Inhibition of Geranylgeranyl Transferase-I Decreases Cell Viability of HTLV-1-Transformed Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
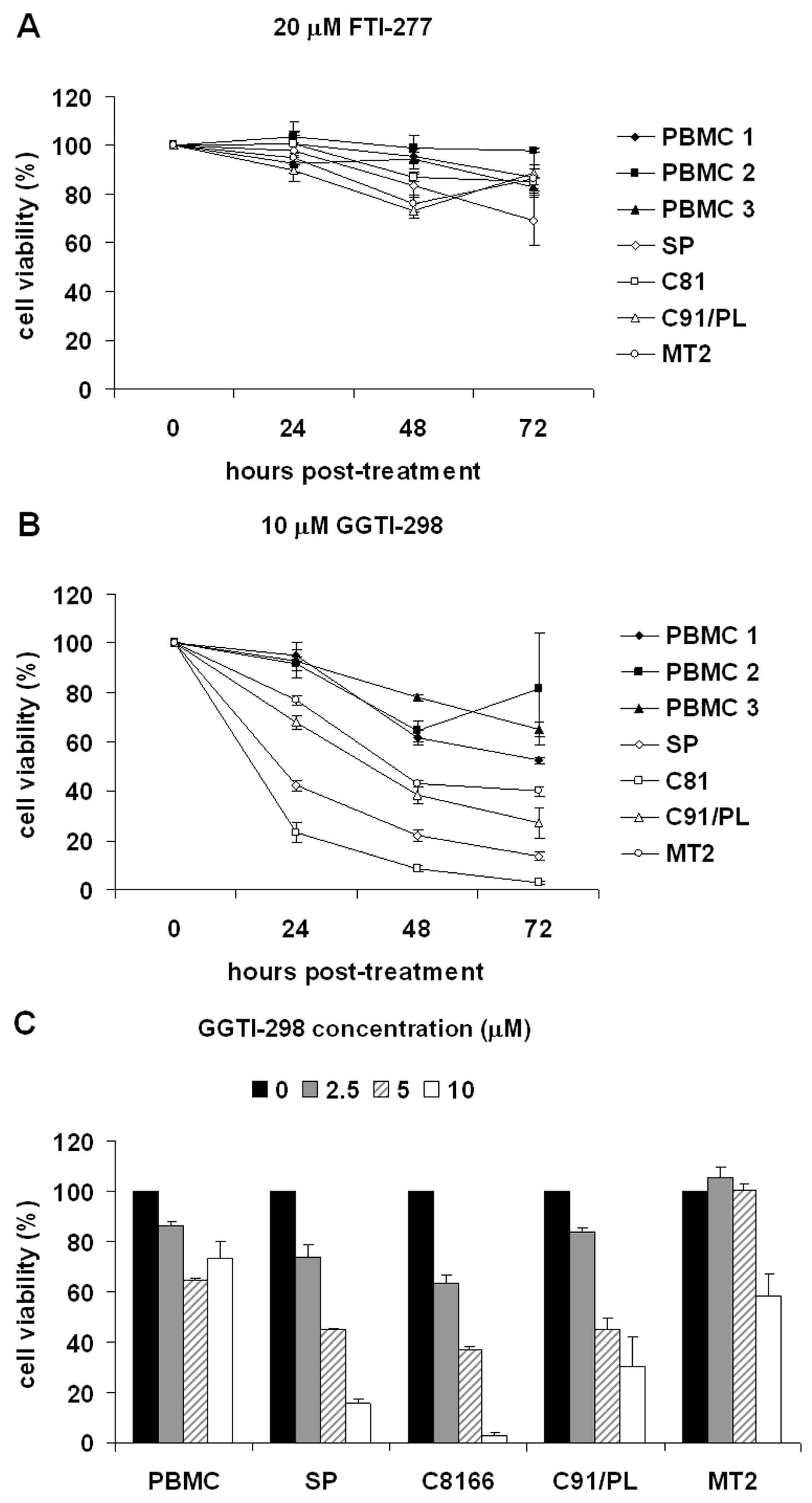
2.1. GGTI-298 Decreases the Viability of HTLV-1-Transformed Cells
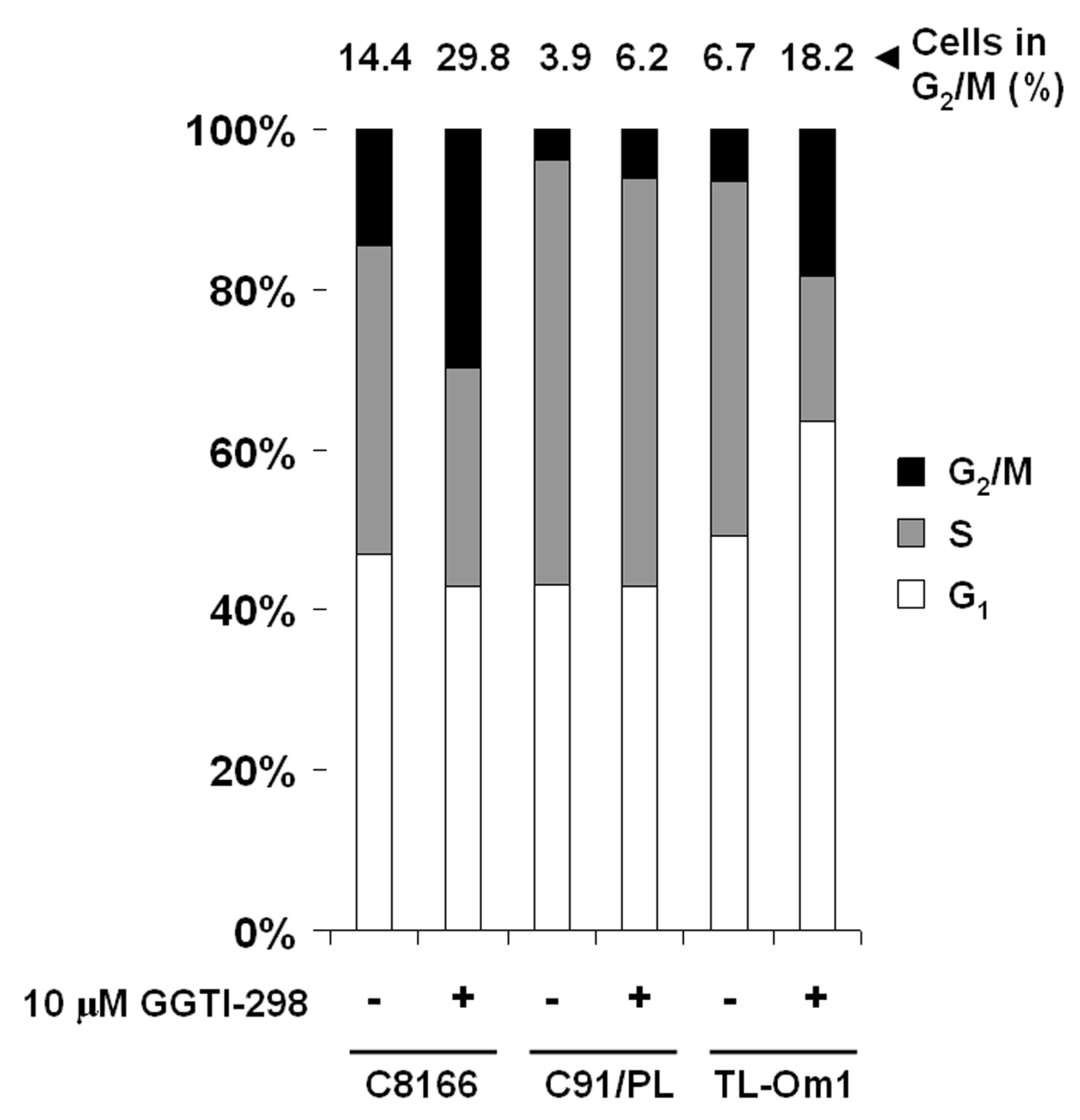
2.2. GGTI-298 Induces G2/M Phase Accumulation in HTLV-1-Transformed Cells
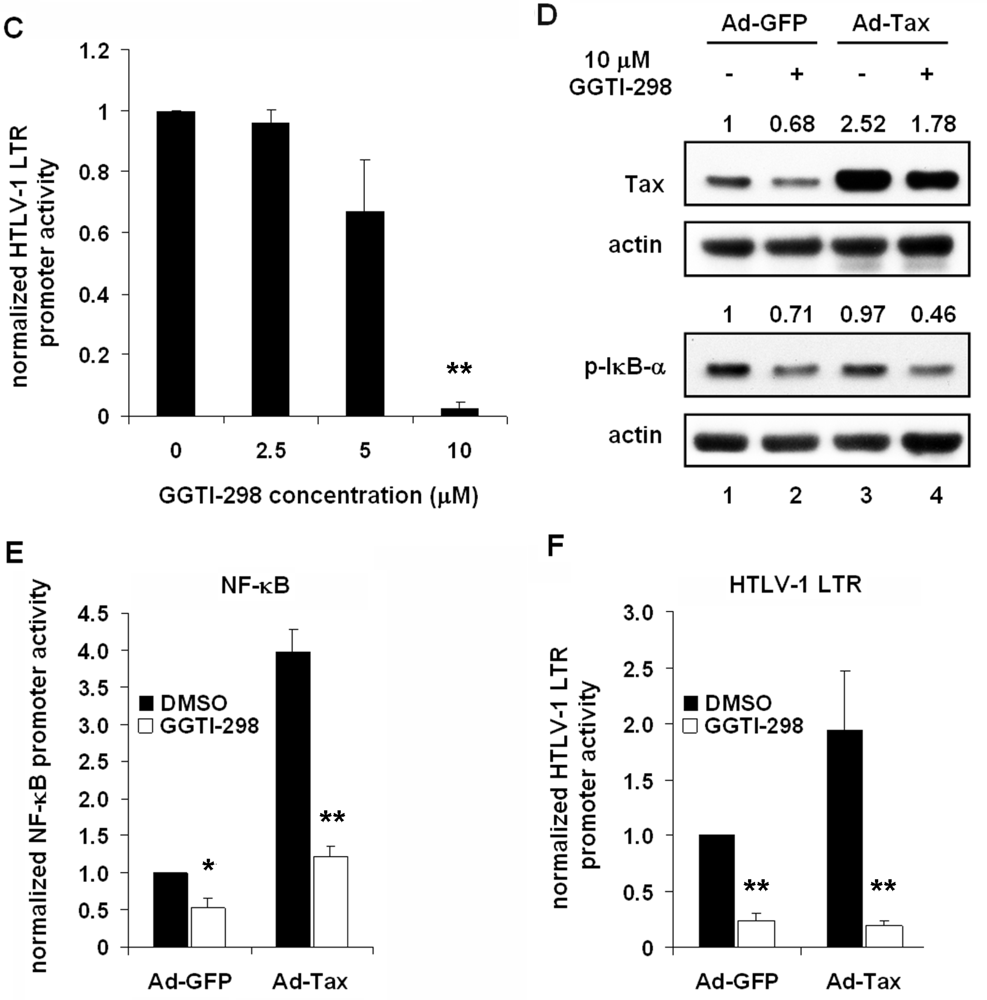
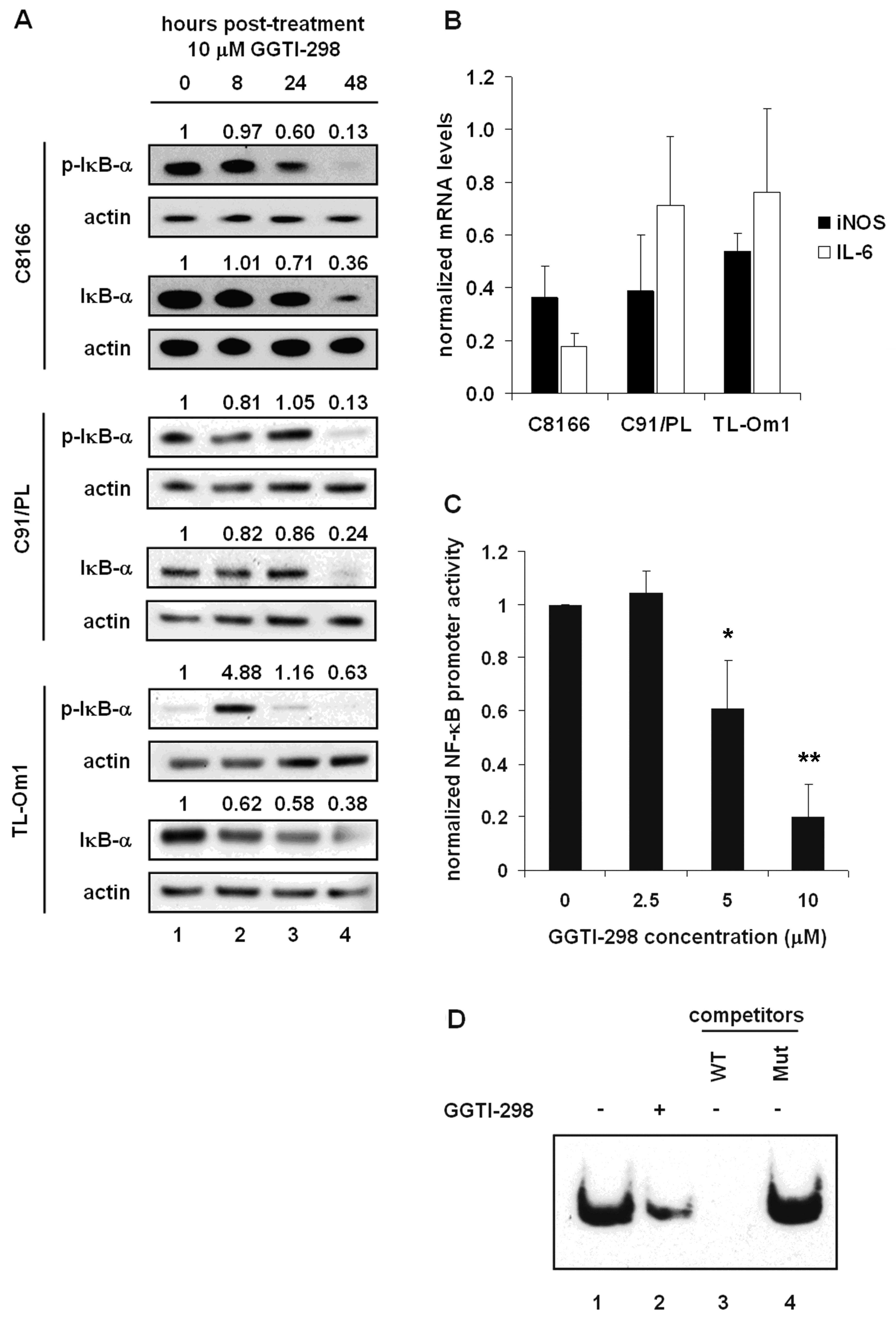
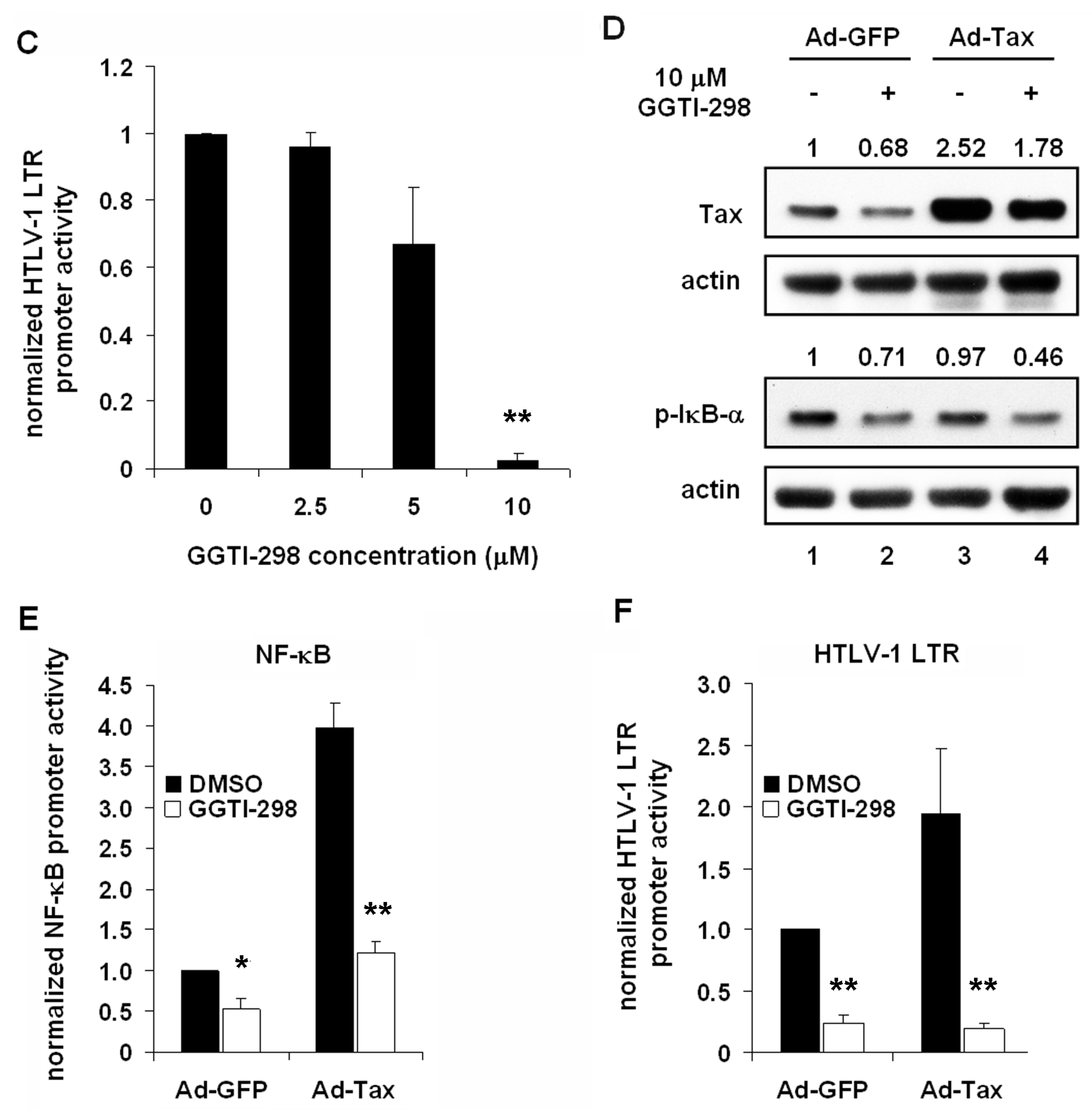
2.3. GGTI-298 Inactivates the NF-κB Pathway
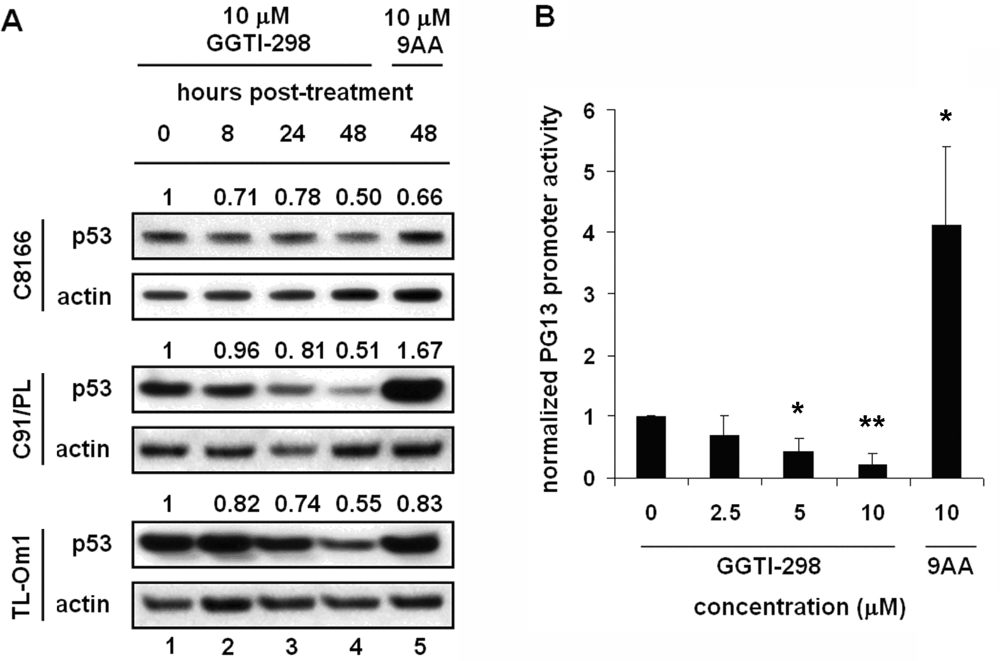
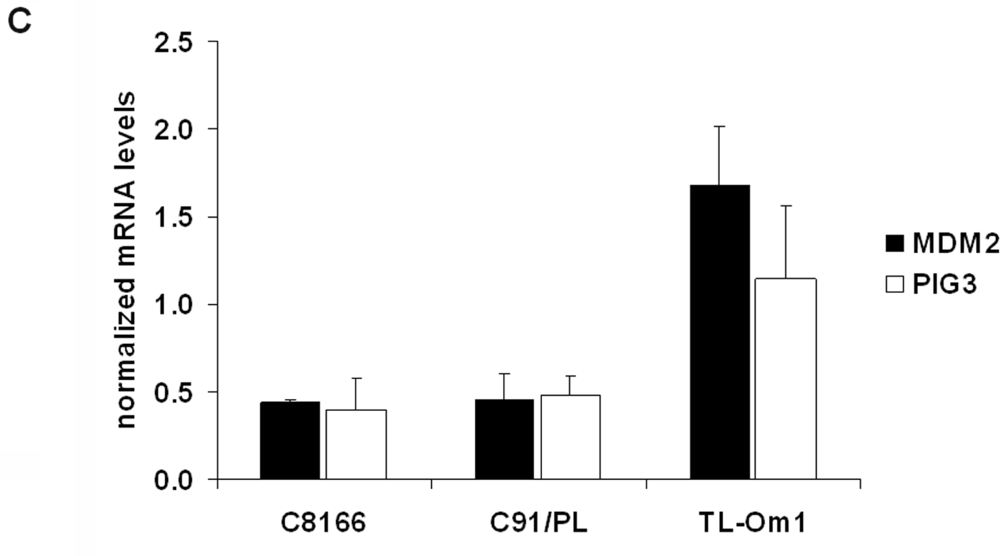
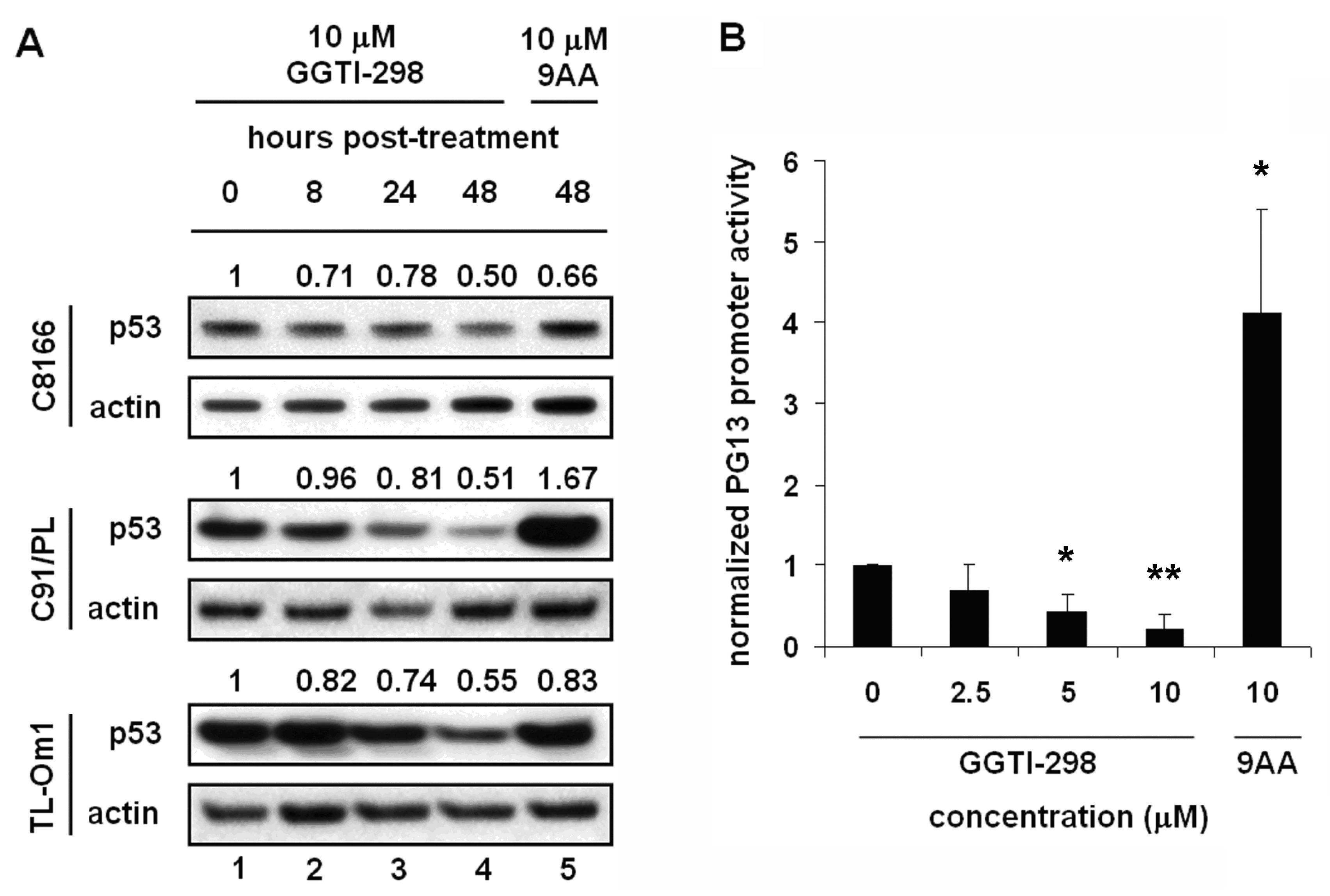
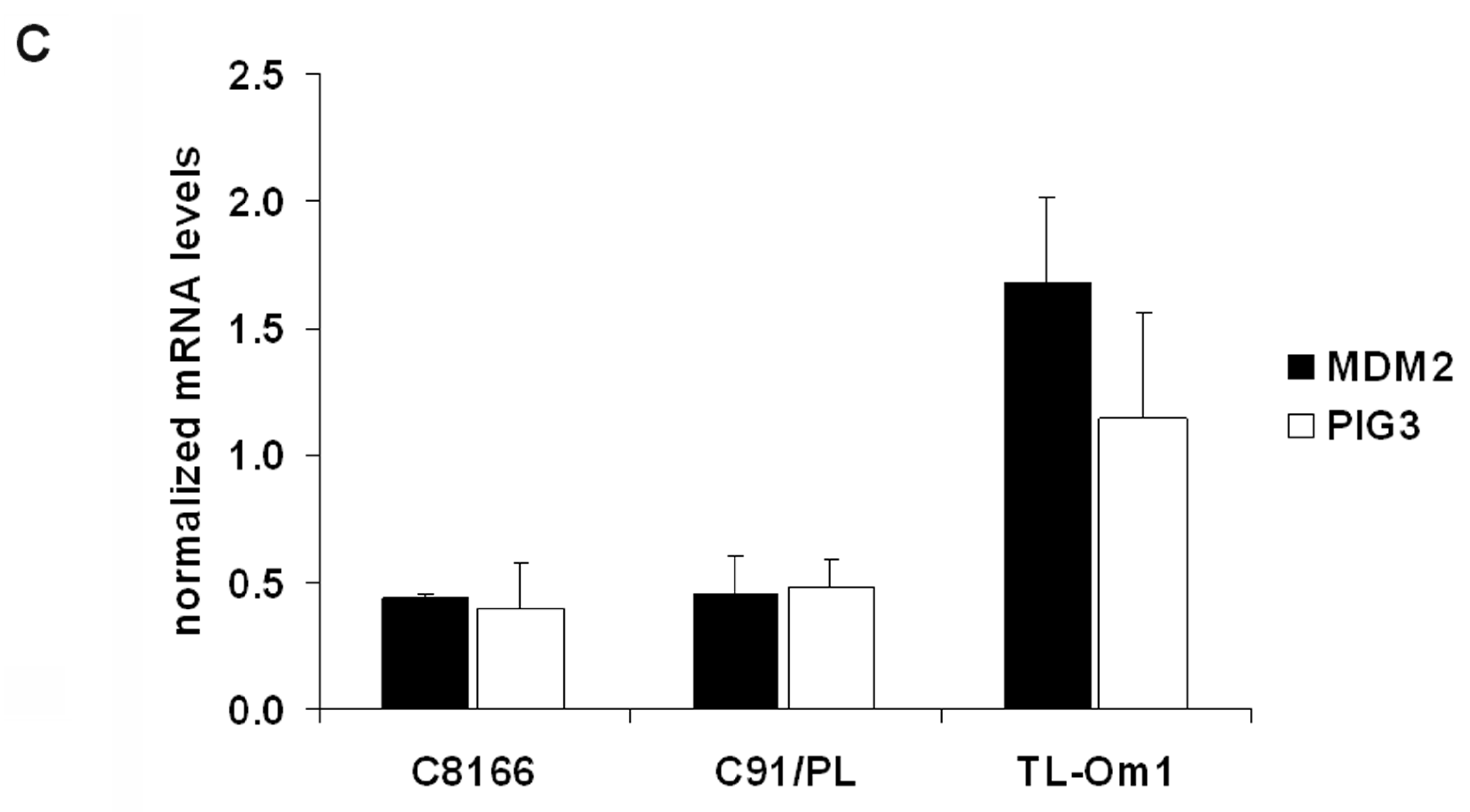
2.4. The Effects of GGTI-298 on HTLV-1-Transformed Cells Are p53-Independent
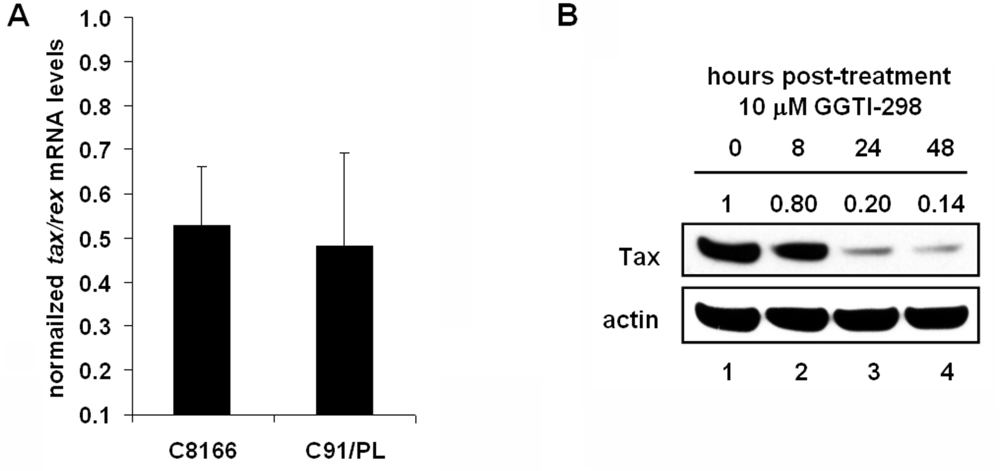
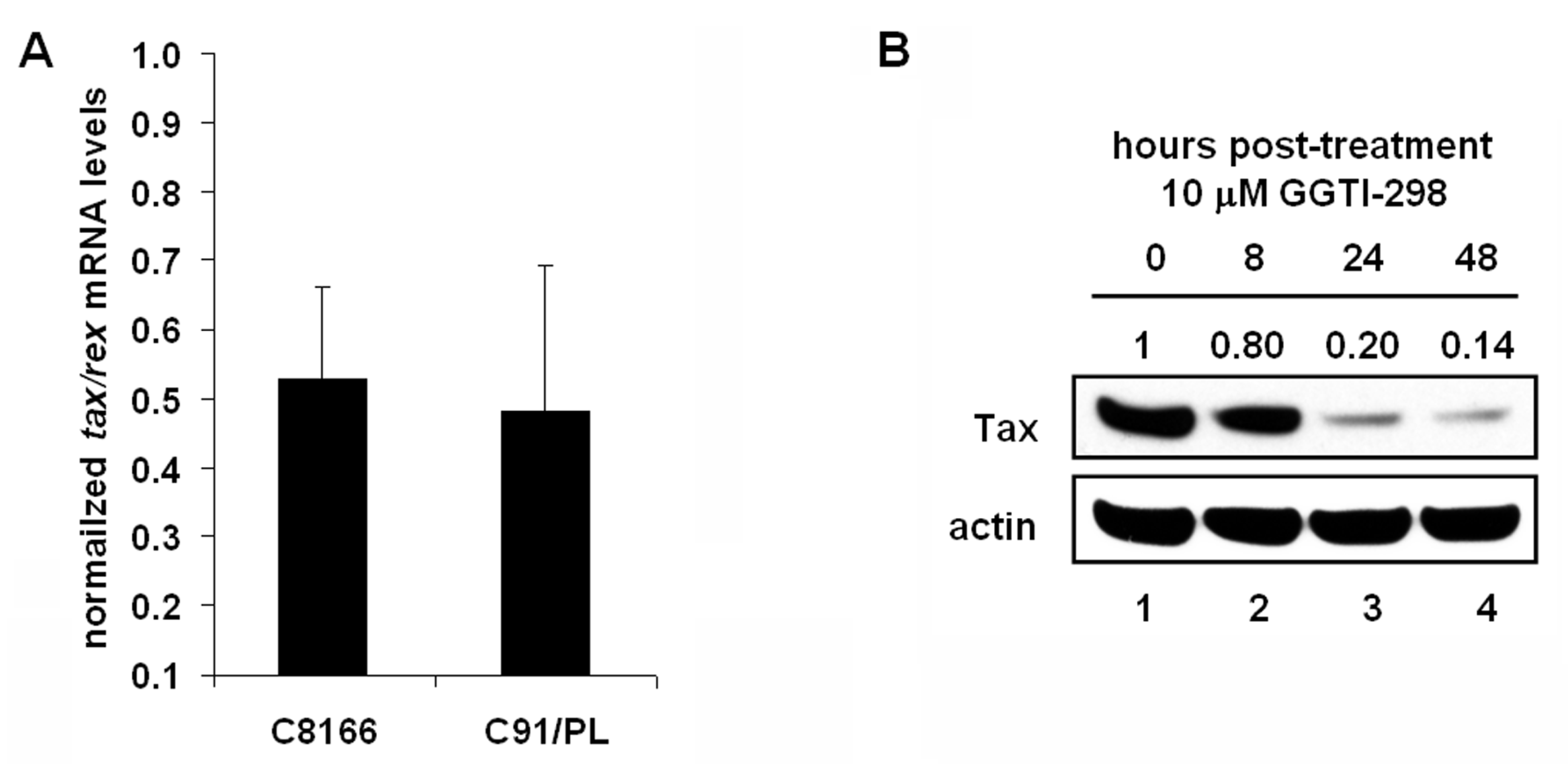
2.5. GGTI-298 Inhibits Transcription of the HTLV-1-LTR and Decreases Tax Expression
2.6. Discussion
3. Experimental Section
3.1. Preparation of PBMCs
3.2. Cell Lines and Drug Treatment
3.3. Whole Cell and Nuclear Extracts Preparation
3.4. Immunoblot Analysis
3.5. Antibodies
3.6. Cell Viability Assays
3.7. Flow Cytometry Analysis of Cell Cycle Progression
3.8. Transfection and Luciferase Assays
3.9. Real-Time PCR
3.10. Electrophoretic Mobility Shift Assays (EMSAs)
3.11. Adenovirus Infection of Cells
3.12. Statistical Analysis
4. Conclusions
Acknowledgements
References and Notes
- Edlich, R.F.; Arnette, J.A.; Williams, F.M. Global epidemic of human T-cell lymphotropic virus type-I (HTLV-I). J. Emerg. Med. 2000, 18, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Poiesz, B.J.; Ruscetti, F.W.; Gazdar, A.F.; Bunn, P.A.; Minna, J.D.; Gallo, R.C. Detection and isolation of type c retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. U. S. A. 1980, 77, 7415–7419. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Miyoshi, I.; Hinuma, Y. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc. Natl. Acad. Sci. U. S. A. 1982, 79, 2031–2035. [Google Scholar] [CrossRef]
