Molecular Aspects of Dopaminergic Neurodegeneration: Gene-Environment Interaction in Parkin Dysfunction

Abstract
:1. Introduction
2. Gene-Environment Interplay
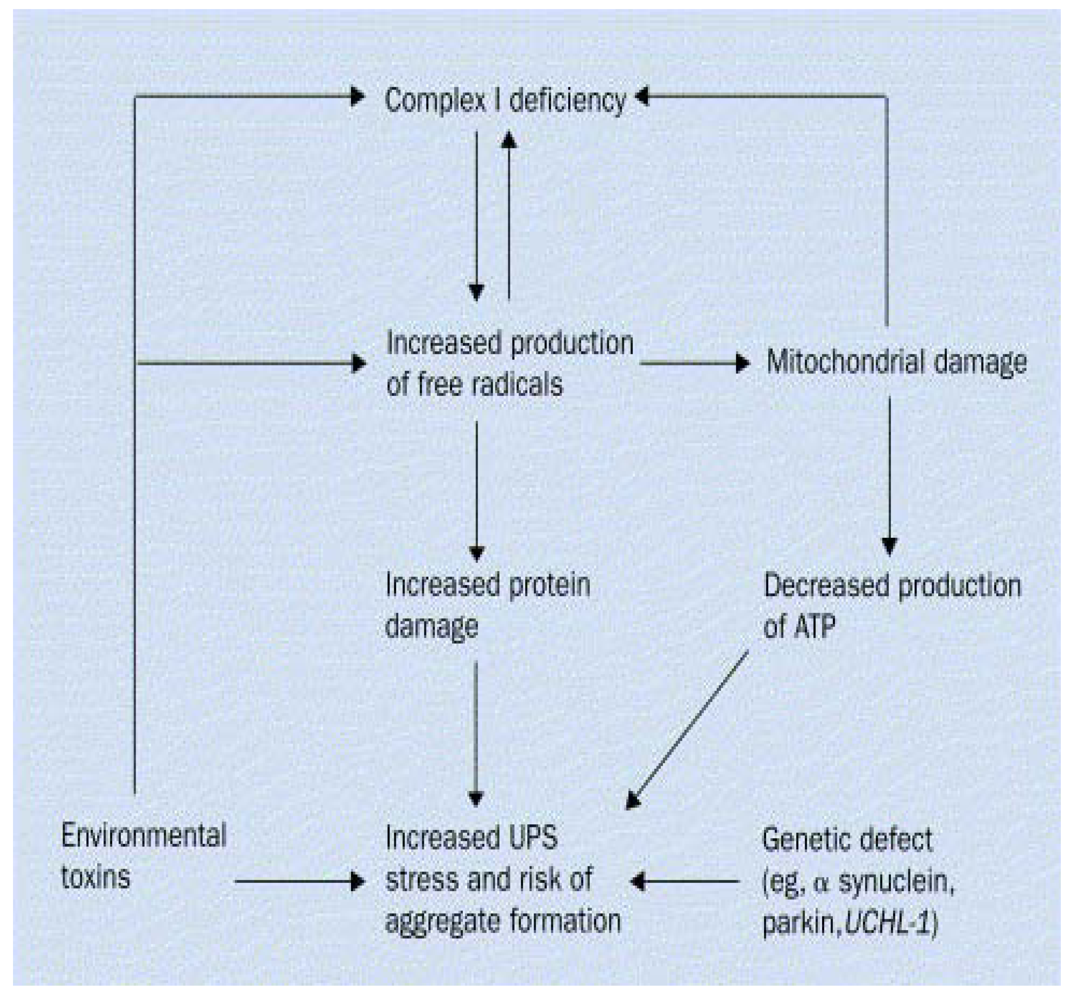

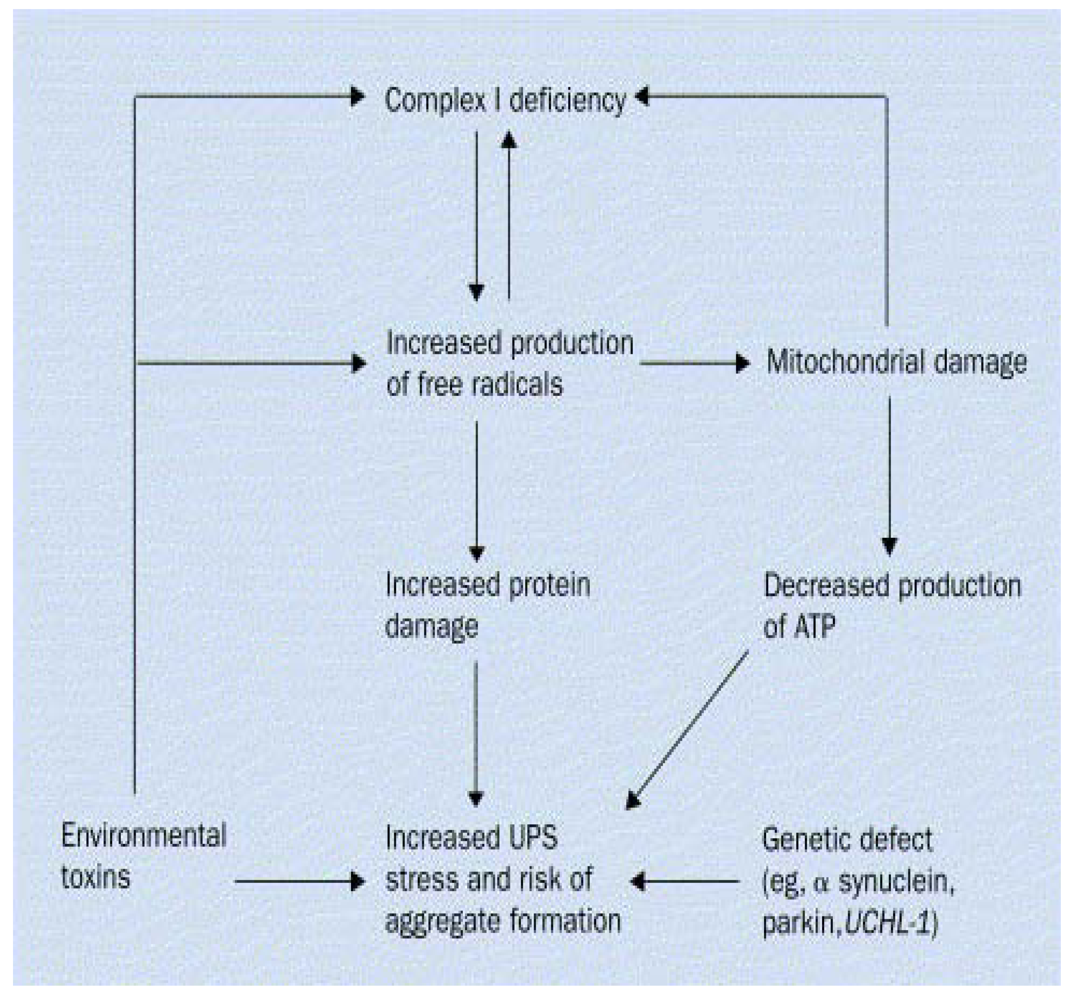{kind=link}
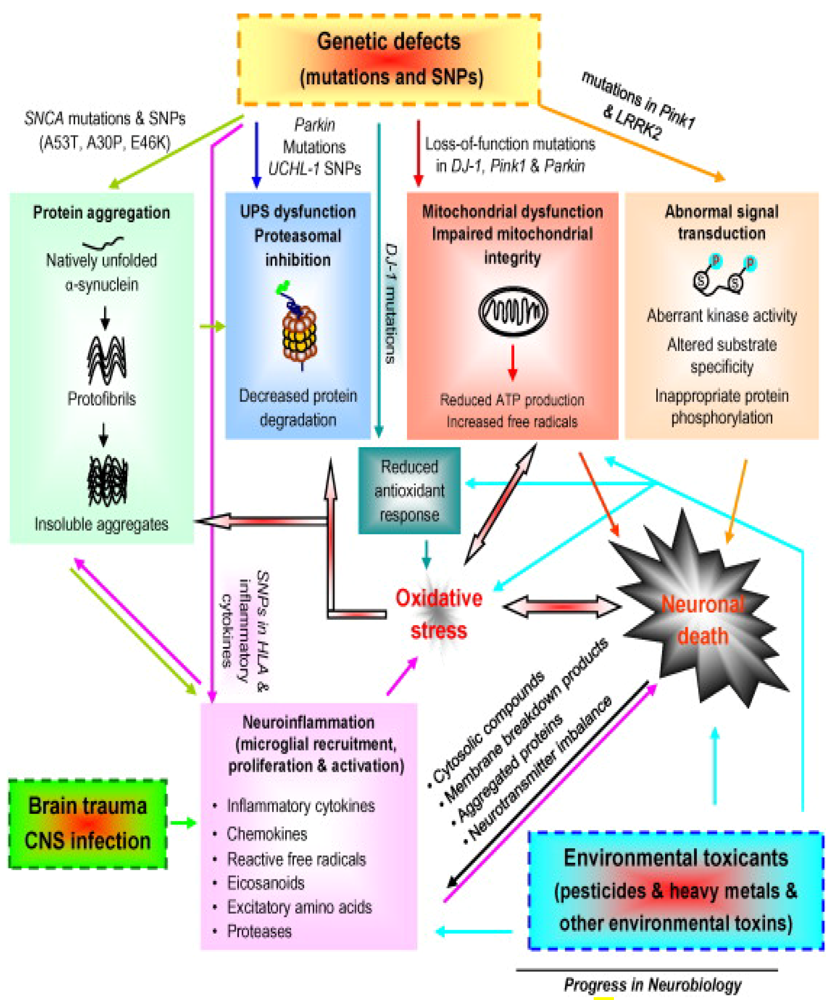{kind=link}
{kind=link}
| Locus | Chromosome Location | Gene | Inheritance Pattern | Typical Phenotype |
|---|---|---|---|---|
| PARK1& PARK4 | 4q21-q23 | α-synuclein | AD | Earlier onset, features of DLB common |
| PARK2 | 6q25.2-q27 | parkin | Usually AR | Early onset with slow progression |
| PARK3 | 2p13 | unknown | AD, IP | Classic PD, sometimes dementia |
| PARK5 | 4p14 | UCH-L1 | unlcear | Classic PD |
| PARK6 | 1p35-p36 | PINK1 | AR | Early onset with slow progression |
| PARK7 | 1p36 | DJ-1 | AR | Early onset with slow progression |
| PARK8 | 12p11.2-q13.1 | LRRK2 | AD | Classic PD |
| PARK10 | 1p32 | unknown | unclear | Classic PD |
| PARK11 | 2q36-q37 | unknown | unclear | Classic PD |
| N/A | 5q23.1-q23.3 | Synphilin-1 | unclear | Classic PD |
| N/A | 2q22-q23 | NR4A2 | unclear | Classic PD |
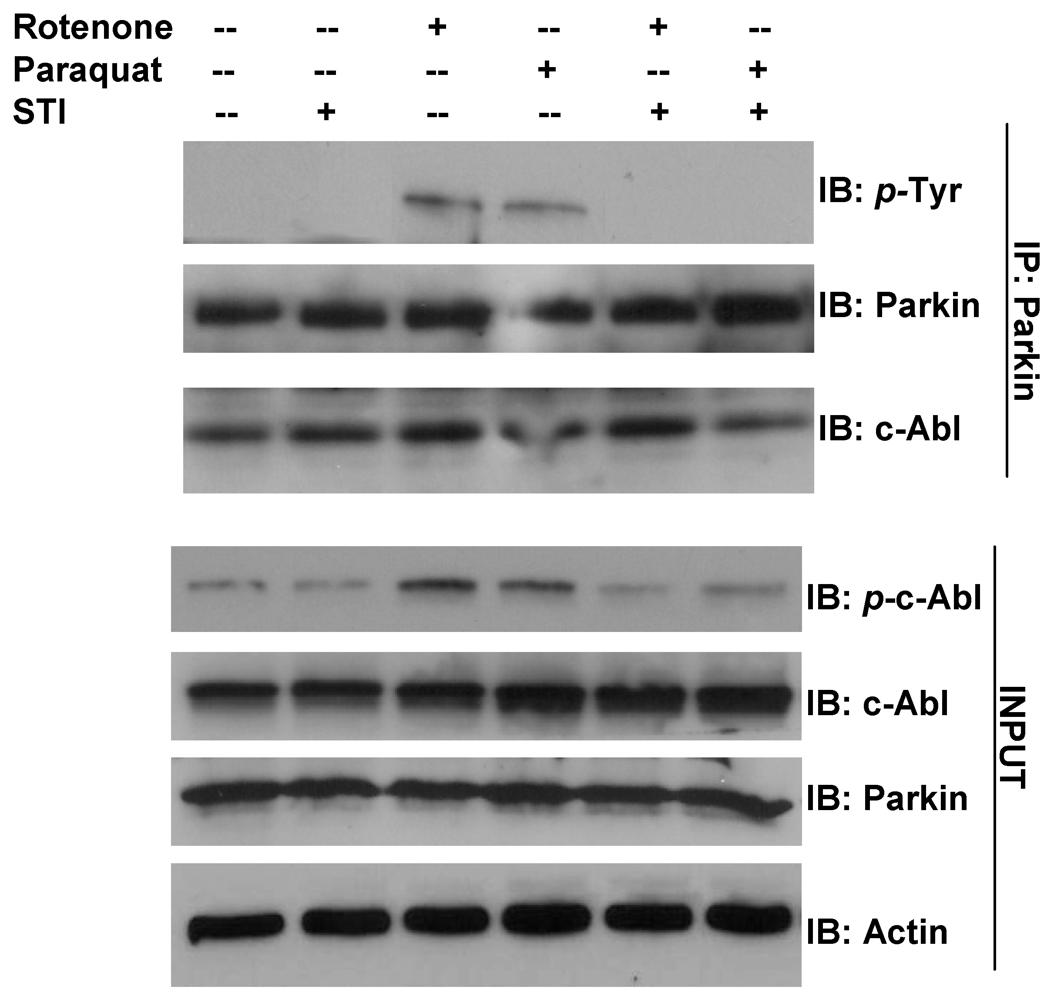
3. Parkin Modification as an Example of Gene-Environment Interaction in PD


4. Conclusions
References
- Siderowf, A.; Stern, M. Update on Parkinson disease. Ann. Intern. Med. 2003, 138, 651–658. [Google Scholar]
- Goedert, M. Parkinson’s disease and other alpha-synucleinopathies. Clin. Chem. Lab Med. 2001, 39, 308–312. [Google Scholar]
- Schlossmacher, M.G.; Frosch, M.P.; Gai, W.P.; Medina, M.; Sharma, N.; Forno, L.; Ochiishi, T.; Shimura, H.; Sharon, R.; Hattori, N.; et al. Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am. J. Pathol. 2002, 160, 1655–1667. [Google Scholar]
