Expanding the Applicability of Poly(Ionic Liquids) in Solid Phase Microextraction: Pyrrolidinium Coatings

 , ,
, ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
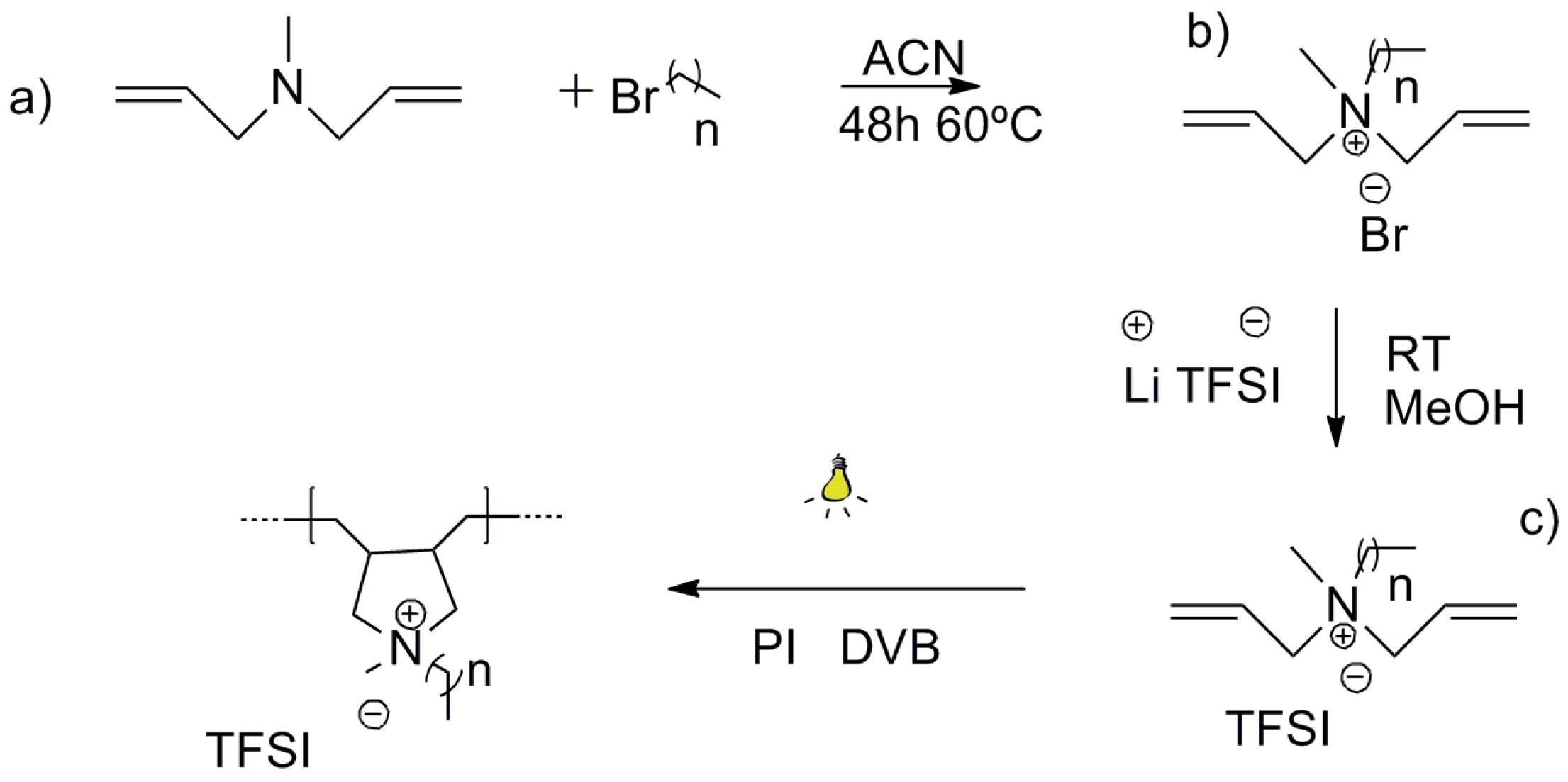
2.1. Synthesis of Ionic Liquid Monomers and Poly(Ionic Liquids)
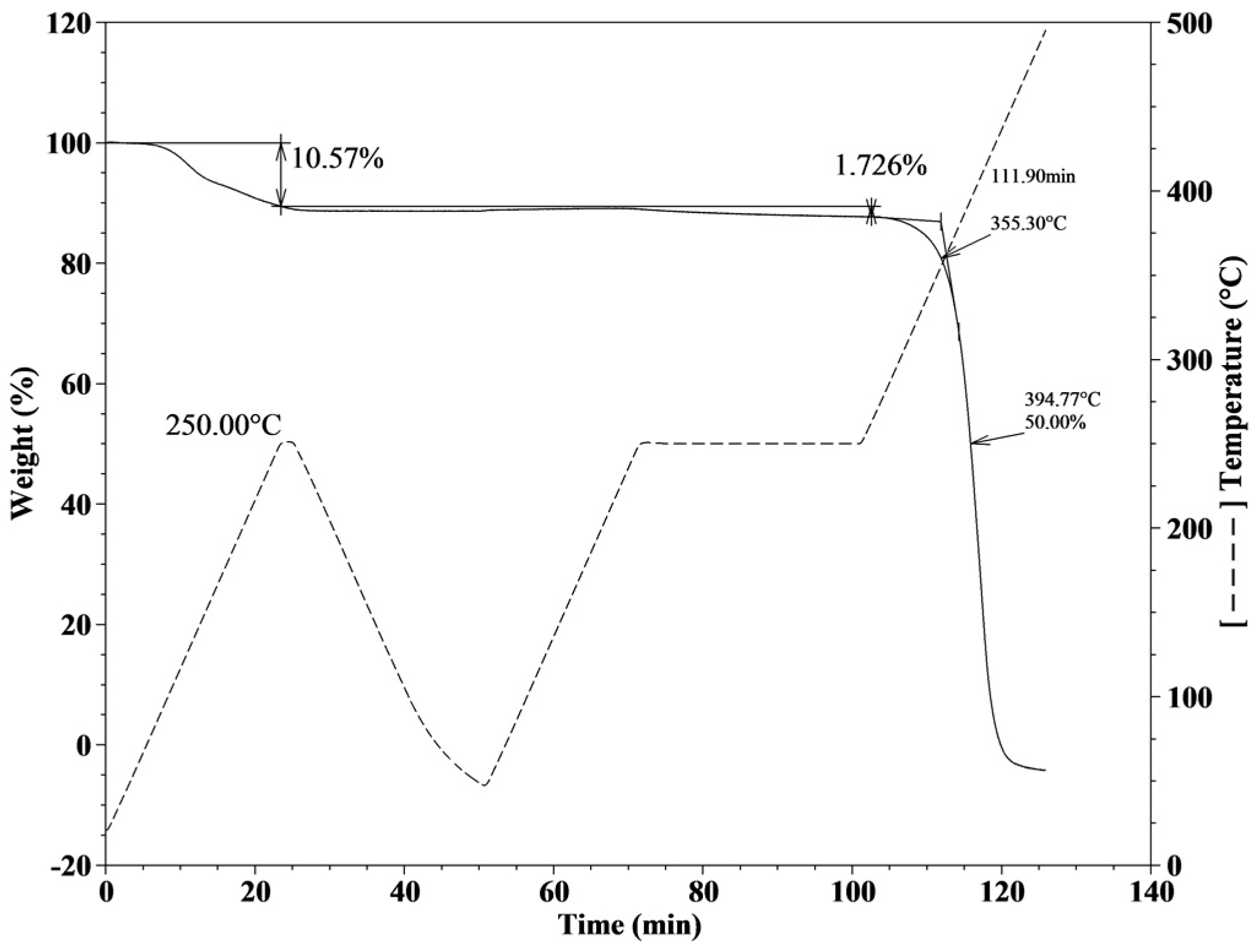
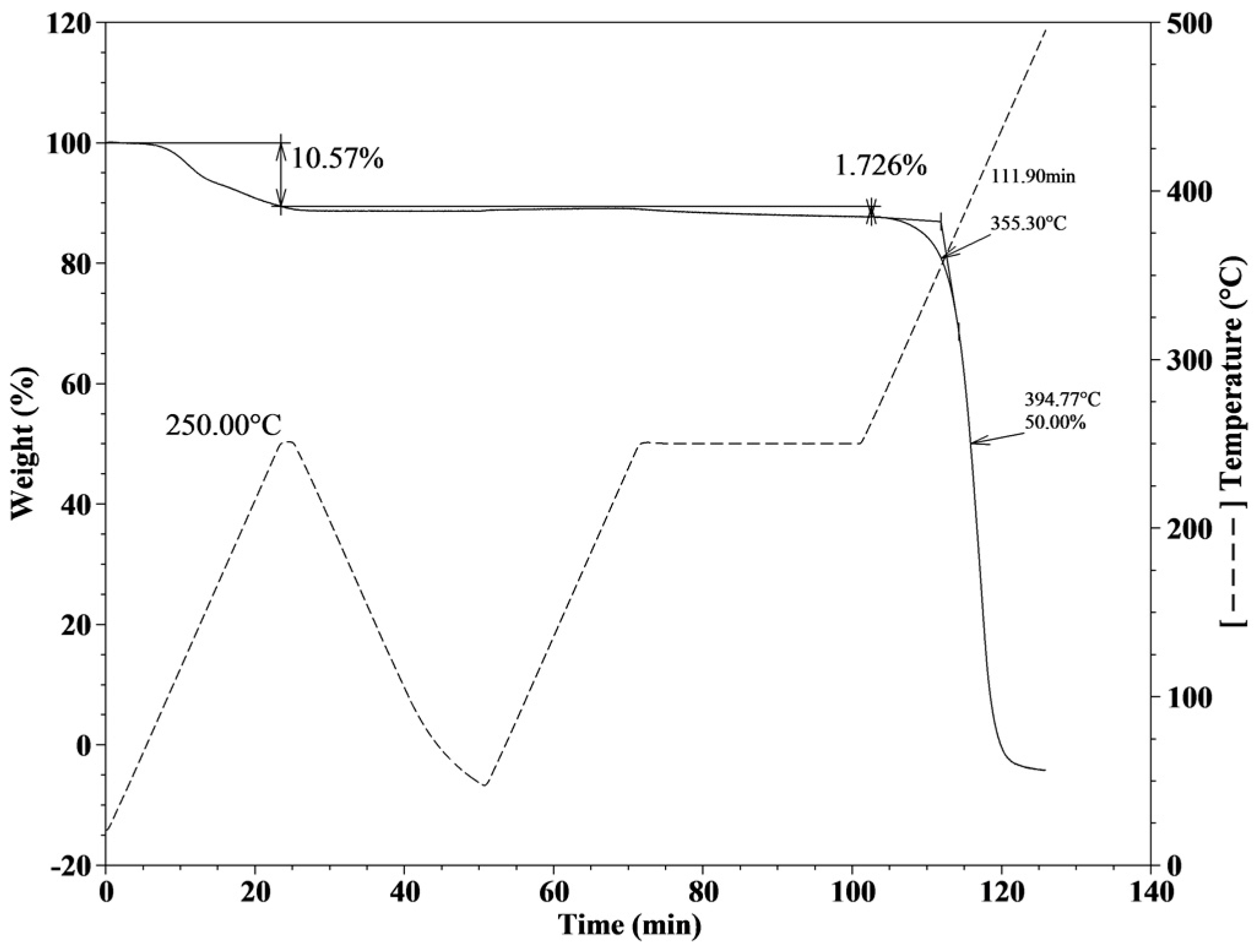
2.2. Thermal Characterization of the Poly(Ionic Liquids)
2.3. Fiber Coating Analysis
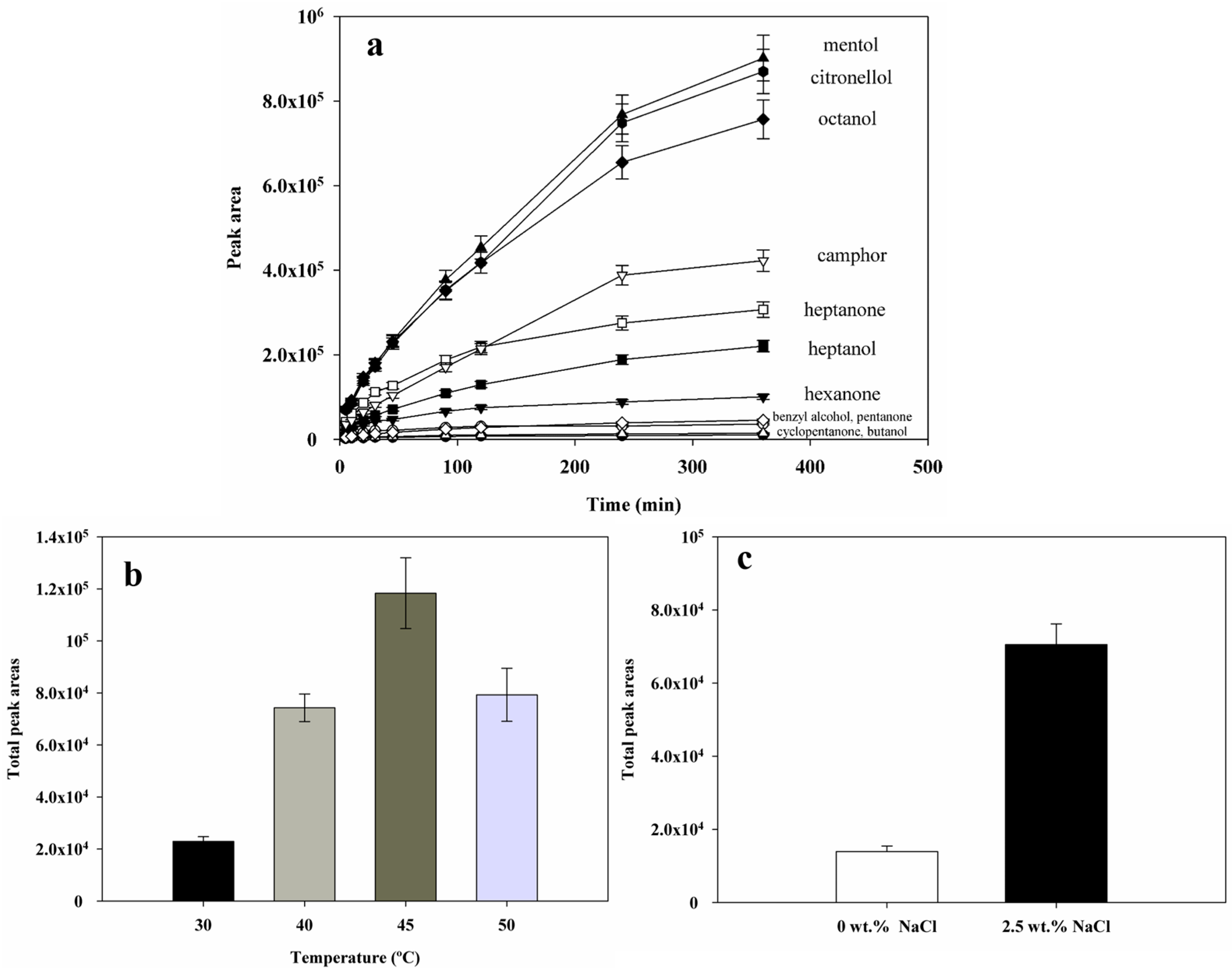
2.4. Optimization of Headspace-SPME Parameters
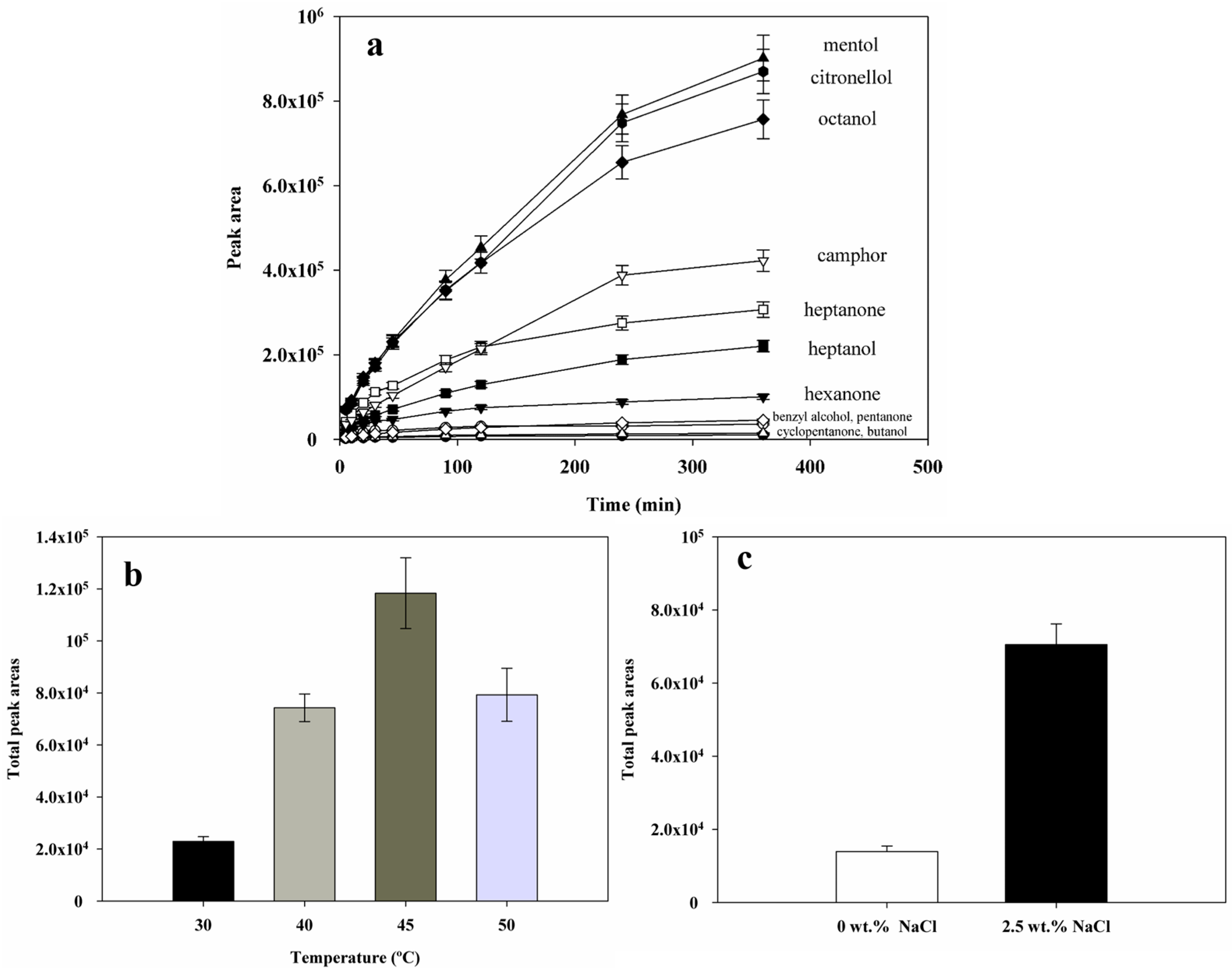
2.4.1. Sorption Time
2.4.2. Extraction Temperature
2.4.3. Salt Addition
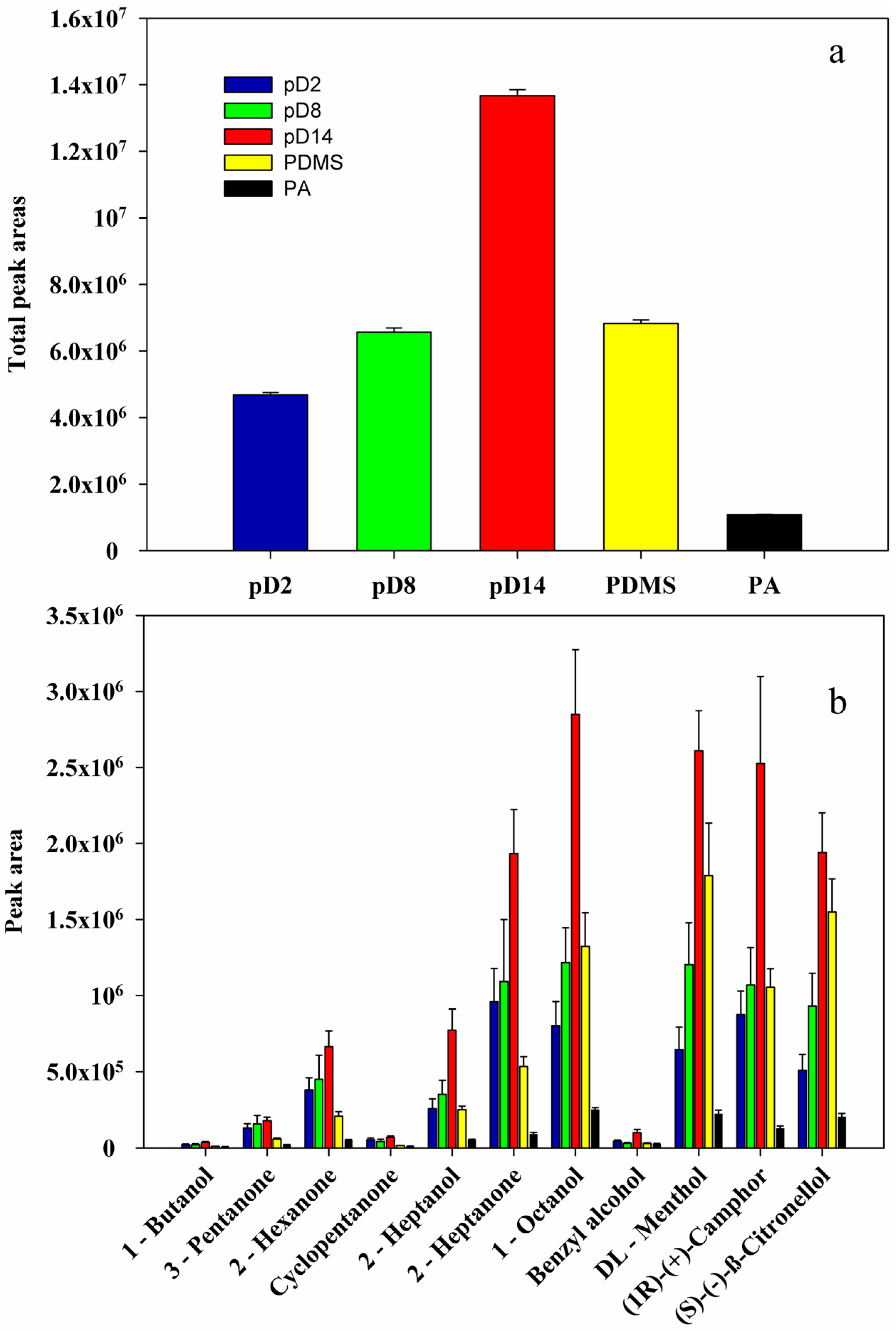
2.5. Performance of the Prepared Fibers
2.6. Validation, Matrix Effect and Recovery Using Fiber pD14
3. Materials and Methods
3.1. Synthesis of Ionic Liquid Monomers
3.2. TGA and DSC Experiments
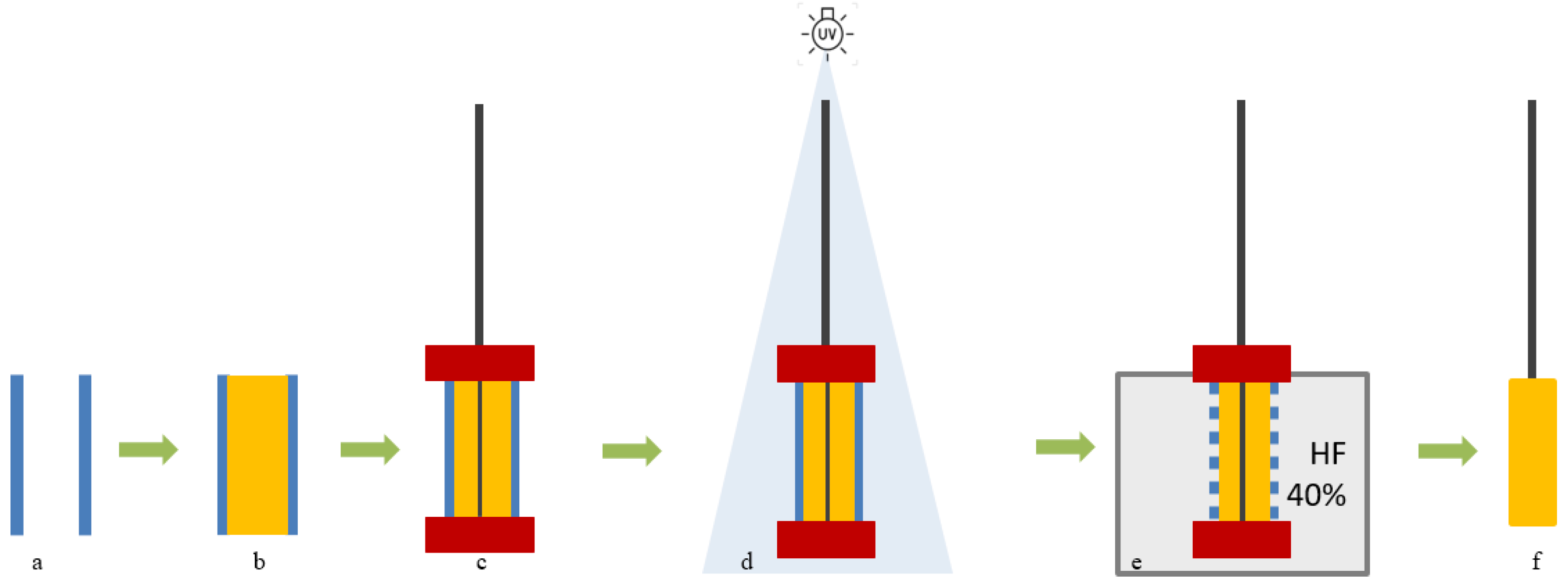
3.3. SPME Fiber Coating Preparation
3.4. Headspace—SPME Methodology
3.5. Matrix Effect, Validation and Recovery Tests
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Arthur, C.L.; Pawliszyn, J. Solid-phase microextraction with thermal-desorption using fused-silica optical fibers. Anal. Chem. 1990, 62, 2145–2148. [Google Scholar] [CrossRef]