- Gessain, A.; Barin, F.; Vernant, J.C.; Gout, O.; Maurs, L.; Calender, A.; de The, G. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet 1985, 2, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, F.; Kajihara, H.; Nakamura, M.; Sasaki, H.; Kumamoto, T.; Okada, K. HTLV-1 associated myelopathy in an HTLV-1 and HBV double carrier family: Report of a case and the mode of vertical transmission of both viruses. J. Gastroenterol. Hepatol. 1989, 4, 387–390. [Google Scholar] [CrossRef]
- Osame, M.; Usuku, K.; Izumo, S.; Ijichi, N.; Amitani, H.; Igata, A.; Matsumoto, M.; Tara, M. HTLV-I associated myelopathy, a new clinical entity. Lancet 1986, 1, 1031–1032. [Google Scholar] [CrossRef]
- Murphy, E.L.; Hanchard, B.; Figueroa, J.P.; Gibbs, W.N.; Lofters, W.S.; Campbell, M.; Goedert, J.J.; Blattner, W.A. Modelling the risk of adult T-cell leukemia/lymphoma in persons infected with human t-lymphotropic virus type I. Int. J. Cancer 1989, 43, 250–253. [Google Scholar] [CrossRef]
- Marriott, S.J.; Semmes, O.J. Impact of HTLV-I tax on cell cycle progression and the cellular DNA damage repair response. Oncogene 2005, 24, 5986–5995. [Google Scholar] [CrossRef]
- Grassmann, R.; Aboud, M.; Jeang, K.T. Molecular mechanisms of cellular transformation by HTLV-1 tax. Oncogene 2005, 24, 5976–5985. [Google Scholar] [CrossRef]
- Jeong, S.J.; Pise-Masison, C.A.; Radonovich, M.F.; Park, H.U.; Brady, J.N. Activated AKT regulates NF-kappaB activation, p53 inhibition and cell survival in HTLV-1-transformed cells. Oncogene 2005, 24, 6719–6728. [Google Scholar] [CrossRef]
