The Evolution of HIV-1 Diversity in Rural Cameroon and its Implications in Vaccine Design and Trials

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
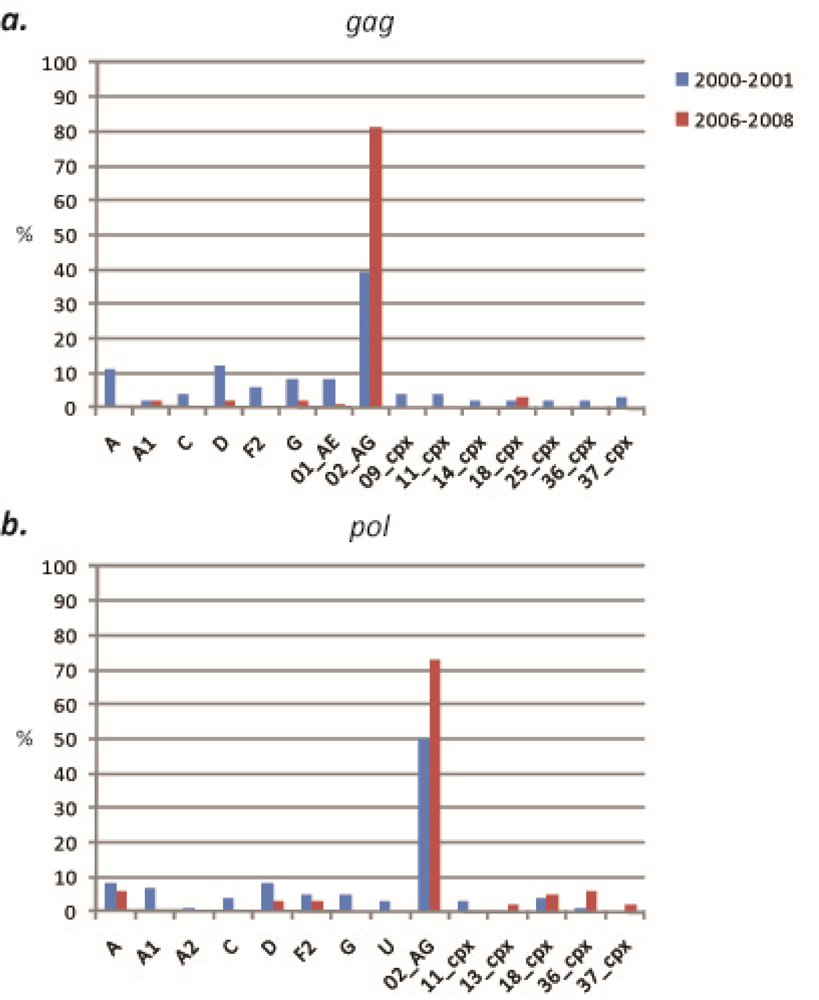
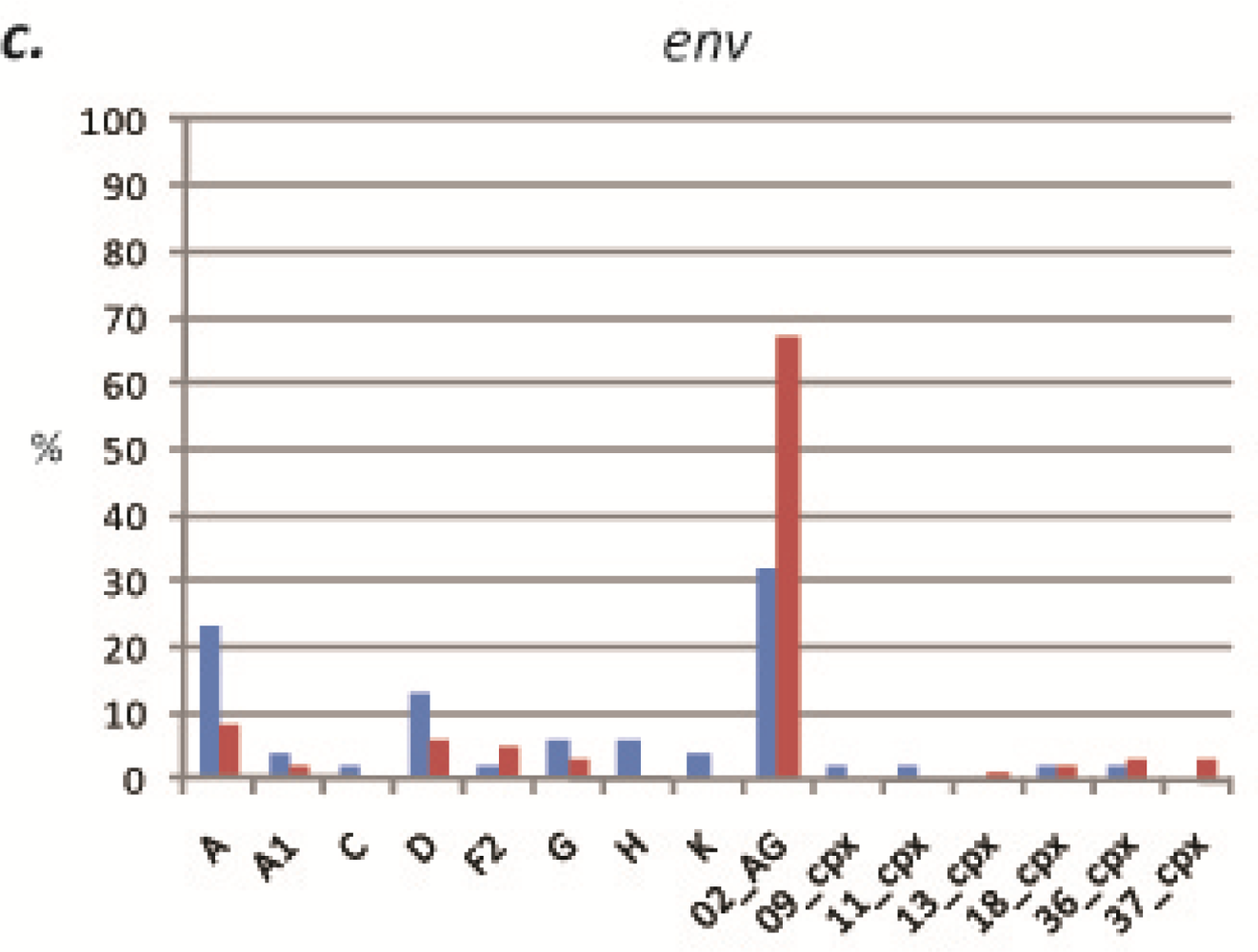
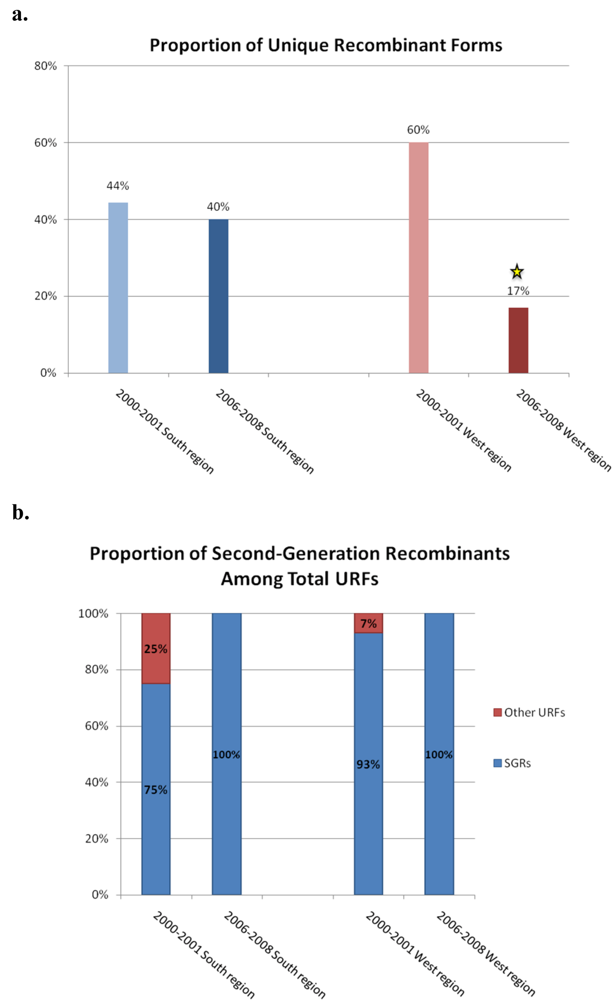
2.1. HIV-1 genetic diversity by geographic region identified in calendar year 2000–2001
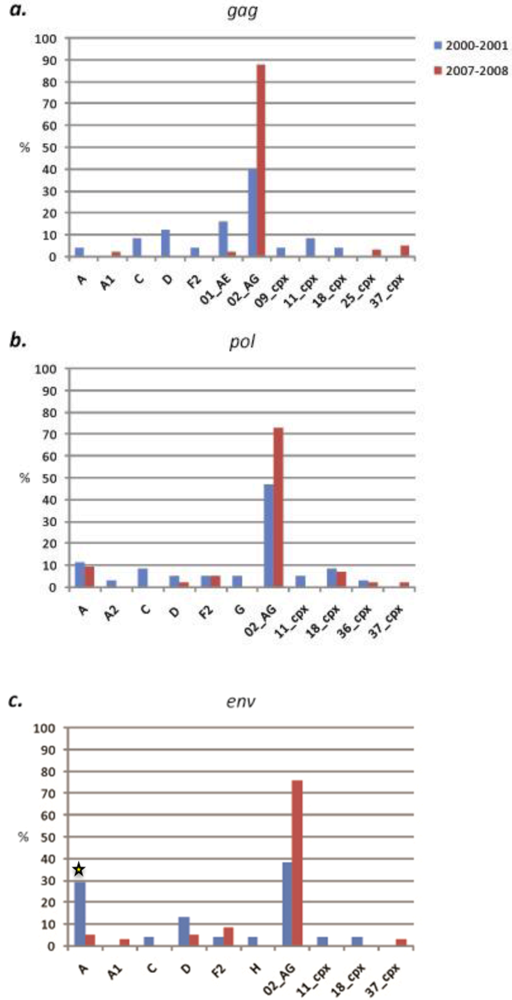
2.1.1. South Region

2.1.2. West Province
2.2. HIV-1 genetic diversity by geographic region identified in calendar year 2006-2008
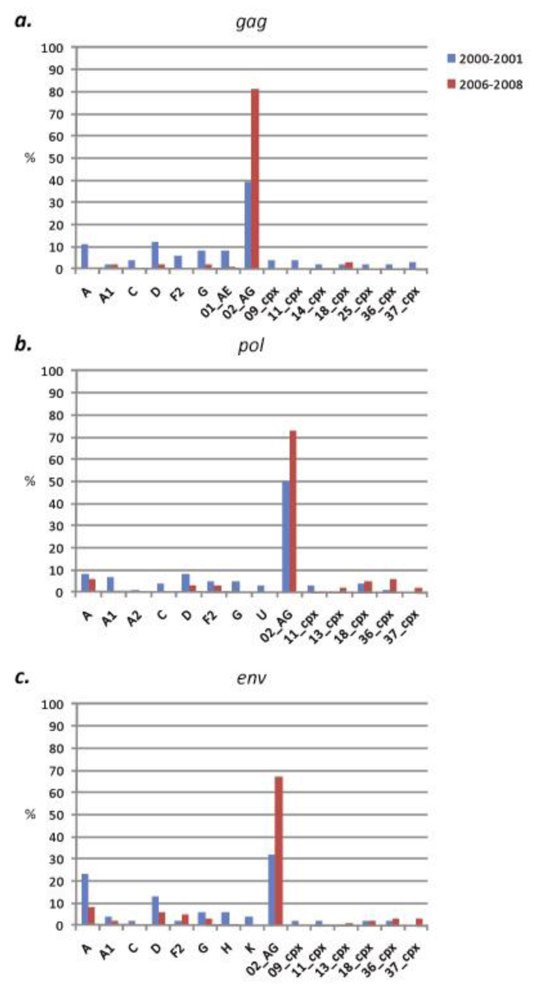
2.2.1. South Region
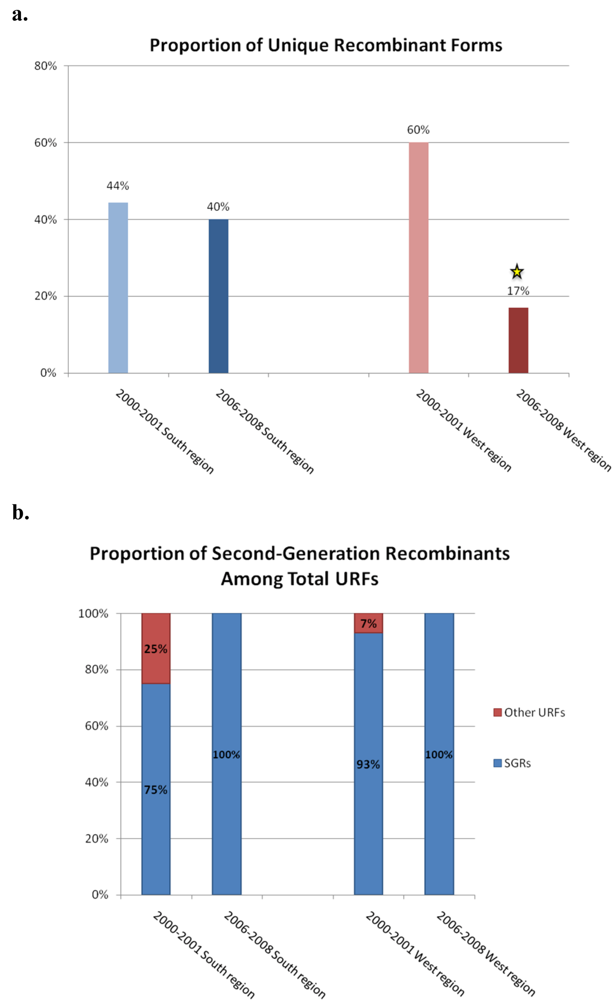
2.2.2. West Region
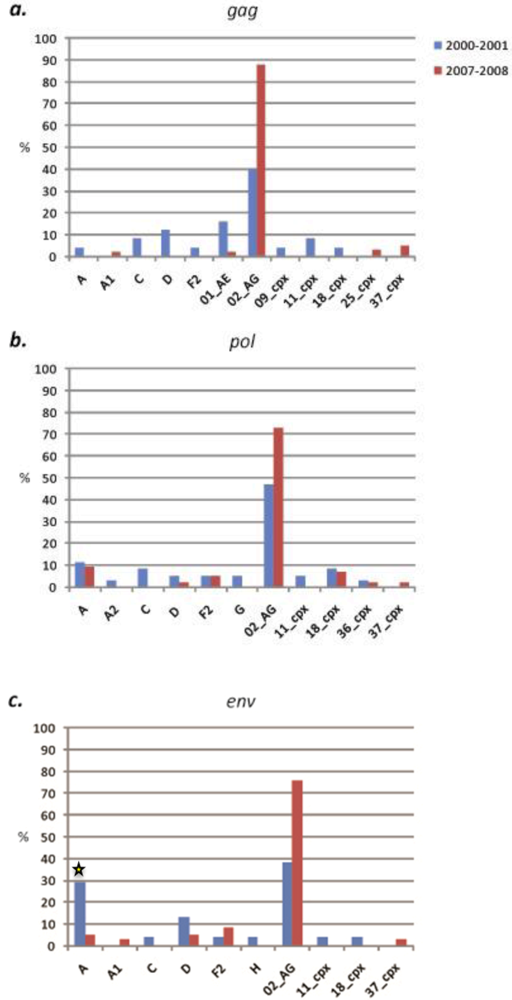
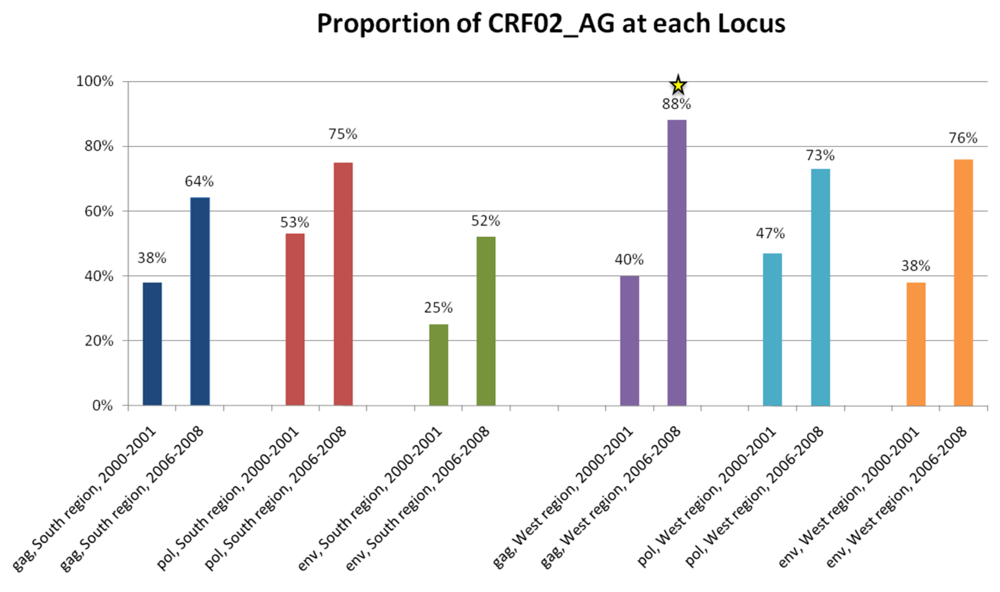
2.3. Discussion
3. Experimental Section
3.1. Study subjects