- Petrucelli, L.; Dawson, T.M. Mechanism of neurodegenerative disease: Role of the ubiquitin proteasome system. Ann. Med. 2004, 36, 315–320. [Google Scholar]
- Moore, D.J.; Dawson, V.L.; Dawson, T.M. Role for the ubiquitin-proteasome system in Parkinson’s disease and other neurodegenerative brain amyloidoses. Neuromolecular Med. 2003, 4, 95–108. [Google Scholar]
- Gu, Z.; Nakamura, T.; Yao, D.; Shi, Z.Q.; Lipton, S.A. Nitrosative and oxidative stress links dysfunctional ubiquitination to Parkinson’s disease. Cell Death Differ. 2005, 12, 1202–1204. [Google Scholar]
- Vance, J.M.; Ali, S.F.; Bradley, W.G.; Singer, C.; Di Monte, D.A. Gene-environment interaction in Parkinson’s disease and other forms of parkinsonism. Neurotoxicology 2010, 31, 598–602. [Google Scholar]
- Dawson, T.M.; Dawson, V.L. Molecular pathways of neurodegeneration in Parkinson’s disease. Science 2003, 302, 819–822. [Google Scholar]
- Gao, H.M.; Hong, J.S. Gene-environment interactions: Key to unraveling the mystery of Parkinson’s disease. Prog. Neurobiol. 2011, 94, 1–19. [Google Scholar]
- Kahle, P.J.; Haass, C. How does parkin ligate ubiquitin to Parkinson’s disease? EMBO Rep. 2004, 5, 681–685. [Google Scholar]
- Shimura, H.; Hattori, N.; Kubo, S.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; Suzuki, T. Familial Parkinson disease gene product, parkin, is a ubiquitin- protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [PubMed]
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular pathophysiology of Parkinson’s disease. Ann. Rev. Neurosci. 2005, 28, 57–87. [Google Scholar]
- Ko, H.S.; von Coelln, R.; Sriram, S.R.; Kim, S.W.; Chung, K.K.; Pletnikova, O.; Troncoso, J.; Johnson, B.; Saffary, R.; Goh, E.L.; et al. Accumulation of the authentic parkin substrate aminoacyl-tRNA synthetase cofactor, p38/JTV-1, leads to catecholaminergic cell death. J. Neurosci. 2005, 25, 7968–7978. [Google Scholar] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar]