- Tabakin-Fix, Y.; Azran, I.; Schavinky-Khrapunsky, Y.; Levy, O.; Aboud, M. Functional inactivation of p53 by human T-cell leukemia virus type 1 tax protein: Mechanisms and clinical implications. Carcinogenesis 2006, 27, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M.; Nicot, C. HTLV-1 and apoptosis: Role in cellular transformation and recent advances in therapeutic approaches. Apoptosis 2008, 13, 733–747. [Google Scholar] [CrossRef]
- Dasgupta, A.; Jung, K.J.; Jeong, S.J.; Brady, J.N. Inhibition of methyltransferases results in induction of G2/M checkpoint and programmed cell death in human T-lymphotropic virus type 1-transformed cells. J. Virol. 2008, 82, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.J.; Dasgupta, A.; Huang, K.; Jeong, S.J.; Pise-Masison, C.; Gurova, K.V.; Brady, J.N. Small-molecule inhibitor which reactivates p53 in human T-cell leukemia virus type 1-transformed cells. J. Virol. 2008, 82, 8537–8547. [Google Scholar] [CrossRef]
- Vogt, A.; Sun, J.; Qian, Y.; Hamilton, A.D.; Sebti, S.M. The geranylgeranyltransferase-I inhibitor GGTI-298 arrests human tumor cells in G0/G1 and induces p21(WAF1/CIP1/SDI1) in a p53-independent manner. J. Biol. Chem. 1997, 272, 27224–27229. [Google Scholar] [CrossRef] [PubMed]
- Vitale, M.; Di Matola, T.; Rossi, G.; Laezza, C.; Fenzi, G.; Bifulco, M. Prenyltransferase inhibitors induce apoptosis in proliferating thyroid cells through a p53-independent CrmA-sensitive, and caspase-3-like protease-dependent mechanism. Endocrinology 1999, 140, 698–704. [Google Scholar] [CrossRef]
- Dan, H.C.; Jiang, K.; Coppola, D.; Hamilton, A.; Nicosia, S.V.; Sebti, S.M.; Cheng, J.Q. Phosphatidylinositol-3-OH kinase/AKT and survivin pathways as critical targets for geranylgeranyltransferase I inhibitor-induced apoptosis. Oncogene 2004, 23, 706–715. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Karamouzis, M.V.; Papavassiliou, A.G. Post-translational modifications and regulation of the Ras superfamily of GTPases as anticancer targets. Nat. Rev. Drug Discov. 2007, 6, 541–555. [Google Scholar] [CrossRef]
