Revisiting the Role of Transcription Factors in Coordinating the Defense Response Against Citrus Bark Cracking Viroid Infection in Commercial Hop (Humulus Lupulus L.)

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of Hop Transcription Factors
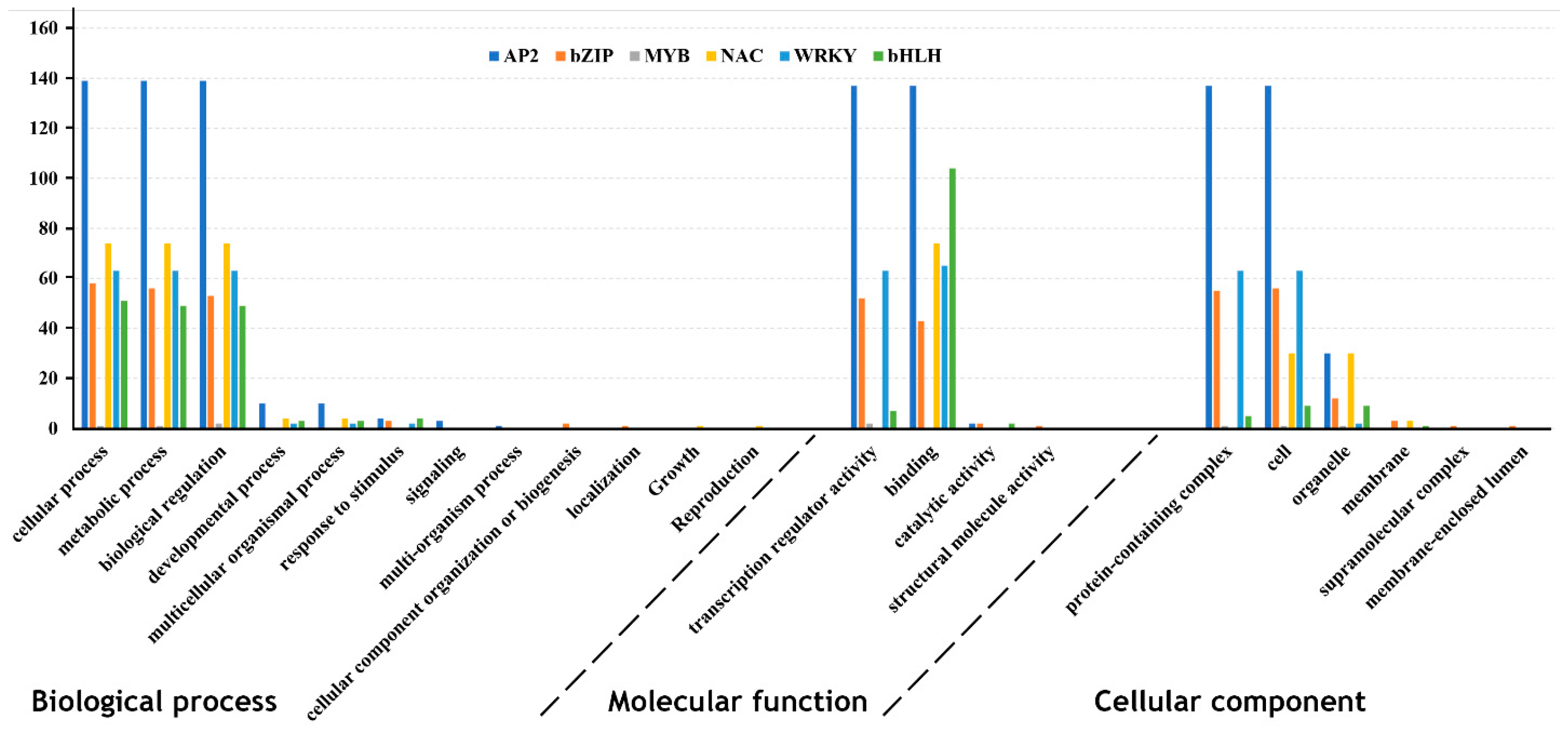
2.2. Protein Characterization, Conserved Motifs and Gene Ontology Identification
2.3. Phylogenetic Affinity of Hop TFs
2.4. The CBCVd-Responsive Expression Datasets Availability and Analyses of Differentially Regulated TFs
2.5. Identification of Orthologous Genes and Protein Interactions
2.6. RNA Extraction and qRT-PCR Validation of Selected Hop TFs
3. Results
3.1. Identification and Classification of Hop TFs
3.2. Protein Characterization, Conserved Motifs and Gene Ontology Identification
3.3. Phylogenetic Affinity of Hop TFs
3.4. Differential Modulation of Hop TFs
3.5. Identification of Orthologous Genes and Protein Interactions
3.6. qRT-PCR Validation of Selected Hop TFs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moore, J.W.; Loake, G.J.; Spoel, S.H. Transcription Dynamics in Plant Immunity. Plant Cell 2011, 23, 2809–2820. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.B.; Foley, R.C.; Oñate-Sánchez, L. Transcription factors in plant defense and stress responses. Curr. Opin. Plant Biol. 2002, 5, 430–436. [Google Scholar] [CrossRef]
- Tsuda, K.; Somssich, I.E. Tansley review Transcriptional networks in plant immunity. New Phytol. 2015, 206, 932–947. [Google Scholar] [CrossRef]
- Eulgem, T.; Somssich, I.E. Networks of WRKY transcription factors in defense signaling. Curr. Opin. Plant Biol. 2007, 10, 366–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stracke, R.; Werber, M.; Weisshaar, B. The R2R3-MYB gene family in Arabidopsis thaliana. Curr. Opin. Plant Biol. 2001, 4, 447–456. [Google Scholar] [CrossRef]
- Toledo-Ortiz, G.; Huq, E.; Quail, P.H. The Arabidopsis Basic/Helix-Loop-Helix Transcription Factor Family. Plant Cell 2003, 15, 1749–1770. [Google Scholar] [CrossRef]
- Jakoby, M.; Weisshaar, B.; Dröge-Laser, W.; Vicente-Carbajosa, J.; Tiedemann, J.; Kroj, T.; Parcy, F. bZIP transcription factors in Arabidopsis. Trends Plant Sci. 2002, 7, 106–111. [Google Scholar] [CrossRef]
- Muthamilarasan, M.; Bonthala, V.S.; Mishra, A.K.; Khandelwal, R.; Khan, Y.; Roy, R.; Prasad, M. C2H2 type of zinc finger transcription factors in foxtail millet define response to abiotic stresses. Funct. Integr. Genom. 2014, 14, 531–543. [Google Scholar] [CrossRef]
- Muthamilarasan, M.; Bonthala, V.S.; Khandelwal, R.; Jaishankar, J.; Shweta, S.; Nawaz, K.; Prasad, M. Global analysis of WRKY transcription factor superfamily in Setaria identifies potential candidates involved in abiotic stress signaling. Front. Plant Sci. 2015, 6, 1–15. [Google Scholar] [CrossRef]