- Lucking, C.B.; Durr, A.; Bonifati, V.; Vaughan, J.; De, M.G.; Gasser, T.; Harhangi, B.S.; Meco, G.; Denefle, P.; Wood, N.W.; et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. French Parkinson’s Disease Genetics Study Group. N. Engl. J. Med. 2000, 342, 1560–1567. [Google Scholar]
- Mata, I.F.; Lockhart, P.J.; Farrer, M.J. Parkin genetics: One model for Parkinson’s disease. Hum. Mol. Genet. 2004, 1, R127–R133. [Google Scholar]
- McNaught, K.S.; Mytilineou, C.; Jnobaptiste, R.; Yabut, J.; Shashidharan, P.; Jennert, P.; Olanow, C.W. Impairment of the ubiquitin-proteasome system causes dopaminergic cell death and inclusion body formation in ventral mesencephalic cultures. J. Neurochem. 2002, 81, 301–306. [Google Scholar]
- Foroud, T.; Uniacke, S.K.; Liu, L.; Pankratz, N.; Rudolph, A.; Halter, C.; Shults, C.; Marder, K.; Conneally, P.M.; Nichols, W.C. Heterozygosity for a mutation in the parkin gene leads to later onset Parkinson disease. Neurology 2003, 60, 796–801. [Google Scholar]
- West, A.B.; Maraganore, D.; Crook, J.; Lesnick, T.; Lockhart, P.J.; Wilkes, K.M.; Kapatos, G.; Hardy, J.A.; Farrer, M.J. Functional association of the parkin gene promoter with idiopathic Parkinson’s disease. Hum. Mol. Genet. 2002, 11, 2787–2792. [Google Scholar]
- Feany, M.B.; Pallanck, L.J. Parkin: A multipurpose neuroprotective agent? Neuron 2003, 38, 13–16. [Google Scholar]
- Higashi, Y.; Asanuma, M.; Miyazaki, I.; Hattori, N.; Mizuno, Y.; Ogawa, N. Parkin attenuates manganese-induced dopaminergic cell death. J. Neurochem. 2004, 89, 1490–1497. [Google Scholar]