- Khwaja, A.; Sharpe, C.C.; Noor, M.; Hendry, B.M. The role of geranylgeranylated proteins in human mesangial cell proliferation. Kidney Int. 2006, 70, 1296–1304. [Google Scholar] [CrossRef]
- Forsberg, M.; Blomgran, R.; Lerm, M.; Sarndahl, E.; Sebti, S.M.; Hamilton, A.; Stendahl, O.; Zheng, L. Differential effects of invasion by and phagocytosis of salmonella typhimurium on apoptosis in human macrophages: Potential role of Rho-GTPases and Akt. J. Leuk. Biol. 2003, 74, 620–629. [Google Scholar] [CrossRef]
- Ramirez, S.; Ait-Si-Ali, S.; Robin, P.; Trouche, D.; Harel-Bellan, A. The CREB-binding protein (CBP) cooperates with the serum response factor for transactivation of the c-fos serum response element. J. Biol. Chem. 1997, 272, 31016–31021. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S.; Wynne, J.; Treisman, R. The Rho family GTPases RhoA, Rac1, and Cdc42Hs regulate transcriptional activation by SRF. Cell 1995, 81, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Minden, A.; Lin, A.; Claret, F.X.; Abo, A.; Karin, M. Selective activation of the JNK signaling cascade and c-jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell 1995, 81, 1147–1157. [Google Scholar] [CrossRef]
- Perona, R.; Montaner, S.; Saniger, L.; Sanchez-Perez, I.; Bravo, R.; Lacal, J.C. Activation of the nuclear factor-kappaB by Rho, Cdc42, and Rac-1 proteins. Genes Dev. 1997, 11, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Kashanchi, F.; Brady, J.N. Transcriptional and post-transcriptional gene regulation of HTLV-1. Oncogene 2005, 24, 5938–5951. [Google Scholar] [CrossRef]
- Casey, P.J.; Seabra, M.C. Protein prenyltransferases. J. Biol. Chem. 1996, 271, 5289–5292. [Google Scholar] [CrossRef]