- Xu, J.; Zheng, J.; Tian, J.; Zhu, F.; Zeng, F.; Su, C.; Ouyang, G. New materials in solid-phase microextraction. Trends Anal. Chem. 2013, 47, 68–83. [Google Scholar] [CrossRef]
- Spietelun, A.; Pilarczyk, M.; Kloskowski, A.; Namieśnik, J. Current trends in solid-phase microextraction (SPME) fibre coatings. Chem. Soc. Rev. 2010, 39, 4524–4537. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef] [PubMed]
- Rocío-Bautista, P.; Andez, I.P.; An, J.P.; Pino, V.O. Are metal-organic frameworks able to provide a new generation of solid-phase microextraction coatings?—A review. Anal. Chim. Acta 2016, 939, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.N.; Simão, V.; Merib, J.; Carasek, E. Use of green coating (cork) in solid-phase microextraction for the determination of organochlorine pesticides in water by gas chromatography-electron capture detection. Talanta 2015, 134, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.D.; Canestraro, A.J.; Anderson, J.L. Ionic liquids in solid-phase microextraction: A review. Anal. Chim. Acta 2011, 695, 18–43. [Google Scholar] [CrossRef] [PubMed]
- Patinha, D.J.S.; Tomé, L.C.; Garcia, H.; Ferreira, R.; Pereira, C.S.; Rebelo, L.P.N.; Marrucho, I.M. The role of water in cholinium carboxylate ionic liquid’s aqueous solutions. J. Chem. Thermodyn. 2015, 84, 93–100. [Google Scholar] [CrossRef]