- Petrucelli, L.; O’Farrell, C.; Lockhart, P.J.; Baptista, M.; Kehoe, K.; Vink, L.; Choi, P.; Wolozin, B.; Farrer, M.; Hardy, J.; Cookson, M.R. Parkin protects against the toxicity associated with mutant alpha-synuclein: Proteasome dysfunction selectively affects catecholaminergic neurons. Neuron 2002, 36, 1007–1019. [Google Scholar]
- Tsai, Y.C.; Fishman, P.S.; Thakor, N.V.; Oyler, G.A. Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J. Biol. Chem. 2003, 278, 22044–22055. [Google Scholar]
- Takahashi, R.; Imai, Y.; Hattori, N.; Mizuno, Y. Parkin and endoplasmic reticulum stress. Ann. NY Acad. Sci. 2003, 991, 101–106. [Google Scholar]
- Yang, Y.; Nishimura, I.; Imai, Y.; Takahashi, R.; Lu, B. Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila. Neuron 2003, 37, 911–924. [Google Scholar]
- Staropoli, J.F.; McDermott, C.; Martinat, C.; Schulman, B.; Demireva, E.; Abeliovich, A. Parkin is a component of an SCF-like ubiquitin ligase complex and protects postmitotic neurons from kainate excitotoxicity. Neuron 2003, 37, 735–749. [Google Scholar]
- Darios, F.; Corti, O.; Lucking, C.B.; Hampe, C.; Muriel, M.P.; Abbas, N.; Gu, W.J.; Hirsch, E.C.; Rooney, T.; Ruberg, M.; Brice, A. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum. Mol. Genet. 2003, 12, 517–526. [Google Scholar]
- Jiang, H.; Ren, Y.; Zhao, J.; Feng, J. Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis. Hum. Mol. Genet. 2004, 13, 1745–1754. [Google Scholar]
- Zhang, Y.; Gao, J.; Chung, K.K.; Huang, H.; Dawson, V.L.; Dawson, T.M. Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc. Natl. Acad. Sci. USA 2000, 97, 13354–13359. [Google Scholar]
- Lim, K.L.; Chew, K.C.; Tan, J.M.; Wang, C.; Chung, K.K.; Zhang, Y.; Tanaka, Y.; Smith, W.; Engelender, S.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: Implications for Lewy body formation. J. Neurosci. 2005, 25, 2002–2009. [Google Scholar]