- Stark, W.W., Jr.; Blaskovich, M.A.; Johnson, B.A.; Qian, Y.; Vasudevan, A.; Pitt, B.; Hamilton, A.D.; Sebti, S.M.; Davies, P. Inhibiting geranylgeranylation blocks growth and promotes apoptosis in pulmonary vascular smooth muscle cells. Am. J. Physiol. 1998, 275, L55–L63. [Google Scholar] [CrossRef]
- Nonaka, M.; Uota, S.; Saitoh, Y.; Takahashi, M.; Sugimoto, H.; Amet, T.; Arai, A.; Miura, O.; Yamamoto, N.; Yamaoka, S. Role for protein geranylgeranylation in adult T-cell leukemia cell survival. Exp. Cell Res. 2009, 315, 141–150. [Google Scholar] [CrossRef]
- Miquel, K.; Pradines, A.; Sun, J.; Qian, Y.; Hamilton, A.D.; Sebti, S.M.; Favre, G. GGTI-298 induces G0-G1 block and apoptosis whereas FTI-277 causes G2-M enrichment in A549 cells. Cancer Res. 1997, 57, 1846–1850. [Google Scholar]
- Sun, J.; Qian, Y.; Chen, Z.; Marfurt, J.; Hamilton, A.D.; Sebti, S.M. The geranylgeranyltransferase I inhibitor GGTI-298 induces hypophosphorylation of retinoblastoma and partner switching of cyclin-dependent kinase inhibitors. A potential mechanism for GGTI-298 antitumor activity. J. Biol. Chem. 1999, 274, 6930–6934. [Google Scholar] [CrossRef]
- Li, X.; Liu, L.; Tupper, J.C.; Bannerman, D.D.; Winn, R.K.; Sebti, S.M.; Hamilton, A.D.; Harlan, J.M. Inhibition of protein geranylgeranylation and RhoA/RhoA kinase pathway induces apoptosis in human endothelial cells. J. Biol. Chem. 2002, 277, 15309–15316. [Google Scholar] [CrossRef]
- van de Donk, N.W.; Schotte, D.; Kamphuis, M.M.; van Marion, A.M.; van Kessel, B.; Bloem, A.C.; Lokhorst, H.M. Protein geranylgeranylation is critical for the regulation of survival and proliferation of lymphoma tumor cells. Clin. Cancer Res. 2003, 9, 5735–5748. [Google Scholar]
- van de Donk, N.W.; Kamphuis, M.M.; van Kessel, B.; Lokhorst, H.M.; Bloem, A.C. Inhibition of protein geranylgeranylation induces apoptosis in myeloma plasma cells by reducing Mcl-1 protein levels. Blood 2003, 102, 3354–3362. [Google Scholar] [CrossRef]
