Resonance in Interacting Induced-Dipole Polarizing Force Fields: Application to Force-Field Derivatives


Abstract
:1. Introduction
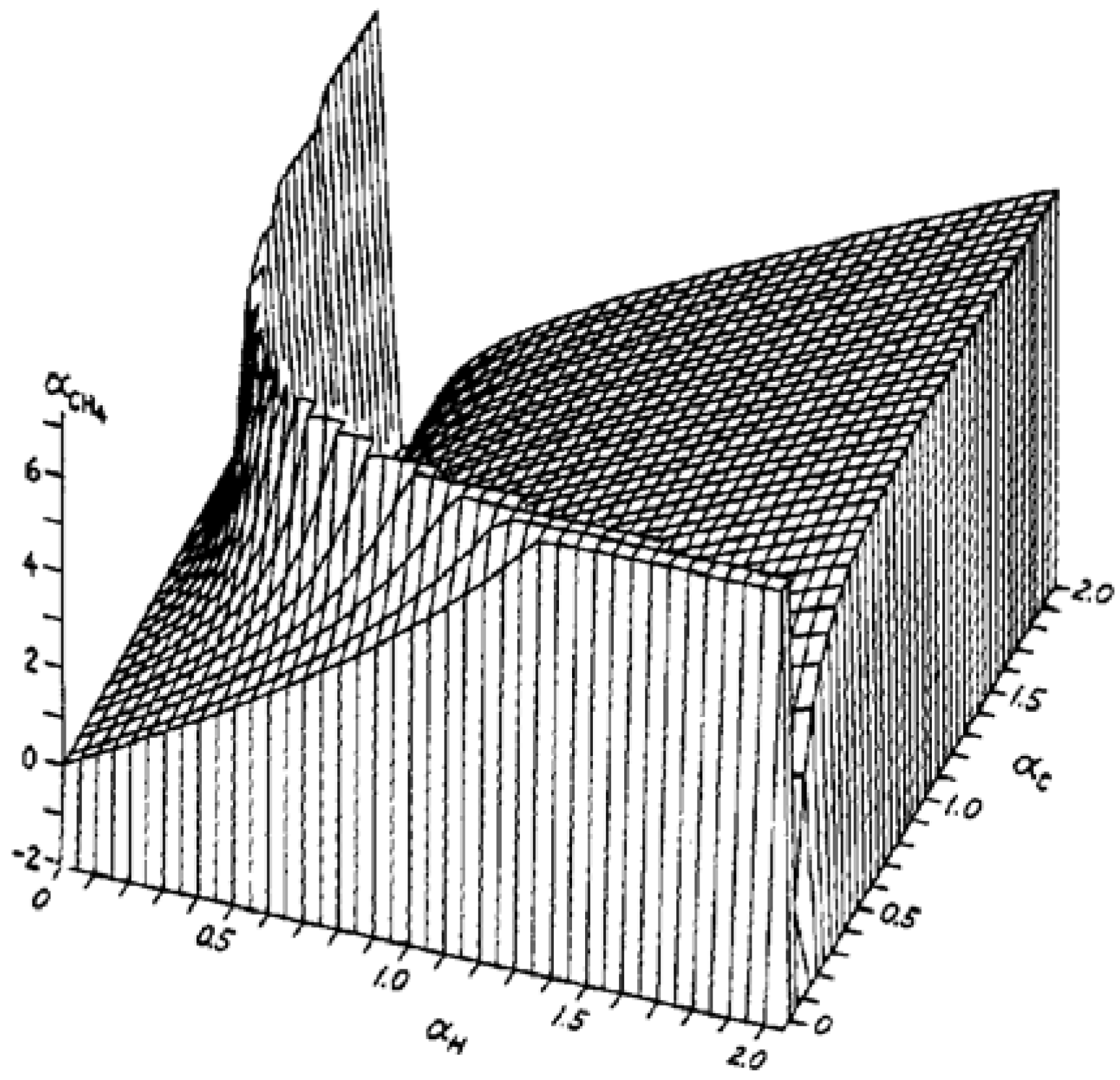
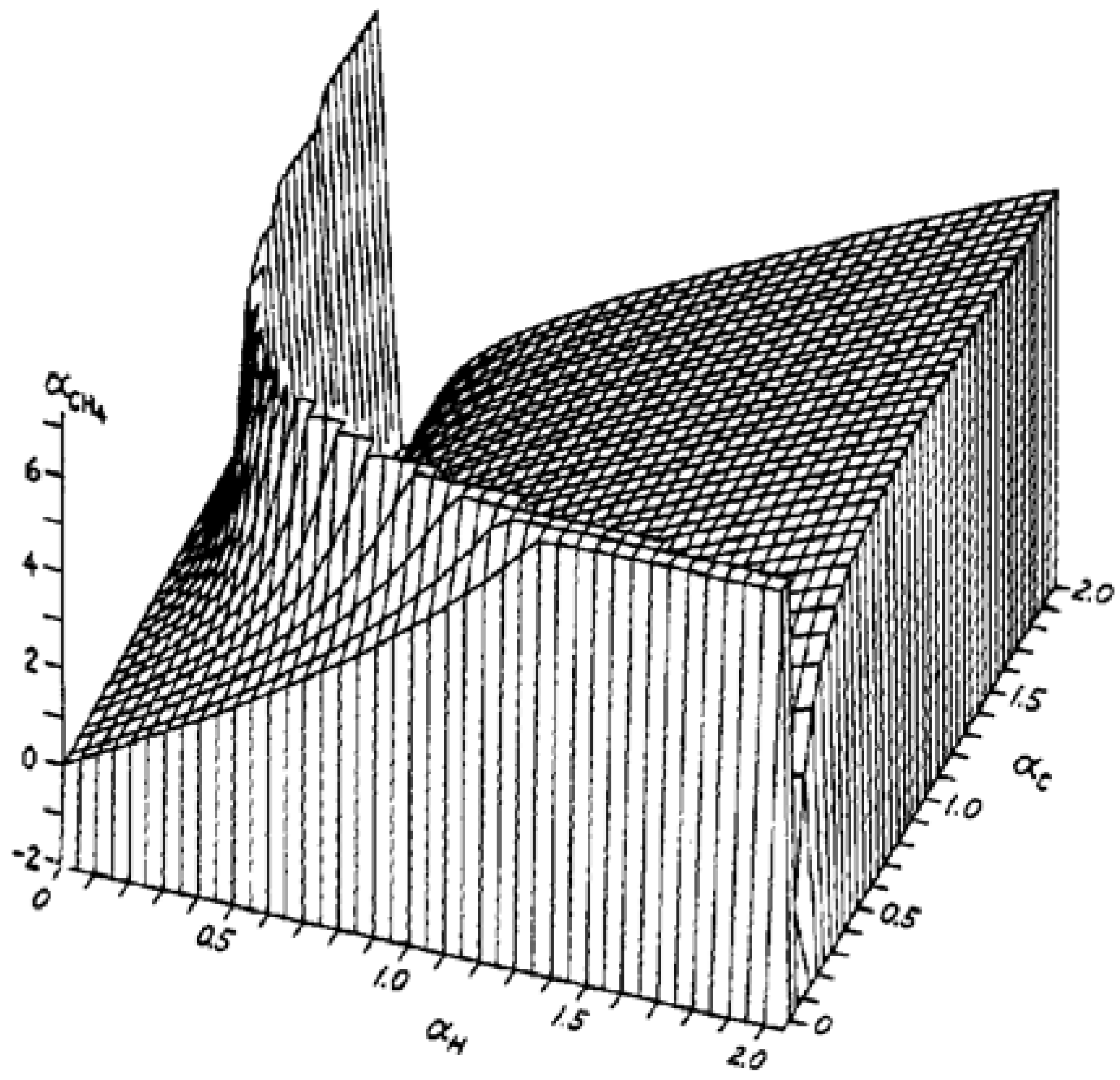
2. Results and Discussion
2.1. Molecular Dipolar Polarizability Model