- Muthamilarasan, M.; Mangu, V.R.; Zandkarimi, H.; Prasad, M.; Baisakh, N. Structure, organization and evolution of ADP-ribosylation factors in rice and foxtail millet, and their expression in rice. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Saidi, M.N.; Mergby, D.; Brini, F. Identification and expression analysis of the NAC transcription factor family in durum wheat (Triticum turgidum L. ssp. durum). Plant Physiol. Biochem. 2017, 112, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Gao, M.; Huang, L.; Wang, Y.; van Nocker, S.; Wan, R.; Guo, C.; Wang, X.; Gao, H. Identification and expression analysis of the apple (Malus × domestica) basic helix-loop-helix transcription factor family. Sci. Rep. 2017, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.L.; Cheng, Q.; Zhao, L.; Mao, A.; Yang, J.; Yu, S.; Weng, Y.; Xu, Y. Identification and characterisation of Dof transcription factors in the cucumber genome. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Hu, W.; Yang, H.; Yan, Y.; Wei, Y.; Tie, W.; Ding, Z.; Zuo, J.; Peng, M.; Li, K. Genome-wide characterization and analysis of bZIP transcription factor gene family related to abiotic stress in cassava. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Ding, B. The Biology of Viroid-Host Interactions. Annu. Rev. Phytopathol. 2009, 47, 105–131. [Google Scholar] [CrossRef]
- Hernández, C.; Flores, R.; de Alba, A.E.M.; Daròs, J.-A.; Serio, F. Di Viroids and Viroid-Host Interactions. Annu. Rev. Phytopathol. 2005, 43, 117–139. [Google Scholar] [CrossRef]
- Wilson, C.R.; Hay, F.S.; Eastwell, K.C.; Pethybridge, S.J.; Barbara, D.J. Viruses and Viroids Infecting Hop: Significance, Epidemiology, and Management. Plant Dis. 2008, 92, 324–338. [Google Scholar] [CrossRef]
- Pokorn, T.; Radišek, S.; Javornik, B.; Štajner, N.; Jakše, J. Development of hop transcriptome to support research into host-viroid interactions. PLoS ONE 2017, 12, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Jakse, J.; Radisek, S.; Pokorn, T.; Matousek, J.; Javornik, B. Deep-sequencing revealed Citrus bark cracking viroid (CBCVd) as a highly aggressive pathogen on hop. Plant Pathol. 2015, 64, 831–842. [Google Scholar] [CrossRef]
- Mishra, A.K.; Duraisamy, G.S.; Matoušek, J.; Radisek, S.; Javornik, B.; Jakse, J. Identification and characterization of microRNAs in Humulus lupulus using high-throughput sequencing and their response to Citrus bark cracking viroid (CBCVd) infection. BMC Genomics 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Mishra, K.A.; Kumar, A.; Mishra, D.; Nath, S.V.; Jakše, J.; Kocábek, T.; Killi, K.U.; Morina, F.; Matoušek, J. Genome-Wide Transcriptomic Analysis Reveals Insights into the Response to Citrus bark cracking viroid (CBCVd) in Hop (Humulus lupulus L.). Viruses 2018, 10, 570. [Google Scholar] [CrossRef]
- Natsume, S.; Takagi, H.; Shiraishi, A.; Murata, J.; Toyonaga, H.; Patzak, J.; Takagi, M.; Yaegashi, H.; Uemura, A.; Mitsuoka, C.; et al. The draft genome of hop (Humulus lupulus), an essence for brewing. Plant Cell Physiol. 2015, 56, 428–441. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Sinharoy, S.; Udvardi, M.; Zhao, P.X. PlantTFcat: An online plant transcription factor and transcriptional regulator categorization and analysis tool. BMC Bioinform. 2013, 14. [Google Scholar] [CrossRef]