- Kalia, S.K.; Lee, S.; Smith, P.D.; Liu, L.; Crocker, S.J.; Thorarinsdottir, T.E.; Glover, J.R.; Fon, E.A.; Park, D.S.; Lozano, A.M. BAG5 inhibits parkin and enhances dopaminergic neuron degeneration. Neuron 2004, 44, 931–945. [Google Scholar]
- Yamada, M.; Mizuno, Y.; Mochizuki, H. Parkin gene therapy for alpha-synucleinopathy: A rat model of Parkinson’s disease. Hum. Gene Ther. 2005, 16, 262–270. [Google Scholar]
- Lo, B.C.; Schneider, B.L.; Bauer, M.; Sajadi, A.; Brice, A.; Iwatsubo, T.; Aebischer, P. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 17510–17515. [Google Scholar]
- Rosen, K.M.; Veereshwarayya, V.; Moussa, C.E.; Fu, Q.; Goldberg, M.S.; Schlossmacher, M.G.; Shen, J.; Querfurth, H.W. Parkin protects against mitochondrial toxins and beta-amyloid accumulation in skeletal muscle cells. J. Biol. Chem. 2006, 281, 12809–12816. [Google Scholar]
- Chung, K.K.; Thomas, B.; Li, X.; Pletnikova, O.; Troncoso, J.C.; Marsh, L.; Dawson, V.L.; Dawson, T.M. S-nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science 2004, 304, 1328–1331. [Google Scholar]
- Yamamoto, A.; Friedlein, A.; Imai, Y.; Takahashi, R.; Kahle, P.J.; Haass, C. Parkin phosphorylation and modulation of its E3 ubiquitin ligase activity. J. Biol. Chem. 2005, 280, 3390–3399. [Google Scholar]
- Lavoie, M.J.; Ostaszewski, B.L.; Weihofen, A.; Schlossmacher, M.G.; Selkoe, D.J. Dopamine covalently modifies and functionally inactivates parkin. Nat. Med. 2005, 11, 1214–1221. [Google Scholar]
- Tieu, K.; Ischiropoulos, H.; Przedborski, S. Nitric oxide and reactive oxygen species in Parkinson’s disease. IUBMB Life 2003, 55, 329–335. [Google Scholar]