3.2. RNA Extraction, Reverse Transcriptase-Polymerase Chain Reaction and Sequencing
3.3. Phylogenetic Analysis
3.4. Accession numbers
4. Conclusion
Supplementary Tables
Acknowledgments
References
- Nyambi, P.; Zekeng, L.; Kenfack, H.; Tongo, M.; Nanfack, A.; Nkombe, I.; Ndonko, F.; Shang, J.; Burda, S.; Mbah, H.; Agyingi, L.; Zhong, P.; Nadas, A.; Zolla-Pazner, S.; Marmor, M. HIV infection in rural villages of Cameroon. J. Acquir. Immune Defic. Syndr. 2002, 31, 506–513. [Google Scholar] [PubMed]
- Zhong, P.; Burda, S.; Urbanski, M.; Kenfack, H.; Tongo, M.; Heyndrickx, L.; Nanfack, A.; Shang, J.; Agyingi, L.; Zolla-Pazner, S.; Zekeng, L.; Nyambi, P. HIV type 1 group M clades infecting subjects from rural villages in equatorial rain forests of Cameroon. J. Acquir. Immune Defic. Syndr. 2002, 31, 495–505. [Google Scholar] [PubMed]
- Zhong, P.; Burda, S.; Konings, F.; Urbanski, M.; Ma, L.Y.; Zekeng, L.; Ewane, L.; Agyingi, L.; Agwara, M.; Saa, D.; E., A.Z.; Kinge, T.; Zolla-Pazner, S.; Nyambi, P. Genetic and biological properties of HIV type 1 isolates prevalent in villagers of the Cameroon equatorial rain forests and grass fields: further evidence of broad HIV type 1 genetic diversity. AIDS Res. Hum. Retroviruses 2003, 19, 1167–1178. [Google Scholar] [PubMed]
- Konings, F.A.; Nyambi, P.N. V118I substitution in the reverse transcriptase gene of HIV type 1 CRF02_AG strains infecting drug-naive individuals in Cameroon. AIDS Res. Hum. Retroviruses 2004, 20, 673–678. [Google Scholar] [PubMed]
- Ndongmo, C.B.; Pieniazek, D.; Holberg-Petersen, M.; Holm-Hansen, C.; Zekeng, L.; Jeansson, S.L.; Kaptue, L.; Kalish, M.L. HIV genetic diversity in Cameroon: possible public health importance. AIDS Res. Hum. Retroviruses 2006, 22, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Machuca, A.; Tang, S.; Hu, J.; Lee, S.; Wood, O.; Vockley, C.; Vutukuri, S.G.; Deshmukh, R.; Awazi, B.; Hewlett, I. Increased genetic diversity and intersubtype recombinants of HIV-1 in blood donors from urban Cameroon. J. Acquir. Immune Defic. Syndr. 2007, 45, 361–363. [Google Scholar] [CrossRef] [PubMed]
- Fonjungo, P.N.; Mpoudi, E.N.; Torimiro, J.N.; Alemnji, G.A.; Eno, L.T.; Nkengasong, J.N.; Gao, F.; Rayfield, M.; Folks, T.M.; Pieniazek, D.; Lal, R.B. Presence of diverse human immunodeficiency virus type 1 viral variants in Cameroon. AIDS Res. Hum. Retroviruses 2000, 16, 1319–1324. [Google Scholar] [PubMed]
- Heyndrickx, L.; Janssens, W.; Ndumbe, P.M.; Vereecken, K.; Coppens, S.; De Houwer, K.; Fransen, K.; Van der Auwera, G.; van der Groen, G. HIV-1 genetic variability in Cameroon. Aids 2000, 14, 1862–1864. [Google Scholar] [CrossRef] [PubMed]
- Montavon, C.; Toure-Kane, C.; Nkengasong, J.N.; Vergne, L.; Hertogs, K.; Mboup, S.; Delaporte, E.; Peeters, M. CRF06-cpx: a new circulating recombinant form of HIV-1 in West Africa involving subtypes A, G, K, and J. J. Acquir. Immune Defic. Syndr. 2002, 29, 522–530. [Google Scholar] [PubMed]
- Powell, R.L.; Zhao, J.; Konings, F.A.; Tang, S.; Ewane, L.; Burda, S.; Urbanski, M.M.; Saa, D. R.; Hewlett, I.; Nyambi, P.N. Circulating recombinant form (CRF) 37_cpx: an old strain in Cameroon composed of diverse, genetically distant lineages of subtypes A and G. AIDS Res. Hum. Retroviruses 2007, 23, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Powell, R.L.; Zhao, J.; Konings, F.A.; Tang, S.; Nanfack, A.; Burda, S.; Urbanski, M.M.; Saa, D.R.; Hewlett, I.; Nyambi, P.N. Identification of a novel circulating recombinant form (CRF) 36_cpx in Cameroon that combines two CRFs (01_AE and 02_AG) with ancestral lineages of subtypes A and G. AIDS Res. Hum. Retroviruses 2007, 23, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Wilbe, K.; Casper, C.; Albert, J.; Leitner, T. Identification of two CRF11-cpx genomes and two preliminary representatives of a new circulating recombinant form (CRF13-cpx) of HIV type 1 in Cameroon. AIDS Res. Hum. Retroviruses 2002, 18, 849–856. [Google Scholar] [PubMed]
- HIV Immunology and Sequence Databases. Los Alamos National Laboratory: Los Alamos, NM, USA, 2004. And: http://www.hiv.lanl.gov/content/sequence/HIV/CRFs/CRFs.html (accessed 2009).