- Freire, M.G.; Claudio, A.F.M.; Araujo, J.M.M.; Coutinho, J.A.P.; Marrucho, I.M.; Lopes, J.N.C.; Rebelo, L.P.N.; Lopes, J.N.C.; Rebelo, L.P.N. Aqueous biphasic systems: A boost brought about by using ionic liquids. Chem. Soc. Rev. 2012, 41, 4966–4995. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Li, N.; Jiang, G.B.; Li, J.M.; Jonsson, J.A.; Wen, M.J. Disposable ionic liquid coating for headspace solid-phase microextraction of benzene, toluene, ethylbenzene, and xylenes in paints followed by gas chromatography-flame ionization detection. J. Chromatogr. A 2005, 1066, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Marcilla, R.; Blazquez, J.A.; Fernandez, R.; Grande, H.; Pomposo, J.A.; Mecerreyes, D. Synthesis of novel polycations using the chemistry of ionic liquids. Macromol. Chem. Phys. 2005, 206, 299–304. [Google Scholar] [CrossRef]
- Zhao, F.; Meng, Y.; Anderson, J.L. Polymeric ionic liquids as selective coatings for the extraction of esters using solid-phase microextraction. J. Chromatogr. A 2008, 1208, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.D.; Ho, T.D.; Cole, W.T.S.; Anderson, J.L. Determination of polychlorinated biphenyls in ocean water and bovine milk using crosslinked polymeric ionic liquid sorbent coatings by solid-phase microextraction. Talanta 2014, 118, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Merib, J.; Yu, H.; Carasek, E.; Anderson, J.L. Determination