- Ischiropoulos, H.; Beckman, J.S. Oxidative stress and nitration in neurodegeneration: Cause, effect, or association? J. Clin. Invest. 2003, 111, 163–169. [Google Scholar] [PubMed]
- Chung, K.K.; Thomas, B.; Li, X.; Pletnikova, O.; Troncoso, J.C.; Marsh, L.; Dawson, V.L.; Dawson, T.M. S-nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science 2004, 304, 1328–1331. [Google Scholar]
- Yao, D.; Gu, Z.; Nakamura, T.; Shi, Z.Q.; Ma, Y.; Gaston, B.; Palmer, L.A.; Rockenstein, E.M.; Zhang, Z.; Masliah, E.; et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10810–10814. [Google Scholar]
- Brooks, A.I.; Chadwick, C.A.; Gelbard, H.A.; Cory-Slechta, D.A.; Federoff, H.J. Paraquat elicited neurobehavioral syndrome caused by dopaminergic neuron loss. Brain Res. 1999, 823, 1–10. [Google Scholar]
- McCormack, A.L.; Thiruchelvam, M.; Manning-Bog, A.B.; Thiffault, C.; Langston, J.W.; Cory-Slechta, D.A.; Di Monte, D.A. Environmental risk factors and Parkinson’s disease: Selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol. Dis. 2002, 10, 119–127. [Google Scholar]
- Liou, H.H.; Tsai, M.C.; Chen, C.J.; Jeng, J.S.; Chang, Y.C.; Chen, S.Y.; Chen, R.C. Environmental risk factors and Parkinson’s disease: A case-control study in Taiwan. Neurology 1997, 48, 1583–1588. [Google Scholar]
- Liu, B.; Gao, H.M.; Hong, J.S. Parkinson’s disease and exposure to infectious agents and pesticides and the occurrence of brain injuries: Role of neuroinflammation. Environ. Health Perspect. 2003, 111, 1065–1073. [Google Scholar]
- Sherer, T.B.; Kim, J.H.; Betarbet, R.; Greenamyre, J.T. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp. Neurol. 2003, 179, 9–16. [Google Scholar]
- Gao, H.M.; Hong, J.S.; Zhang, W.; Liu, B. Synergistic dopaminergic neurotoxicity of the pesticide rotenone and inflammogen lipopolysaccharide: Relevance to the etiology of Parkinson’s disease. J. Neurosci. 2003, 23, 1228–1236. [Google Scholar]
