Proteolysis of the Ebola Virus Glycoprotein Is Necessary for Infection. Science 2005, 308, 1643–1645. [Google Scholar]
- Schornberg, K.; Matsuyama, S.; Kabsch, K.; Delos, S.; Bouton, A.; White, J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 2006, 80, 4174–4178. [Google Scholar]
- Chan, S.Y.; Speck, R.F.; Ma, M.C.; Goldsmith, M.A. Distinct mechanisms of entry by envelope glycoproteins of Marburg and Ebola (Zaire) viruses. J. Virol. 2000, 74, 4933–4937. [Google Scholar]
- Hoenen, T.; Shabman, R.S.; Groseth, A.; Herwig, A.; Weber, M.; Schudt, G.; Dolnik, O.; Basler, C.F.; Becker, S.; Feldmann, H. Inclusion bodies are a site of Ebola virus replication. J. Virol. 2012. [Google Scholar]
- Hoenen, T.; Jung, S.; Herwig, A.; Groseth, A.; Becker, S. Both matrix proteins of Ebola virus contribute to the regulation of viral genome replication and transcription. Virology 2010, 403, 56–66. [Google Scholar] [CrossRef]
- Watanabe, S.; Noda, T.; Halfmann, P.; Jasenosky, L.; Kawaoka, Y. Ebola virus (EBOV) VP24 inhibits transcription and replication of the EBOV genome. J. Infect. Dis. 2007, 196, S284–S290. [Google Scholar] [CrossRef]
- Yamayoshi, S.; Noda, T.; Ebihara, H.; Goto, H.; Morikawa, Y.; Lukashevich, I.S.; Neumann, G.; Feldmann, H.; Kawaoka, Y. Ebola virus matrix protein VP40 uses the COPII transport system for its intracellular transport. Cell Host Microbe. 2008, 3, 168–177. [Google Scholar] [CrossRef]
- Kolesnikova, L.; Bohil, A.B.; Cheney, R.E.; Becker, S. Budding of Marburgvirus is associated with filopodia. Cell Microbiol. 2007, 9, 939–951. [Google Scholar] [CrossRef]
- Welsch, S.; Kolesnikova, L.; Krahling, V.; Riches, J.D.; Becker, S.; Briggs, J. A. Electron tomography reveals the steps in filovirus budding. PLoS Pathog. 2010, 6, e1000875. [Google Scholar]
- Schmidt, K.M.; Schümann, M.; Olejnik, J.; Krahling, V.; Mühlberger, E. Recombinant Marburg virus expressing EGFP allows rapid screening of virus growth and real-time visualization of virus spread. J. Infect. Dis. 2011, 204, S861–S870. [Google Scholar]
- Kolesnikova, L.; Ryabchikova, E.; Shestopalov, A.; Becker, S. Basolateral budding of Marburg virus: VP40 retargets viral glycoprotein GP to the basolateral surface. J. Infect. Dis. 2007, 196, S232–S236. [Google Scholar]
- Skripchenko, A.A.; Riabchikova, E.I.; Vorontsova, L.A.; Shestopalov, A.M.; Viazunov, S.A. (Marburg virus and mononuclear phagocytes: study of interactions). Vopr. Virusol. 1994, 39, 214–218. [Google Scholar]
- Fritz, E.A.; Geisbert, J.B.; Geisbert, T.W.; Hensley, L.E.; Reed, D.S. Cellular immune response to Marburg virus infection in cynomolgus macaques. Viral Immunol. 2008, 21, 355–363. [Google Scholar]
- Hensley, L.E.; Alves, D.A.; Geisbert, J.B.; Fritz, E.A.; Reed, C.; Larsen, T.; Geisbert, T.W. Pathogenesis of Marburg hemorrhagic fever in cynomolgus macaques. J. Infect. Dis. 2011, 204, S1021–S1031. [Google Scholar]
- Warfield, K.L.; Alves, D.A.; Bradfute, S.B.; Reed, D.K.; VanTongeren, S.; Kalina, W.V.; Olinger, G.G.; Bavari, S. Development of a model for marburgvirus based on severe-combined immunodeficiency mice. Virol. J. 2007, 4, 108. [Google Scholar]