- Yu, C.-S.; Chen, Y.-C.; Lu, C.-H.; Hwang, J.-K. Prediction of protein subcellular localization. Proteins Struct. Funct. Bioinforma. 2006, 64, 643–651. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S. Blast2GO: A Comprehensive Suite for Functional Analysis in Plant Genomics. Int. J. Plant Genom. 2008, 2008, 1–12. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; Von Mering, C.; Bork, P. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. EGGNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef]
- Morris, J.H.; Huerta-Cepas, J.; Junge, A.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Lyon, D.; Gable, A.L.; Wyder, S.; Simonovic, M.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2018, 47, D607–D613. [Google Scholar] [CrossRef]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Marshall, O.J. PerlPrimer: Cross-platform, graphical primer design for standard, bisulphite and real-time PCR. Bioinformatics 2004, 20, 2471–2472. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Štajner, N.; Cregeen, S.; Javornik, B. Evaluation of reference genes for RT-qPCR expression studies in hop (Humulus lupulus L.) during infection with vascular pathogen verticillium albo-atrum. PLoS ONE 2013, 8, e68228. [Google Scholar] [CrossRef]
- McGrath, K.C.; Dombrecht, B.; Manners, J.M.; Schenk, P.M.; Edgar, C.I.; Maclean, D.J.; Scheible, W.-R.; Udvardi, M.K.; Kazan, K. Repressor- and Activator-Type Ethylene Response Factors Functioning in Jasmonate Signaling and Disease Resistance Identified via a Genome-Wide Screen of Arabidopsis Transcription Factor Gene Expression. Plant Physiol. 2005, 139, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.; Choi, D. Functional studies of transcription factors involved in plant defenses in the genomics era. Brief. Funct. Genom. 2015, 14, 260–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakuma, Y.; Liu, Q.; Dubouzet, J.G.; Abe, H.; Shinozaki, K.; Yamaguchi- Shinozaki, K. DNA-binding specificity of the ERF/AP2 domain of Arabidopsis DREBs, transcription factors involved in dehydration- and cold-inducible gene expression. Biochem. Biophys. Res. Commun. 2002, 290, 998–1009. [Google Scholar] [CrossRef]
- Eulgem, T. Regulation of the Arabidopsis defense transcriptome. Trends Plant Sci. 2005, 10, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Nevo, E.; Sun, D.; Peng, J. Phylogenetic analyses unravel the evolutionary history of nac proteins in plants. Evolution 2012, 66, 1833–1848. [Google Scholar] [CrossRef]
- Buscaill, P.; Rivas, S. Transcriptional control of plant defence responses. Curr. Opin. Plant Biol. 2014, 20, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Riechmann, J.L.; Heard, J.; Martin, G.; Reuber, L.; Jiang, C.-Z.; Keddie, J.; Adam, L.; Pineda, O.; Ratcliffe, O.J.; Samaha, R.R.; et al. Transcription Factors: Genome-Wide Comparative Analysis Among Eukaryotes. Science 2000, 290, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Suzuki, K.; Fujimura, T.; Shinshi, H. Genome-Wide Analysis of the ERF Gene Family in Arabidopsis and Rice. Plant Physiol. 2006, 140, 411–432. [Google Scholar] [CrossRef]
