1. Introduction
During a respiratory infection, multiple viruses may be present and working in concert to cause disease [
1,
2,
3,
4,
5]. Several respiratory viruses, including rhinovirus (RV), respiratory syncytial virus (RSV), influenza A and B viruses (IAV and IBV), human metapneumovirus (HMPV), human parainfluenza viruses (PIV), adenoviruses (ADV), and coronaviruses (CoVs), have been found concurrently within hosts with pneumonia [
6,
7,
8,
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20]. Children are more likely to be concurrently infected with multiple respiratory viruses, where up to 30% have more than one respiratory virus present when admitted to the hospital for severe clinical disease [
16,
19,
21,
22,
23,
24,
25]. Patients with coinfection have shown diverse disease outcomes that ranged from mild to severe, with severity increasing compared to that of patients who were infected with a single virus.
Data on viral–viral coinfections are somewhat limited, but some studies have begun evaluating the outcomes of coinfection with commonly observed viral pairs (e.g., IAV and RSV [
26,
27,
28,
29,
30,
31], IAV and PIV [
32], IAV and RV [
28,
33,
34,
35], RSV and RV [
28], RSV and HMPV [
36], IAV and severe acute respiratory syndrome (SARS)-CoV-2 [
37,
38,
39,
40,
41,
42,
43], RSV and SARS-CoV-2 [
43], and RV and SARS-CoV-2 [
44]). Collectively, these studies observed diverse outcomes of viral–viral coinfections with some interactions resulting in enhanced spread of one or both viruses within the respiratory tract, while other mechanisms seem to work in an inhibitory manner. For example, PIV-2 enhanced cell-to-cell fusion through the expression of its surface glycoproteins, which boosted viral spread between cells, and increased IAV titers but not PIV titers [
32]. On the other hand, infection with IAV limited a concurrent RSV infection by promoting intracellular competition for proteins or amino acids needed for the successful replication of both viruses within cell cultures [
11,
26]. In vivo infections in animal models support the exclusion of RSV by IAV and suggest that RSV prior to IAV decreases disease severity [
27,
29,