- Ageberg, M.; Rydstrom, K.; Linden, O.; Linderoth, J.; Jerkeman, M.; Drott, K. Inhibition of geranylgeranylation mediates sensitivity to CHOP-induced cell death of DLBCL cell lines. Exp. Cell Res. 2011, 317, 1179–1191. [Google Scholar] [CrossRef]
- Liu, L.; Moesner, P.; Kovach, N.L.; Bailey, R.; Hamilton, A.D.; Sebti, S.M.; Harlan, J.M. Integrin-dependent leukocyte adhesion involves geranylgeranylated protein(s). J. Biol. Chem. 1999, 274, 33334–33340. [Google Scholar] [CrossRef] [PubMed]
- Efuet, E.T.; Keyomarsi, K. Farnesyl and geranylgeranyl transferase inhibitors induce G1 arrest by targeting the proteasome. Cancer Res. 2006, 66, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- Allal, C.; Pradines, A.; Hamilton, A.D.; Sebti, S.M.; Favre, G. Farnesylated Rhob prevents cell cycle arrest and actin cytoskeleton disruption caused by the geranylgeranyltransferase I inhibitor GGTI-298. Cell Cycle 2002, 1, 430–437. [Google Scholar] [CrossRef]
- Nomura, M.; Ohashi, T.; Nishikawa, K.; Nishitsuji, H.; Kurihara, K.; Hasegawa, A.; Furuta, R.A.; Fujisawa, J.; Tanaka, Y.; Hanabuchi, S.; Harashima, N.; Masuda, T.; Kannagi, M. Repression of tax expression is associated both with resistance of human T-cell leukemia virus type 1-infected T cells to killing by tax-specific cytotoxic T lymphocytes and with impaired tumorigenicity in a rat model. J. Virol. 2004, 78, 3827–3836. [Google Scholar] [CrossRef] [PubMed]
- Aznar, S.; Fernandez-Valeron, P.; Espina, C.; Lacal, J.C. Rho GTPases: Potential candidates for anticancer therapy. Cancer Lett. 2004, 206, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Bottazzi, M.E.; de la Fuente, C.; Deng, L.; Gitlin, S.D.; Maddukuri, A.; Dadgar, S.; Li, H.; Vertes, A.; Pumfery, A.; Kashanchi, F. Protein profile of tax-associated complexes. J. Biol. Chem. 2004, 279, 495–508. [Google Scholar] [CrossRef]
