The Liver-Brain Axis of Alcohol-Mediated Neurodegeneration: Role of Toxic Lipids

Abstract
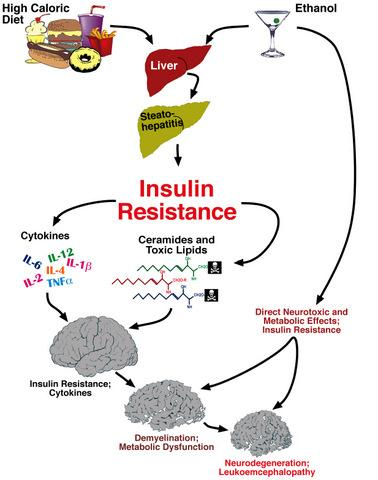
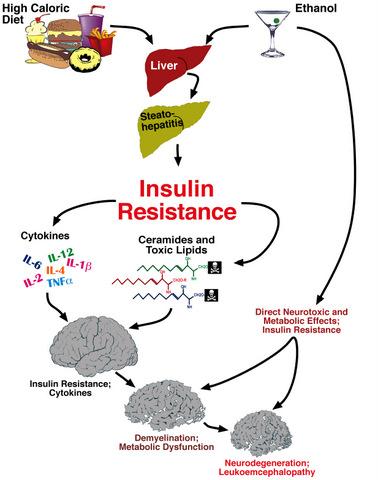
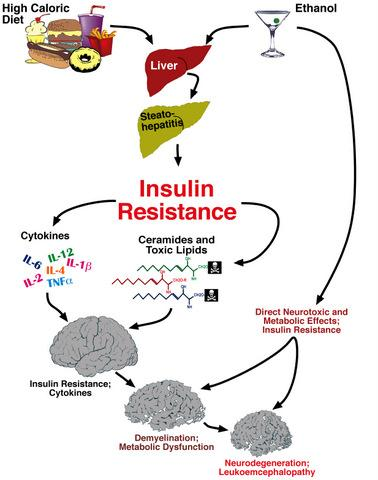
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction-Alcoholic Degeneration in Liver and Brain
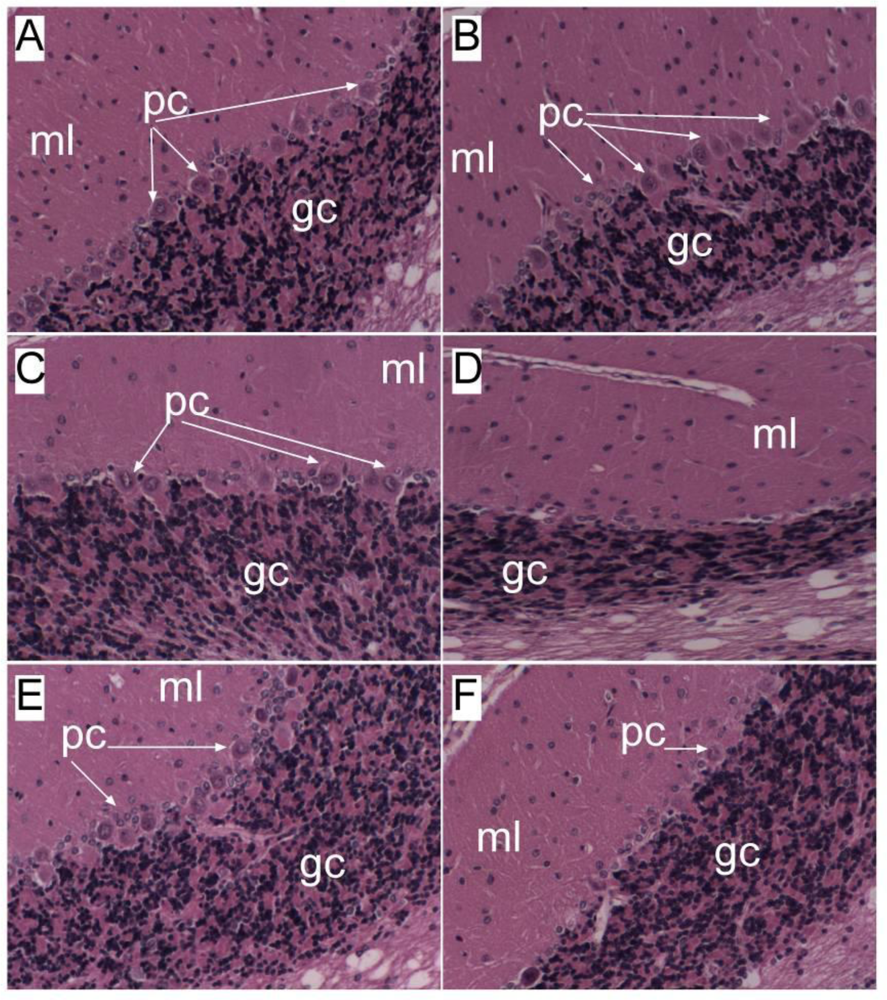
Alcoholic Liver and Brain Diseases
2. Effects of Alcohol on Insulin and Insulin-like Growth Factor Signaling
Insulin and IGF-1 Actions in Liver and Brain:
Ethanol-Mediated Liver Injury and Degeneration Linked to Inhibition of Insulin and IGF Signaling
3. Ethanol-Mediated Insulin Resistance-Mechanisms and Consequences
Chronic Ethanol Abuse Causes Brain Insulin Resistance
Signaling Pathways Mediating Hepatic and Brain Insulin Resistance
Ethanol-Induced Brain Insulin/IGF Resistance Linked to Altered Membrane Lipid Composition