- Fan, K.; Wang, M.; Miao, Y.; Ni, M.; Bibi, N.; Yuan, S.; Li, F.; Wang, X. Molecular evolution and expansion analysis of the NAC transcription factor in Zea mays. PLoS ONE 2014, 9, e111837. [Google Scholar] [CrossRef] [PubMed]
- Shiu, S.-H.; Shih, M.-C.; Li, W.-H. Transcription factor families have much higher expansion rates in plants than in animals. Plant Physiol. 2005, 139, 18–26. [Google Scholar] [CrossRef]
- Wen, J.; Zhang, J.-Q.; Nie, Z.-L.; Zhong, Y.; Sun, H. Evolutionary diversifications of plants on the Qinghai-Tibetan Plateau. Front. Genet. 2014, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Kaessmann, H. Origins, evolution, and phenotypic impact of new genes. Genome Res. 2010, 20, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Muthamilarasan, M.; Khandelwal, R.; Yadav, C.B.; Bonthala, V.S.; Khan, Y.; Prasad, M. Identification and molecular characterization of MYB Transcription Factor Superfamily in C4 model plant foxtail millet (Setaria italica L.). PLoS ONE 2014, 9, e109920. [Google Scholar] [CrossRef]
- Tompa, P.; Davey, N.E.; Gibson, T.J.; Babu, M.M. A Million peptide motifs for the molecular biologist. Mol. Cell 2014, 55, 161–169. [Google Scholar] [CrossRef]
- Wu, K.; Wu, K.; Guo, Z.; Guo, Z.; Wang, H.; Wang, H.; Li, J.; Li, J. The WRKY Family of Transcription Factors in Rice and. Gene 2005, 26, 9–26. [Google Scholar] [CrossRef]
- Xie, T.; Chen, C.; Li, C.; Liu, J.; Liu, C.; He, Y. Genome-wide investigation of WRKY gene family in pineapple: Evolution and expression profiles during development and stress. BMC Genom. 2018, 19, 1–18. [Google Scholar] [CrossRef]
- Nath, V.S.; Koyyappurath, S.; Alex, T.E.; Geetha, K.A.; Augustine, L.; Nasser, A.; Thomas, G. Transcriptome-based mining and expression profiling of Pythium responsive transcription factors in Zingiber sp. Funct. Integr. Genom. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A genomic perspective on genomic families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef]
- Thornton, J.W.; DeSalle, R. Gene Family Evolution and Homology: Genomics Meets Phylogenetics. Annu. Rev. Genom. Hum. Genet. 2000, 1, 41–73. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Jeong, S.S. Functional prediction: Identification of protein orthologs and paralogs. Protein Sci. 2000, 9, 2344–2353. [Google Scholar] [CrossRef]
- Jiang, Z.; Dong, X.; Zhang, Z. Network-Based Comparative Analysis of Arabidopsis Immune Responses to Golovinomyces orontii and Botrytis cinerea Infections. Sci. Rep. 2016, 6, 19149. [Google Scholar] [CrossRef]
- Reményi, A.; Good, M.C.; Bhattacharyya, R.P.; Lim, W.A. The Role of Docking Interactions in Mediating Signaling Input, Output, and Discrimination in the Yeast MAPK Network. Mol. Cell 2005, 20, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.S.; Dadalto, S.P.; Gonçalves, A.B.; de Souza, G.B.; Barros, V.A.; Fietto, L.G. Transcription Factor Functional Protein-Protein Interactions in Plant Defense Responses. Proteomes 2014, 2, 85–106. [Google Scholar] [CrossRef] [Green Version]
- Geisler-Lee, J.; O’Toole, N.; Ammar, R.; Provart, N.J.; Millar, A.H.; Geisler, M. A predicted interactome for Arabidopsis. Plant Physiol. 2007, 145, 317–329. [Google Scholar] [CrossRef]