- Ryabchikova, E.; Kolesnikova, L.; Smolina, M.; Tkachev, V.; Pereboeva, L.; Baranova, S.; Grazhdantseva, A.; Rassadkin, Y. Ebola virus infection in guinea pigs: Presumable role of granulomatous inflammation in pathogenesis. Arch. Virol. 1996, 141, 909–921. [Google Scholar]
- Geisbert, T.W.; Jaax, N.K. Marburg hemorrhagic fever: report of a case studied by immunohistochemistry and electron microscopy. Ultrastruct. Pathol. 1998, 22, 3–17. [Google Scholar]
- Feldmann, H.; Bugany, H.; Mahner, F.; Klenk, H.D.; Drenckhahn, D.; Schnittler, H.J. Filovirus-induced endothelial leakage triggered by infected monocytes/macrophages. J. Virol. 1996, 70, 2208–2214. [Google Scholar]
- Bockeler, M.; Stroher, U.; Seebach, J.; Afanasieva, T.; Suttorp, N.; Feldmann, H.; Schnittler, H.J. Breakdown of paraendothelial barrier function during Marburg virus infection is associated with early tyrosine phosphorylation of platelet endothelial cell adhesion molecule-1. J. Infect. Dis. 2007, 196, S337–S346. [Google Scholar]
- Ströher, U.; West, E.; Bugany, H.; Klenk, H.D.; Schnittler, H.J.; Feldmann, H. Infection and activation of monocytes by Marburg and Ebola viruses. J. Virol. 2001, 75, 11025–11033. [Google Scholar]
- Geisbert, T.W.; Hensley, L.E.; Gibb, T.R.; Steele, K.E.; Jaax, N.K.; Jahrling, P.B. Apoptosis induced in vitro and in vivo during infection by Ebola and Marburg viruses. Lab. Invest. 2000, 80, 171–186. [Google Scholar] [CrossRef]
- Gedigk, P.; Bechtelsheimer, H.; Korb, G. The morbid anatomy of Marburg virus disease. Ger. Med. Mon. 1969, 14, 68–77. [Google Scholar]
- Schnittler, H.J.; Feldmann, H. Marburg and Ebola hemorrhagic fevers: does the primary course of infection depend on the accessibility of organ-specific macrophages? Clin. Infect. Dis. 1998, 27, 404–406. [Google Scholar] [CrossRef]
- Schnittler, H.J.; Feldmann, H. Viral hemorrhagic fever-a vascular disease? Thromb. Haemost. 2003, 89, 967–972. [Google Scholar]
- Geisbert, T.W.; Hensley, L.E.; Geisbert, J.B.; Leung, A.; Johnson, J.C.; Grolla, A.; Feldmann, H. Postexposure treatment of Marburg virus infection. Emerg. Infect. Dis. 2010, 16, 1119–1122. [Google Scholar]
- Simpson, D.I. Marburg agent disease: In monkeys. Trans. R. Soc. Trop. Med. Hyg. 1969, 63, 303–309. [Google Scholar]
- Ryabchikova, E.; Strelets, L.; Kolesnikova, L.; Pyankov, O.; Sergeev, A. Respiratory Marburg virus infection in guinea pigs. Arch. Virol. 1996, 141, 2177–2190. [Google Scholar]
- Zlotnik, I. Marburg agent disease: pathology. Trans R Soc Trop Med Hyg 1969, 63(3), 310–327. [Google Scholar] [CrossRef]
- Ignat'ev, G. M.; Strel'tsova, M. A.; Kashentseva, E. A.; Patrushev, N. A. [Effects of tumor necrosis factor antiserum of the course of Marburg hemorrhagic fever]. Vestn Ross Akad Med Nauk 1998, 3, 35–38. [Google Scholar]
- Ignatyev, G.M.; Agafonov, A.P.; Streltsova, M.A.; Kashentseva, E.A. Inactivated Marburg virus elicits a nonprotective immune response in Rhesus monkeys. J Biotechnol 1996, 44(1-3), 111–118. [Google Scholar]