of compounds with varied volatilities from aqueous samples using a polymeric ionic liquid sorbent coating by direct immersion-headspace solid-phase microextraction. Anal. Methods 2016, 8, 4108–4118. [Google Scholar] [CrossRef]
- Feng, J.; Sun, M.; Li, J.; Liu, X.; Jiang, S. A novel aromatically functional polymeric ionic liquid as sorbent material for solid-phase microextraction. J. Chromatogr. A 2012, 1227, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Wajert, J.C.; Anderson, J.L. Polymeric Ionic Liquids as CO2 Selective Sorbent Coatings for Solid-Phase Microextraction. Anal. Chem. 2010, 82, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Sun, M.; Wang, X.; Liu, X.; Jiang, S. Ionic liquids-based crosslinked copolymer sorbents for headspace solid-phase microextraction of polar alcohols. J. Chromatogr. A 2012, 1245, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Pino, V.; Anderson, J.L. Role of counteranions in polymeric ionic liquid-based solid-phase microextraction coatings for the selective extraction of polar compounds. Anal. Chim. Acta 2010, 687, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.D.; Joshi, M.D.; Silver, M.A.; Anderson, J.L. Selective extraction of genotoxic impurities and structurally alerting compounds using polymeric ionic liquid sorbent coatings in solid-phase microextraction: Alkyl halides and aromatics. J. Chromatogr. A 2012, 1240, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Liu, J.