- Horowitz, M.P.; Greenamyre, J.T. Gene-environment interactions in Parkinson’s disease: The importance of animal modeling. Clin. Pharmacol. Ther. 2010, 88, 467–474. [Google Scholar]
- Imam, S.Z.; Zhou, Q.; Yamamoto, A.; Valente, A.J.; Ali, S.F.; Bains, M.; Roberts, J.L.; Kahle, P.J.; Clark, R.A.; Li, S. Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: Implications for Parkinson’s disease. J. Neurosci. 2011, 31, 157–163. [Google Scholar]
- He, Y.; Imam, S.Z.; Dong, Z.; Jankovic, J.; Ali, S.F.; Appel, S.H.; Le, W. Role of nitric oxide in rotenone-induced nigro-striatal injury. J. Neurochem. 2003, 86, 1338–1345. [Google Scholar]
- Meng, F.; Yao, D.; Shi, Y.; Kabakoff, J.; Wu, W.; Reicher, J.; Ma, Y.; Moosmann, B.; Masliah, E.; Lipton, S.A.; Gu, Z. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol. Neurodegener. 2011, 6. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ali, S.F.; Binienda, Z.K.; Imam, S.Z. Molecular Aspects of Dopaminergic Neurodegeneration: Gene-Environment Interaction in Parkin Dysfunction. Int. J. Environ. Res. Public Health 2011, 8, 4702-4713. https://doi.org/10.3390/ijerph8124702
Ali SF, Binienda ZK, Imam SZ. Molecular Aspects of Dopaminergic Neurodegeneration: Gene-Environment Interaction in Parkin Dysfunction. International Journal of Environmental Research and Public Health. 2011; 8(12):4702-4713. https://doi.org/10.3390/ijerph8124702
Chicago/Turabian StyleAli, Syed F., Zbigniew K. Binienda, and Syed Z. Imam. 2011. "Molecular Aspects of Dopaminergic Neurodegeneration: Gene-Environment Interaction in Parkin Dysfunction" International Journal of Environmental Research and Public Health 8, no. 12: 4702-4713. https://doi.org/10.3390/ijerph8124702