Application of CRISPR/Cas9 Gene Editing System on MDV-1 Genome for the Study of Gene Function

 ,
, 
Abstract
:1. Introduction
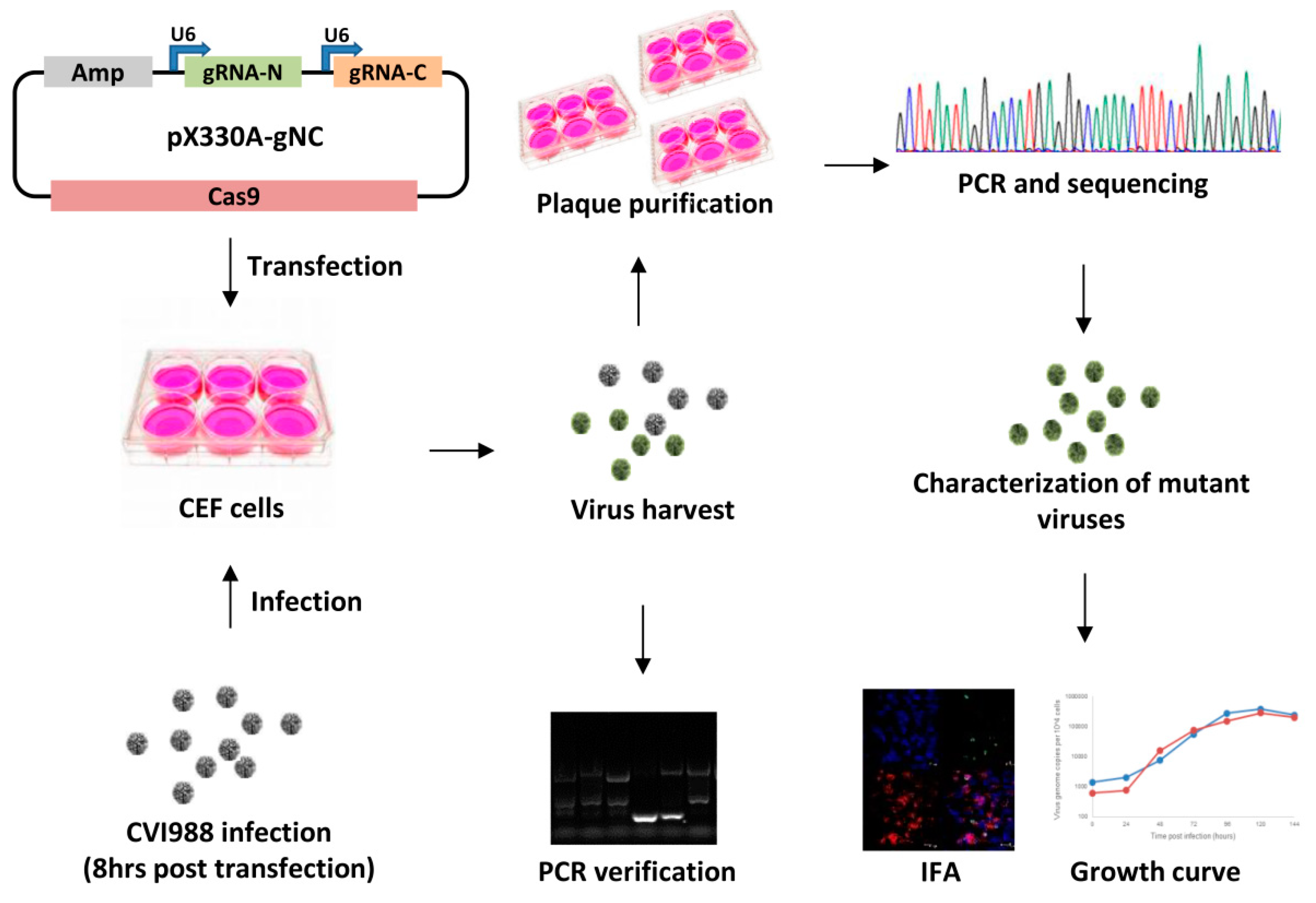
2. Materials and Methods
2.1. Cell Culture and Viruses
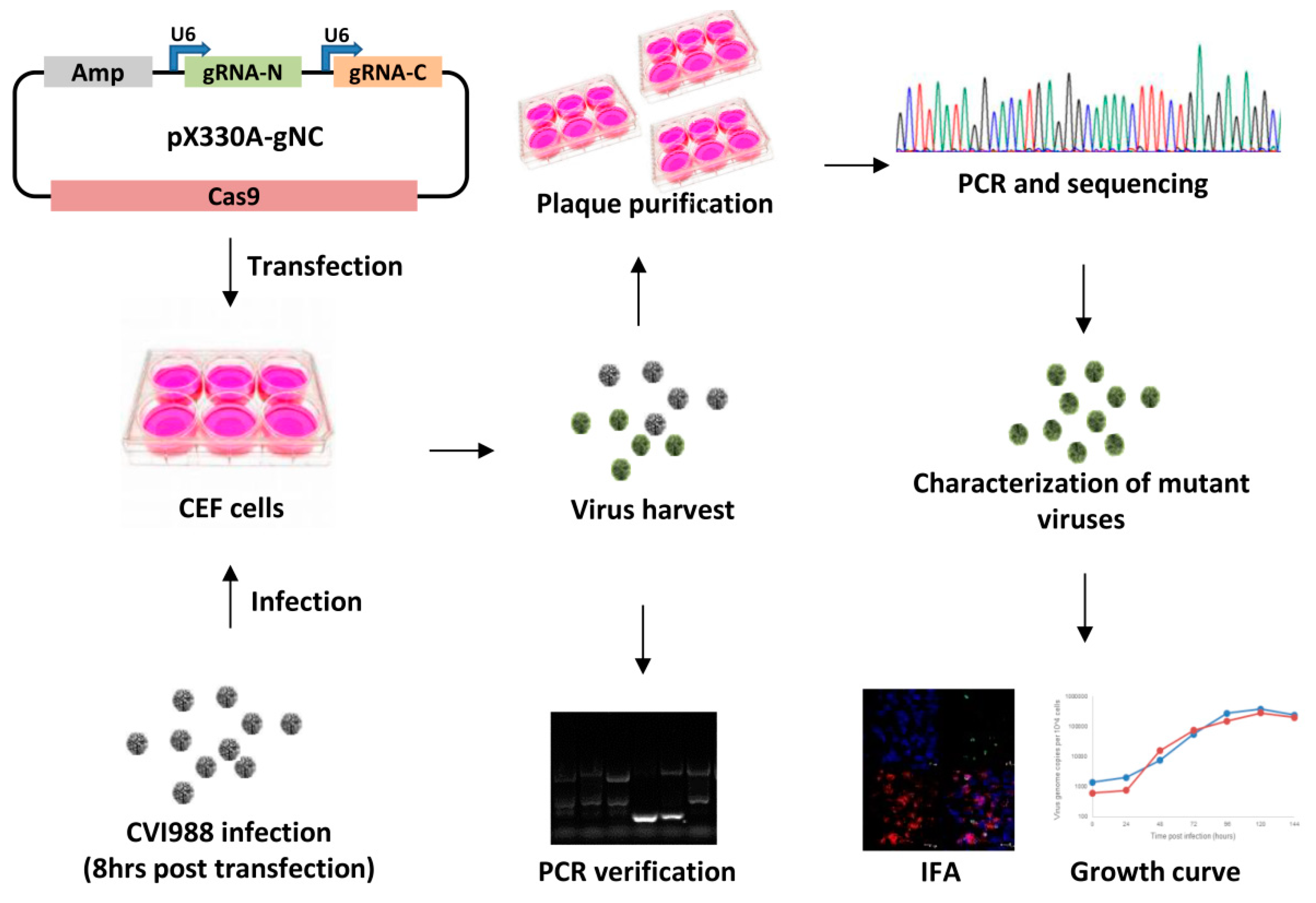
2.2. Construction of Guide RNA Constructs
2.3. Generation of Meq and pp38 Gene Deletion CVI988 Virus
2.4. Characterization of Gene Knockout CV988 Virus
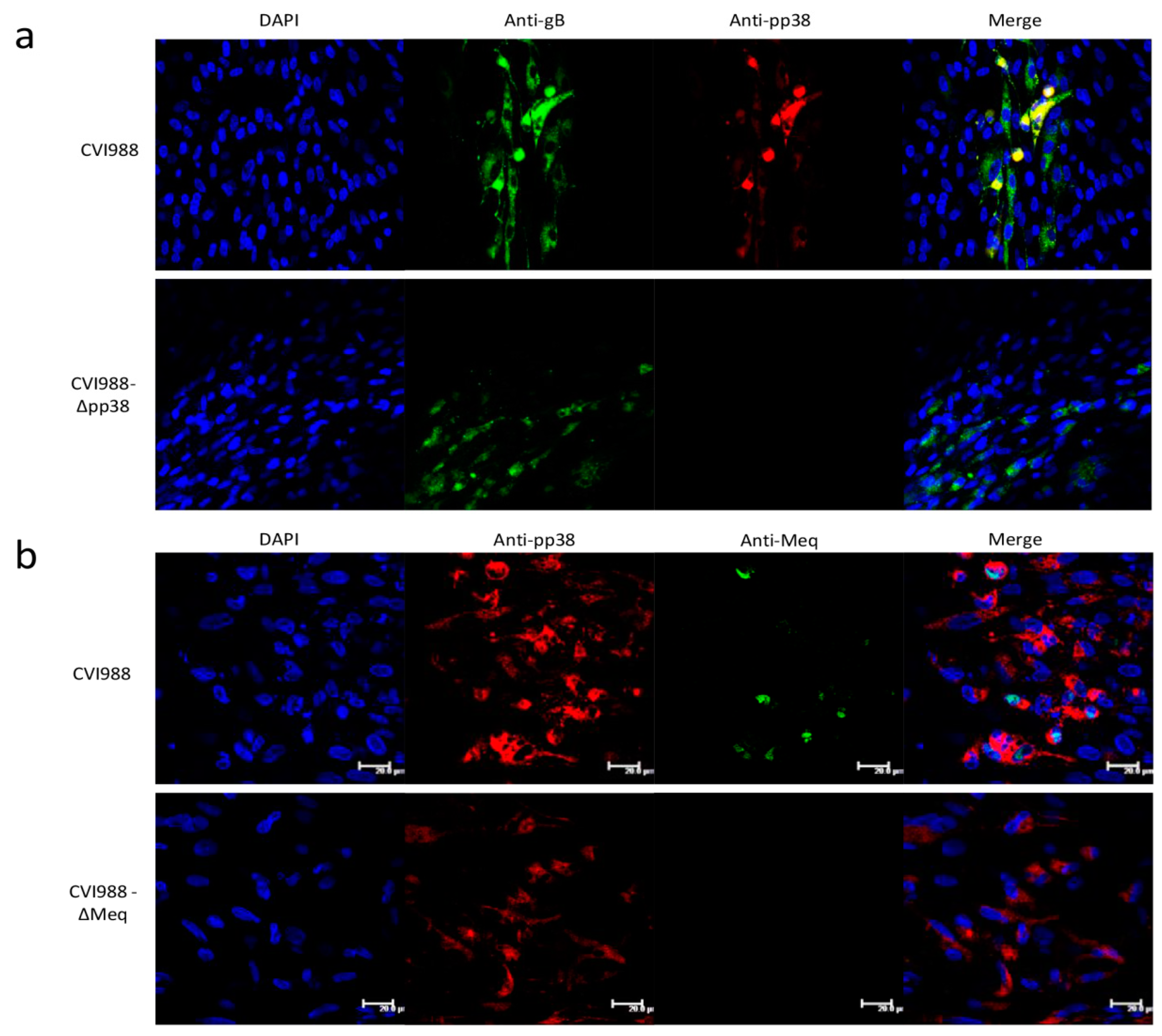
2.5. Immunofluorescence Assay (IFA)
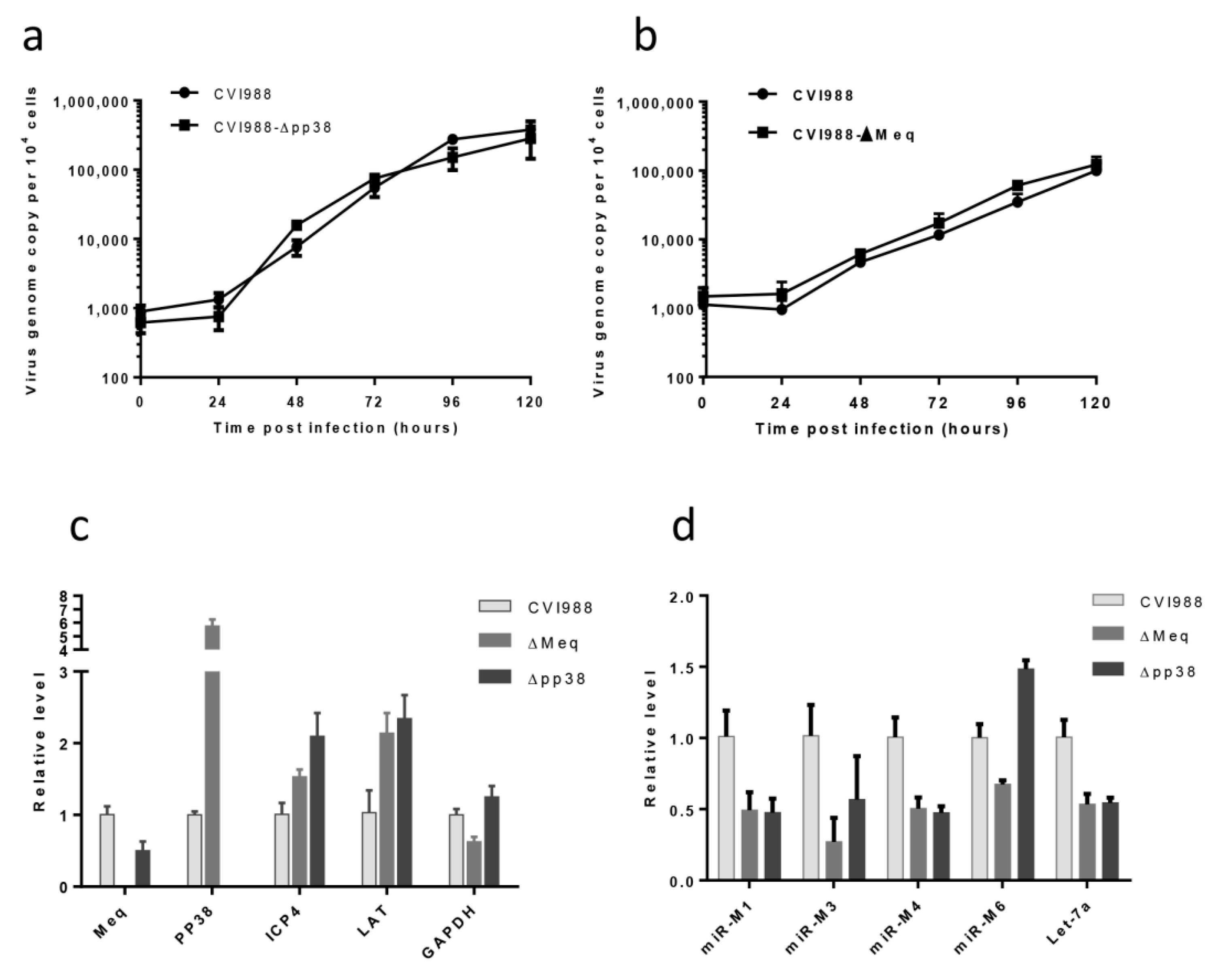
2.6. The Growth Kinetics of the Gene Knockout Viruses
2.7. qRT-PCR for MDV Transcripts and miRNAs
3. Results
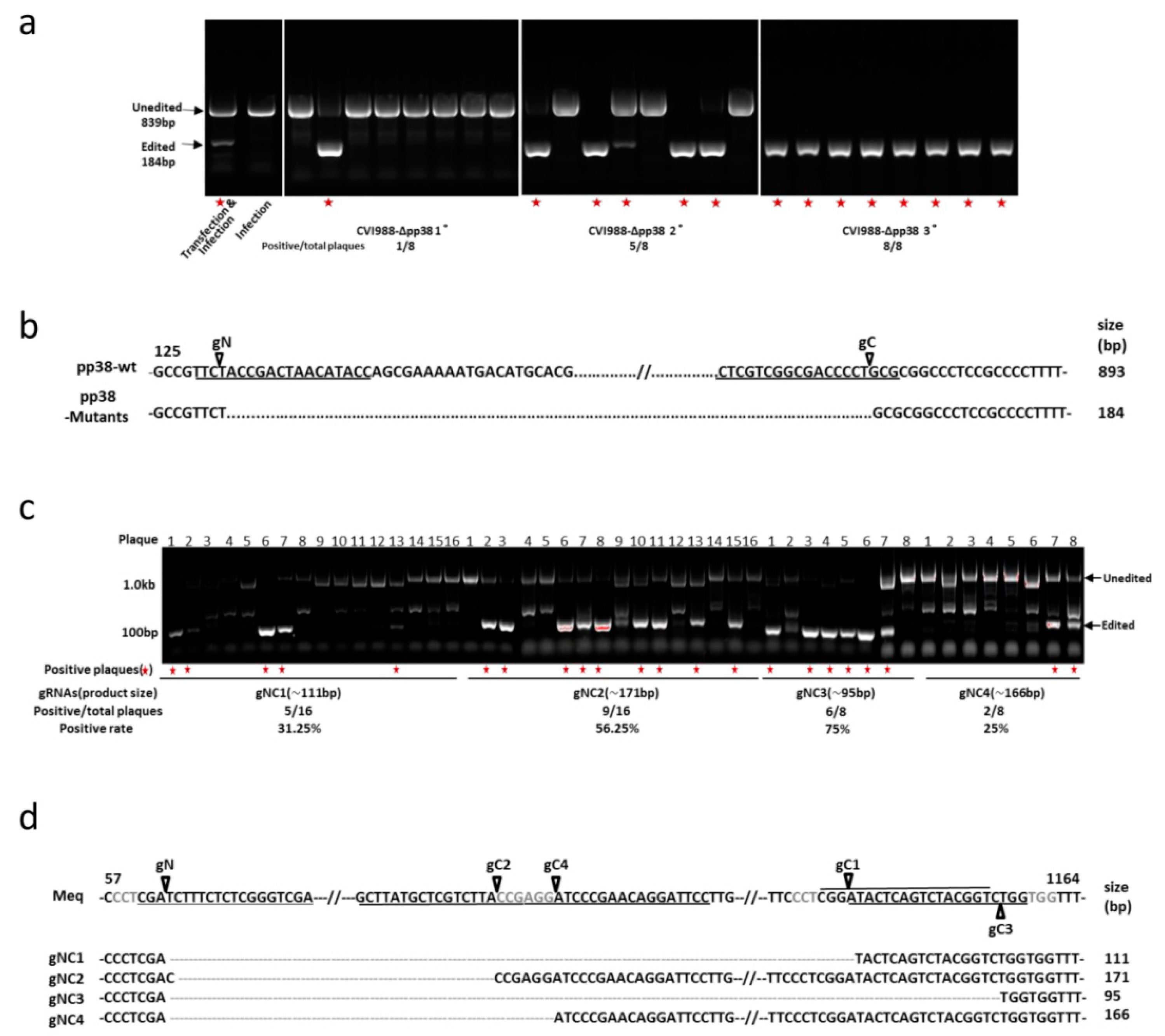
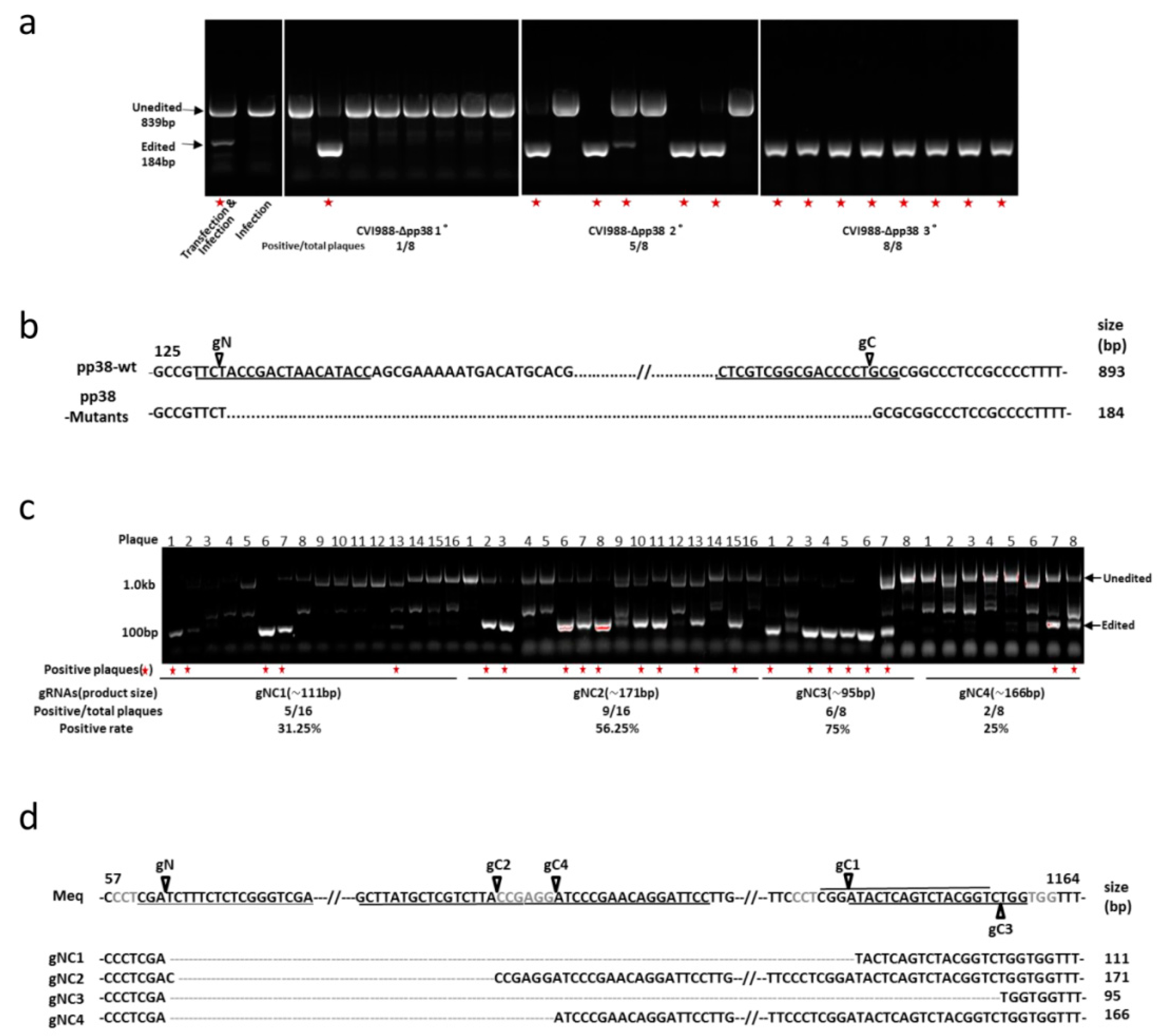
3.1. Construction of pp38 Deletion Mutant CVI988-Δpp38 Using CRISPR/Cas9 System
3.2. Construction of Meq Deletion Mutant CVI988-ΔMeq Using CRISPR/Cas9 System
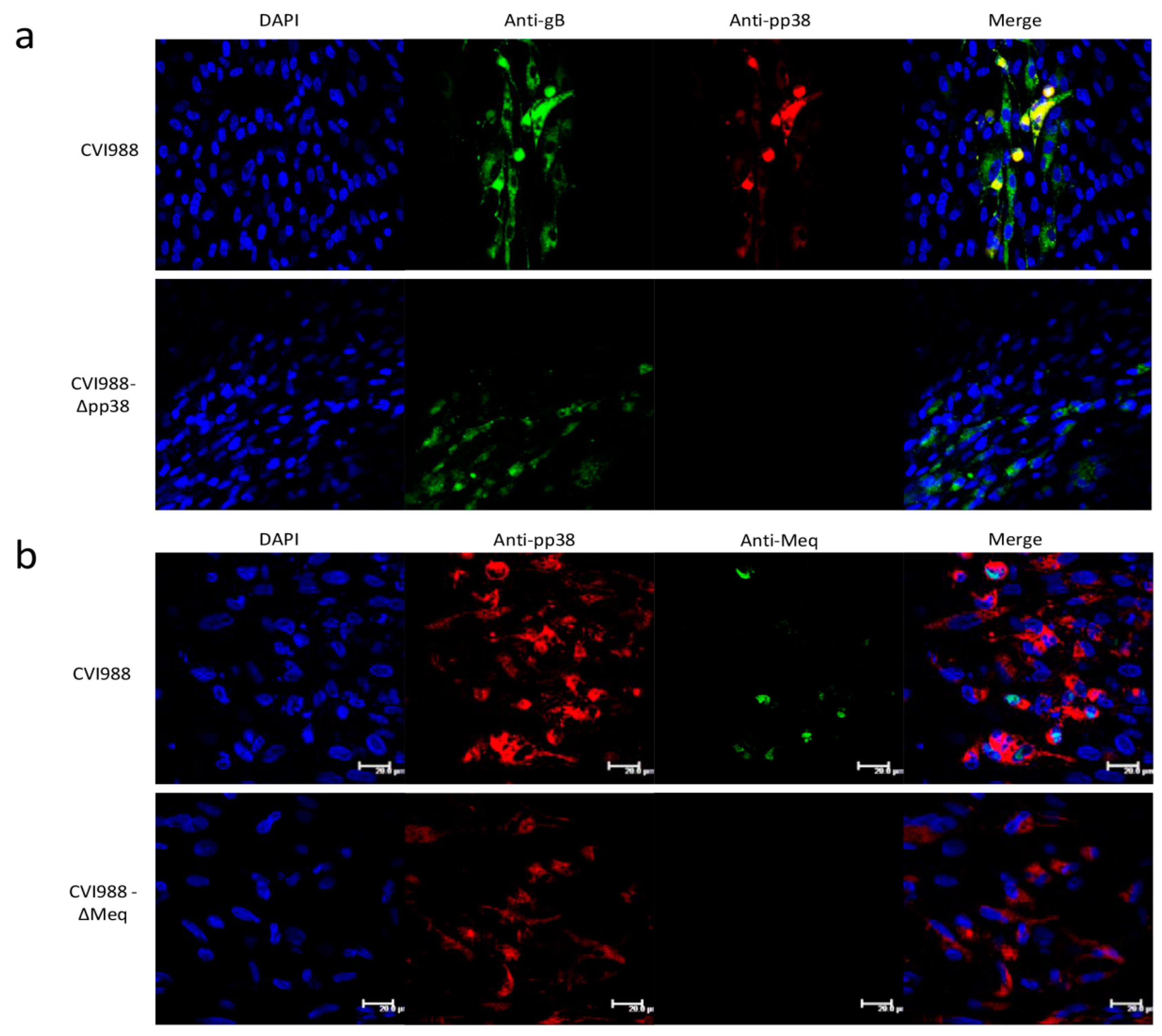
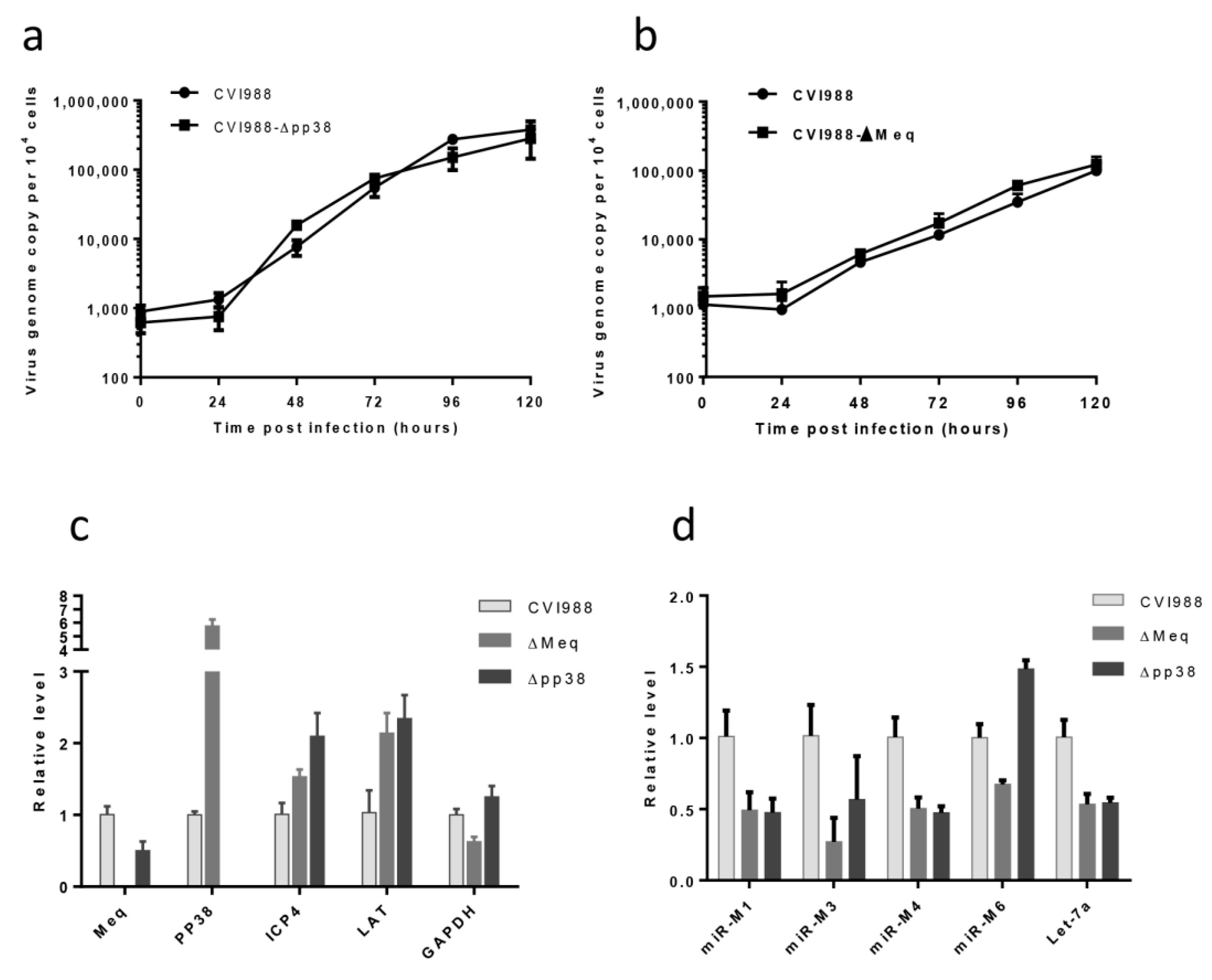
3.3. Characterization of Mutant CVI988 Viruses
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Okazaki, W.; Purchase, H.G.; Burmester, B.R. Protection against mareks disease by vaccination with a herpesvirus of turkeys. Avian Dis. 1970, 14, 413–429. [Google Scholar] [CrossRef] [PubMed]
- Schat, K.A.; Calnek, B.W. Characterization of an apparently nononcogenic mareks-disease virus. J. Natl. Cancer Inst. 1978, 60, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Rispens, B.H.; Vanvlote, H.; Schat, K.A.; Mastenbroek, N.; Maas, H.J.L. Control of mareks-disease in netherlands 1. Isolation of an avirulent mareks-disease virus (strain cvi 988) and its use in laboratory vaccination trials. Avian Dis. 1972, 16, 108. [Google Scholar] [CrossRef] [PubMed]
- Fragnet, L.; Blasco, M.A.; Klapper, W.; Rasschaert, D. The rna subunit of telomerase is encoded by marek’s disease virus. J. Virol. 2003, 77, 5985–5996. [Google Scholar] [CrossRef] [PubMed]