- Ignat'ev, G.M.; Strel'tsova, M.A.; Kashentseva, E.A. Induction of immune mediators in human cultured mononuclear cells by Marburg virus. Vopr. Virusol. 1998, 43, 169–173. [Google Scholar]
- Villinger, F.; Rollin, P.E.; Brar, S.S.; Chikkala, N.F.; Winter, J.; Sundstrom, J.B.; Zaki, S.R.; Swanepoel, R.; Ansari, A.A.; Peters, C.J. Markedly elevated levels of interferon (IFN)-gamma, IFN-alpha, interleukin (IL)-2, IL-10, and tumor necrosis factor-alpha associated with fatal Ebola virus infection. J. Infect. Dis. 1999, 179, S188–S191. [Google Scholar]
- Wauquier, N.; Becquart, P.; Padilla, C.; Baize, S.; Leroy, E.M. Human fatal zaire ebola virus infection is associated with an aberrant innate immunity and with massive lymphocyte apoptosis. PLoS Negl. Trop. Dis. 2010, 4, e837. [Google Scholar]
- Gupta, M.; Mahanty, S.; Ahmed, R.; Rollin, P.E. Monocyte-derived human macrophages and peripheral blood mononuclear cells infected with ebola virus secrete MIP-1alpha and TNF-alpha and inhibit poly-IC-induced IFN-alpha in vitro. Virology 2001, 284, 20–25. [Google Scholar] [CrossRef]
- Gupta, M.; MacNeil, A.; Reed, Z.D.; Rollin, P.E.; Spiropoulou, C.F. Serology and cytokine profiles in patients infected with the newly discovered Bundibugyo ebolavirus. Virology 2012, 423, 119–124. [Google Scholar] [CrossRef]
- Bosio, C.M.; Moore, B.D.; Warfield, K.L.; Ruthel, G.; Mohamadzadeh, M.; Aman, M.J.; Bavari, S. Ebola and Marburg virus-like particles activate human myeloid dendritic cells. Virology 2004, 326, 280–287. [Google Scholar]
- Leung, L.W.; Martinez, O.; Reynard, O.; Volchkov, V.E.; Basler, C.F. Ebola virus failure to stimulate plasmacytoid dendritic cell interferon responses correlates with impaired cellular entry. J. Infect. Dis. 2011, 204, S973–S977. [Google Scholar]
- Simpson, D.I.; Zlotnik, I.; Rutter, D.A. Vervet monkey disease. Experiment infection of guinea pigs and monkeys with the causative agent. Br. J. Exp. Pathol. 1968, 49, 458–464. [Google Scholar]
- Lub, M.; Sergeev, A.N.; P'Iankov, O.V.; P'Iankova, O.G.; Petrishchenko, V.A.; Kotliarov, L.A. Certain pathogenetic characteristics of a disease in monkeys in infected with the Marburg virus by an airborne route. Vopr. Virusol. 1995, 40, 158–161. [Google Scholar]
- Gonchar, N.I.; Pshenichnov, V.A.; Pokhodiaev, V.A.; Lopatov, K.L.; Firsova, I.V. The sensitivity of different experimental animals to the Marburg virus. Vopr. Virusol. 1991, 36, 435–437. [Google Scholar]
- Carrion, R., Jr.; Ro, Y.; Hoosien, K.; Ticer, A.; Brasky, K.; de la Garza, M.; Mansfield, K.; Patterson, J.L. A small nonhuman primate model for filovirus-induced disease. Virology 2011, 420, 117–124. [Google Scholar]
- Lofts, L.L.; Ibrahim, M.S.; Negley, D.L.; Hevey, M.C.; Schmaljohn, A.L. Genomic differences between guinea pig lethal and nonlethal Marburg virus variants. J. Infect. Dis. 2007, 196, S305–S312. [Google Scholar]
- Bente, D.; Gren, J.; Strong, J. E.; Feldmann, H. Disease modeling for Ebola and Marburg viruses. Dis. Model. Mech. 2009, 2, 12–17. [Google Scholar]