30]. A similar competitive exclusion was observed in 3D tissue cultures during coinfection with RSV and HMPV, where HMPV was inhibited without any effect on RSV [
36]. The reduction was accompanied by higher Type I and III interferon (IFN) responses [
36]. IFN-mediated effects were also implicated in RV–IAV coinfection where RV-induced IFN protected against subsequent IAV infection within differentiated airway cell cultures [
34].
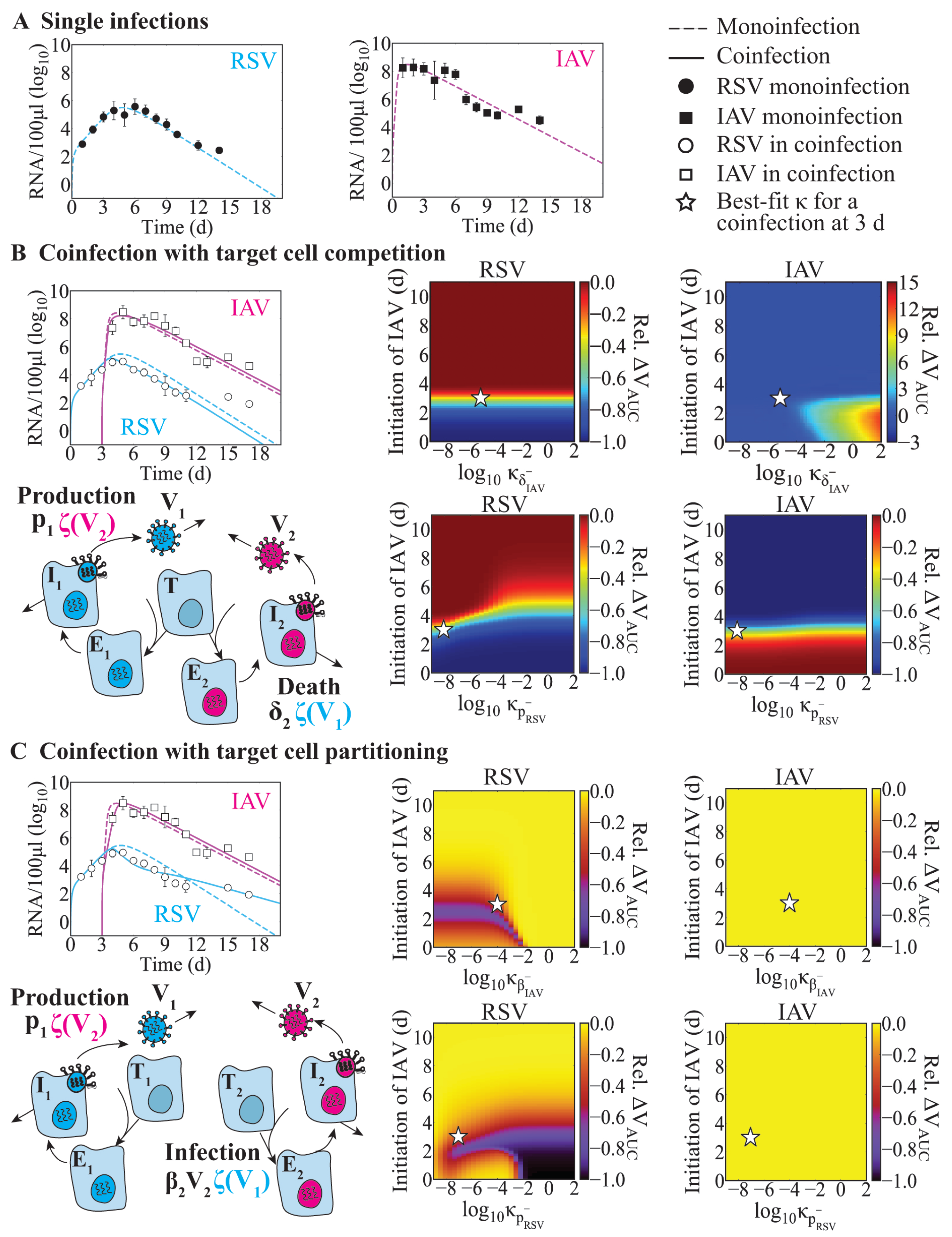
The relative timing between viruses and the order in which the viruses infect the host seem to contribute to differing disease outcomes [
27,
29,
33,
37,
39,
40,
42]. Interestingly, the exclusion effect during RSV–HMPV coinfection was more robust during a concurrent infection compared to that in an infection where HMPV followed RSV after 2 days. This is in contrast to other viral–viral coinfections where infections separated by 2 to 5 days had more robust effects [
33,
37,
42]. For example, RV attenuated IAV-mediated disease severity and reduced IAV titers when RV infection had occurred 2 days before IAV, but the effect was reduced during simultaneous infection [
33]. Conversely, animals coinfected with IAV 2 days before RV experienced greater disease severity [
33]. Similar outcomes occurred in animals coinfected with IAV 3 days before SARS-CoV-2 [
37].
These empirical studies illuminate the breadth of interactions that lead to diverse outcomes of respiratory viral–viral coinfections and the need for mathematical methods that could dissect complex, time-dependent, and potentially nonlinear mechanisms, as they do for viral–bacterial coinfection [
45]. One study on viral–viral coinfections suggested that a faster-replicating virus would outcompete other viruses in a scenario where two viruses were competing for epithelial cells [
46]. However, it is possible that viruses infect different cells or infect different areas of the respiratory tract. In addition, as noted above, they may inhibit or enhance other processes (e.g., replication rates and/or immune responses) and ultimately modulate disease. Thus, expanded modeling infrastructures are needed and are a focus of this