- Trapp, S.; Parcells, M.S.; Kamil, J.P.; Schumacher, D.; Tischer, B.K.; Kumar, P.M.; Nair, V.K.; Osterrieder, N. A virus-encoded telomerase rna promotes malignant t cell lymphomagenesis. J. Exp. Med. 2006, 203, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Cortes, P.L.; Cardona, C.J. Pathogenesis of a marek’s disease virus mutant lacking vil-8 in resistant and susceptible chickens. Avian Dis. 2004, 48, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Lee, L.F.; Hunt, H.D.; Reed, W.M.; Lupiani, B.; Reddy, S.M. A marek’s disease virus vil-8 deletion mutant has attenuated virulence and confers protection against challenge with a very virulent plus strain. Avian Dis. 2005, 49, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Lee, L.F.; Reed, W.M.; Kung, H.J.; Reddy, S.M. Marek’s disease virus-encoded vil-8 gene is involved in early cytolytic infection but dispensable for establishment of latency. J. Virol. 2004, 78, 4753–4760. [Google Scholar] [CrossRef] [PubMed]
- Parcells, M.S.; Lin, S.F.; Dienglewicz, R.L.; Majerciak, V.; Robinson, D.R.; Chen, H.C.; Wu, Z.; Dubyak, G.R.; Brunovskis, P.; Hunt, H.D.; et al. Marek’s disease virus (mdv) encodes an interleukin-8 homolog (vil-8): Characterization of the vil-8 protein and a vil-8 deletion mutant mdv. J. Virol. 2001, 75, 5159–5173. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.C.; Baigent, S.J.; Smith, L.P.; Chattoo, J.P.; Petherbridge, L.J.; Hawes, P.; Allday, M.J.; Nair, V. Interaction of MEQ protein and C-terminal-binding protein is critical for induction of lymphomas by marek’s disease virus. Proc. Natl. Acad. Sci. USA 2006, 103, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Lupiani, B.; Lee, L.F.; Cui, X.; Gimeno, I.; Anderson, A.; Morgan, R.W.; Silva, R.F.; Witter, R.L.; Kung, H.J.; Reddy, S.M. Marek’s disease virus-encoded meq gene is involved in transformation of lymphocytes but is dispensable for replication. Proc. Natl. Acad. Sci. USA 2004, 101, 11815–11820. