Exploiting a Phage-Bacterium Interaction System as a Molecular Switch to Decipher Macromolecular Interactions in the Living Cell


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Media, and Growth Conditions
2.2. Cloning and Mutagenesis
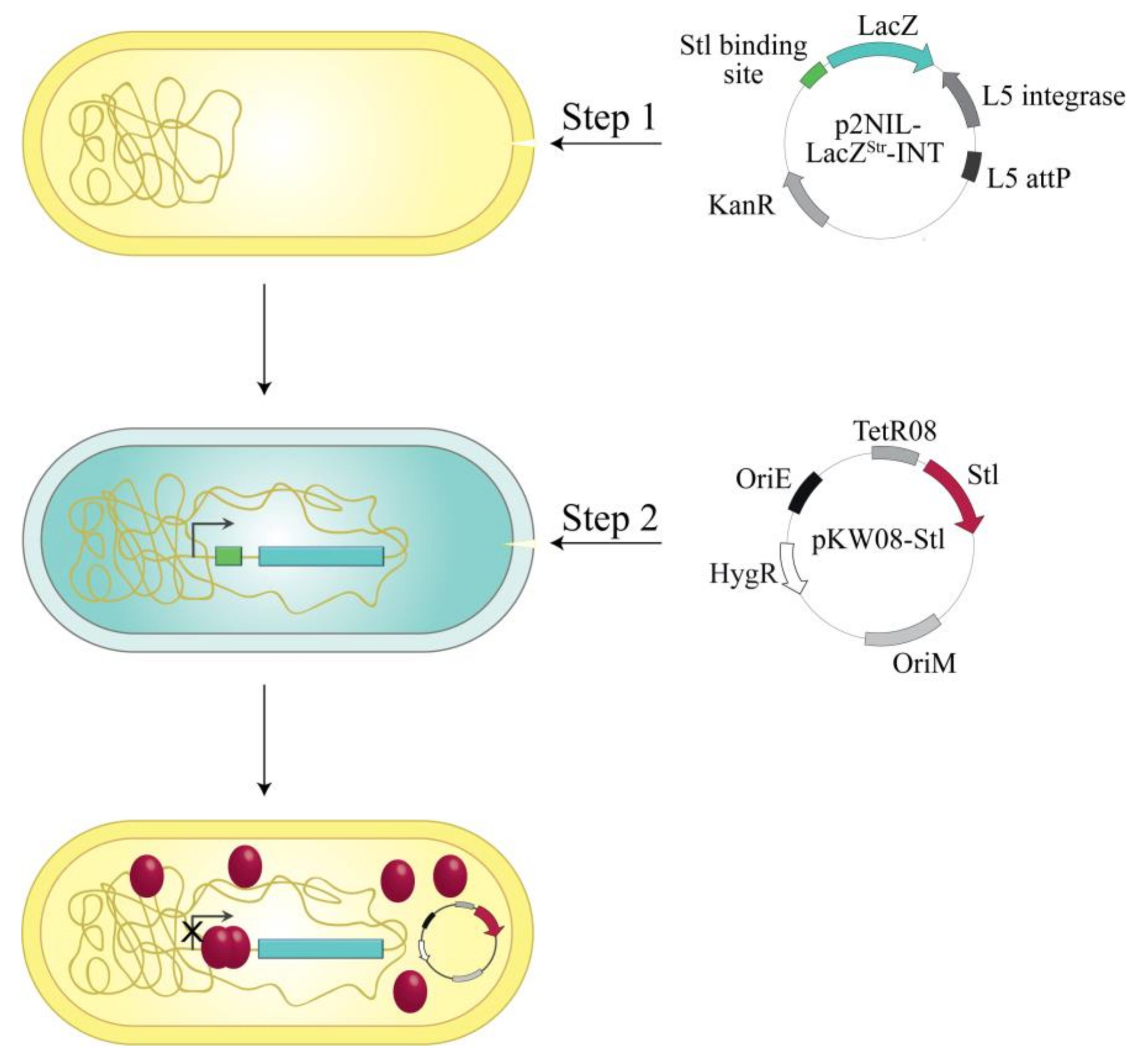
2.3. Construction of the M. smegmatis Reporter Strain
2.4. Analysis of Different Stl Mutants in the Reporter System
2.5. Random Mutagenesis of Stl
2.6. Protein Expression and Purification
2.7. Electrophoretic Mobility Shift Assay (EMSA)
2.8. Construction of the Double Switch System to Test Protein:Protein Interaction
3. Results
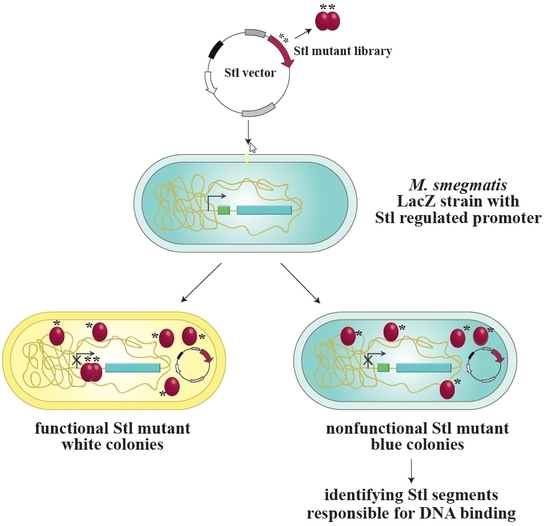
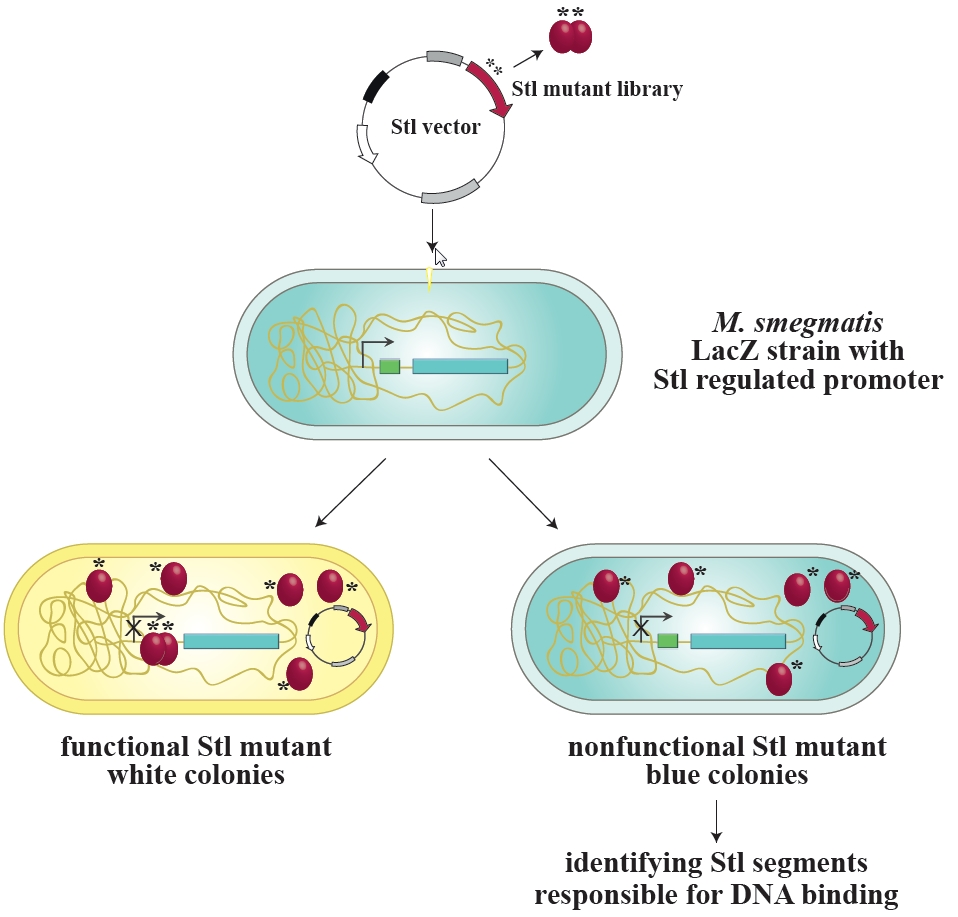
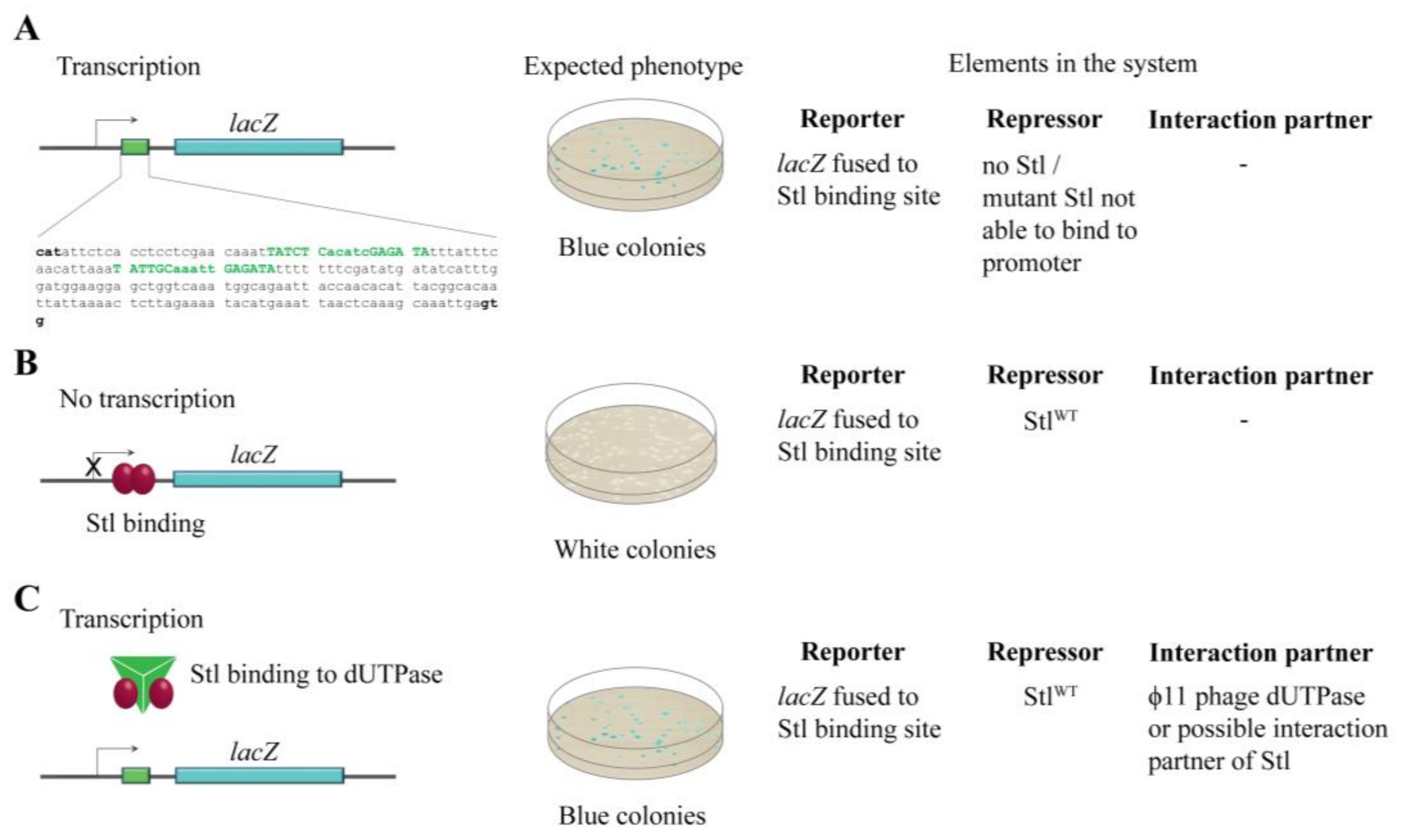
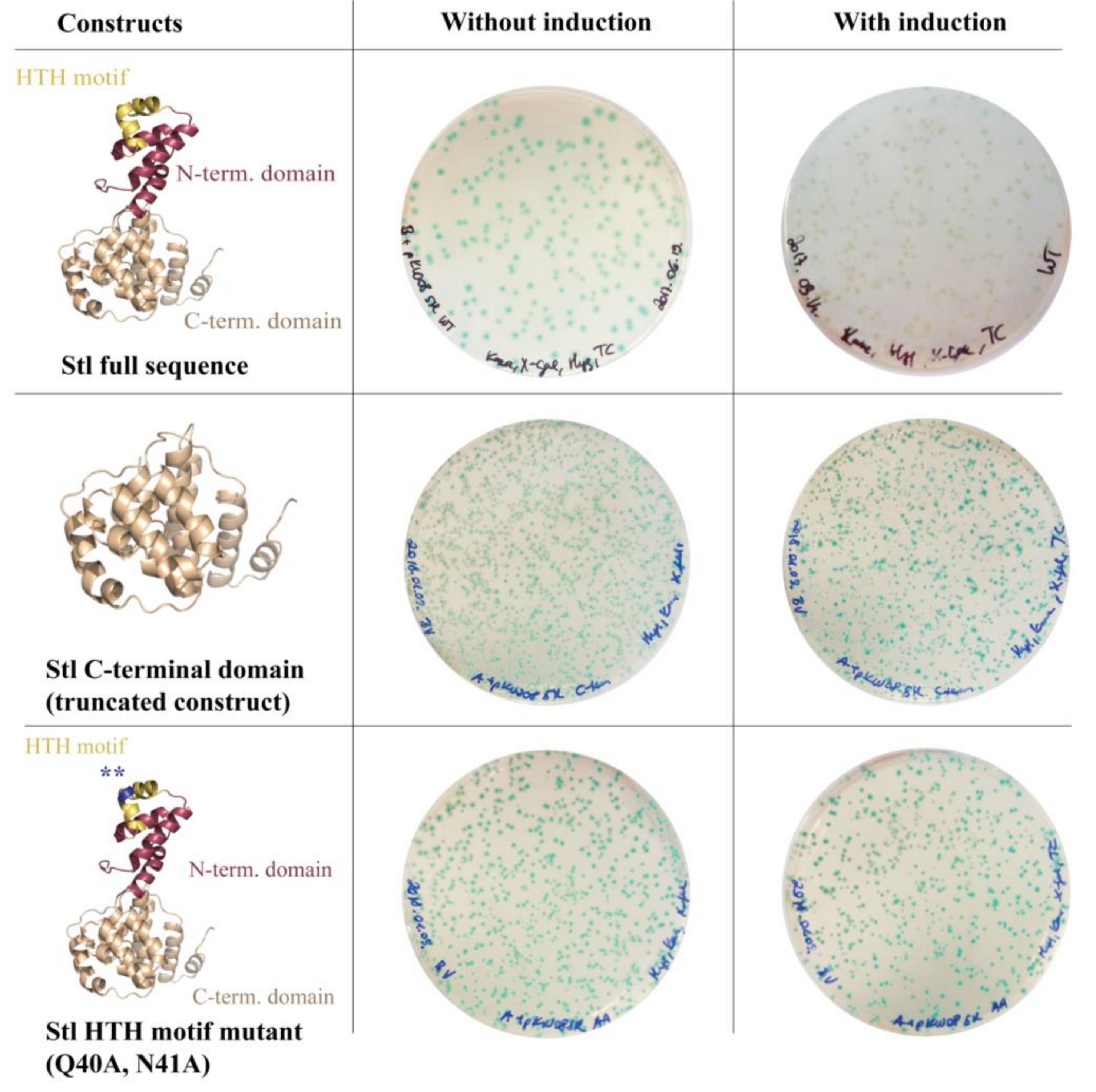
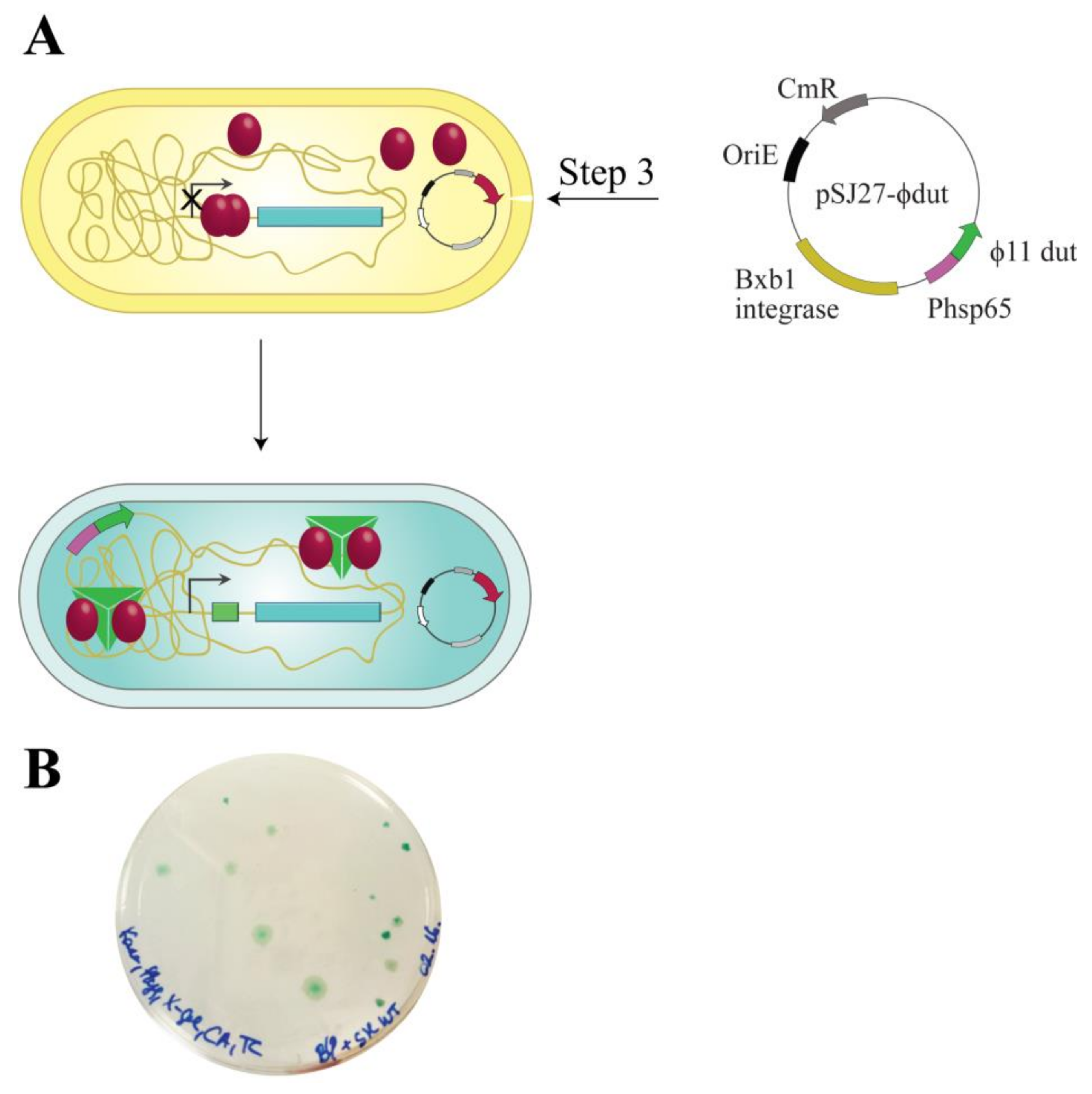
3.1. Design and Construction of a Reporter System to Characterize Different Functions of the Stl Protein
3.2. Validation of the Switch System Using Wild-Type and Mutant Stl Constructs
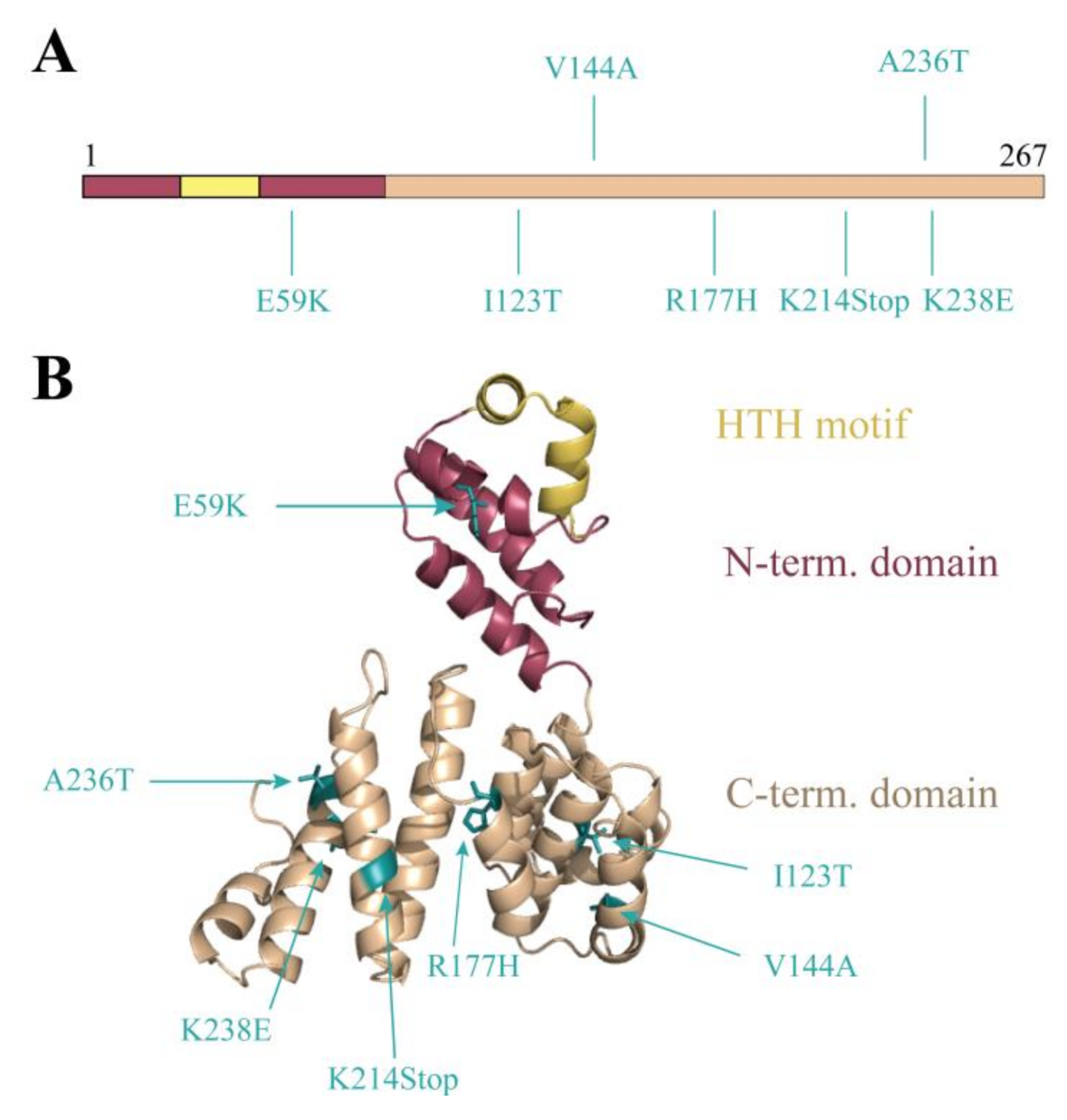
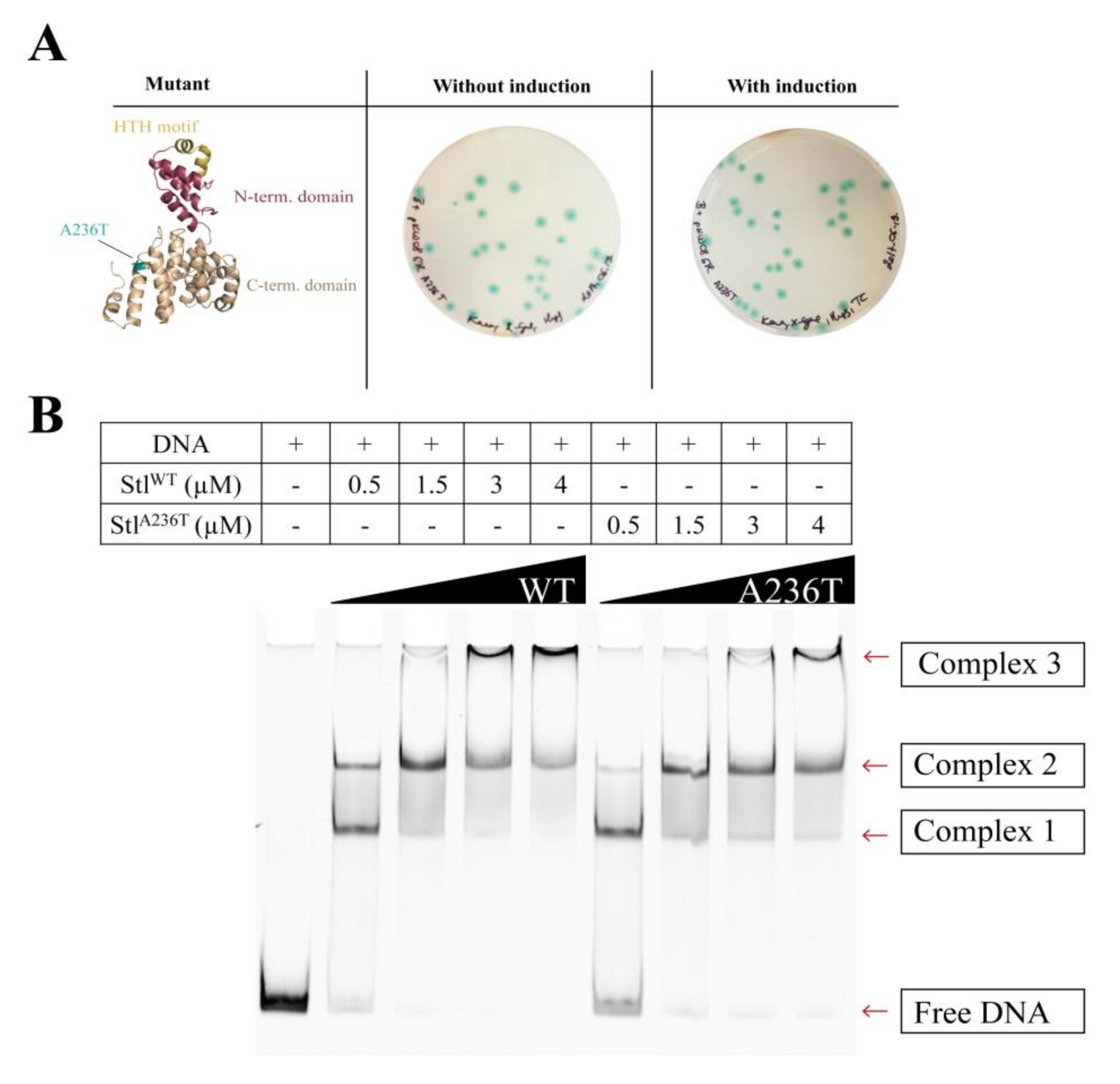
3.3. Random Mutagenesis-Based Search for Stl Mutations Affecting DNA-Binding