study. Unfortunately, most studies on viral–viral coinfections lack quantitative information on viral loads and/or host immune responses that are needed to effectively use mathematical approaches, which typically include fitting a mechanistic model to data. However, weight loss, which is a measure of disease severity, is tracked in most murine studies, and our recent work showed that mathematical models can accurately connect animal weight loss to infection kinetics during monoinfections and viral–bacterial coinfections [
47,
48]. These links allow for us to better interpret weight-loss data, and afford the ability to assess mechanisms with limited data to modeling studies such as this one.
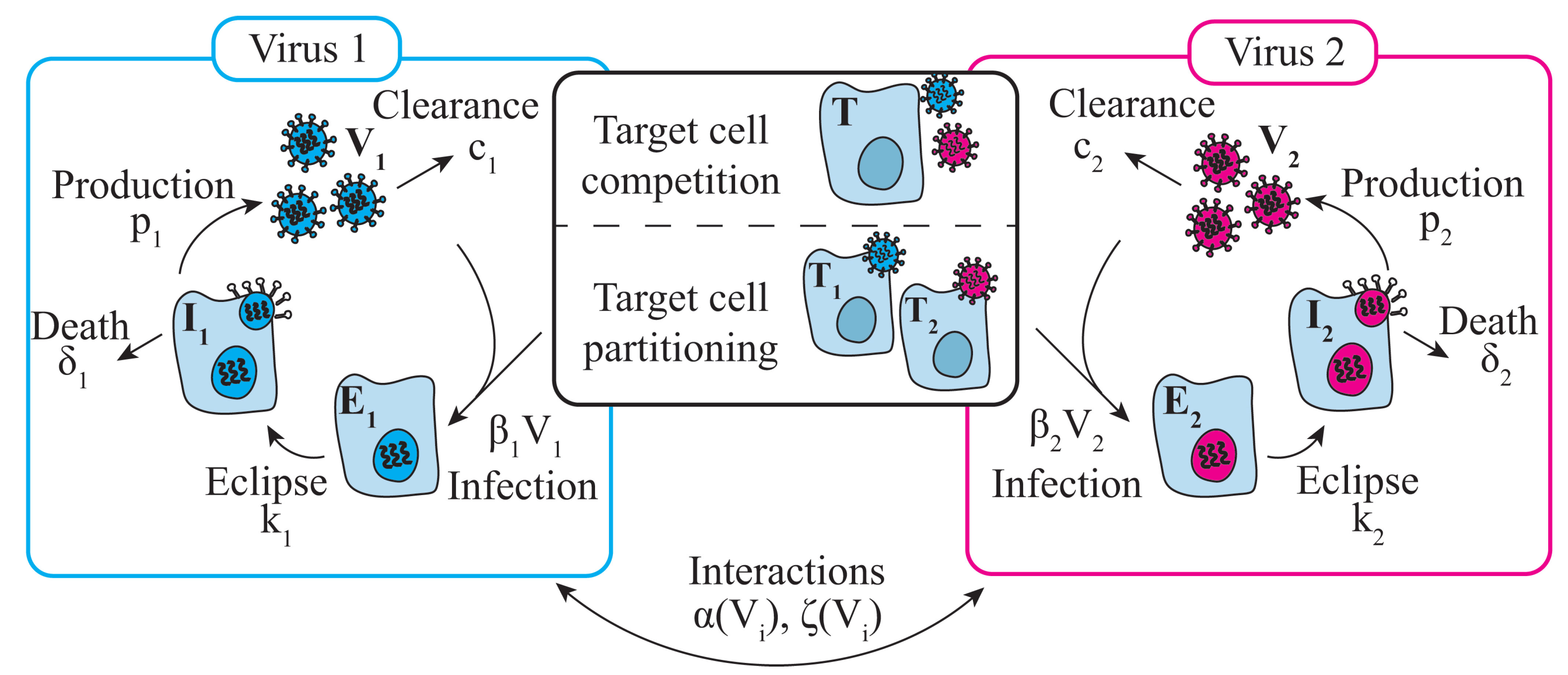
To address gaps in understanding viral–viral coinfections, we assessed RSV, RV, and SARS-CoV-2 coinfections with IAV using two mathematical models. We predicted potential underlying mechanisms of viral interference and cooperation and assessed how different viral orders, timings, and pairings affected the infection dynamics and disease severity. The results provide important insights into divergent outcomes, in addition to generating novel hypotheses regarding why certain viral orders enhance or reduce disease severity.
4. Discussion
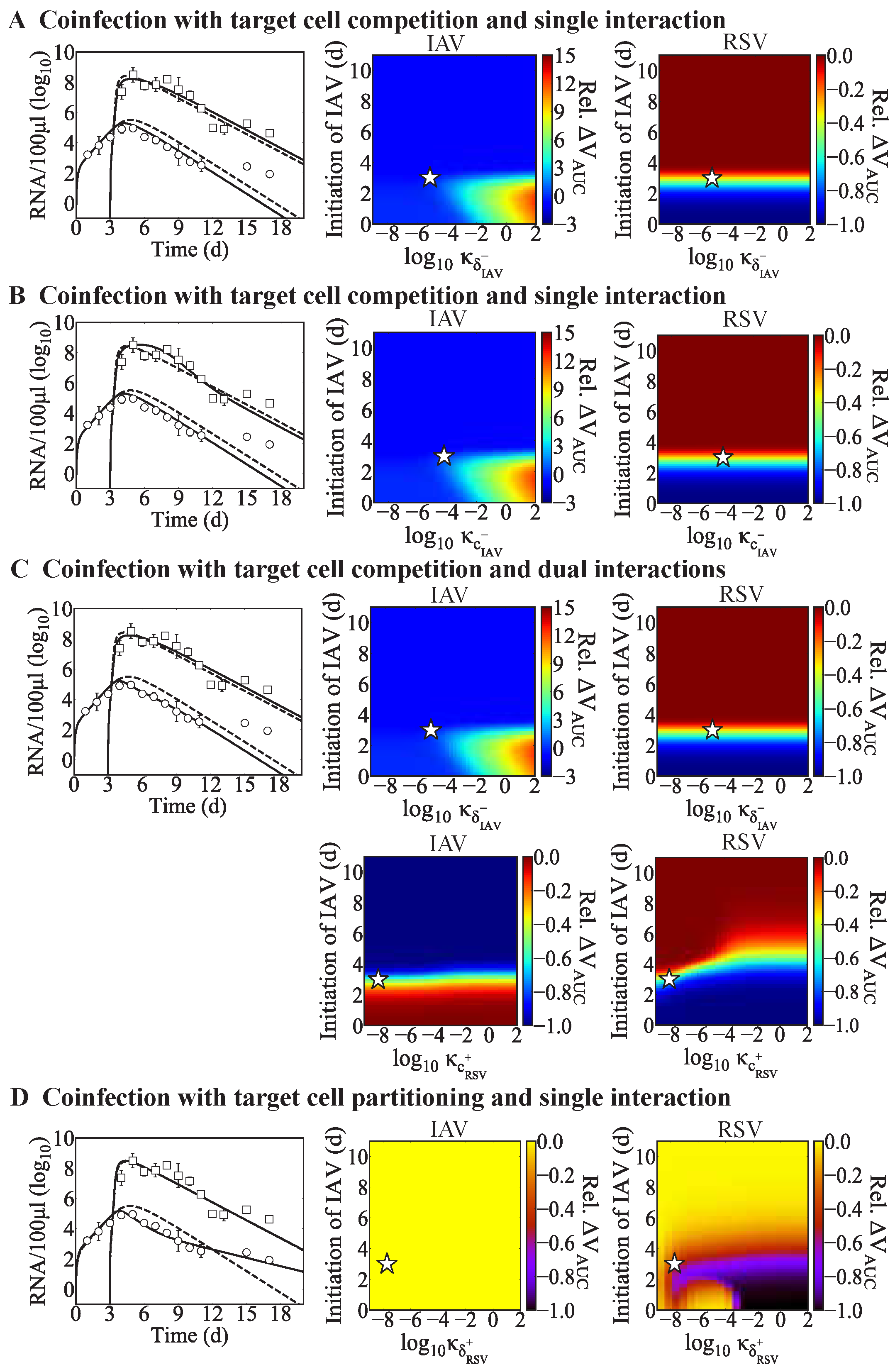
Respiratory coinfections with multiple viruses are becoming more recognized clinically, particularly in light of the SARS-CoV-2 pandemic. Experimental studies have begun illuminating the outcome heterogeneity, which seems to rely on numerous factors such as the viral pairing, order, and timing of each infection, and specific immune factors. Although viral and immune dynamics during viral–viral coinfections is only beginning to be defined, mathematical models are useful to predict and narrow the spectrum of potential mechanisms, guide new experiments, and help in interpreting clinical, experimental, and epidemiological observations. Our analysis of different viral coinfection scenarios suggests that only a small subset of mechanisms could lead to the alterations in viral loads and/or disease severity observed in animal models (summarized in
Table 4).