- Brennan, C.A.; Bodelle, P.; Coffey, R.; Devare, S.G.; Golden, A.; Hackett Jr., J.; Harris, B.; Holzmayer, V.; Luk, K.C.; Schochetman, G.; Swanson, P.; Yamaguchi, J.; Vallari, A.; Ndembi, N.; Ngansop, C.; Makamche, F.; Mbanya, D.; Gurtler, L.G.; Zekeng, L.; Kaptue, L. The Prevalence of Diverse HIV-1 Strains Was Stable in Cameroonian Blood Donors From 1996 to 2004 . J. Acquir. Immune Defic. Syndr. 2008, 49, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Konings, F.A.; Haman, G.R.; Xue, Y.; Urbanski, M.M.; Hertzmark, K.; Nanfack, A.; Achkar, J.M.; Burda, S.T.; Nyambi, P.N. Genetic Analysis of HIV-1 Strains in Rural Eastern Cameroon Indicates the Evolution of Second-Generation Recombinants to Circulating Recombinant Forms. J. Acquir. Immune Defic. Syndr. 2006, 42, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Nyambi, P.; Heyndrickx, L.; Vereecken, K.; Burda, S.; De Houwer, K.; Coppens, S.; Urbanski, M.; Williams, C.; Ndumbe, P.; Janssens, W. Predominance of infection with HIV-1 circulating recombinant form CRF02_AG in major Cameroonian cities and towns. Aids 2002, 16, 295–296. [Google Scholar] [CrossRef] [PubMed]
- Powell, R.L.R.; Urbanski, M.M.; Burda, S.; Kinge, T.; Nyambi, P.N. High Frequency of HIV-1 Dual Infections among HIV-Positive Individuals in Cameroon, West-Central Africa. 2009, 50, 84–92. [Google Scholar] [PubMed]
- West, B. The Globe Pequot Press: Guilford, Connecticut, CT, USA, 2008; p. 248.
- Konings, F.A.; Zhong, P.; Agwara, M.; Agyingi, L.; Zekeng, L.; Achkar, J.M.; Ewane, L. Protease mutations in HIV-1 non-B strains infecting drug-naive villagers in Cameroon. AIDS Res. Hum. Retroviruses 2004, 20, 105–109. [Google Scholar] [PubMed]
- Heyndrickx, L.; Janssens, W.; Zekeng, L.; Musonda, R.; Anagonou, S.; Van der Auwera, G.; Coppens, S.; Vereecken, K.; De Witte, K.; Van Rampelbergh, R.; Kahindo, M.; Morison, L.; McCutchan, F.E.; Carr, J.K.; Albert, J.; Essex, M.; Goudsmit, J.; Asjo, B.; Salminen, M.; Buve, A.; van Der Groen, G. Simplified strategy for detection of recombinant human immunodeficiency virus type 1 group M isolates by gag/env heteroduplex mobility assay. Study Group on Heterogeneity of HIV Epidemics in African Cities. J. Virol. 2000, 74, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Delwart, E.L.; Shpaer, E.G.; Louwagie, J.; McCutchan, F.E.; Grez, M.; Rubsamen-Waigmann, H.; Mullins, J.I. Genetic relationships determined by a DNA heteroduplex mobility assay: analysis of HIV-1 env genes. Science 1993, 262, 1257–1261. [Google Scholar] [PubMed]
- Galtier, N.; Gouy, M.; Gautier, C. SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny. Comput. Appl. Biosci. 1996, 12, 543–548. [Google Scholar] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nei, M.; Dudley, J.; Tamura, K. MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform. 2008, 9, 299–306. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Powell, R.; Barengolts, D.; Mayr, L.; Nyambi, P. The Evolution of HIV-1 Diversity in Rural Cameroon and its Implications in Vaccine Design and Trials. Viruses 2010, 2, 639-654. https://doi.org/10.3390/v2020639
Powell R, Barengolts D, Mayr L, Nyambi P. The Evolution of HIV-1 Diversity in Rural Cameroon and its Implications in Vaccine Design and Trials. Viruses. 2010; 2(2):639-654. https://doi.org/10.3390/v2020639
Chicago/Turabian StylePowell, Rebecca, Denis Barengolts, Luzia Mayr, and Phillipe Nyambi. 2010. "The Evolution of HIV-1 Diversity in Rural Cameroon and its Implications in Vaccine Design and Trials" Viruses 2, no. 2: 639-654. https://doi.org/10.3390/v2020639