-F. Development of a solid-phase microextraction fiber by chemical binding of polymeric ionic liquid on a silica coated stainless steel wire. J. Chromatogr. A 2012, 1230, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.D.; Anderson, J.L. Recent advances of ionic liquids in separation science and mass spectrometry. RSC Adv. 2012, 2, 5470–5484. [Google Scholar] [CrossRef]
- Tomé, L.C.; Marrucho, I.M. Ionic liquid-based materials: A platform to design engineered CO2 separation membranes. Chem. Soc. Rev. 2016, 45, 2785–2824. [Google Scholar] [CrossRef] [PubMed]
- Döbbelin, M.; Azcune, I.; Bedu, M.; de Luzuriaga, A.R.; Genua, A.; Jovanovski, V.; Cabañero, G.; Odriozola, I. Synthesis of pyrrolidinium-based poly(ionic liquid) electrolytes with poly(ethylene glycol) side chains. Chem. Mater. 2012, 24, 1583–1590. [Google Scholar] [CrossRef]
- Zeng, J.; Chen, J.; Wang, Y.; Chen, W.; Chen, X.; Wang, X. Development of relatively selective, chemically and mechanically robust solid-phase microextraction fibers based on methacrylic acid–trimethylolpropanetrimethacrylate co-polymers. J. Chromatogr. A 2008, 1208, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Podmore, I. Current Challenges in Volatile Organic Compounds Analysis as Potential Biomarkers of Cancer. J. Biomark. 