- Musungu, B.; Bhatnagar, D.; Brown, R.L.; Fakhoury, A.M.; Geisler, M. A predicted protein interactome identifies conserved global networks and disease resistance subnetworks in maize. Front. Genet. 2015, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Yang, Y.; Zhou, Y.; Zhou, J.; Fan, B.; Yu, J.-Q.; Chen, Z. Protein–Protein Interactions in the Regulation of WRKY Transcription Factors. Mol. Plant 2013, 6, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Owens, R.A.; Tech, K.B.; Shao, J.Y.; Sano, T.; Baker, C.J. Global Analysis of Tomato Gene Expression During Potato spindle tuber viroid Infection Reveals a Complex Array of Changes Affecting Hormone Signaling. Mol. Plant-Microbe Interact. 2012, 25, 582–598. [Google Scholar] [CrossRef]
- Katsarou, K.; Wu, Y.; Zhang, R.; Bonar, N.; Morris, J.; Hedley, P.E.; Bryan, G.J.; Kalantidis, K.; Hornyik, C. Insight on Genes Affecting Tuber Development in Potato upon Potato spindle tuber viroid (PSTVd) Infection. PLoS One 2016, 11, e0150711. [Google Scholar] [CrossRef] [Green Version]
- Więsyk, A.; Iwanicka-Nowicka, R.; Fogtman, A.; Zagórski-Ostoja, W.; Góra-Sochacka, A. Time-Course Microarray Analysis Reveals Differences between Transcriptional Changes in Tomato Leaves Triggered by Mild and Severe Variants of Potato Spindle Tuber Viroid. Viruses 2018, 10, 257. [Google Scholar] [CrossRef]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef]
- Asai, T.; Tena, G.; Plotnikova, J.; Willmann, M.R.; Chiu, W.-L.; Gomez-Gomez, L.; Boller, T.; Ausubel, F.M.; Sheen, J. MAP kinase signalling cascade in Arabidopsis innate immunity. Nature 2002, 415, 977. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, Y.; Ding, B.; Fei, Z. Comprehensive Transcriptome Analyses Reveal that Potato Spindle Tuber Viroid Triggers Genome-Wide Changes in Alternative Splicing, Inducible trans-Acting Activity of Phased Secondary Small Interfering RNAs, and Immune Responses. J. Virol. 2017, 91, e00247-17. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, A.M.; Amil-Ruiz, F.; Muñoz-Blanco, J.; Encinas-Villarejo, S.; Caballero, J.L.; de los Santos, B.; Romero, F.; Pliego-Alfaro, F. Evidence for a positive regulatory role of strawberry (Fragaria×ananassa) Fa WRKY1 and Arabidopsis At WRKY75 proteins in resistance. J. Exp. Bot. 2009, 60, 3043–3065. [Google Scholar] [CrossRef] [Green Version]
- Bhattarai, K.K.; Atamian, H.S.; Kaloshian, I.; Eulgem, T. WRKY72-type transcription factors contribute to basal immunity in tomato and Arabidopsis as well as gene-for-gene resistance mediated by the tomato R gene Mi-1. Plant J. 2010, 63, 229–240. [Google Scholar] [CrossRef]
- Shi, W.; Hao, L.; Li, J.; Liu, D.; Guo, X.; Li, H. The Gossypium hirsutum WRKY gene GhWRKY39-1 promotes pathogen infection defense responses and mediates salt stress tolerance in transgenic Nicotiana benthamiana. Plant Cell Rep. 2014, 33, 483–498. [Google Scholar] [CrossRef]
- YANG, S.; ZHOU, L.; MIAO, L.; SHI, J.; SUN, C.; FAN, W.; LAN, J.; CHEN, H.; LIU, L.; DOU, S.; et al. The expression and binding properties of the rice WRKY68 protein in the Xa21-mediated resistance response to Xanthomonas oryzae pv. Oryzae. J. Integr. Agric. 2016, 15, 2451–2460. [Google Scholar] [CrossRef]