4. Ceramides Mediate Insulin Resistance
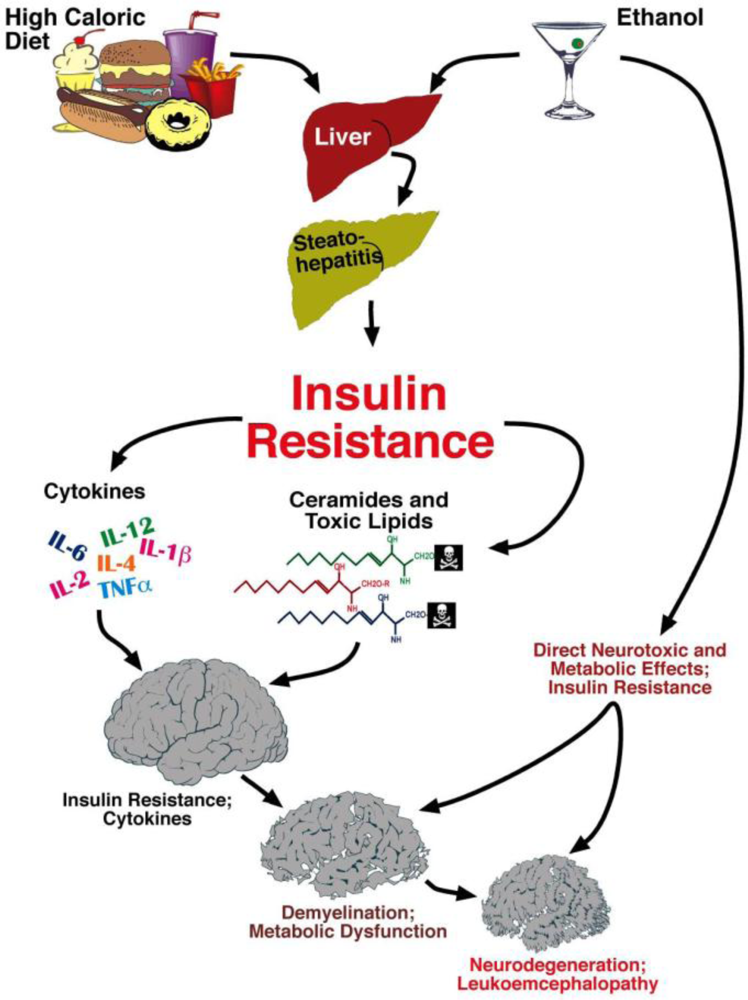
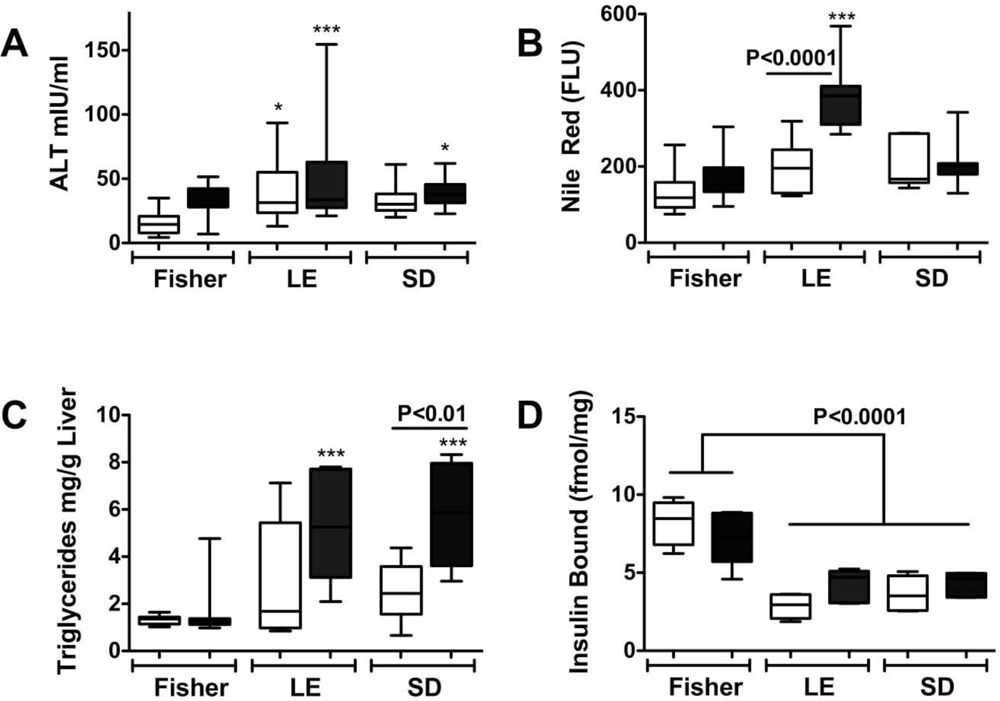
Steatohepatitis, Insulin Resistance, and Toxic Lipid Generation
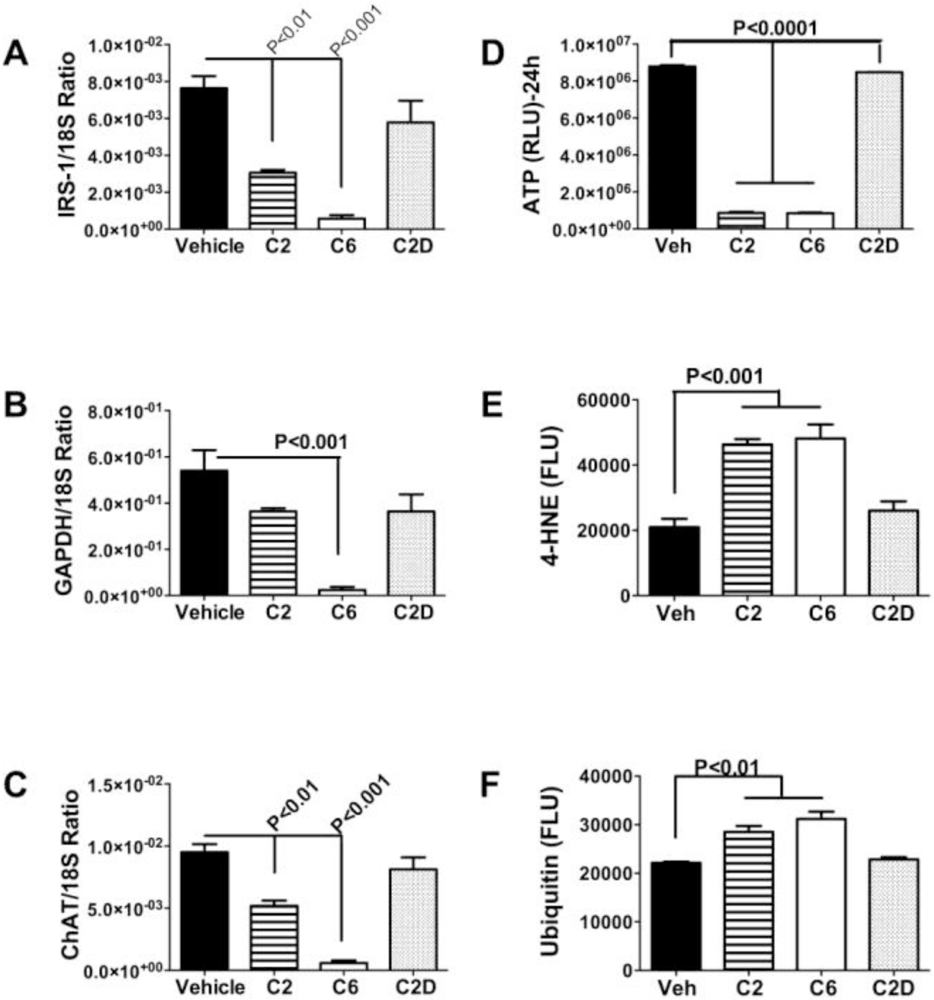
Ceramides as Culprits
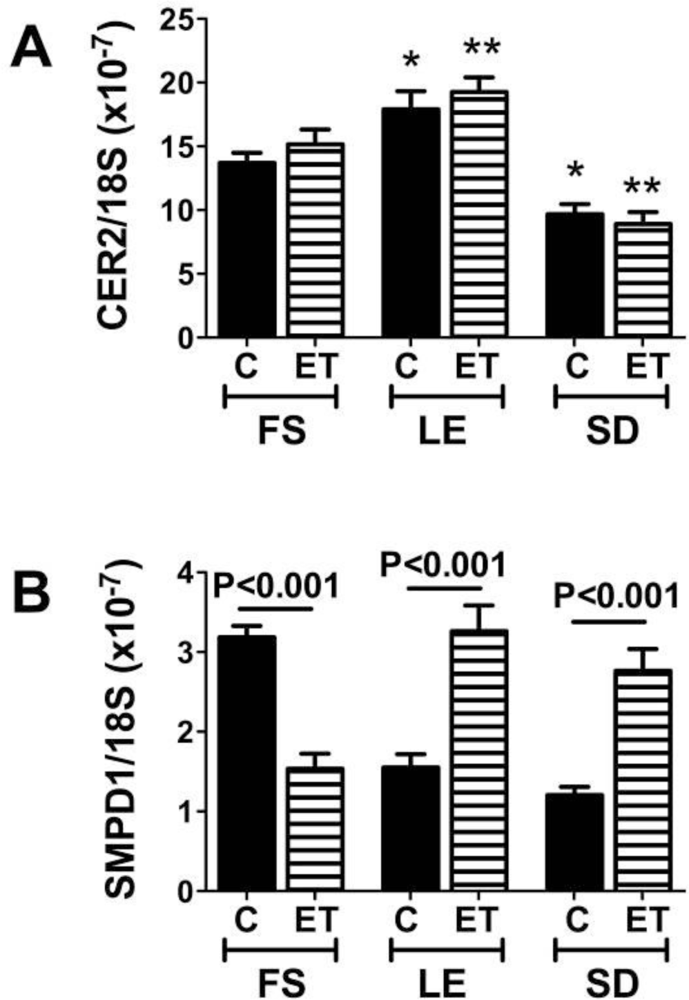
5. Factors Regulating Ceramide Biosynthesis and Accumulation in Liver
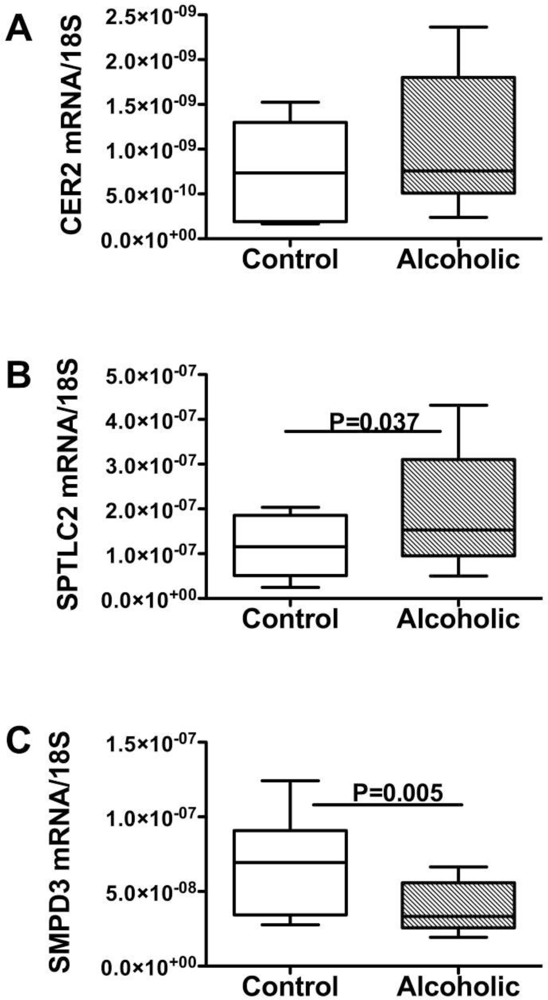
Endogenous Brain Ceramide Production
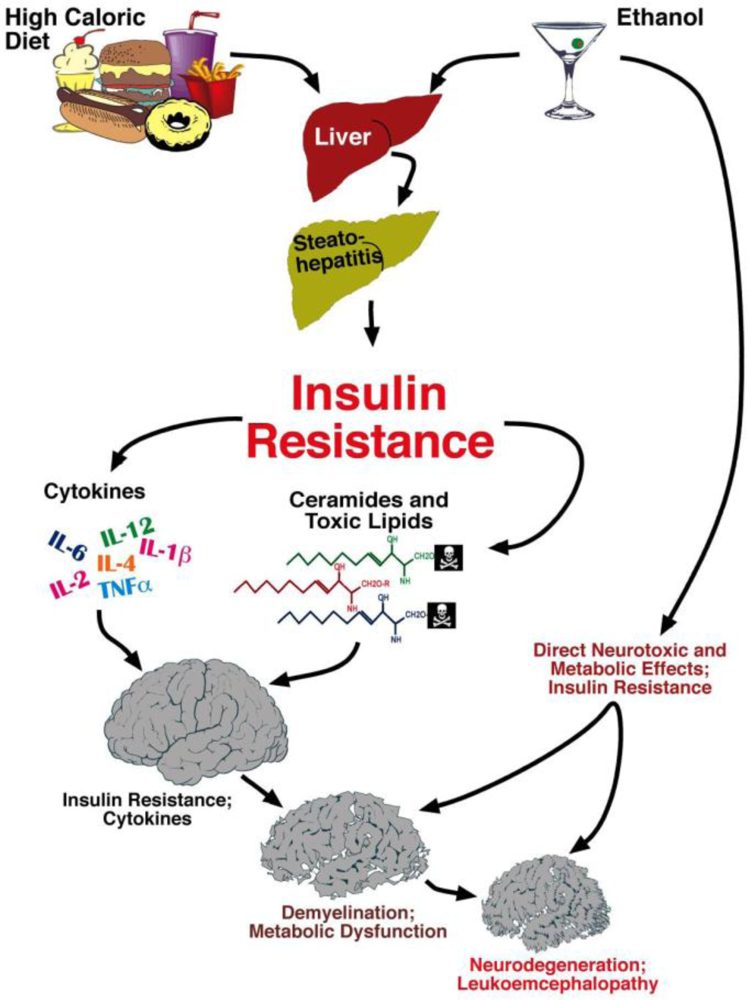
6. Liver Brain Axis of Neurodegeneration
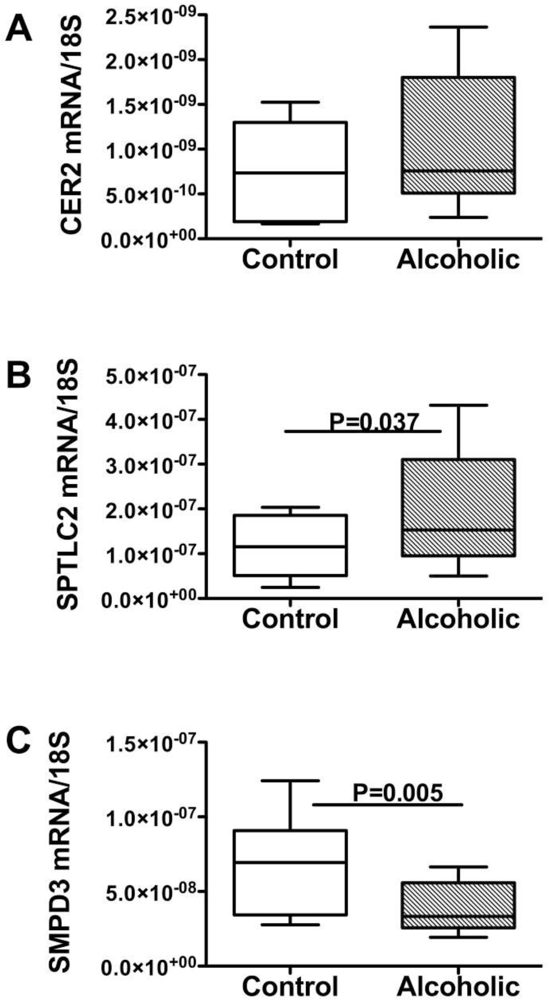
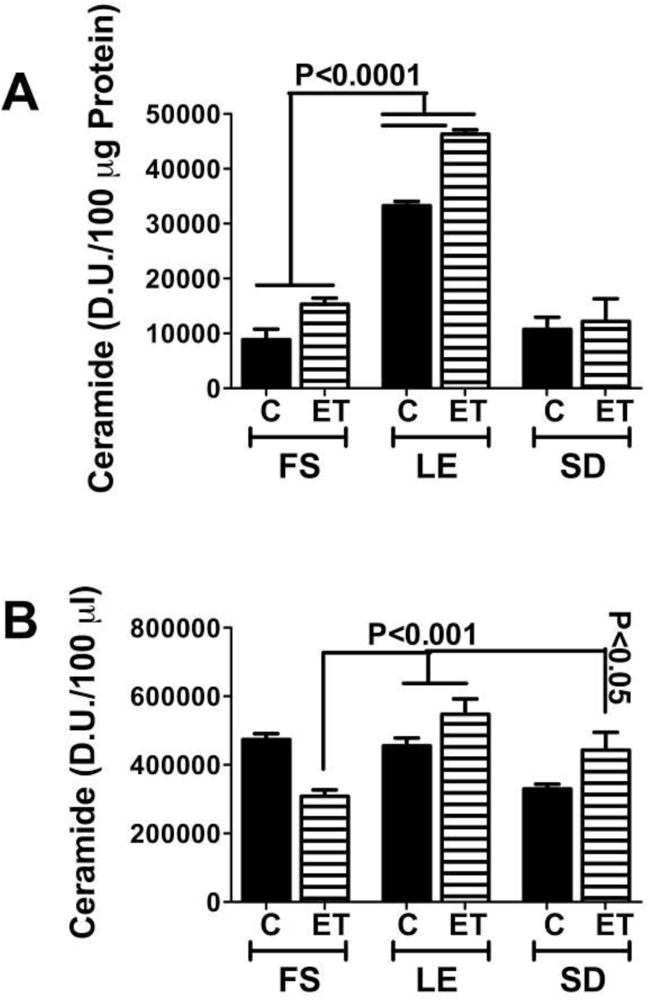
Ceramides and the Liver-Brain Axis of Neurodegeneration
7. Conclusions
References
- Diehl, AM; Thorgeirsson, SS; Steer, CJ. Ethanol inhibits liver regeneration in rats without reducing transcripts of key protooncogenes. Gastroenterology 1990, 99, 1105–1112. [Google Scholar]