2.2. Molecular Dipolar Polarizability of Methane
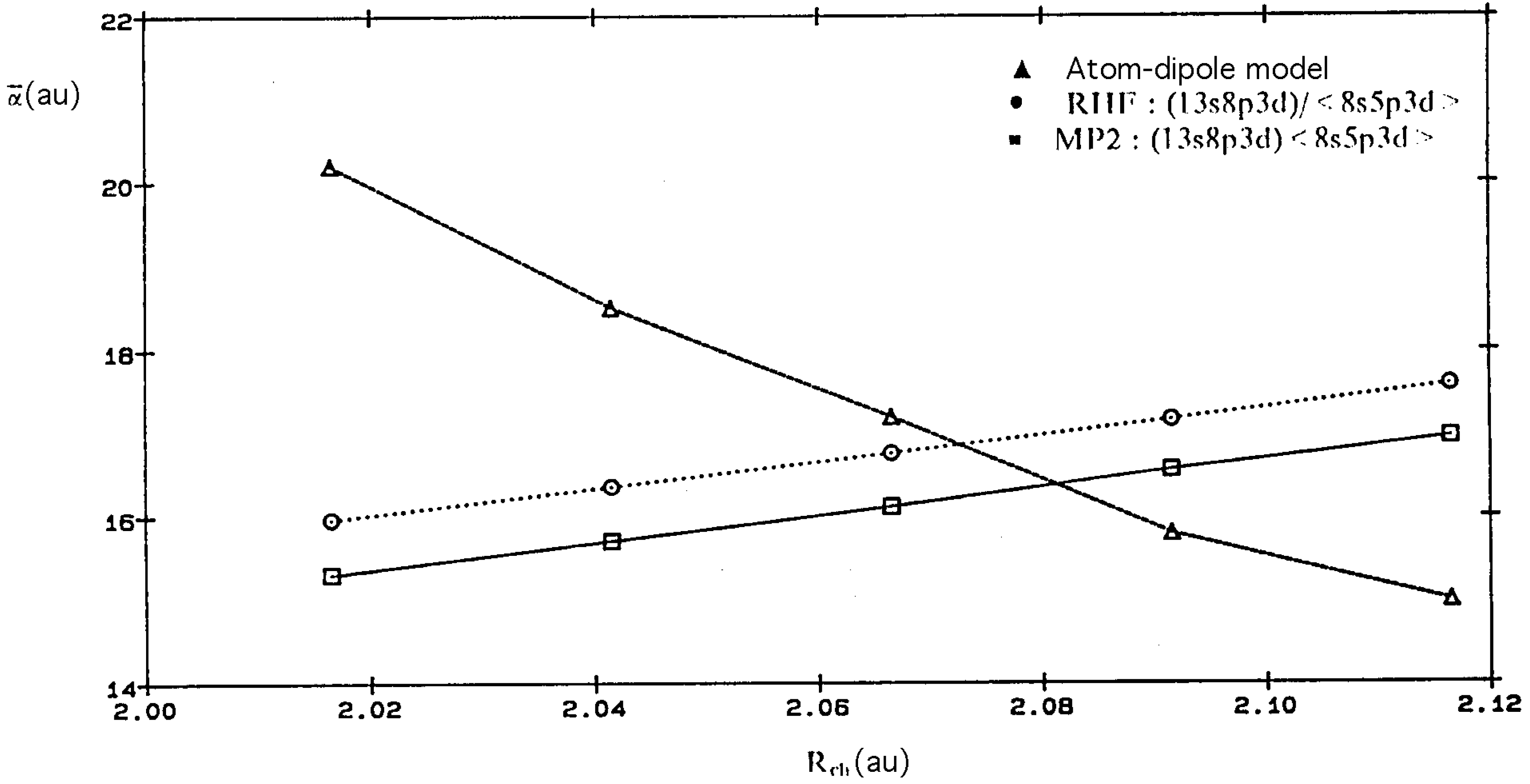

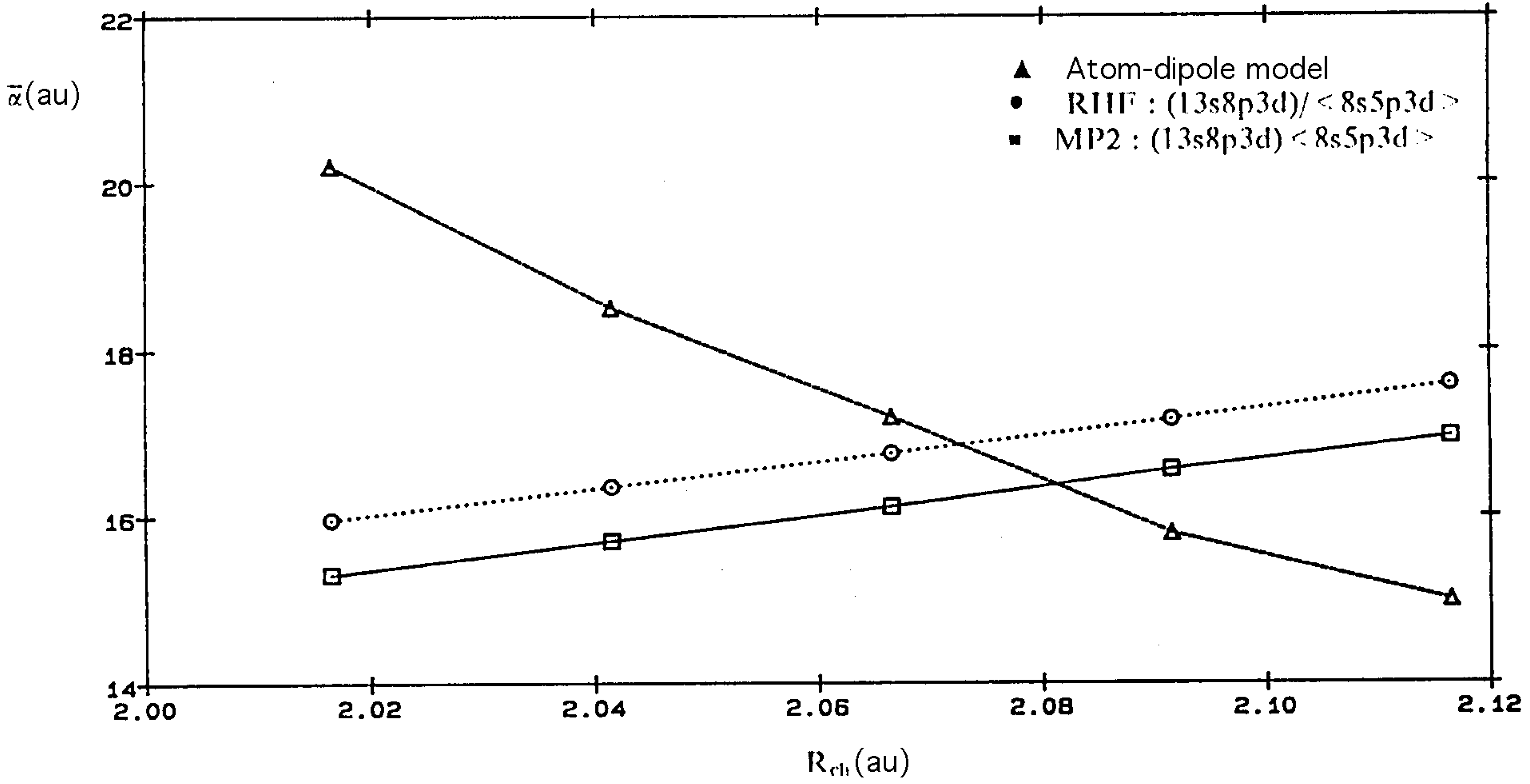
{kind=link}
{kind=link}
| Atom | αa | RH–Cb | α(CH4)c | RvibH–Cd | α’e | αvibf | αvib(CH4)g |
|---|---|---|---|---|---|---|---|
| H | 0.135 | – | 0.123 | – | 1.233 | 0.159 | 0.146 |
| C | 0.878 | – | 1.288 | – | –2.081 | 0.838 | 1.308 |
| CH4 | 1.418 | 1.113 | 1.779 | 1.094 | – | 1.474 | 1.892 |
- a.
- Böttcher proposed [25]:
- b.
- Thole proposed [26]:
- c.
- a.
- test whether matrix B–1 is singular, where B is the many-body polarizability matrix;
- b.
- test whether matrix B–1 is not defined positive;
- c.
- test whether matrix B is not defined positive;
- d.
- test whether some effective matrix Bp is not defined positive.