- Klener, P.; Szynal, M.; Cleuter, Y.; Merimi, M.; Duvillier, H.; Lallemand, F.; Bagnis, C.; Griebel, P.; Sotiriou, C.; Burny, A.; Martiat, P.; Van den Broeke, A. Insights into gene expression changes impacting B-cell transformation: Cross-species microarray analysis of bovine leukemia virus tax-responsive genes in ovine B cells. J. Virol. 2006, 80, 1922–1938. [Google Scholar] [CrossRef]
- Ishikawa, C.; Matsuda, T.; Okudaira, T.; Tomita, M.; Kawakami, H.; Tanaka, Y.; Masuda, M.; Ohshiro, K.; Ohta, T.; Mori, N. Bisphosphonate incadronate inhibits growth of human T-cell leukaemia virus type I-infected T-cell lines and primary adult T-cell leukaemia cells by interfering with the mevalonate pathway. Br. J. Haematol. 2007, 136, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, T.; Ishikawa, T.; Imura, A. Adhesion properties of adult T cell leukemia cells. Leuk. Lymphoma 1995, 16, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Mine, S.; Figdor, C.G.; Wake, A.; Hirano, H.; Tsukada, J.; Aso, M.; Fujii, K.; Saito, K.; van Kooyk, Y.; Eto, S. Constitutive chemokine production results in activation of leukocyte function-associated antigen-1 on adult T-cell leukemia cells. Blood 1998, 91, 3909–3919. [Google Scholar] [CrossRef] [PubMed]
- Infante, E.; Heasman, S.J.; Ridley, A.J. Statins inhibit T-acute lymphoblastic leukemia cell adhesion and migration through Rap1b. J. Leuk. Biol. 2011, 89, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Van Prooyen, N.; Gold, H.; Andresen, V.; Schwartz, O.; Jones, K.; Ruscetti, F.; Lockett, S.; Gudla, P.; Venzon, D.; Franchini, G. Human T-cell leukemia virus type 1 p8 protein increases cellular conduits and virus transmission. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20738–20743. [Google Scholar] [CrossRef]