- Duguay, L; Coutu, D; Hetu, C; Joly, JG. Inhibition of liver regeneration by chronic alcohol administration. Gut 1982, 23, 8–13. [Google Scholar]
- Wands, JR; Carter, EA; Bucher, NL; Isselbacher, KJ. Inhibition of hepatic regeneration in rats by acute and chronic ethanol intoxication. Gastroenterology 1979, 77, 528–531. [Google Scholar]
- Wands, JR; Carter, EA; Bucher, NL; Isselbacher, KJ. Effect of acute and chronic ethanol intoxication on hepatic regeneration. Adv. Exp. Med. Biol 1980, 132, 663–670. [Google Scholar]
- Banerjee, K; Mohr, L; Wands, JR; de la Monte, SM. Ethanol inhibition of insulin signaling in hepatocellular carcinoma cells. Alcohol Clin. Exp. Res 1998, 22, 2093–2101. [Google Scholar]
- Carter, EA; Wands, JR. Ethanol inhibits hormone stimulated hepatocyte DNA synthesis. Biochem. Biophys. Res. Commun 1985, 128, 767–774. [Google Scholar]
- Li, W; Liu, X; Yanoff, M. Phosphatidylcholine hydrolysis and DNA synthesis in cultured retinal capillary pericytes. Microvasc. Res 1995, 49, 350–363. [Google Scholar]
- Mohr, L; Tanaka, S; Wands, JR. Ethanol inhibits hepatocyte proliferation in insulin receptor substrate 1 transgenic mice. Gastroenterology 1998, 115, 1558–1565. [Google Scholar]
- Sasaki, Y; Hayashi, N; Ito, T; Fusamoto, H; Kamada, T; Wands, JR. Influence of ethanol on insulin receptor substrate-1-mediated signal transduction during rat liver regeneration. Alcohol Alcoholism 1994, 1, 99–106. [Google Scholar]
- Sasaki, Y; Wands, JR. Ethanol impairs insulin receptor substrate-1 mediated signal transduction during rat liver regeneration. Biochem. Biophys. Res. Commun 1994, 199, 403–409. [Google Scholar]
- de la Monte, SM; Yeon, JE; Tong, M; Longato, L; Chaudhry, R; Pang, MY; Duan, K; Wands, JR. Insulin resistance in experimental alcohol-induced liver disease. J. Gastroenterol. Hepatol 2008, 23, e477–86. [Google Scholar]
