Preparation and Characterization of Modified Soda Lignin with Polyethylene Glycol

Abstract
:1. Introduction
2. Results
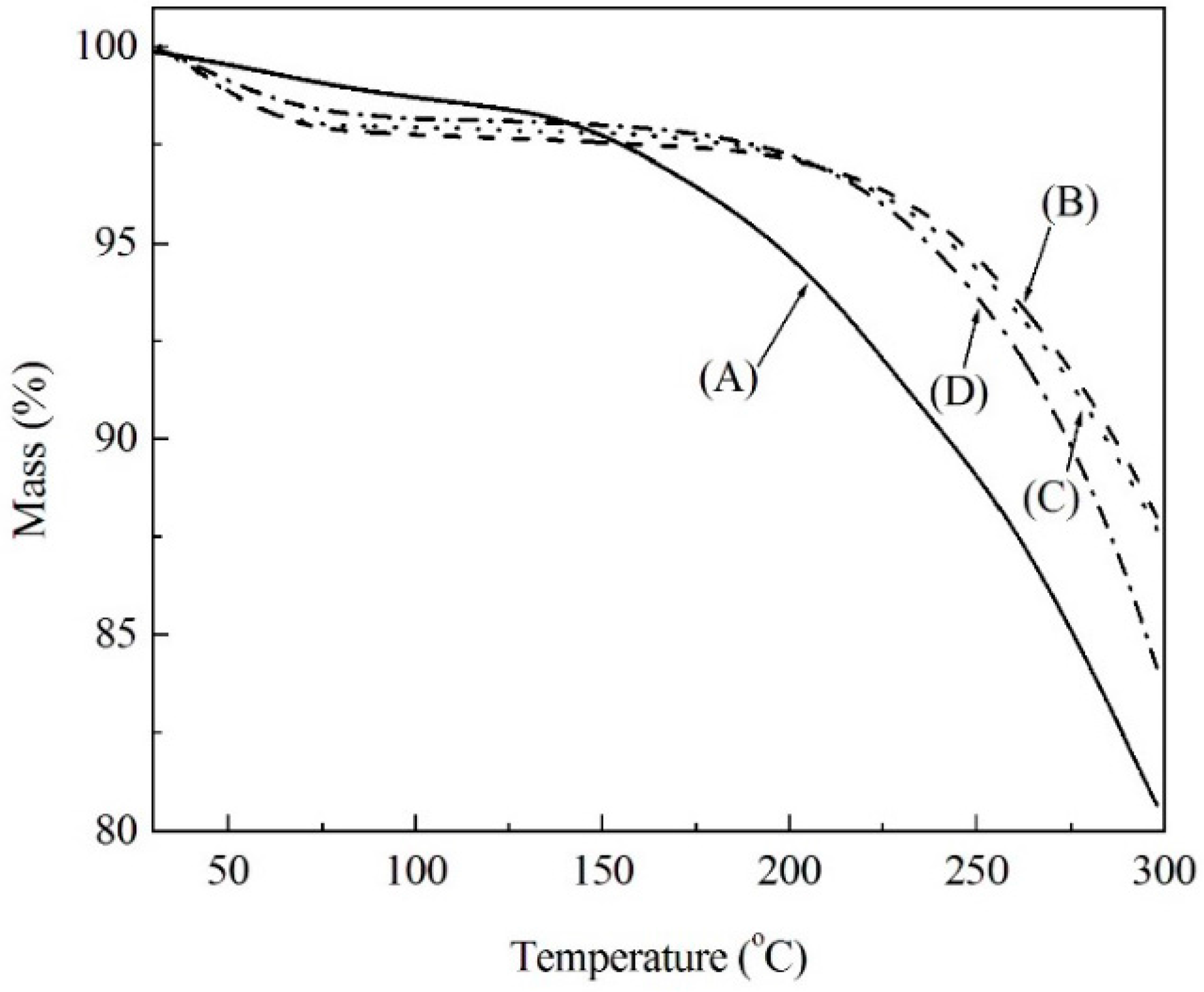
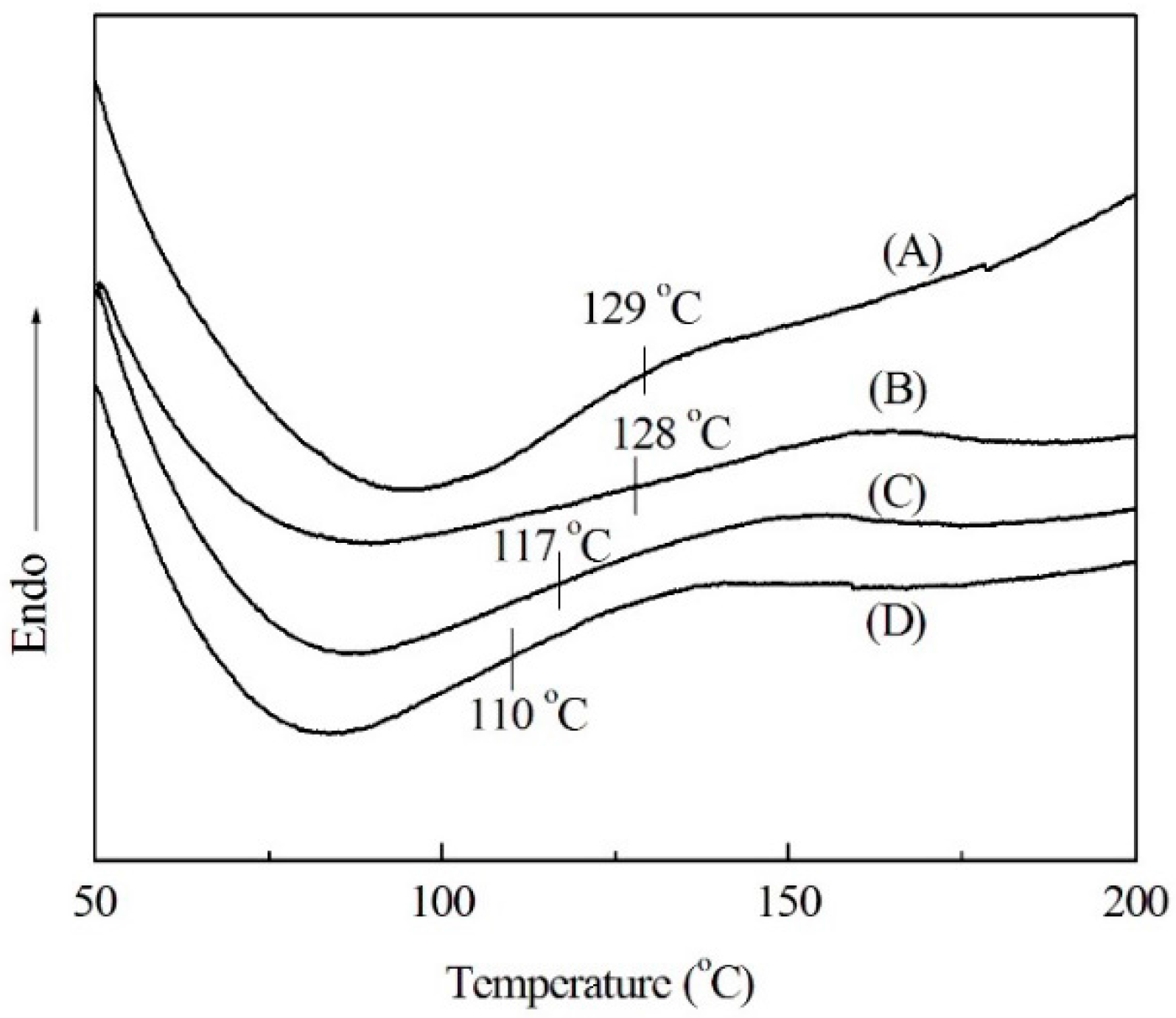
2.1. Thermal Analysis of Soda Lignin
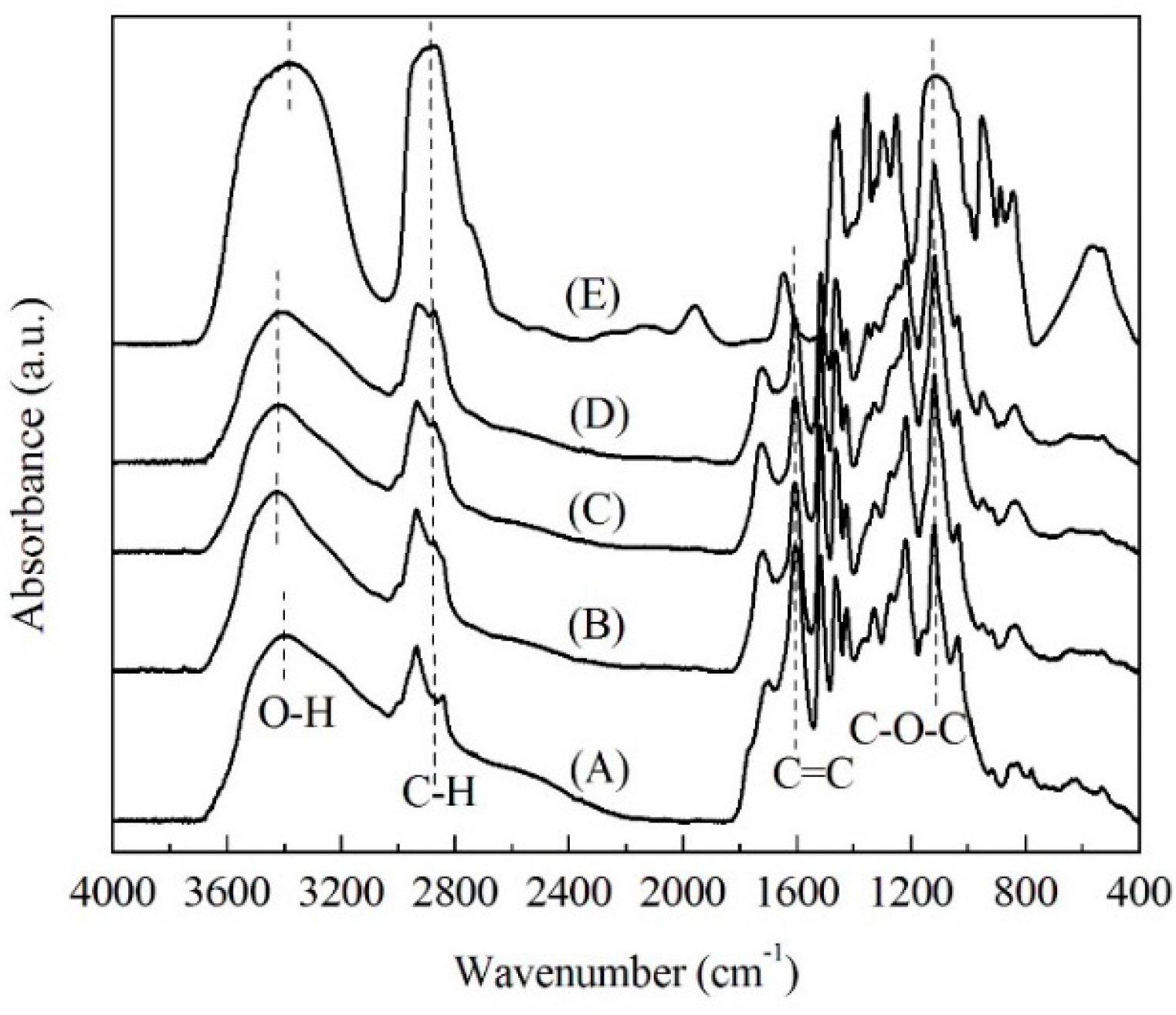
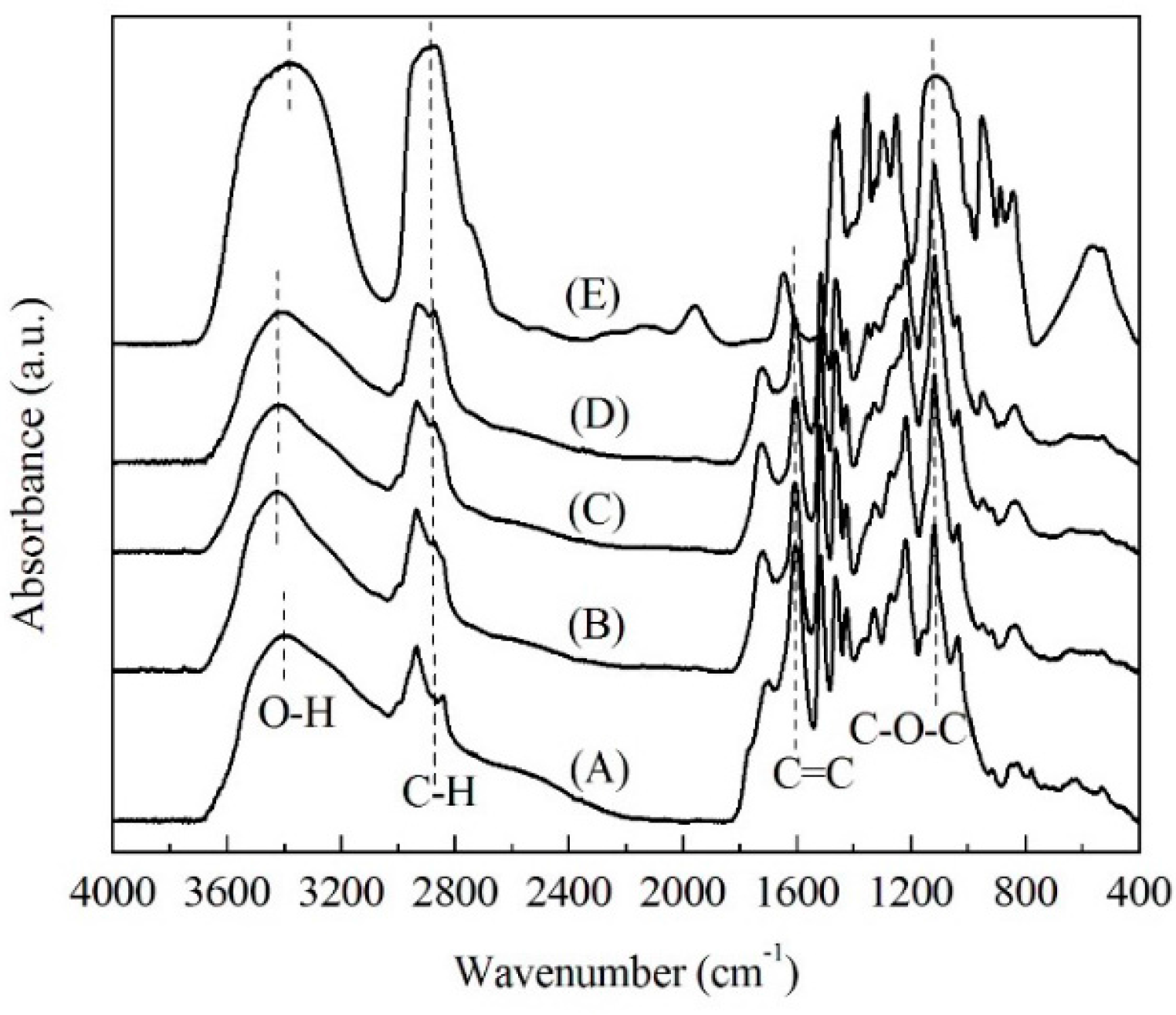
2.2. FTIR Spectra Analysis
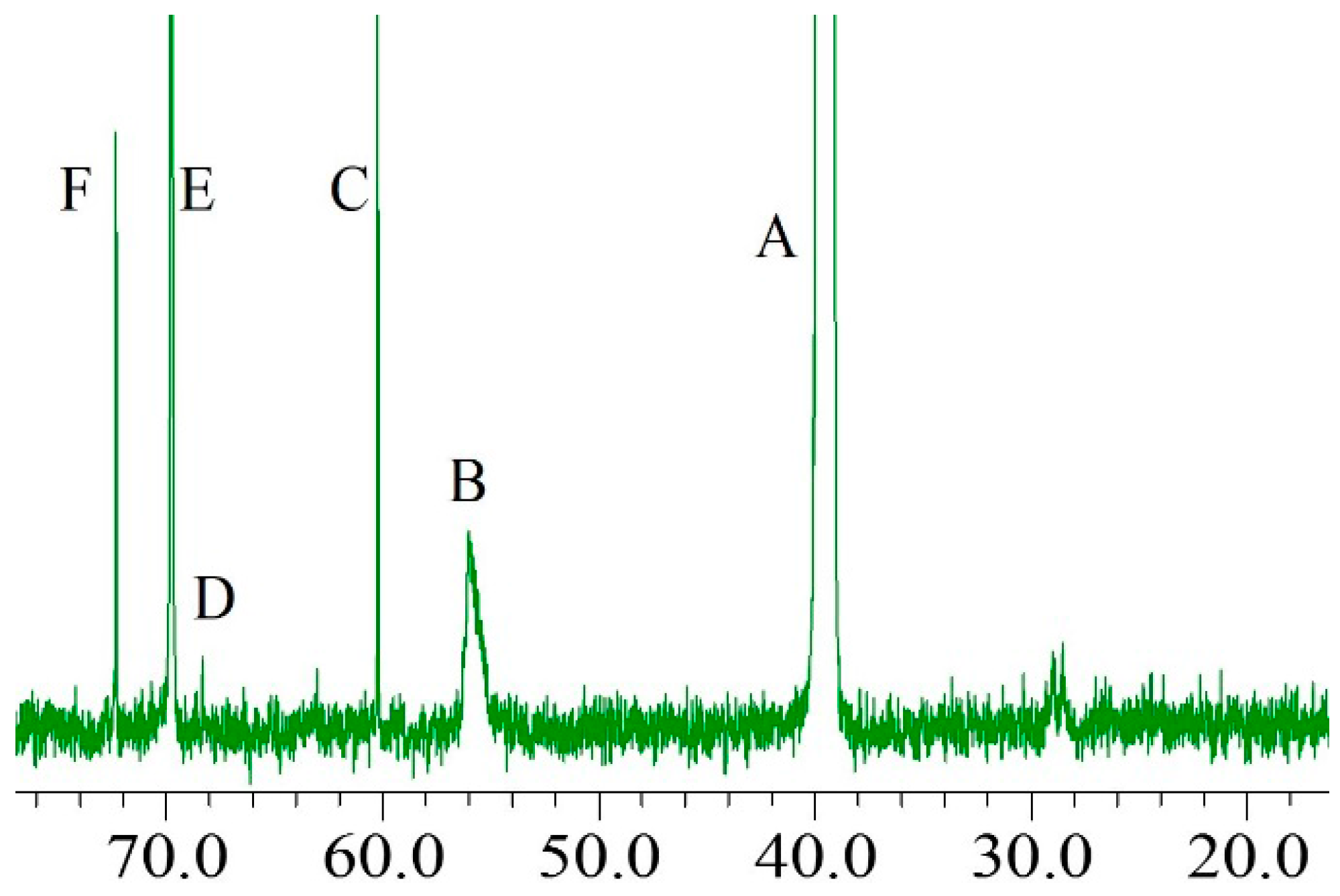
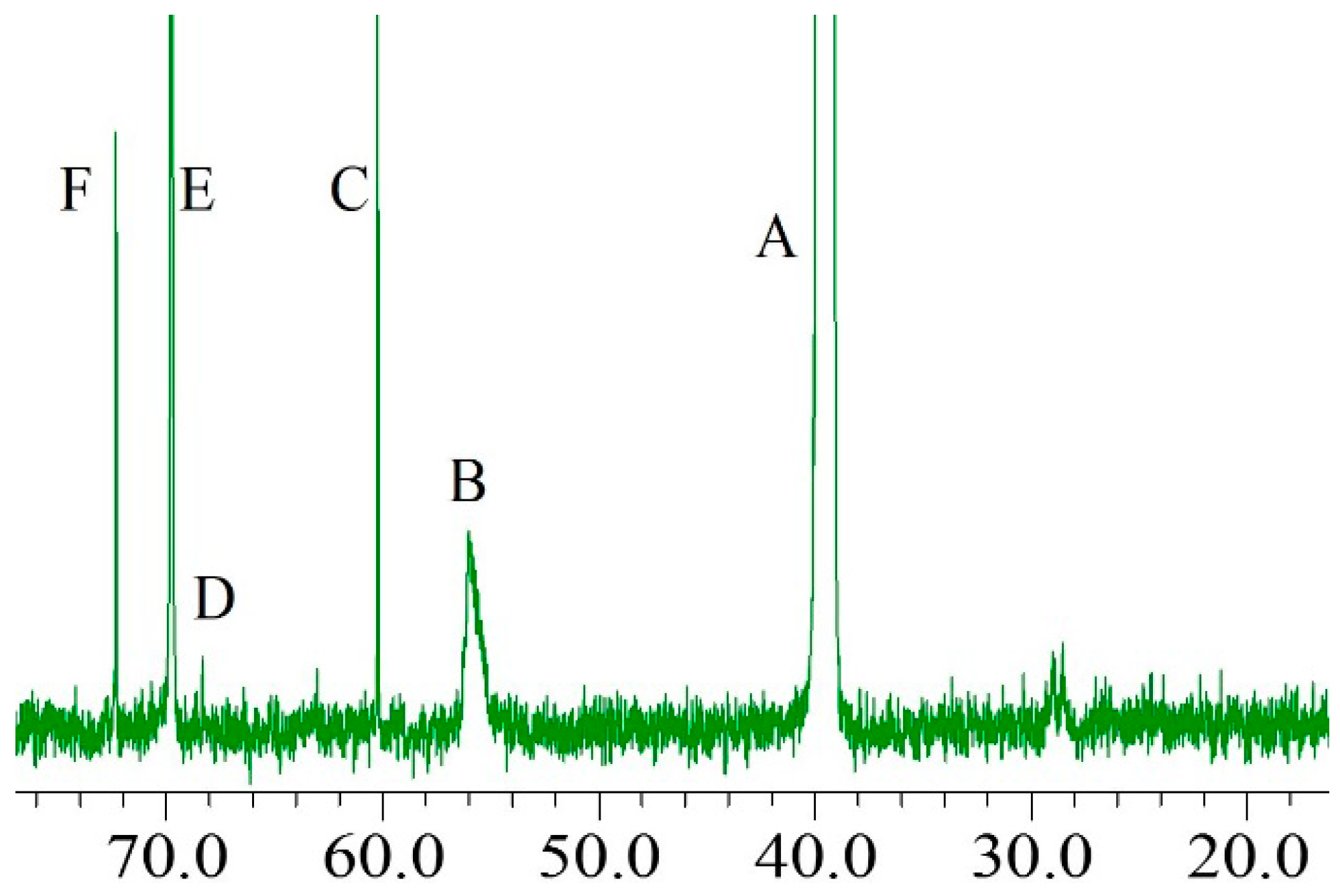
2.3. 13C-NMR Analysis
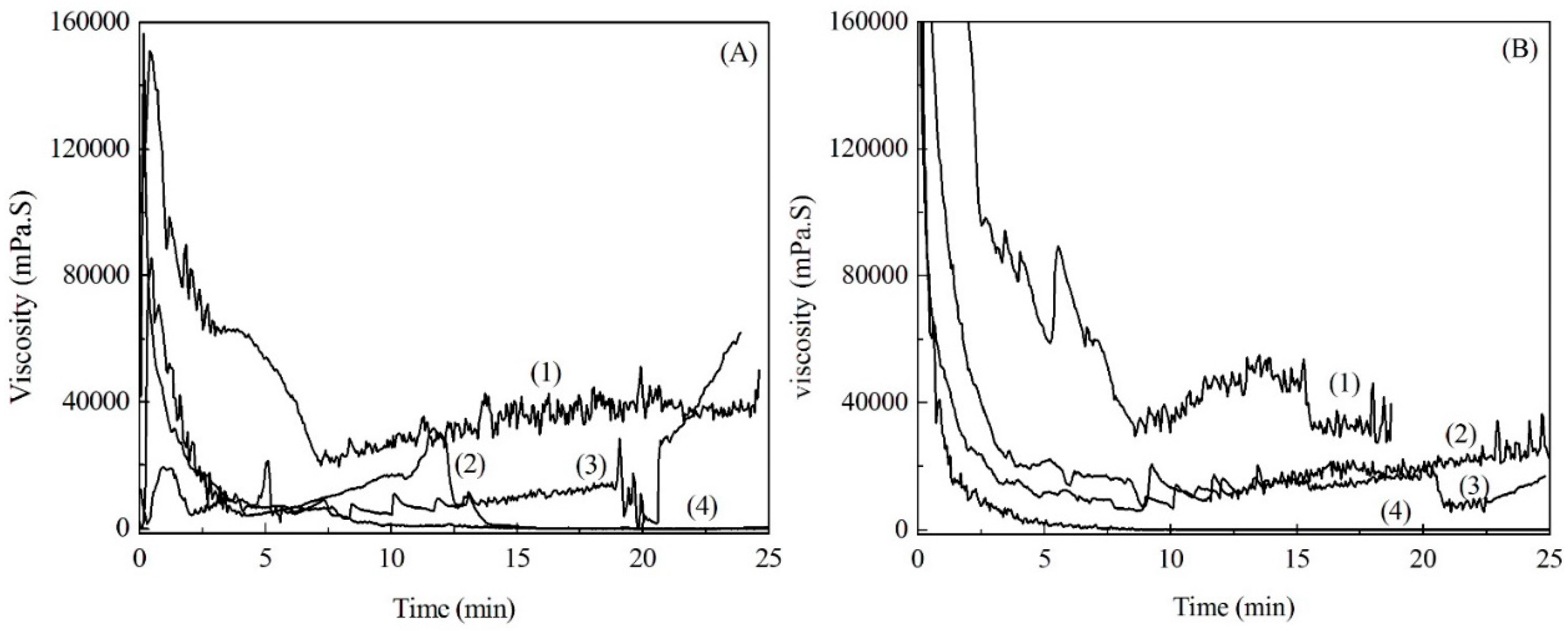
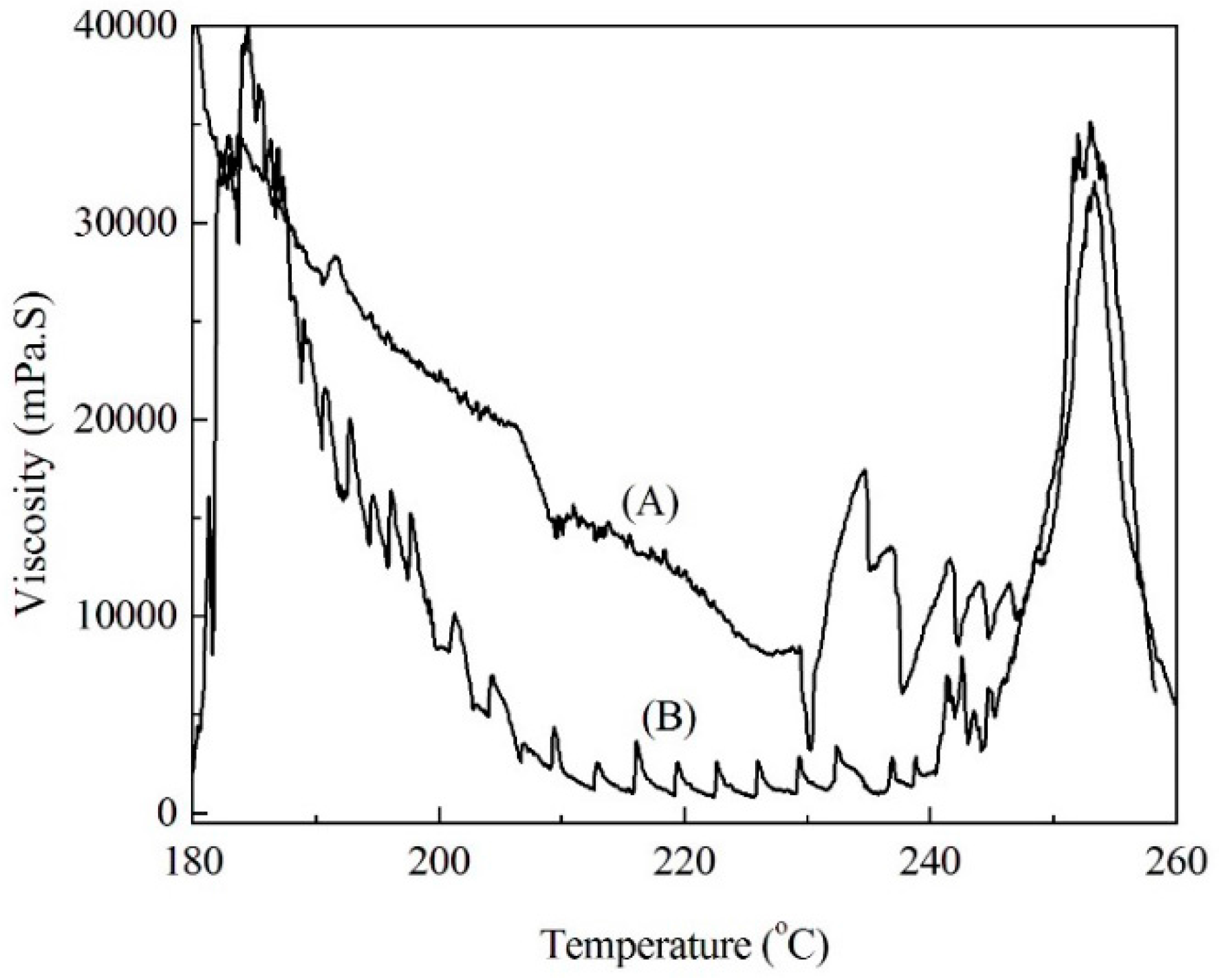
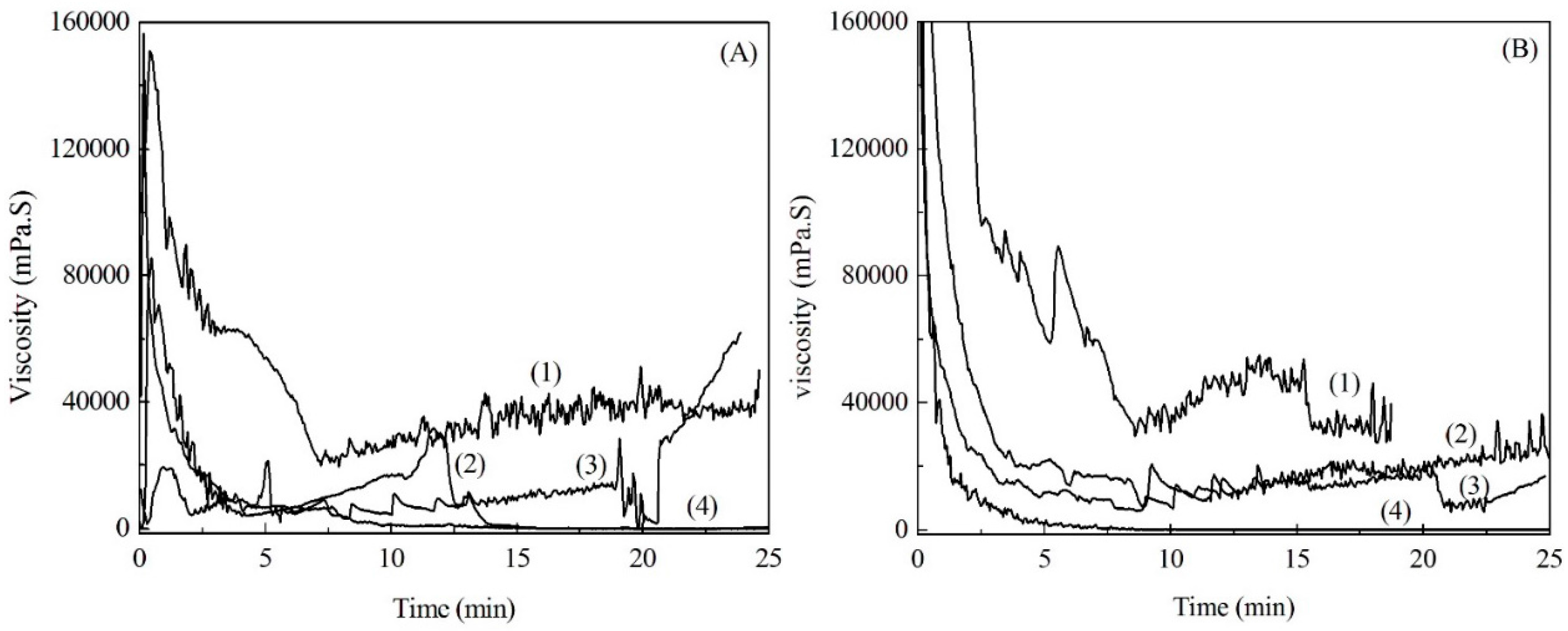
2.4. Viscosity of Soda Lignin and Fiber Preparation
3. Materials and Methods
3.1. Materials
3.2. Modification of Soda Lignin
3.3. Soda Lignin Melt-Spinning
3.4. Characterization
3.4.1. Thermal Analysis
3.4.2. Fourier Transform Infrared Spectroscopy (FTIR)
3.4.3. 13C-Nuclear Magnetic Resonance Analysis (13C-NMR)
3.4.4. Dynamic Viscoelastic Measurement