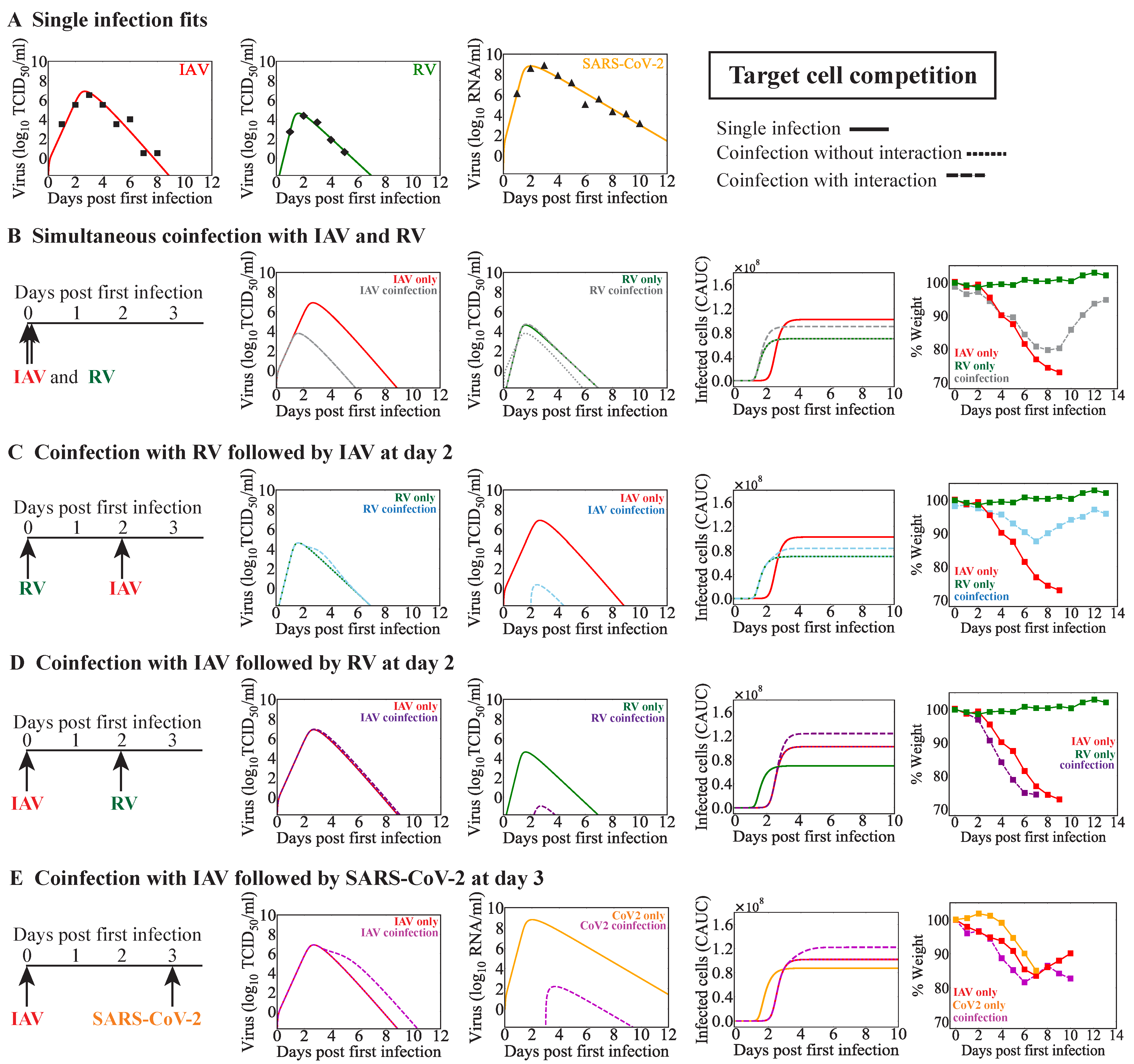
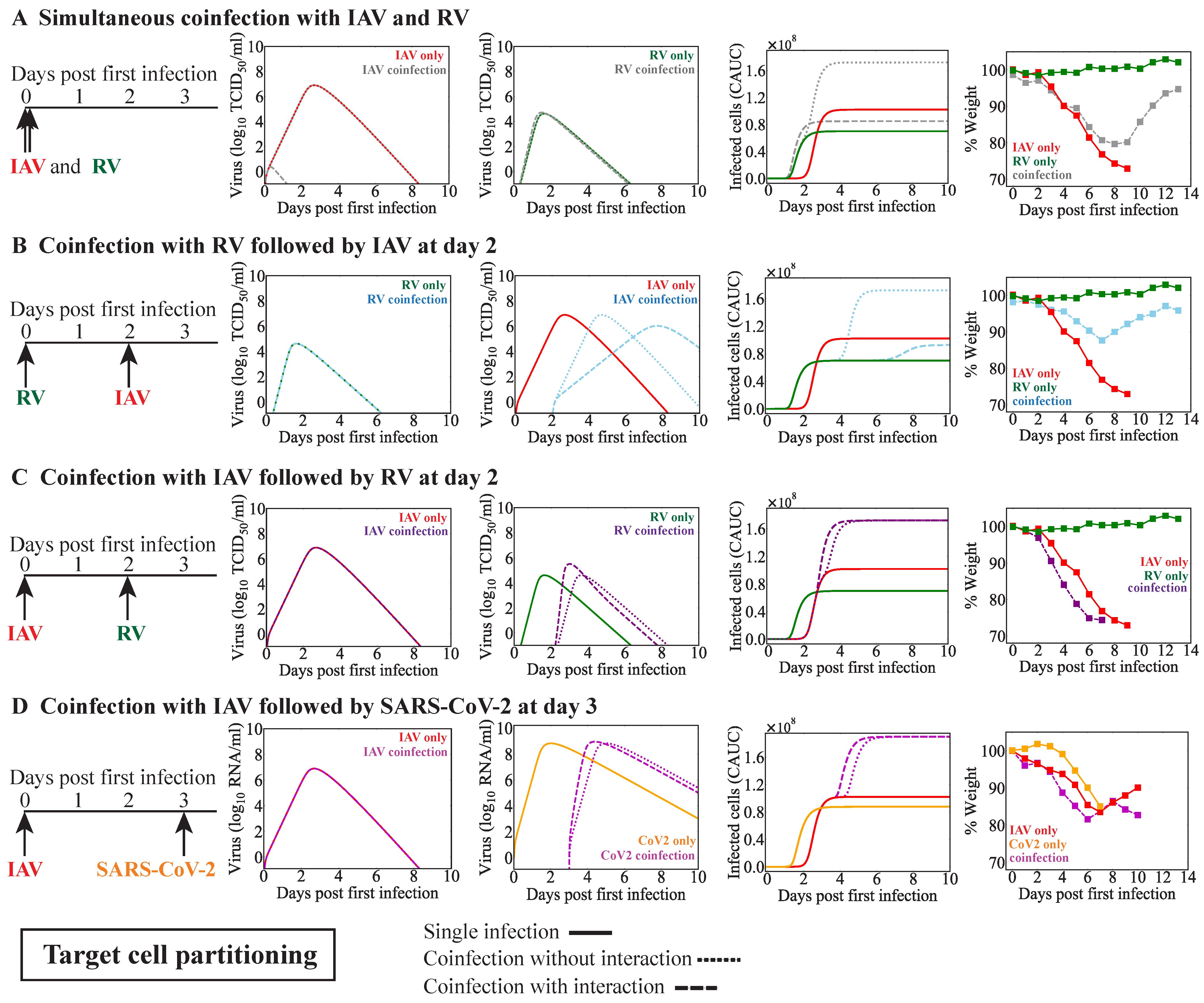
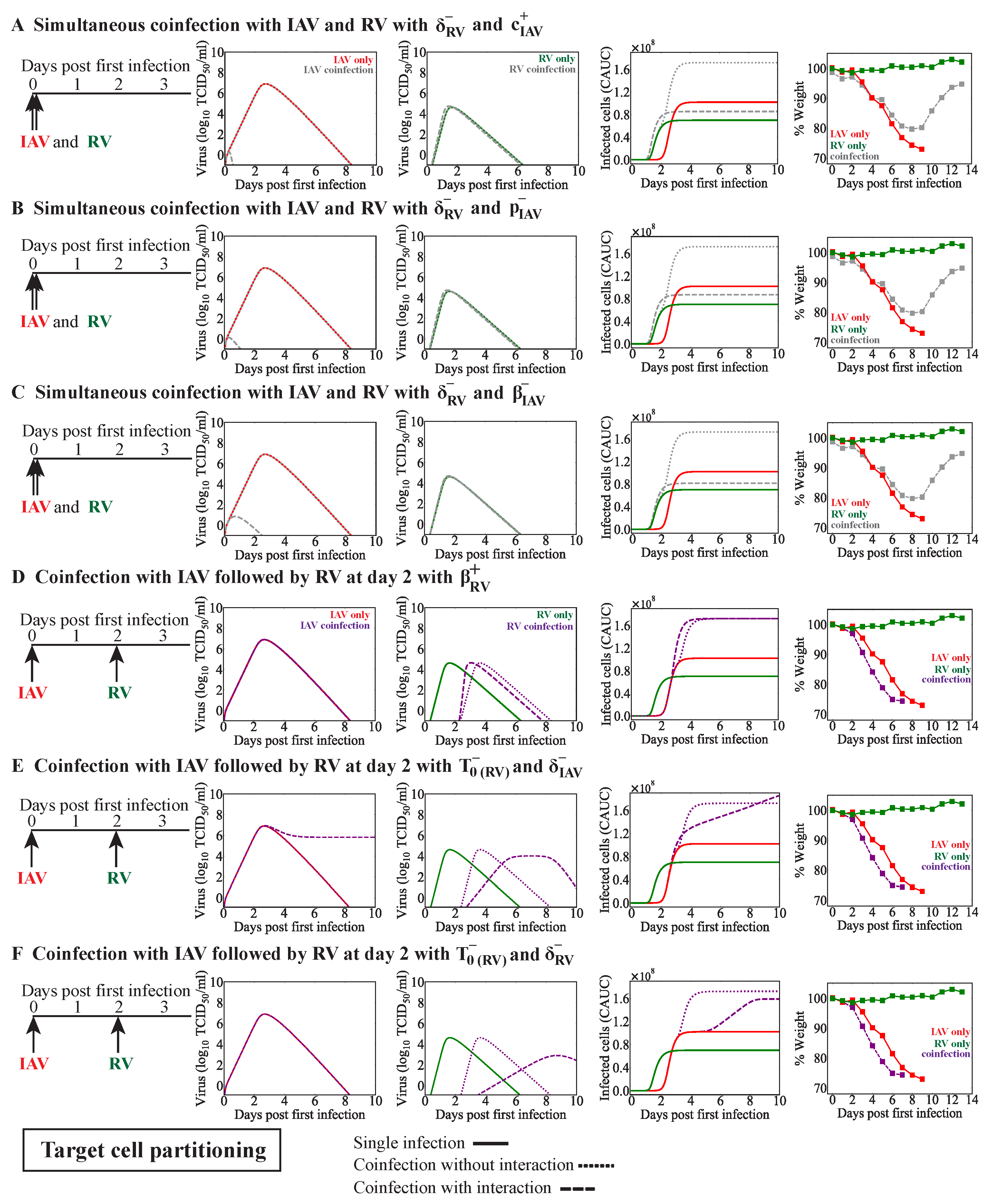
When a new virus invades within a few days before or after influenza, innate immune responses may be disrupted. The early regulation of Type I IFNs, macrophages, and/or neutrophils, among other innate immune responses, by a virus could impact the dynamics of subsequent infections. Although we did not assess these immune responses directly, we modeled them indirectly in various ways. The target cell competition model automatically assumes that fewer cells are available for infection by the second virus, which could emulate a protective mechanism that reduces the possible infection size for the coinfecting virus. In the target cell partitioning model, we decreased the number of target cells available for the second virus, which was used to mimic lower doses [
47,
60]. Both approaches assume that some cells are protected or otherwise unavailable for infection, which can be interpreted as the IFN-mediated protection of susceptible cells or an immediate clearance of the virus upon infection due to the activation of macrophages and/or neutrophils by the first virus. We observed the latter phenomenon in an experiment where CD8 T cells were depleted before infection [