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, A.; Su, S.; Zhao, P.; Cui, Z.; Zhu, H. Deletion of the MEQ gene significantly decreases immunosuppression in chickens caused by pathogenic marek’s disease virus. Virol. J. 2011, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.Z.; Lee, L.F.; Liu, J.L.; Kung, H.J. Structural analysis and transcriptional mapping of the marek’s disease virus gene encoding pp38, an antigen associated with transformed cells. J. Virol. 1991, 65, 6509–6515. [Google Scholar] [PubMed]
- Reddy, S.M.; Lupiani, B.; Gimeno, I.M.; Silva, R.F.; Lee, L.F.; Witter, R.L. Rescue of a pathogenic marek’s disease virus with overlapping cosmid dnas: Use of a pp38 mutant to validate the technology for the study of gene function. Proc. Natl. Acad. Sci. USA 2002, 99, 7054–7059. [Google Scholar] [CrossRef] [PubMed]
- Jarosinski, K.W.; Osterrieder, N.; Nair, V.K.; Schat, K.A. Attenuation of marek’s disease virus by deletion of open reading frame RLORF4 but not RLORF5a. J. Virol. 2005, 79, 11647–11659. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Lee, L.F.; Khan, O.A.; Heidari, M.; Zhang, H.; Lupiani, B.; Reddy, S.M. Deletion of marek’s disease virus large subunit of ribonucleotide reductase impairs virus growth in vitro and in vivo. Avian Dis. 2013, 57, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Dorange, F.; Tischer, B.K.; Vautherot, J.F.; Osterrieder, N. Characterization of marek’s disease virus serotype 1 (MDV-1) deletion mutants that lack UL46 to UL49 genes: MDV-1 UL49, encoding VP22, is indispensable for virus growth. J. Virol. 2002, 76, 1959–1970. [Google Scholar] [CrossRef] [PubMed]