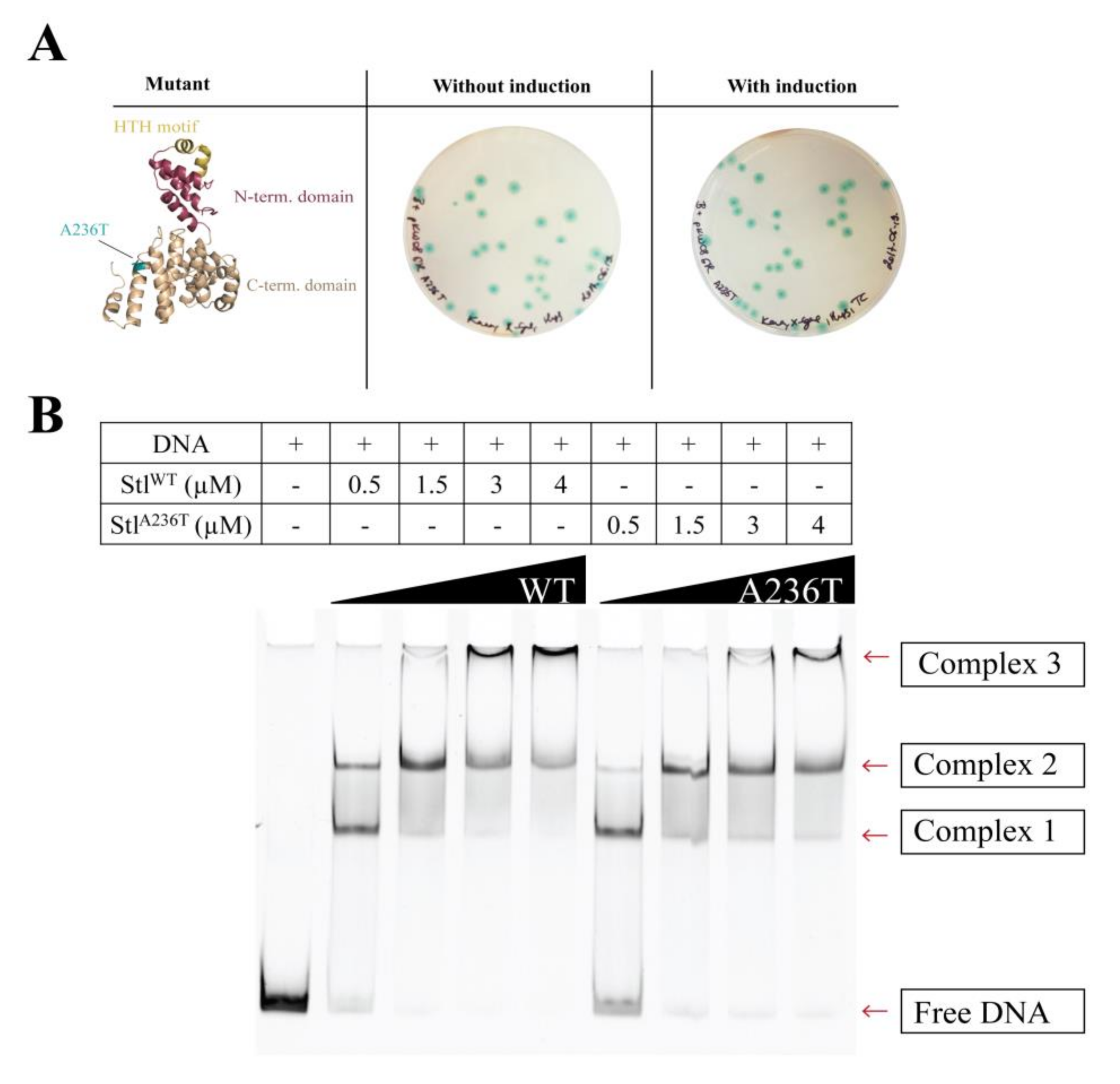
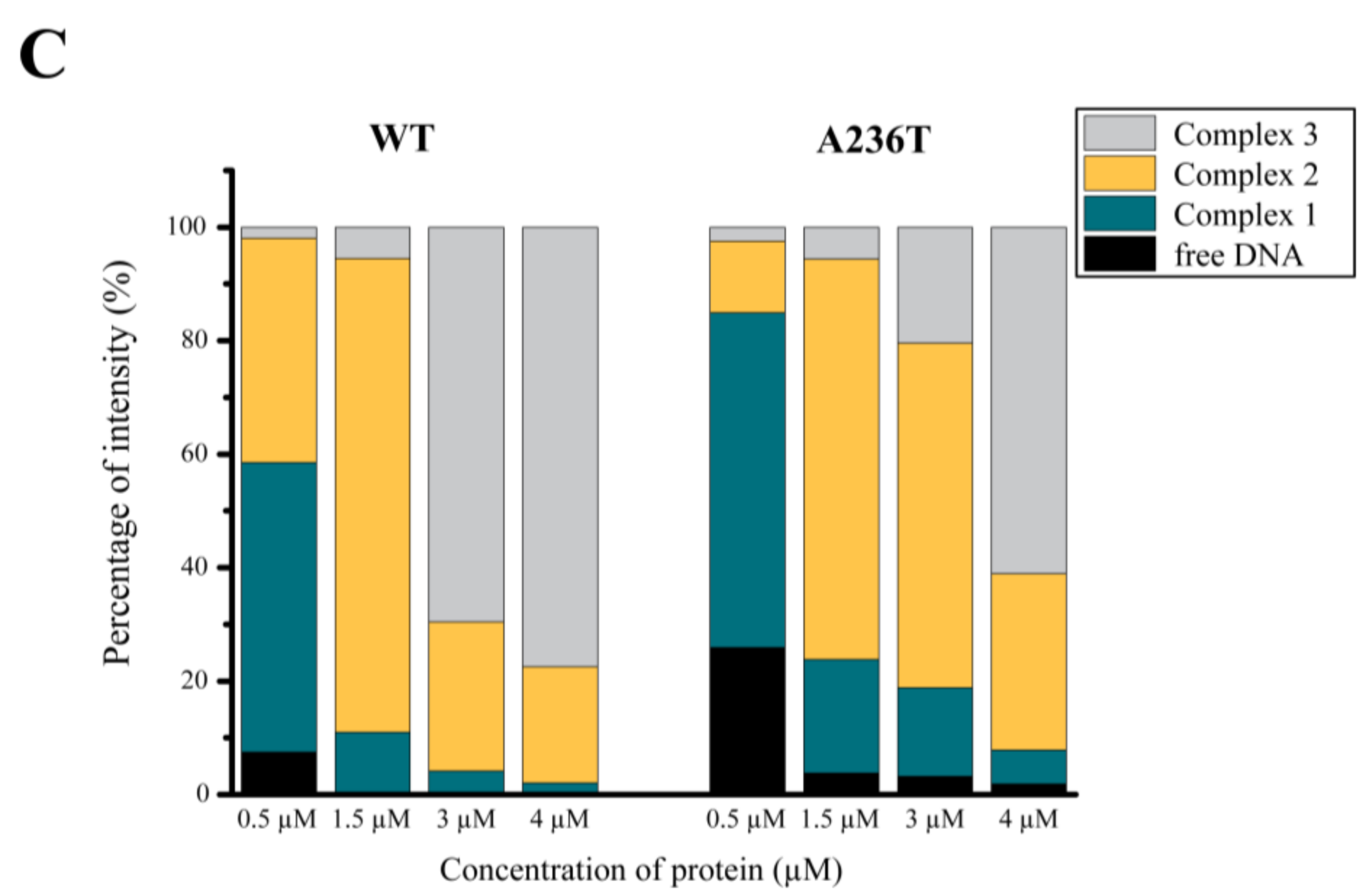
3.4. Analysis of the StlA236T DNA Binding Properties In Vivo and In Vitro
3.5. The Investigation of Protein:Protein Complexation Using the LacZStr Reporter System
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Malachowa, N.; DeLeo, F.R. Mobile genetic elements of Staphylococcus aureus. Cell. Mol. Life Sci. 2010, 67, 3057–3071. [Google Scholar] [CrossRef] [PubMed]
- Mir-Sanchis, I.; Martínez-Rubio, R.; Martí, M.; Chen, J.; Lasa, Í.; Novick, R.P.; Tormo-Más, M.Á.; Penadés, J.R. Control of Staphylococcus aureus pathogenicity island excision. Mol. Microbiol. 2012, 85, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Papp-Kádár, V.; Szabó, J.E.; Nyíri, K.; Vertessy, B.G. In Vitro Analysis of Predicted DNA-Binding Sites for the Stl Repressor of the Staphylococcus aureus SaPIBov1 Pathogenicity Island. PLoS ONE 2016, 11, e0158793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tormo-Más, M.A.; Mir, I.; Shrestha, A.; Tallent, S.M.; Campoy, S.; Lasa, I.; Barbé, J.; Novick, R.P.; Christie, G.E.; Penadés, J.R. Moonlighting