3. Conclusions
4. Experimental Section
References and Notes
- Silberstein, L. Molecular refractivity and atomic interaction. Philos. Mag. 1917, 33, 92–128. [Google Scholar] [CrossRef]
- Silberstein, L. Dispersion and the size of molecules of hydrogen, oxygen, and nitrogen. Philos. Mag. 1917, 33, 215–222. [Google Scholar] [CrossRef]
- Silberstein, L. Molecular refractivity and atomic interaction. II. Philos. Mag. 1917, 33, 521–533. [Google Scholar] [CrossRef]
- Applequist, J.; Carl, J.R.; Fung, K.-K. An atom dipole interaction model for molecular polarizability. Application to polyatomic molecules and determination of atom polarizabilities. J. Am. Chem. Soc. 1972, 94, 2952–2960. [Google Scholar] [CrossRef]
- Kauzmann, W. Quantum Chemistry; Academic Press: New York, 1957; p. 568. [Google Scholar]
- Mahan, G.D. Davydov splittings in anthracene. J. Chem. Phys. 1964, 41, 2930–2933. [Google Scholar] [CrossRef]
- Rhodes, W.; Chase, M. Generalized susceptibility theory I. Theories of hypochromism. Rev. Mod. Phys. 1967, 39, 348–361. [Google Scholar] [CrossRef]
- Philpott, M.R. Dipole Davydov splittings in crystalline anthracene, tetracene, naphthalene, and phenanthrene. J. Chem. Phys. 1969, 50, 5117–5128. [Google Scholar] [CrossRef]
- Torrens, F.; Sánchez-Marín, J.; Nebot-Gil, I. Torsional effects on the molecular polarizabilities of the benzothiazole (A)-benzobisthiazole (B) oligomer A-B13-A. J. Mol. Graphics 1996, 14, 245–259. [Google Scholar] [CrossRef]
- Torrens, F.; Sánchez-Marín, J.; Nebot-Gil, I. Interacting induced dipoles polarization model for molecular polarizabilities. Application to benzothiazole (A)-benzobisthiazole (B) oligomers: A-B13-A. J. Mol. Struct. (Theochem) 1998, 426, 105–116. [Google Scholar] [CrossRef]
- Torrens, F.; Sánchez-Marín, J; Nebot-Gil, I. Interacting induced dipoles polarization model for molecular polarizabilities. Reference molecules, amino acids and model peptides. J. Mol. Struct. (Theochem) 1999, 463, 27–39. [Google Scholar] [CrossRef]
- Torrens, F.; Sánchez-Marín, J.; Nebot-Gil, I. Polarization by the effect of a small torsional change in the benzothiazole (A)-benzobisthiazole (B) oligomer A-B13-A. Molecules 1999, 4, 28–51. [Google Scholar] [CrossRef]
- Torrens, F. Molecular polarizability of Scn, Cn and endohedral Scn@Cm clusters. Microelectron. Eng. 2000, 51-52, 613–626. [Google Scholar] [CrossRef]
- Torrens, F. Molecular polarizability of Sc and C (fullerene and graphite) clusters. Molecules 2001, 6, 496–509. [Google Scholar] [CrossRef]
- Torrens, F. Molecular polarizability of semionductor clusters and nanostructures. Physica E 2002, 13, 67–71. [Google Scholar] [CrossRef]
- Torrens, F. Molecular polarizability of fullerenes and endohedral metallofullerenes. J. Phys. Org. Chem. 2002, 15, 742–749. [Google Scholar] [CrossRef]
- Torrens, F. Effect of elliptical deformation on molecular polarizabilities of model carbon nanotubes from atomic increments. J. Nanosci. Nanotechnol. 2003, 3, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Torrens, F. Molecular polarizability of Si/Ge/GaAs semiconductor clusters. J. Comput. Method. Sci. Eng. 2004, 4, 439–450. [Google Scholar]
- Torrens, F. Effect of size and deformation on polarizabilities of carbon nanotubes from atomic increments. Future Gener. Comput. Syst. 2004, 20, 763–772. [Google Scholar] [CrossRef]
- Vogel, A.I. Physical properties and chemical constitution. XXIII. Miscellaneous compounds. Investigation of the so-called co-ordinate or dative link in esters of oxy acids and in nitro paraffins by molecular refractivity determinations. Atomic, structural, and group parachors and refractivities. J. Chem. Soc. 1948, 1833–1855. [Google Scholar]
- Gresh, N.; Claverie, P.; Pullman, A. Intermolecular interactions: Reproduction of the results of ab initio supermolecule computations by an additive procedure. Int. J. Quantum Chem., Symp. 1979, 13, 243–253. [Google Scholar] [CrossRef]