3.4.5. Scanning Electronic Microscopy (SEM)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mai, C.; Milstein, O.; Huttermann, A. Chemoenzymatical grafting of acrylamide onto lignin. J. Biotechnol. 2000, 79, 173–183. [Google Scholar] [CrossRef]
- Zimovets, I.P.; Imarov, A.K. Production and utilization of generator gas from lignin. Chem. Tech. Fuels Oils 1996, 32, 288–289. [Google Scholar] [CrossRef]
- Dilling, P. Low electrolyte sodium lignosulphonate(s) are used as dispersants in dyestuffs and additives in printing gels. U.S. Patent 4,590,262, 20 May 1986. [Google Scholar]
- Ouyang, X.P.; Ke, L.X.; Qiu, X.Q.; Guo, Y.X.; Pang, Y.X. Sulfonation of alkali lignin and its potential use in dispersant for cement. J. Disper. Sci. Technol. 2009, 30, 1–6. [Google Scholar] [CrossRef]
- Oiivares, M.; Guzmfin, J.A.; Natho, A.; Saavedra, A. Kraft lignin utilization in adhesives. Wood Sci. Tech. 1988, 22, 157–165. [Google Scholar] [CrossRef]
- Pearl, I.A. The Chemistry of Lignin; Marcel Dekker: New York, NY, USA, 1967; pp. 298–307. [Google Scholar]
- Frank, E.; Hermanutz, F.; Buchmeiser, M.R. Carbon Fibers: Precursors, manufacturing, and properties. Macromol. Mater. Eng. 2012, 297, 493–501. [Google Scholar] [CrossRef]
- Lallave, M.; Bedia, J.; Ruiz-Rosas, R.; Rodriguez-Mirasol, J.; Cordero, T.; Otero, J.C.; Marquez, M.; Barrero, A.; Loscertales, I.G. Filled and hollow carbon nanofibers by coaxial electrospinning of Alcell lignin without binder polymers. Adv. Mater. 2007, 19, 4292–4296. [Google Scholar] [CrossRef]
- Maradur, S.P.; Kim, C.H.; Kim, S.Y.; Kim, B.H.; Kim, W.C.; Yang, K.S. Preparation of carbon fibers from a lignin copolymer with polyacrylonitrile. Synthetic Met. 2012, 162, 453–459. [Google Scholar] [CrossRef]
- Zhang, M.; Ogale, A.A. Carbon fibers from dry-spinning of acetylated softwood kraft lignin. Carbon 2014, 69, 626–629. [Google Scholar] [CrossRef]