bacteriophage proteins derepress staphylococcal pathogenicity islands. Nature 2010, 465, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Novick, R.P.; Christie, G.E.; Penadés, J.R. The phage-related chromosomal islands of Gram-positive bacteria. Nat. Rev. Microbiol. 2010, 8, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.L.L.; Dokland, T. The Type 2 dUTPase of Bacteriophage PhiNM1 Initiates Mobilization of Staphylococcus aureus Bovine Pathogenicity Island 1. J. Mol. Biol. 2016, 428, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Donderis, J.; Bowring, J.; Maiques, E.; Ciges-Tomas, J.R.; Alite, C.; Mehmedov, I.; Tormo-Mas, M.A.; Penadés, J.R.; Marina, A. Convergent evolution involving dimeric and trimeric dUTPases in pathogenicity island mobilization. PLoS Pathog. 2017, 13, e1006581. [Google Scholar] [CrossRef] [PubMed]
- Nyíri, K.; Kőhegyi, B.; Micsonai, A.; Kardos, J.; Vertessy, B.G. Evidence-Based Structural Model of the Staphylococcal Repressor Protein: Separation of Functions into Different Domains. PLoS ONE 2015, 10, e0139086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, R.G.; Matthews, B.W. The helix-turn-helix DNA binding motif. J. Biol. Chem. 1989, 264, 1903–1906. [Google Scholar] [PubMed]