47]. In that case, innate responses were activated by the CD8 T cell death, which resulted in the partial clearance of the inoculum that emulated a reduced dose, and led to fewer infected cells and thus less virus. This automatically reduced inflammation and weight loss [
47], which was similar to the reduced weight loss observed during RV–IAV coinfection [
33]. Allowing for fewer susceptible cells within the target cell partitioning model was sufficient to explain the reduced weight loss in RV-IAV coinfected animals, potentially indicating a similar mechanism. While neither our model nor the data were sophisticated enough to specify the exact mechanism, higher IFN-
was detected at 2 days post coinfection, and IAV titers trended slightly lower compared to in the monoinfection [
33]. A follow-up study suggested that the protection of IAV-mediated disease by RV was dependent on IFN and that this contributed to the control of neutrophilic inflammation [
35]. These data align nicely with our predictions, which may help in connecting the underlying reasoning (i.e., fewer cells becoming infected) with downstream consequences (i.e., reduced immune activation and inflammation).
In addition to potential effects on early host responses, one of the most common mechanisms defined by our analysis was altered rates of infected cell clearance, which may indicate an effect on virus-specific CD8 T-cell responses [
47]. In some cases, there was a negative effect, while in others, there was a positive effect (
Table 4). Further, there were some scenarios where each virus affected the other virus’ infected cell clearance rate. This may indicate variation in the number, composition, and/or function of epitope-specific T cells, which was observed for other viral pairs (e.g., lymphocytic choriomeningitis (LCMV) and Pichinde (PICV) viruses) [
62]. For scenarios where the rates are reduced, it could also indicate that having fewer available target cells from preinfection would have automatically reduced the number of T cells needed to clear the infection. Data defining host responses are important to in helping to distinguish between these possibilities, particularly because CD8 T cells have to be significantly reduced to have a robust impact on viral loads, and their efficacy is dependent on the number of infected cells [
47].