- Pang, M; de la Monte, SM; Longato, L; Tong, M; He, J; Chaudhry, R; Duan, K; Ouh, J; Wands, JR. PPARdelta agonist attenuates alcohol-induced hepatic insulin resistance and improves liver injury and repair. J. Hepatol 2009, 50, 1192–1201. [Google Scholar]
- Harper, C. The neuropathology of alcohol-specific brain damage, or does alcohol damage the brain? J. Neuropathol. Exp. Neurol 1998, 57, 101–110. [Google Scholar]
- Harper, C; Dixon, G; Sheedy, D; Garrick, T. Neuropathological alterations in alcoholic brains. Studies arising from the New South Wales Tissue Resource Centre. Prog. Neuropsychopharmacol. Biol. Psychiat 2003, 27, 951–961. [Google Scholar]
- de la Monte, SM. Disproportionate atrophy of cerebral white matter in chronic alcoholics. Arch. Neurol 1988, 45, 990–992. [Google Scholar]
- Cohen, AC; Tong, M; Wands, JR; de la Monte, SM. Insulin and insulin-like growth factor resistance with neurodegeneration in an adult chronic ethanol exposure model. Alcohol Clin. Exp. Res 2007, 31, 1558–1573. [Google Scholar]
- de la Monte, SM; Wands, JR. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer’s disease. J. Alzheimers Dis 2005, 7, 45–61. [Google Scholar]
- Chang, L; Chiang, SH; Saltiel, AR. Insulin signaling and the regulation of glucose transport. Mol. Med 2004, 10, 65–71. [Google Scholar]
- Giovannone, B; Scaldaferri, ML; Federici, M; Porzio, O; Lauro, D; Fusco, A; Sbraccia, P; Borboni, P; Lauro, R; Sesti, G. Insulin receptor substrate (IRS) transduction system: distinct and overlapping signaling potential. Diabetes Metab. Res. Rev 2000, 16, 434–441. [Google Scholar]
- Gammeltoft, S; Fehlmann, M; Van, OE. Insulin receptors in the mammalian central nervous system: binding characteristics and subunit structure. Biochimie 1985, 67, 1147–1153. [Google Scholar]