- Fan, S.; Dong, L.; Han, D.; Zhang, F.; Wu, J.; Jiang, L.; Cheng, Q.; Li, R.; Lu, W.; Meng, F.; et al. GmWRKY31 and GmHDL56 Enhances Resistance to Phytophthora sojae by Regulating Defense-Related Gene Expression in Soybean. Front. Plant Sci. 2017, 8, 781. [Google Scholar] [CrossRef]
- Shearer, H.L.; Wang, L.; DeLong, C.; Despres, C.; Fobert, P.R. NPR1 enhances the DNA binding activity of the Arabidopsis bZIP transcription factor TGA7This paper is one of a selection of papers published in a Special Issue from the National Research Council of Canada – Plant Biotechnology Institute. Botany 2009, 87, 561–570. [Google Scholar] [CrossRef]
- Kan, J.; Liu, T.; Ma, N.; Li, H.; Li, X.; Wang, J.; Zhang, B.; Chang, Y.; Lin, J. Transcriptome analysis of Callery pear (Pyrus calleryana) reveals a comprehensive signalling network in response to Alternaria alternata. PLoS ONE 2017, 12, e0184988. [Google Scholar] [CrossRef]
- Berens, M.L.; Berry, H.M.; Mine, A.; Argueso, C.T.; Tsuda, K. Evolution of Hormone Signaling Networks in Plant Defense. Annu. Rev. Phytopathol. 2017, 55, 401–425. [Google Scholar] [CrossRef] [PubMed]
- Dang, F.; Wang, Y.; She, J.; Lei, Y.; Liu, Z.; Eulgem, T.; Lai, Y.; Lin, J.; Yu, L.; Lei, D.; et al. Overexpression of CaWRKY27, a subgroup IIe WRKY transcription factor of Capsicum annuum, positively regulates tobacco resistance to Ralstonia solanacearum infection. Physiol. Plant. 2014, 150, 397–411. [Google Scholar] [CrossRef]
- Li, J.; Brader, G.; Kariola, T.; Tapio Palva, E. WRKY70 modulates the selection of signaling pathways in plant defense. Plant J. 2006, 46, 477–491. [Google Scholar] [CrossRef] [Green Version]
- Phukan, U.J.; Jeena, G.S.; Shukla, R.K. WRKY Transcription Factors: Molecular Regulation and Stress Responses in Plants. Front. Plant Sci. 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Cai, X.-T.; Xu, P.; Zhao, P.-X.; Liu, R.; Yu, L.-H.; Xiang, C.-B. Arabidopsis ERF109 mediates cross-talk between jasmonic acid and auxin biosynthesis during lateral root formation. Nat. Commun. 2014, 5, 5833. [Google Scholar] [CrossRef]
- Scheideler, M.; Schlaich, N.L.; Fellenberg, K.; Beissbarth, T.; Hauser, N.C.; Vingron, M.; Slusarenko, A.J.; Hoheisel, J.D. Monitoring the Switch from Housekeeping to Pathogen Defense Metabolism in Arabidopsis thaliana Using cDNA Arrays. J. Biol. Chem. 2002, 277, 10555–10561. [Google Scholar] [CrossRef]
- Windram, O.; Madhou, P.; McHattie, S.; Hill, C.; Hickman, R.; Cooke, E.; Jenkins, D.J.; Penfold, C.A.; Baxter, L.; Breeze, E.; et al. Arabidopsis Defense against Botrytis cinerea: Chronology and Regulation Deciphered by High-Resolution Temporal Transcriptomic Analysis. Plant Cell 2012, 24, 3530–3557. [Google Scholar] [CrossRef]
- Shi, J.X.; Malitsky, S.; de Oliveira, S.; Branigan, C.; Franke, R.B.; Schreiber, L.; Aharoni, A. SHINE transcription factors act redundantly to pattern the archetypal surface of arabidopsis flower organs. PLoS Genet. 2011, 7. [Google Scholar] [CrossRef]