2015, 2015, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zimudzi, T.J.; Li, N.; Pan, J.; Lin, B.; Hickner, M.A. Crosslinking of comb-shaped polymer anion exchange membranes via thiol–ene click chemistry. Polym. Chem. 2016, 7, 2464–2475. [Google Scholar] [CrossRef]
- Jovanovski, V.; Marcilla, R.; Mecerreyes, D. Tuning the properties of functional pyrrolidinium polymers by (Co)polymerization of diallyldimethylammonium ionic liquids. Macromol. Rapid Commun. 2010, 31, 1646–1651. [Google Scholar] [CrossRef] [PubMed]
- Butler, G.B.; Angelo, R.J. Preparation and Polymerization of Unsaturated Quaternary Ammonium Compounds. VIII. A Proposed Alternating Intramolecular-Intermolecular Chain Propagation. J. Am. Chem. Soc. 1957, 79, 3128–3131. [Google Scholar] [CrossRef]
- Hall, A.W.; Blackwood, K.M.; Milne, P.E.Y.; Goodby, J.W. Novel UV cured coatings and adhesives based on the photoinitiated cyclopolymerization of derivatives of diallylamine. Chem. Commun. 2003, 2530–2531. [Google Scholar] [CrossRef]
- Ojovan, M.I. Ordering and structural changes at the glass-liquid transition. J. Non-Cryst. Solids 2013, 382, 79–86. [Google Scholar] [CrossRef]
- George, S.C.; Thomas, S. Transport phenomena through polymeric systems. Prog. Polym. Sci. 2001, 26, 985–1017. [Google Scholar] [CrossRef]