- Bechtelsheimer, H.; Jacob, H.; Solcher, H. On the neuropathology of the green monkey (Cercopithecus aethiops) transmitted infectious diseases in Marburg. Dtsch. Med. Wochenschr. 1968, 93, 602–604. [Google Scholar]
- Kissling, R.E.; Murphy, F.A.; Henderson, B.E. Marburg virus. Ann. N. Y. Acad. Sci. 1970, 174, 932–945. [Google Scholar]
- Bray, M. The role of the Type I interferon response in the resistance of mice to filovirus infection. J. Gen. Virol. 2001, 82, 1365–1373. [Google Scholar]
- Lever, M.S.; Piercy, T.J.; Steward, J.A.; Eastaugh, L.; Smither, S.J.; Taylor, C.; Salguero, F.J.; Phillpotts, R.J. Lethality and pathogenesis of airborne infection with filoviruses in A129 alpha/beta -/- interferon receptor-deficient mice. J. Med. Microbiol. 2011, 61, 8–15. [Google Scholar]
- Raymond, J.; Bradfute, S.; Bray, M. Filovirus infection of STAT-1 knockout mice. J. Infect. Dis. 2011, 204, S986–S990. [Google Scholar]
- Saijo, M.; Niikura, M.; Morikawa, S.; Ksiazek, T.G.; Meyer, R.F.; Peters, C.J.; Kurane, I. Enzyme-linked immunosorbent assays for detection of antibodies to Ebola and Marburg viruses using recombinant nucleoproteins. J. Clin. Microbiol. 2001, 39, 1–7. [Google Scholar]
- Saijo, M.; Georges-Courbot, M.C.; Fukushi, S.; Mizutani, T.; Philippe, M.; Georges, A.J.; Kurane, I.; Morikawa, S. Marburgvirus nucleoprotein-capture enzyme-linked immunosorbent assay using monoclonal antibodies to recombinant nucleoprotein: Detection of authentic Marburgvirus. Jpn. J. Infect. Dis. 2006, 59, 323–325. [Google Scholar]
- Sherwood, L.J.; Osborn, L.E.; Carrion, R., Jr.; Patterson, J.L.; Hayhurst, A. Rapid assembly of sensitive antigen-capture assays for Marburg virus, using in vitro selection of llama single-domain antibodies, at biosafety level 4. J. Infect. Dis. 2007, 196, S213–S219. [Google Scholar] [CrossRef]
- Nakayama, E.; Yokoyama, A.; Miyamoto, H.; Igarashi, M.; Kishida, N.; Matsuno, K.; Marzi, A.; Feldmann, H.; Ito, K.; Saijo, M.; et al. Enzyme-linked immunosorbent assay for detection of filovirus species-specific antibodies. Clin. Vaccine Immunol. 2010, 17, 1723–1728. [Google Scholar] [CrossRef]
- Ogawa, H.; Miyamoto, H.; Ebihara, H.; Ito, K.; Morikawa, S.; Feldmann, H.; Takada, A. Detection of all known filovirus species by reverse transcription-polymerase chain reaction using a primer set specific for the viral nucleoprotein gene. J. Virol. Methods. 2010, 171, 310–313. [Google Scholar]
- Drosten, C.; Gottig, S.; Schilling, S.; Asper, M.; Panning, M.; Schmitz, H.; Gunther, S. Rapid detection and quantification of RNA of Ebola and Marburg viruses, Lassa virus, Crimean-Congo hemorrhagic fever virus, Rift Valley fever virus, dengue virus, and yellow fever virus by real-time reverse transcription-PCR. J. Clin. Microbiol. 2002, 40, 2323–2330. [Google Scholar] [CrossRef]
- Gibb, T.R.; Norwood, D.A., Jr.; Woollen, N.; Henchal, E.A. Development and evaluation of a fluorogenic 5'-nuclease assay to identify Marburg virus. Mol. Cell. Probes. 2001, 15, 259–266. [Google Scholar]