- Ayyathan, D.M.; Ilic, N.; Gil-Henn, H.; Blank, M. Generation of SMURF2 knockout human cells using the CRISPR/Cas9 system. Anal. Biochem. 2017, 531, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wei, D.; Zhu, X.; Pan, J.; Zhang, P.; Huo, L.; Zhu, X. A ‘suicide’ crispr-cas9 system to promote gene deletion and restoration by electroporation in cryptococcus neoformans. Sci. Rep. 2016, 6, 31145. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Peng, J.; Yan, Y.; Cao, P.; Wang, J.; Qiu, C.; Tang, L.; Liu, D.; Tang, L.; Jin, J.; et al. Efficient gene editing in adult mouse livers via adenoviral delivery of CRISPR/Cas9. FEBS Lett. 2014, 588, 3954–3958. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Chen, W.; Ma, C.; Zhang, Z.; Zhao, P.; Du, Y.; Zhang, Y.; Duan, L.; Fang, J.; Li, S.; et al. Transcriptional activity comparison of different sites in recombinant marek’s disease virus for the expression of the H9N2 avian influenza virus hemagglutinin gene. J. Virol. Methods 2014, 207, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Suenaga, T.; Kohyama, M.; Hirayasu, K.; Arase, H. Engineering large viral DNA genomes using the CRISPR-Cas9 system. Microbiol. Immunol. 2014, 58, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.P.; Cai, X.H.; Tan, M.H.; Schaffert, S.; Arnold, C.P.; Gong, X.; Chen, C.Z.; Huang, S.L. Precise gene deletion and replacement using the CRISPR/Cas9 system in human cells. Biotechniques 2014, 57, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Garneau, J.E.; Dupuis, M.E.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadan, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Horvath, P.; Barrangou, R. Crispr/cas, the immune system of bacteria and archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.N.; Wang, Y.J.; Zuo, Q.S.; Li, D.; Zhang, W.H.; Wang, F.; Ji, Y.Q.; Jin, J.; Lu, Z.Y.; Wang, M.; et al. CRISPR/Cas9 mediated chicken stra8 gene knockout and inhibition of male germ cell differentiation. PLoS ONE 2017, 12, e0172207. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Esvelt, K.M.; Church, G.M. Cas9 as a versatile tool for engineering biology. Nat. Methods 2013, 10, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Sun, L.; Gao, D.; Ding, C.; Li, Z.; Li, Y.; Cun, W.; Li, Q. High-efficiency targeted editing of large viral genomes by RNA-guided nucleases. PLoS Pathog. 2014, 10, e1004090. [Google Scholar] [CrossRef] [PubMed]
- Bierle, C.J.; Anderholm, K.M.; Wang, J.B.; McVoy, M.A.; Schleiss, M.R. Targeted mutagenesis of guinea pig cytomegalovirus using CRISPR/Cas9-mediated gene editing. J. Virol. 2016, 90, 6989–6998. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Qin, C.; Lang, Y.; Wang, M.; Lin, M.; Li, C.; Zhang, R.; Tang, J. A simple and rapid approach to manipulate pseudorabies virus genome by CRISPR/Cas9 system. Biotechnol. Lett. 2015, 37, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Zhang, W.; Wang, J.; Al Yaghchi, C.; Ahmed, J.; Chard, L.; Lemoine, N.R.; Wang, Y. Efficiently editing the vaccinia virus genome by using the CRISPR-Cas9 system. J. Virol. 2015, 89, 5176–5179. [Google Scholar] [CrossRef] [PubMed]
- Yuen, K.S.; Chan, C.P.; Wong, N.H.; Ho, C.H.; Ho, T.H.; Lei, T.; Deng, W.; Tsao, S.W.; Chen, H.; Kok, K.H.; et al. CRISPR/Cas9-mediated genome editing of Epstein-Barr virus in human cells. J. Gen. Virol. 2015, 96, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Sun, L.; Yu, T.; Pan, Y.; Wang, D.; Hu, X.; Fu, Z.; He, Q.; Cao, G. A CRISPR/Cas9 and cre/lox system-based express vaccine development strategy against re-emerging pseudorabies virus. Sci. Rep. 2016, 6, 19176. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Huang, K.; Wei, Y.; Chen, H.; Liu, Z.; Jin, M. Construction of a highly efficient CRISPR/Cas9-mediated duck enteritis virus-based vaccine against H5N1 avian influenza virus and duck tembusu virus infection. Sci. Rep. 2017, 7, 1478. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Ouyang, T.; Pang, D.; Ma, T.; Chen, X.; Guo, N.; Chen, F.; Yuan, L.; Ouyang, H.; Ren, L. Pseudorabies virus can escape from CRISPR-Cas9-mediated inhibition. Virus Res. 2016, 223, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.D.; Liu, J.T.; Wang, T.Y.; An, T.Q.; Sun, M.X.; Wang, S.J.; Fang, Q.Q.; Hou, L.L.; Tian, Z.J.; Cai, X.H. Live attenuated pseudorabies virus developed using the CRISPR/Cas9 system. Virus Res. 2016, 225, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Bassett, A.; Nair, V. Targeted editing of avian herpesvirus vaccine vector using CRISPR/Cas9 nucleases. J. Vaccine Technol. 2016, 1–7. [Google Scholar]
- Tang, N.; Zhang, Y.; Pedrera, M.; Chang, P.; Baigent, S.; Moffat, K.; Shen, Z.; Nair, V.; Yao, Y. A simple and rapid approach to develop recombinant avian herpesvirus vectored vaccines using CRISPR/Cas9 system. Vaccine 2018, 36, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.F.; Wu, P.; Sui, D.; Ren, D.; Kamil, J.; Kung, H.J.; Witter, R.L. The complete unique long sequence and the overall genomic organization of the ga strain of marek’s disease virus. Proc. Natl. Acad. Sci. USA 2000, 97, 6091–6096. [Google Scholar] [CrossRef] [PubMed]
- Tulman, E.R.; Afonso, C.L.; Lu, Z.; Zsak, L.; Rock, D.L.; Kutish, G.F. The genome of a very virulent marek’s disease virus. J. Virol. 2000, 74, 7980–7988. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Kung, H.J. Marek’s disease virus oncogenicity: Molecular mechanisms. In Marek’s Disease: An Evolving Problem, 1st ed.; Davison, F., Nair, V., Eds.; Elsevier Academic Press: London, UK, 2004; pp. 32–48. [Google Scholar]