- Kim, S.J.; Nair, A.M.; Fernandez, S.; Mathes, L.; Lairmore, M.D. Enhancement of LFA-1-mediated T cell adhesion by human T lymphotropic virus type 1 p12I1. J. Immunol. 2006, 176, 5463–5470. [Google Scholar] [CrossRef] [PubMed]
- AlphaEase FC, 6.0.0. 2006; Alpha Innotech: Santa Clara, CA, USA.
- Modfit lt, 3.2. 2008; Verity Software House: Topsham, ME, USA.
- Flowjo, 9.3.2. 2008; Tree Star: Ashland, OR, USA.
- Rende, F.; Cavallari, I.; Corradin, A.; Silic-Benussi, M.; Toulza, F.; Toffolo, G.M.; Tanaka, Y.; Jacobson, S.; Taylor, G.P.; D'Agostino, D.M.; Bangham, C.R.; Ciminale, V. Kinetics and intracellular compartmentalization of HTLV-1 gene expression: Nuclear retention of HBZ mRNAs. Blood 2011, 117, 4855–4859. [Google Scholar] [CrossRef]
- Cho, W.K.; Zhou, M.; Jang, M.K.; Huang, K.; Jeong, S.J.; Ozato, K.; Brady, J.N. Modulation of the Brd4/P-TEFb interaction by the human T-lymphotropic virus type 1 tax protein. J. Virol. 2007, 81, 11179–11186. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2011 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Edwards, D.C.; McKinnon, K.M.; Fenizia, C.; Jung, K.-J.; Brady, J.N.; Pise-Masison, C.A. Inhibition of Geranylgeranyl Transferase-I Decreases Cell Viability of HTLV-1-Transformed Cells. Viruses 2011, 3, 1815-1835. https://doi.org/10.3390/v3101815
Edwards DC, McKinnon KM, Fenizia C, Jung K-J, Brady JN, Pise-Masison CA. Inhibition of Geranylgeranyl Transferase-I Decreases Cell Viability of HTLV-1-Transformed Cells. Viruses. 2011; 3(10):1815-1835. https://doi.org/10.3390/v3101815
Chicago/Turabian StyleEdwards, Dustin C., Katherine M. McKinnon, Claudio Fenizia, Kyung-Jin Jung, John N. Brady, and Cynthia A. Pise-Masison. 2011. "Inhibition of Geranylgeranyl Transferase-I Decreases Cell Viability of HTLV-1-Transformed Cells" Viruses 3, no. 10: 1815-1835. https://doi.org/10.3390/v3101815