- Trombley, A.R.; Wachter, L.; Garrison, J.; Buckley-Beason, V.A.; Jahrling, J.; Hensley, L.E.; Schoepp, R.J.; Norwood, D.A.; Goba, A.; Fair, J.N.; Kulesh, D.A. Comprehensive panel of real-time TaqMan polymerase chain reaction assays for detection and absolute quantification of filoviruses, arenaviruses, and New World hantaviruses. Am. J. Trop. Med. Hyg. 2010, 82, 954–960. [Google Scholar] [CrossRef]
- Weidmann, M.; Mühlberger, E.; Hufert, F.T. Rapid detection protocol for filoviruses. J. Clin. Virol. 2004, 30, 94–99. [Google Scholar]
- Weidmann, M.; Hufert, F.T.; Sall, A.A. Viral load among patients infected with Marburgvirus in Angola. J. Clin. Virol. 2007, 39, 65–66. [Google Scholar]
- Towner, J.S.; Sealy, T.K.; Ksiazek, T.G.; Nichol, S.T. High-throughput molecular detection of hemorrhagic fever virus threats with applications for outbreak settings. J. Infect. Dis. 2007, 196, S205–S212. [Google Scholar]
- Panning, M.; Laue, T.; Olschlager, S.; Eickmann, M.; Becker, S.; Raith, S.; Courbot, M.C.; Nilsson, M.; Gopal, R.; Lundkvist, A.; et al. Diagnostic reverse-transcription polymerase chain reaction kit for filoviruses based on the strain collections of all European biosafety level 4 laboratories. J. Infect. Dis. 2007, 196, S199–S204. [Google Scholar] [CrossRef]
- Kurosaki, Y.; Grolla, A.; Fukuma, A.; Feldmann, H.; Yasuda, J. Development and evaluation of a simple assay for Marburg virus detection using a reverse transcription-loop-mediated isothermal amplification method. J. Clin. Microbiol. 2010, 48, 2330–2336. [Google Scholar] [CrossRef]
- Hevey, M.; Negley, D.; Geisbert, J.; Jahrling, P.; Schmaljohn, A. Antigenicity and vaccine potential of Marburg virus glycoprotein expressed by baculovirus recombinants. Virology 1997, 239, 206–216. [Google Scholar]
- Hevey, M.; Negley, D.; VanderZanden, L.; Tammariello, R.F.; Geisbert, J.; Schmaljohn, C.; Smith, J.F.; Jahrling, P.B.; Schmaljohn, A.L. Marburg virus vaccines: comparing classical and new approaches. Vaccine 2001, 20, 586–593. [Google Scholar]
- Warfield, K.L.; Swenson, D.L.; Negley, D.L.; Schmaljohn, A.L.; Aman, M.J.; Bavari, S. Marburg virus-like particles protect guinea pigs from lethal Marburg virus infection. Vaccine 2004, 22, 3495–3502. [Google Scholar]
- Geisbert, T.W.; Bausch, D.G.; Feldmann, H. Prospects for immunisation against Marburg and Ebola viruses. Rev. Med. Viro. l 2010, 20, 344–357. [Google Scholar]
- Grant-Klein, R.J.; Altamura, L.A.; Schmaljohn, C.S. Progress in recombinant DNA-derived vaccines for Lassa virus and filoviruses. Virus Res. 2011, 162, 148–161. [Google Scholar]
- Riemenschneider, J.; Garrison, A.; Geisbert, J.; Jahrling, P.; Hevey, M.; Negley, D.; Schmaljohn, A.; Lee, J.; Hart, M.K.; Vanderzanden, L.; et al. Comparison of individual and combination DNA vaccines for B. anthracis, Ebola virus, Marburg virus and Venezuelan equine encephalitis virus. Vaccine 2003, 21, 4071–4080. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Bailey, M.; Geisbert, J.B.; Asiedu, C.; Roederer, M.; Grazia-Pau, M.; Custers, J.; Jahrling, P.; Goudsmit, J.; Koup, R.; Sullivan, N.J. Vector choice determines immunogenicity and potency of genetic vaccines against Angola Marburg virus in nonhuman primates. J. Virol. 2010, 84, 10386–10394. [Google Scholar]