- Pabo, C.O.; Lewis, M. The operator-binding domain of λ repressor: Structure and DNA recognition. Nature 1982, 298, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Rohs, R.; Jin, X.; West, S.M.; Joshi, R.; Honig, B.; Mann, R.S. Origins of Specificity in Protein-DNA Recognition. Annu. Rev. Biochem. 2010, 79, 233–269. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961, 3, 318–356. [Google Scholar] [CrossRef]
- Hillen, W.; Berens, C. Mechanisms Underlying Expression of TN10 Encoded Tetracycline Resistance. Annu. Rev. Microbiol. 1994, 48, 345–369. [Google Scholar] [CrossRef] [PubMed]
- Berens, C.; Hillen, W. Gene regulation by tetracyclines: Constraints of resistance regulation in bacteria shape TetR for application in eukaryotes. Eur. J. Biochem. 2003, 270, 3109–3121. [Google Scholar] [CrossRef] [PubMed]
- Guzman, L.M.; Belin, D.; Carson, M.J.; Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995, 177, 4121–4130. [Google Scholar] [CrossRef] [PubMed]
- De Boer, H.A.; Comstock, L.J.; Vasser, M. The tac promoter: A functional hybrid derived from the trp and lac promoters. Biochemistry 1983, 80, 21–25. [Google Scholar] [CrossRef]
- Brosius, J.; Erfle, M.; Storella, J. Spacing of the −10 and −35 Regions in the tac Promoter. J. Biol. Chem. 1985, 260, 3539–3541. [Google Scholar] [PubMed]
- Tabor, S.; Richardson, C.C. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes (T7 DNA polymerase/T7 gene 5 protein/proteolysis/13-lactamase/rifampicin). Biochemistry 1985, 82, 1074–1078. [Google Scholar]
- Giesecke, A.V.; Joung, J.K. The bacterial two-hybrid system as a reporter system for analyzing protein-protein interactions. Cold Spring Harb. Protoc. 2007. [Google Scholar] [CrossRef] [PubMed]
- Hirmondó, R.; Szabó, J.E.; Nyíri, K.; Tarjányi, S.; Dobrotka, P.; Tóth, J.; Vértessy, B.G. Cross-species inhibition of dUTPase via the Staphylococcal Stl protein perturbs dNTP pool and colony formation in Mycobacterium. DNA Repair 2015, 30, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parish, T.; Brown, A.C. (Eds.) Mycobacteria Protocols; Humana Press: Totowa, NJ, USA, 2009; ISBN 978-1-58829-889-8. [Google Scholar]