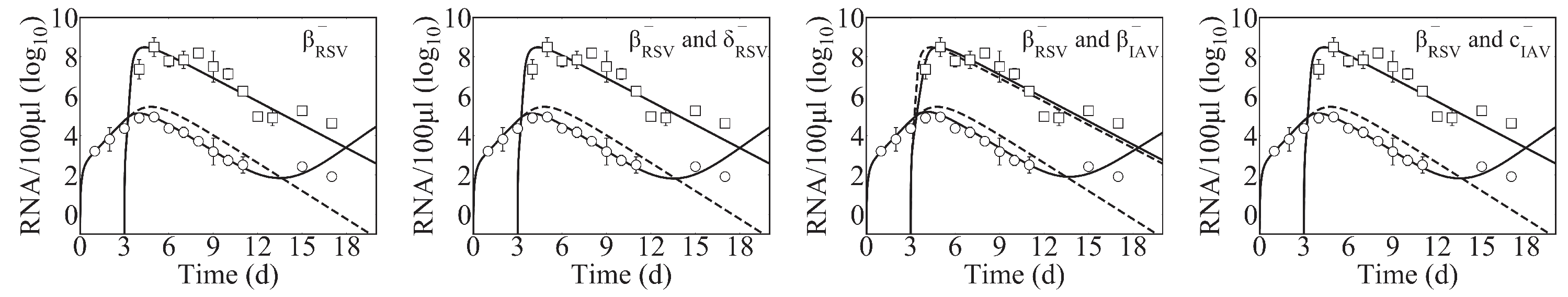
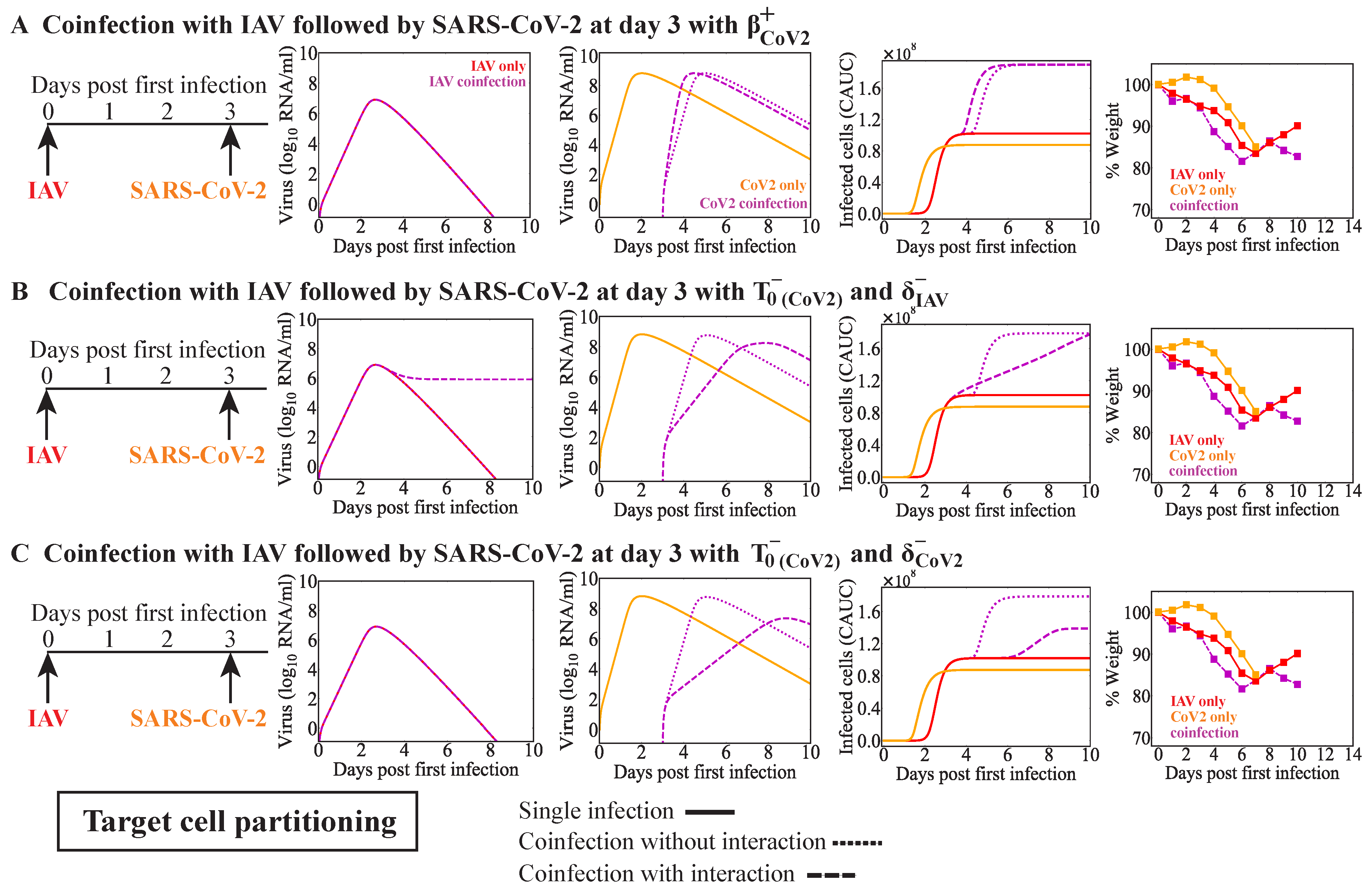
Effects on other infection processes such as the rates of viral infectivity or production were detected for some coinfection scenarios, although this was rarely the sole mechanism (
Table 4). Only in IAV-RV and IAV-CoV2 coinfections within the target cell partitioning model were these potential single interactions, and they each resulted in similar viral load dynamics (
Figure 4C,D,
Figure A3D, and
Figure A4A). This is because the type of model used here cannot typically distinguish between the effects of these two processes [
63]. There is some evidence that the infectivity of SARS-CoV-2 is increased by IAV but not RSV within cell cultures [
41,
43]. The underlying mechanism driving this remains unknown, but other studies with IAV and PIV have shown enhanced infection rates where PIV increased cell-to-cell fusion and, thus, spread of IAV [
32]. However, another study found that IAV titers were increased while SARS-CoV-2 titers were decreased during IAV-CoV2 coinfection in mice [
42]. The discrepancy between these studies, in which one found there was an increase in the number of infected cells, while the other found reduced viral loads, may have been due to the results being obtained in vitro vs. in vivo or to another interaction (e.g., IFN suppression of SARS-CoV-2 or clearance by macrophages). Our analyses suggest that SARS-CoV-2 titers would be reduced within the target cell competition model (
Figure A3E,F) or when the number of susceptible cells was reduced within the target cell partitioning model (
Figure A4B,C). This may indicate a role for the IAV-activated innate immune response and/or a lower effective dose of SARS-CoV-2.
A limitation of this work is that we used minimal data that were of different types (i.e., viral loads or weight loss) to predict potential mechanisms during viral coinfection. The quantity and type of data used are important because altered viral loads (e.g., as in RSV–IAV coinfection), immune cells, or cytokines do not always directly equate to differences in disease severity. We previously showed this phenomenon where we found that the number of infected cells and inflammation were nonlinearly correlated to disease severity [
47]. This was particularly evident in one experiment where CD8 T cells were depleted, which resulted in only small reductions in viral loads, yet large reductions in weight loss. However, as mentioned above, the early immune activation led to a predicted lower effective dose (i.e., fewer cells becoming infected) and, thus, reduced disease severity. Similarly, in RSV–IAV coinfection, IAV titers were slightly increased, yet less weight loss occurred [