- Otani, S.; Fukuoka, Y.; Igarashi, B.; Sasaki, K. Method for producing carbonized lignin fiber. U.S. Patent 3,461,082, 12 August 1969. [Google Scholar]
- Kadla, J.F.; Kubo, S.; Venditti, R.A.; Gilbert, R.D.; Compere, A.L.; Griffith, W. Lignin-based carbon fiber for composite fiber applications. Carbon 2002, 40, 2913–2920. [Google Scholar] [CrossRef]
- Uraki, Y.; Kubo, S.; Nigo, N.; Sano, Y.; Sasaya, T. Preparation of carbon fibers from organosolv lignin obtained by aqueous acetic acid pulping. Holzforschung 1995, 49, 343–350. [Google Scholar] [CrossRef]
- Lin, J.; Kubo, S.; Yamada, T.; Koda, K.; Uraki, Y. Chemical thermostabilization for the preparation of carbon fibers from softwood lignin. Bioresources 2012, 7, 5634–5646. [Google Scholar] [CrossRef]
- Sudo, K.; Shimizu, K. A new carbon fiber from lignin. J. Appl. Polym. Sci. 1992, 44, 127–134. [Google Scholar] [CrossRef]
- Sudo, K.; Shimizu, K.; Nakashima, N.; Yokoyama, A. A new modification method of exploded lignin for the preparation of a carbon fiber precursor. J. Appl. Polym. Sci. 1993, 48, 1485–1491. [Google Scholar] [CrossRef]
- Kubo, S.; Kadla, J.F. Lignin-based carbon fibers: Effect of synthetic polymer blending on fiber properties. J. Polym. Environ. 2005, 13, 97–105. [Google Scholar] [CrossRef]
- Kadla, J.F.; Kubo, S.; Venditti, R.A.; Gilbert, R.D. Novel hollow core fibers prepared from lignin polypropylene blends. J. Appl. Polym. Sci. 2002, 85, 1353–1355. [Google Scholar] [CrossRef]
- Kubo, S.; Uraki, Y.; Sano, Y. Preparation of carbon fibers from softwood lignin by atmospheric acetic acid pulping. Carbon 1998, 36, 1119–1124. [Google Scholar] [CrossRef]
- Baker, D.A.; Gallego, N.C.; Baker, F.S. On the characterization and spinning of an organic-purified lignin toward the manufacture of low-cost carbon fiber. J. Appl. Polym. Sci. 2012, 124, 227–234. [Google Scholar] [CrossRef]