- Pawliszyn, J. Applications of Solid Phase Microextraction, 1st ed.; Royal Society of Chemistry: Cambridge, UK, 1999. [Google Scholar]
- Zhang, C.; Anderson, J.L. Polymeric ionic liquid bucky gels as sorbent coatings for solid-phase microextraction. J. Chromatogr. A 2014, 1344, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Pawliszyn, J. Handbook of Solid Phase Microextraction, 1st ed.; Elsevier: Oxford, UK, 2012. [Google Scholar] [CrossRef]
- Lord, H.; Pawliszyn, J. Evolution of solid-phase microextraction technology. J. Chromatogr. A 2000, 885, 153–193. [Google Scholar] [CrossRef]
- Hou, J.G.; Ma, Q.; Du, X.Z.; Deng, H.L.; Gao, J.Z. Inorganic/organic mesoporous silica as a novel fiber coating of solid-phase microextraction. Talanta 2004, 62, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Cowie, J.M.G.; Arrighi, V. Polymers: Chemistry and Physics of Modern Materials, 3rd ed.; CRC Press: New York, NY, USA, 2007. [Google Scholar] [CrossRef]
- Meng, Y.; Anderson, J.L. Tuning the selectivity of polymeric ionic liquid sorbent coatings for the extraction of polycyclic aromatic hydrocarbons using solid-phase microextraction. J. Chromatogr. A 2010, 1217, 6143–6152. [Google Scholar] [CrossRef] [PubMed]