27]. Thus, the higher viral loads later in infection may be insignificant with respect to severity. This highlights that, while some mechanisms may occur and alter viral loads, they could be distinct from those that yield disease outcomes. Our results from matching the qualitative differences in weight-loss data, which is a measure of disease severity, for IAV coinfection with RV or SARS-CoV-2 may better represent potential mechanisms with measurable differences in outcome. However, many of these also led to predicted differences in viral loads. Some information about mechanism may be able to be deduced from the timing of when weight loss begins to deviate from the monoinfection. In several scenarios, this occurred directly after the initiation of the secondary infection, which suggests that the environment created by the first virus had immediate effects.
The mechanisms suggested by our analysis occasionally differed depending on the underlying model hypothesis (i.e., whether viruses competed for epithelial cells) and, in some cases, resulted in different predicted viral load kinetics. Because most respiratory viruses can infect various types of airway epithelial cells, and replicate in the upper and lower respiratory tracts, it is conceivable that each virus would have ample cells to infect. However, by chance or due to the airway structure, they may enter the same region and interact on a local level. This may lead to cells being coinfected with both viruses, which we did not model explicitly. We indirectly modeled the potential effects of coinfected cells by assuming that the rates of infection and/or production could be different. Interestingly, cellular coinfection was detected during simultaneous infection with RSV and HMPV, where coinfected cells were possible but in small numbers and less likely in the presence of IFN [
36]. The same may be true during other coinfections with viruses that are sensitive to IFN antiviral responses. To model the impact of coinfected cells and potential heterogeneity in their prevalence, agent-based models may be better suited than those used here. However, it is important to determine whether and for how long these local effects have a measurable impact during an in vivo infection.
Examining data from viral–viral coinfections using mathematical models allowed for us to reduce the number of possible underlying mechanisms that could result in altered viral load kinetics and/or disease severity. Although the models were relatively simple and lacked investigation into specific host immune responses, the analysis provides the infrastructure to integrate immunological models of higher complexity once data becomes available. Models for some immune responses during respiratory virus infections are already being developed and validated with experimental data (reviewed in [
45,
64]). Some of the insight from those studies was integrated here and aided our ability to interpret the small amount of experimental data currently available. However, establishing better methods that could predict disease severity (e.g., as in [
47]) is critical. Our ability to assess the contribution and timescales of different mechanisms to infection kinetics and outcome should increase as more temporal viral load, immunologic, and pathologic data become available. In addition, the generated hypotheses should aid in experimental design, ultimately leading to a more complete understanding of respiratory virus coinfection.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