- Parish, T.; Stoker, N.G. Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology 2000, 146, 1969–1975. [Google Scholar] [CrossRef] [PubMed]
- Mahenthiralingam, E.; Marklund, B.I.; Brooks, L.; Smith, D.; Bancroft, G.J.; Stokes, R.W. Site-directed mutagenesis of the 19-kilodalton lipoprotein antigen reveals No essential role for the protein in the growth and virulence of Mycobacterium intracellulare. Infect. Immun. 1998, 66, 3626–3634. [Google Scholar] [PubMed]
- Szabó, J.E.; Németh, V.; Papp-Kádár, V.; Nyíri, K.; Leveles, I.; Bendes, A.Á.; Zagyva, I.; Róna, G.; Pálinkás, H.L.; Besztercei, B.; et al. Highly potent dUTPase inhibition by a bacterial repressor protein reveals a novel mechanism for gene expression control. Nucleic Acids Res. 2014, 42, 11912–11920. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.I.; Ghosh, P.; Aaron, M.A.; Bibb, L.A.; Jain, S.; Hatfull, G.F. Mycobacteriophage Bxb1 integrates into the Mycobacterium smegmatis groEL1 gene. Mol. Microbiol. 2003, 50, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.S.; Keefe, A.D. Random Mutagenesis by PCR. In Current Protocols in Molecular Biology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001. [Google Scholar]