- Trujillo-Rodríguez, M.J.; Yu, H.; Cole, W.T.S.; Ho, T.D.; Pino, V.; Anderson, J.L.; Afonso, A.M. Polymeric ionic liquid coatings versus commercial solid-phase microextraction coatings for the determination of volatile compounds in cheeses. Talanta 2014, 121, 153–162. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patinha, D.J.S.; Tomé, L.C.; Isik, M.; Mecerreyes, D.; Silvestre, A.J.D.; Marrucho, I.M. Expanding the Applicability of Poly(Ionic Liquids) in Solid Phase Microextraction: Pyrrolidinium Coatings. Materials 2017, 10, 1094. https://doi.org/10.3390/ma10091094
Patinha DJS, Tomé LC, Isik M, Mecerreyes D, Silvestre AJD, Marrucho IM. Expanding the Applicability of Poly(Ionic Liquids) in Solid Phase Microextraction: Pyrrolidinium Coatings. Materials. 2017; 10(9):1094. https://doi.org/10.3390/ma10091094
Chicago/Turabian StylePatinha, David J. S., Liliana C. Tomé, Mehmet Isik, David Mecerreyes, Armando J. D. Silvestre, and Isabel M. Marrucho. 2017. "Expanding the Applicability of Poly(Ionic Liquids) in Solid Phase Microextraction: Pyrrolidinium Coatings" Materials 10, no. 9: 1094. https://doi.org/10.3390/ma10091094
APA StylePatinha, D. J. S., Tomé, L. C., Isik, M., Mecerreyes, D., Silvestre, A. J. D., & Marrucho, I. M. (2017). Expanding the Applicability of Poly(Ionic Liquids) in Solid Phase Microextraction: Pyrrolidinium Coatings. Materials, 10(9), 1094. https://doi.org/10.3390/ma10091094