- Joachim, C.; Treboux, G.; Tang, H. A model conformational flip-flop molecular switch. AIP Conf. Proc. 1992, 262, 107–117. [Google Scholar]
- Voisin, C.; Cartier, A.; Rivail, J.-L. Computation of accurate electronic molecular polarizabilities. J. Phys. Chem. 1992, 96, 7966–7971. [Google Scholar] [CrossRef]
- Voisin, C.; Cartier, A. Determination of distributed polarizabilities to be used for peptide modeling. J. Mol. Struct. (Theochem) 1993, 286, 35–45. [Google Scholar] [CrossRef]
- Birge, R.R. Calculation of molecular polarizabilities using an anisotropic atom point dipole interaction model which includes the effect of electron repulsion. J. Chem. Phys. 1980, 72, 5312–5319. [Google Scholar] [CrossRef]
- Thole, B.T. Molecular polarizabilities calculated with a modified dipole interaction. Chem. Phys. 1981, 59, 341–350. [Google Scholar] [CrossRef]
- Miller, K.J. Additivity methods in molecular polarizability. J. Am. Chem. Soc. 1990, 112, 8533–8542. [Google Scholar] [CrossRef]
- Miller, K.J. Calculation of the molecular polarizability tensor. J. Am. Chem. Soc. 1990, 112, 8543–8551. [Google Scholar] [CrossRef]
- Applequist, J. An atom dipole interaction model for molecular optical properties. Acc. Chem. Res. 1977, 10, 79–85. [Google Scholar] [CrossRef]
- Allinger, N.L. Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J. Am. Chem. Soc. 1977, 99, 8127–8134. [Google Scholar] [CrossRef]
- Deiters, J.A.; Gallucci, J.C.; Clark, T.E.; Holmes, R.R. Computer simulation of phosphorane structures. J. Am. Chem. Soc. 1977, 99, 5461–5471. [Google Scholar] [CrossRef]
- Némethy, G.; Pottle, M.S.; Scheraga, H.A. Energy parameters in polypeptides. 9. Updating of geometric parameters, nonbonded interactions, and hydrogen bond interactions for the naturally occurring amino acids. J. Phys. Chem. 1983, 87, 1883–1887. [Google Scholar] [CrossRef]
- Torrens, F.; Ruiz-López, M.; Cativiela, C.; García, J.I.; Mayoral, J.A. Conformational aspects of some asymmetric Diels-Alder reactions. A molecular mechanics + polarization study. Tetrahedron 1992, 48, 5209–5218. [Google Scholar]
- Torrens, F.; Sánchez-Marín, J.; Rivail, J.-L. Interacting induced dipoles polarization in a force field for dipeptide models (glycine derivative). An. Fís. 1994, 90, 197–204. [Google Scholar]
- Torrens, F. Nature of FeIII–O2, FeII–CO and FeIII–CN complexes of hemoprotein models. Polyhedron 2003, 22, 1091–1098. [Google Scholar] [CrossRef]
- Torrens, F. Nature of O2, CO, and CN binding to hemoprotein models. Int. J. Quantum Chem. 2004, 99, 963–971. [Google Scholar] [CrossRef]
- Torrens, F. An improved force field for O2, CO and CN binding to metalloporphyrins. J. Inclusion Phenom. Mol. Recognit. Chem. 2004, 49, 37–46. [Google Scholar] [CrossRef]
- Torrens, F. A comparative study of O2, CO and CN binding to heme-IX protein models. Molecules 2004, 9, 632–649. [Google Scholar] [CrossRef] [PubMed]
- Torrens, F. Polarization force fields for peptides implemented in ECEPP2 and MM2. Mol. Simul. 2000, 24, 391–410. [Google Scholar] [CrossRef]
- Torrens, F. Peptide potential energy surfaces and protein folding. J. Argent. Chem. Soc. 2006, 94, 27–47. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Torrens, F.; Castellano, G. Resonance in Interacting Induced-Dipole Polarizing Force Fields: Application to Force-Field Derivatives. Algorithms 2009, 2, 437-447. https://doi.org/10.3390/a2010437
Torrens F, Castellano G. Resonance in Interacting Induced-Dipole Polarizing Force Fields: Application to Force-Field Derivatives. Algorithms. 2009; 2(1):437-447. https://doi.org/10.3390/a2010437
Chicago/Turabian StyleTorrens, Francisco, and Gloria Castellano. 2009. "Resonance in Interacting Induced-Dipole Polarizing Force Fields: Application to Force-Field Derivatives" Algorithms 2, no. 1: 437-447. https://doi.org/10.3390/a2010437
APA StyleTorrens, F., & Castellano, G. (2009). Resonance in Interacting Induced-Dipole Polarizing Force Fields: Application to Force-Field Derivatives. Algorithms, 2(1), 437-447. https://doi.org/10.3390/a2010437