- Parcells, M.S.; Burnside, J.; Morgan, R.W. Marek’s disease virus-induced t-cell lymphomas. In Cancer Associated Viruses; Robertson, E.S., Ed.; Springer: New York, NY, USA, 2012; pp. 307–335. [Google Scholar]
- Gimeno, I.M.; Witter, R.L.; Hunt, H.D.; Reddy, S.M.; Lee, L.F.; Silva, R.F. The pp38 gene of marek’s disease virus (MDV) is necessary for cytolytic infection of b cells and maintenance of the transformed state but not for cytolytic infection of the feather follicle epithelium and horizontal spread of MDV. J. Virol. 2005, 79, 4545–4549. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.F.; Cui, X.P.; Cui, Z.Z.; Gimeno, I.; Lupiani, B.; Reddy, S.M. Characterization of a very virulent marek’s disease virus mutant expressing the pp38 protein from the serotype 1 vaccine strain cvi988/rispens. Virus Genes 2005, 31, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.B.; Cui, Z.Z.; Lee, L.F.; Cui, X.P.; Reddy, S.M. The role of pp38 in regulation of marek’s disease virus bi-directional promoter between pp38 and 1.8-kb mRNA. Virus Genes 2006, 32, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Baigent, S.J.; Petherbridge, L.J.; Smith, L.P.; Zhao, Y.; Chesters, P.M.; Nair, V.K. Herpesvirus of turkey reconstituted from bacterial artificial chromosome clones induces protection against marek’s disease. J. Gen. Virol. 2006, 87, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Baigent, S.J.; Petherbridge, L.J.; Howes, K.; Smith, L.P.; Currie, R.J.; Nair, V.K. Absolute quantitation of marek’s disease virus genome copy number in chicken feather and lymphocyte samples using real-time pcr. J. Virol. Methods 2005, 123, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yao, Y.; Zhao, Y.; Smith, L.P.; Baigent, S.J.; Nair, V. Analysis of the expression profiles of marek’s disease virus-encoded micrornas by real-time quantitative pcr. J. Virol. Methods 2008, 149, 201–208. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′-3′) |