- Grant-Klein, R.J.; Van Deusen, N.M.; Badger, C.V.; Hannaman, D.; Dupuy, L.C.; Schmaljohn, C.S. A multiagent filovirus DNA vaccine delivered by intramuscular electroporation completely protects mice from ebola and Marburg virus challenge. Hum. Vaccin. Immunother. 2012, 8. in press.. [Google Scholar]
- Hevey, M.; Negley, D.; Pushko, P.; Smith, J.; Schmaljohn, A. Marburg virus vaccines based upon alphavirus replicons protect guinea pigs and nonhuman primates. Virology 1998, 251, 28–37. [Google Scholar]
- Warfield, K.L.; Aman, M.J. Advances in virus-like particle vaccines for filoviruses. J. Infect. Dis. 2011, 204, S1053–S1059. [Google Scholar]
- Swenson, D.L.; Warfield, K.L.; Negley, D.L.; Schmaljohn, A.; Aman, M.J.; Bavari, S. Virus-like particles exhibit potential as a pan-filovirus vaccine for both Ebola and Marburg viral infections. Vaccine 2005, 23, 3033–3042. [Google Scholar]
- Swenson, D.L.; Warfield, K.L.; Larsen, T.; Alves, D.A.; Coberley, S.S.; Bavari, S. Monovalent virus-like particle vaccine protects guinea pigs and nonhuman primates against infection with multiple Marburg viruses. Expert Rev. Vaccines 2008, 7, 417–429. [Google Scholar]
- Wang, D.; Schmaljohn, A.L.; Raja, N.U.; Trubey, C.M.; Juompan, L.Y.; Luo, M.; Deitz, S.B.; Yu, H.; Woraratanadharm, J.; Holman, D.H.; et al. De novo syntheses of Marburg virus antigens from adenovirus vectors induce potent humoral and cellular immune responses. Vaccine 2006, 24, 2975–2986. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Feldmann, H. Recombinant vesicular stomatitis virus-based vaccines against Ebola and Marburg virus infections. J. Infect. Dis. 2011, 204, S1075–S1081. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Daddario-Dicaprio, K.M.; Geisbert, J.B.; Reed, D.S.; Feldmann, F.; Grolla, A.; Stroher, U.; Fritz, E.A.; Hensley, L.E.; Jones, S.M.; Feldmann, H. Vesicular stomatitis virus-based vaccines protect nonhuman primates against aerosol challenge with Ebola and Marburg viruses. Vaccine 2008, 26, 6894–6900. [Google Scholar]
- Jones, S.M.; Feldmann, H.; Stroher, U.; Geisbert, J.B.; Fernando, L.; Grolla, A.; Klenk, H.D.; Sullivan, N.J.; Volchkov, V. E.; Fritz, E.A.; et al. Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat. Med. 2005, 11, 786–790. [Google Scholar]
- Jones, S.M.; Stroher, U.; Fernando, L.; Qiu, X.; Alimonti, J.; Melito, P.; Bray, M.; Klenk, H.D.; Feldmann, H. Assessment of a vesicular stomatitis virus-based vaccine by use of the mouse model of Ebola virus hemorrhagic fever. J. Infect. Dis. 2007, 196, S404–S412. [Google Scholar]
- Geisbert, T.W.; Daddario-Dicaprio, K.M.; Lewis, M.G.; Geisbert, J.B.; Grolla, A.; Leung, A.; Paragas, J.; Matthias, L.; Smith, M.A.; Jones, S.M.; et al. Vesicular stomatitis virus-based ebola vaccine is well-tolerated and protects immunocompromised nonhuman primates. PLoS Pathog. 2008, 4, e1000225. [Google Scholar] [CrossRef]