- Ubeda, C.; Maiques, E.; Barry, P.; Matthews, A.; Tormo, M.A.; Lasa, I.; Novick, R.P.; Penadés, J.R. SaPI mutations affecting replication and transfer and enabling autonomous replication in the absence of helper phage. Mol. Microbiol. 2008, 67, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Leveles, I.; Németh, V.; Szabó, J.E.; Harmat, V.; Nyíri, K.; Bendes, Á.Á.; Papp-Kádár, V.; Zagyva, I.; Róna, G.; Ozohanics, O.; et al. Structure and enzymatic mechanism of a moonlighting dUTPase. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 2298–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.J.; Noyes, M.B. Understanding DNA-binding specificity by bacteria hybrid selection. Brief. Funct. Genom. 2015, 14, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Muha, V.; Horváth, A.; Békési, A.; Pukáncsik, M.; Hodoscsek, B.; Merényi, G.; Róna, G.; Batki, J.; Kiss, I.; Jankovics, F.; et al. Uracil-Containing DNA in Drosophila: Stability, Stage-Specific Accumulation, and Developmental Involvement. PLoS Genet. 2012, 8, e1002738. [Google Scholar] [CrossRef] [PubMed]
- Békési, A.; Zagyva, I.; Hunyadi-Gulyás, E.; Pongrácz, V.; Kovári, J.; Nagy, A.O.; Erdei, A.; Medzihradszky, K.F.; Vértessy, B.G. Developmental regulation of dUTPase in Drosophila melanogaster. J. Biol. Chem. 2004, 279, 22362–22370. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Surányi, É.V.; Hírmondó, R.; Nyíri, K.; Tarjányi, S.; Kőhegyi, B.; Tóth, J.; Vértessy, B.G. Exploiting a Phage-Bacterium Interaction System as a Molecular Switch to Decipher Macromolecular Interactions in the Living Cell. Viruses 2018, 10, 168. https://doi.org/10.3390/v10040168
Surányi ÉV, Hírmondó R, Nyíri K, Tarjányi S, Kőhegyi B, Tóth J, Vértessy BG. Exploiting a Phage-Bacterium Interaction System as a Molecular Switch to Decipher Macromolecular Interactions in the Living Cell. Viruses. 2018; 10(4):168. https://doi.org/10.3390/v10040168
Chicago/Turabian StyleSurányi, Éva Viola, Rita Hírmondó, Kinga Nyíri, Szilvia Tarjányi, Bianka Kőhegyi, Judit Tóth, and Beáta G. Vértessy. 2018. "Exploiting a Phage-Bacterium Interaction System as a Molecular Switch to Decipher Macromolecular Interactions in the Living Cell" Viruses 10, no. 4: 168. https://doi.org/10.3390/v10040168