|---|---|
| pp38-gN-F | CACCGGGTATGTTAGTCGGTAGAA |
| pp38-gN-R | AAACTTCTACCGACTAACATACCC |
| pp38-gC-F | CACCGCTCGTCGGCGACCCCTGCG |
| pp38-gC-R | AAACCGCAGGGGTCGCCGACGAGC |
| Meq-gN-F | CACCGCGACCCGAGAGAAAGATCG |
| Meq-gN-R | AAACCGATCTTTCTCTCGGGTCGC |
| Meq-gC1-F | CACCGCCGTAGACTGAGTATCCGA |
| Meq-gC1-R | AAACTCGGATACTCAGTCTACGGC |
| Meq-gC2-F | CACCGCTTTATGCTCGTCTTACCG |
| Meq-gC2-R | AAACCGGTAAGACGAGCATAAAGC |
| Meq-gC3-F | CACCGTACTCAGTCTACGGTCTGG |
| Meq-gC3-R | AAACCCAGACCGTAGACTGAGTAC |
| Meq-gC4-F | CACCGGAATCCTGTTCGGGATCCT |
| Meq-gC4-R | AAACAGGATCCCGAACAGGATTCC |
| pp38-N-F | GATTCCACCTCCCCAGAATCC |
| pp38-C-R | TTCGAAGCAGAACACGAAGGG |
| Meq-N-F | ATGTCTCAGGAGCCAGAG |
| Meq-C-R | TCAGGGTCTCCCGTCA |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Tang, N.; Sadigh, Y.; Baigent, S.; Shen, Z.; Nair, V.; Yao, Y. Application of CRISPR/Cas9 Gene Editing System on MDV-1 Genome for the Study of Gene Function. Viruses 2018, 10, 279. https://doi.org/10.3390/v10060279
Zhang Y, Tang N, Sadigh Y, Baigent S, Shen Z, Nair V, Yao Y. Application of CRISPR/Cas9 Gene Editing System on MDV-1 Genome for the Study of Gene Function. Viruses. 2018; 10(6):279. https://doi.org/10.3390/v10060279
Chicago/Turabian StyleZhang, Yaoyao, Na Tang, Yashar Sadigh, Susan Baigent, Zhiqiang Shen, Venugopal Nair, and Yongxiu Yao. 2018. "Application of CRISPR/Cas9 Gene Editing System on MDV-1 Genome for the Study of Gene Function" Viruses 10, no. 6: 279. https://doi.org/10.3390/v10060279
APA StyleZhang, Y., Tang, N., Sadigh, Y., Baigent, S., Shen, Z., Nair, V., & Yao, Y. (2018). Application of CRISPR/Cas9 Gene Editing System on MDV-1 Genome for the Study of Gene Function. Viruses, 10(6), 279. https://doi.org/10.3390/v10060279