- Mire, C.E.; Miller, A.D.; Carville, A.; Westmoreland, S.V.; Geisbert, J.B.; Mansfield, K.G.; Feldmann, H.; Hensley, L.E.; Geisbert, T.W. Recombinant vesicular stomatitis virus vaccine vectors expressing filovirus glycoproteins lack neurovirulence in nonhuman primates. PLoS Negl. Trop. Dis. 2012, 6, e1567. [Google Scholar]
- Geisbert, T.W.; Geisbert, J.B.; Leung, A.; Daddario-DiCaprio, K.M.; Hensley, L.E.; Grolla, A.; Feldmann, H. Single-injection vaccine protects nonhuman primates against infection with marburg virus and three species of ebola virus. J. Virol. 2009, 83, 7296–7304. [Google Scholar]
- Swenson, D.L.; Wang, D.; Luo, M.; Warfield, K.L.; Woraratanadharm, J.; Holman, D.H.; Dong, J.Y.; Pratt, W.D. Vaccine to confer to nonhuman primates complete protection against multistrain Ebola and Marburg virus infections. Clin. Vaccine Immunol. 2008, 15, 460–467. [Google Scholar]
- Geisbert, T.W.; Hensley, L.E.; Jahrling, P.B.; Larsen, T.; Geisbert, J.B.; Paragas, J.; Young, H. A.; Fredeking, T.M.; Rote, W.E.; et al. Treatment of Ebola virus infection with a recombinant inhibitor of factor VIIa/tissue factor: a study in rhesus monkeys. Lancet 2003, 362, 1953–1958. [Google Scholar]
- Warren, T.K.; Warfield, K.L.; Wells, J.; Swenson, D.L.; Donner, K.S.; Van Tongeren, S.A.; Garza, N.L.; Dong, L.; Mourich, D.V.; Crumley, S.; et al. Advanced antisense therapies for postexposure protection against lethal filovirus infections. Nat. Med. 2010, 16, 991–994. [Google Scholar]
- Warren, T.K.; Warfield, K.L.; Wells, J.; Enterlein, S.; Smith, M.; Ruthel, G.; Yunus, A.S.; Kinch, M.S.; Goldblatt, M.; Aman, M.J.; Bavari, S. Antiviral activity of a small-molecule inhibitor of filovirus infection. Antimicrob. Agents Chemother. 2010, 54, 2152–2159. [Google Scholar]
- Daddario-DiCaprio, K.M.; Geisbert, T.W.; Stroher, U.; Geisbert, J.B.; Grolla, A.; Fritz, E.A.; Fernando, L.; Kagan, E.; Jahrling, P.B.; Hensley, L.E.; et al. Postexposure protection against Marburg haemorrhagic fever with recombinant vesicular stomatitis virus vectors in non-human primates: an efficacy assessment. Lancet 2006, 367, 1399–1404. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Brauburger, K.; Hume, A.J.; Mühlberger, E.; Olejnik, J. Forty-Five Years of Marburg Virus Research. Viruses 2012, 4, 1878-1927. https://doi.org/10.3390/v4101878
Brauburger K, Hume AJ, Mühlberger E, Olejnik J. Forty-Five Years of Marburg Virus Research. Viruses. 2012; 4(10):1878-1927. https://doi.org/10.3390/v4101878
Chicago/Turabian StyleBrauburger, Kristina, Adam J. Hume, Elke Mühlberger, and Judith Olejnik. 2012. "Forty-Five Years of Marburg Virus Research" Viruses 4, no. 10: 1878-1927. https://doi.org/10.3390/v4101878
APA StyleBrauburger, K., Hume, A. J., Mühlberger, E., & Olejnik, J. (2012). Forty-Five Years of Marburg Virus Research. Viruses, 4(10), 1878-1927. https://doi.org/10.3390/v4101878