- Nordstrom, Y.; Norberg, I.; Sjoholm, E.; Drougge, R. A New softening agent for melt spinning of softwood kraft lignin. J. Appl. Polym. Sci. 2013, 129, 1274–1279. [Google Scholar] [CrossRef]
- Kubo, S.; Ishikawa, N.; Uraki, Y.; Sano, Y. Preparation of lignin fibers from softwood acetic acid lignin—Relationship between fusibility and the chemical structure of lignin. Mokuzai Gakkaishi 1997, 43, 655–662. [Google Scholar]
- Silverstein, R.M.; Webster, F.X.; Kiemle, D.J. Infrared spectroscopy. In Spectrometric Identification of Organic Compounds; Robert, M.S., Ed.; Wiley: Hoboken, NJ, USA, 2005; pp. 72–108. [Google Scholar]
- Kadla, J.F.; Kubo, S. Lignin-based polymer blends: analysis of intermolecular interaction in lignin-synthetic polymer blends. Compos. Part A-Appl. S. 2004, 35, 395–400. [Google Scholar] [CrossRef]
- Kubo, S.; Kadla, J.F. Kraft lignin/poly(ethylene oxide) blends: Effect of lignin structure on miscibility and hydrogen bonding. J. Appl. Polym. Sci. 2005, 98, 1437–1444. [Google Scholar] [CrossRef]