- Hasson, A.; Plessis, A.; Blein, T.; Adroher, B.; Grigg, S.; Tsiantis, M.; Boudaoud, A.; Damerval, C.; Laufs, P. Evolution and Diverse Roles of the CUP-SHAPED COTYLEDON Genes in Arabidopsis Leaf Development. Plant Cell 2011, 23, 54–68. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcription Factor Families | Count | Transcription Factor Families | Count | Transcription Factor Families | Count |
|---|---|---|---|---|---|
| A20-like | 12 | HD-ZIP | 14 | STY-LRP1 | 8 |
| ABTB | 5 | HMG | 12 | SWIB-Plus-3 | 5 |
| AP2-EREBP* | 140 | Homeodomain-LIKE | 9 | TAZ | 2 |
| ARF | 20 | Homeodomain-PHD | 1 | TCP | 16 |
| ARID | 6 | Homeodomain-TALE-BEL | 11 | Tc-PD | 3 |
| ARID-HMG | 3 | Homeodomain-TALE-KNOX | 9 | Tesmin | 2 |
| AS2-LOB | 48 | Homobox-WOX | 82 | TIFY | 12 |
| AUX-IAA | 21 | HSF-type-DNA-binding | 19 | TTF-type(Zn) | 18 |
| B3-Domain | 48 | ISWI | 1 | TUBBY | 8 |
| BED-type(Zn) | 23 | JmjC | 20 | WD40-like | 276 |
| BES/BZR | 8 | JmjC-ARID | 1 | WRKY* | 66 |
| bHLH* | 106 | JmjN | 7 | YEATS | 3 |
| Bromodomain | 22 | JUMONJI | 7 | ZF-HD | 11 |
| BTB-POZ | 32 | Lambda-DB | 4 | Znf-B | 28 |
| BTB-POZ-MATH | 5 | LFY | 1 | Znf-LSD | 7 |
| bZIP* | 64 | LIM | 16 | Grand Total | 3818 |
| C2C2-CO-like | 27 | LisH | 21 | ||
| C2C2-Dof | 26 | MADS-MIKC | 17 | ||
| C2C2-GATA | 24 | MADS-type1 | 30 | ||
| C2C2-YABBY | 8 | MYB* | 16 | ||
| C2H2 | 428 | MYB/SANT | 24 | ||
| C3H | 67 | MYB-HB-like | 218 | ||
| C3H-WRC/GRF | 17 | MYB-related | 1 | ||
| CCHC(Zn) | 893 | NAC* | 74 | ||
| CG1-CAMTA | 5 | Nin-like | 6 | ||
| CHROMO-DOMAIN | 114 | NOZZLE | 1 | ||
| CW-Zn | 6 | PAZ-Argonaute | 11 | ||
| CW-Zn-B3/VAL | 2 | PHD | 107 | ||
| DDT | 7 | PLATZ | 13 | ||
| E2F-DP | 7 | RAV | 4 | ||
| EIL | 6 | RB | 1 | ||
| FAR | 39 | RR-A-type | 31 | ||
| FHA-SMAD | 19 | RR-B-type | 4 | ||
| FYR | 6 | S1Fa-like | 1 | ||
| GAGA-Binding-like | 4 | SAP | 9 | ||
| GARP-G2-like | 6 | SBP | 23 | ||
| GeBP | 2 | SET | 29 | ||
| GRAS | 39 | SNF2 | 41 | ||
| GRF | 7 | SPK | 1 | ||
| Hap2/NF-YA | 9 | ssDNA-binding-TF | 8 | ||
| Hap3/NF-YB | 102 | SSXT | 2 | ||
| HD-SAD | 12 | STAT | 1 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sukumari Nath, V.; Kumar Mishra, A.; Kumar, A.; Matoušek, J.; Jakše, J. Revisiting the Role of Transcription Factors in Coordinating the Defense Response Against Citrus Bark Cracking Viroid Infection in Commercial Hop (Humulus Lupulus L.). Viruses 2019, 11, 419. https://doi.org/10.3390/v11050419
Sukumari Nath V, Kumar Mishra A, Kumar A, Matoušek J, Jakše J. Revisiting the Role of Transcription Factors in Coordinating the Defense Response Against Citrus Bark Cracking Viroid Infection in Commercial Hop (Humulus Lupulus L.). Viruses. 2019; 11(5):419. https://doi.org/10.3390/v11050419
Chicago/Turabian StyleSukumari Nath, Vishnu, Ajay Kumar Mishra, Atul Kumar, Jaroslav Matoušek, and Jernej Jakše. 2019. "Revisiting the Role of Transcription Factors in Coordinating the Defense Response Against Citrus Bark Cracking Viroid Infection in Commercial Hop (Humulus Lupulus L.)" Viruses 11, no. 5: 419. https://doi.org/10.3390/v11050419