- Kishimoto, T.; Ueki, A.; Sano, Y. Delignification mechanism during high-boiling solvent pulping. Part 3: Structural changes in lignin analyzed by 13C-NMR spectroscopy. Holzforschung 2003, 57, 602–610. [Google Scholar] [CrossRef]
- Kishimoto, T.; Ueki, A.; Takamori, H.; Uraki, Y.; Ubukata, M. Delignification mechanism during high-boiling solvent pulping. Part 6: Changes in lignin structure analyzed by 1H-13C correlation 2-D NMR spectroscopy. Holzforschung 2004, 58, 355–362. [Google Scholar] [CrossRef]
- Adler, E.; Sven, H. Estimation of phenolic hydroxyl groups in lignin. I: Periodate oxidation of guaiacol compounds. Acta Chem. Scand. 1955, 9, 319–334. [Google Scholar] [CrossRef]
- Kubo, S.; Yamada, T.; Hashida, K.; Ono, H. Grafting of ethylene glycol chains in lignin during the solvolysis for biomass conversion using ethylene carbonate/ethylene glycol system. Chem. Lett. 2007, 36, 502–503. [Google Scholar] [CrossRef]
- Qin, W.; Kadla, J.F. Carbon fibers based on pyrolytic lignin. J. Appl. Polym. Sci. 2012, 126, 203–212. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intensity Ratio | (A) | (B) | (C) | (D) |
|---|---|---|---|---|
| IC-O/IC=C | 1.08 | 1.57 | 1.91 | 2.03 |
| IC-H/IC=C | 0.46 | 0.69 | 0.84 | 1.04 |
| IO-H/IC=C | 0.67 | 0.94 | 0.95 | 1.03 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, F.; Lin, J.; Zhao, G. Preparation and Characterization of Modified Soda Lignin with Polyethylene Glycol. Materials 2016, 9, 822. https://doi.org/10.3390/ma9100822
Zhang F, Lin J, Zhao G. Preparation and Characterization of Modified Soda Lignin with Polyethylene Glycol. Materials. 2016; 9(10):822. https://doi.org/10.3390/ma9100822
Chicago/Turabian StyleZhang, Fangda, Jian Lin, and Guangjie Zhao. 2016. "Preparation and Characterization of Modified Soda Lignin with Polyethylene Glycol" Materials 9, no. 10: 822. https://doi.org/10.3390/ma9100822