- Hill, JM; Lesniak, MA; Pert, CB; Roth, J. Autoradiographic localization of insulin receptors in rat brain: prominence in olfactory and limbic areas. Neuroscience 1986, 17, 1127–1138. [Google Scholar]
- Broughton, SK; Chen, H; Riddle, A; Kuhn, SE; Nagalla, S; Roberts, CT, Jr; Back, SA. Large-scale generation of highly enriched neural stem-cell-derived oligodendroglial cultures: maturation-dependent differences in insulin-like growth factor-mediated signal transduction. J. Neurochem 2007, 100, 628–638. [Google Scholar]
- Freude, S; Leeser, U; Muller, M; Hettich, MM; Udelhoven, M; Schilbach, K; Tobe, K; Kadowaki, T; Kohler, C; Schroder, H; Krone, W; Bruning, JC; Schubert, M. IRS-2 branch of IGF-1 receptor signaling is essential for appropriate timing of myelination. J. Neurochem 2008, 107, 907–917. [Google Scholar]
- Joseph D’Ercole, A; Ye, P. Expanding the mind: insulin-like growth factor I and brain development. Endocrinology 2008, 149, 5958–5962. [Google Scholar]
- Schubert, M; Brazil, DP; Burks, DJ; Kushner, JA; Ye, J; Flint, CL; Farhang-Fallah, J; Dikkes, P; Warot, XM; Rio, C; Corfas, G; White, MF. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J. Neurosci 2003, 23, 7084–7092. [Google Scholar]
- Chesik, D; de Keyser, J; Wilczak, N. Insulin-like growth factor system regulates oligodendroglial cell behavior: therapeutic potential in CNS. J. Mol. Neurosci 2008, 35, 81–90. [Google Scholar]
- Gong, X; Xie, Z; Zuo, H. Invivo insulin deficiency as a potential etiology for demyelinating disease. Med. Hypotheses 2008, 71, 399–403. [Google Scholar]
- Liang, G; Cline, GW; Macica, CM. IGF-1 stimulates de novo fatty acid biosynthesis by Schwann cells during myelination. Glia 2007, 55, 632–641. [Google Scholar]
- Ye, P; Kollias, G; D’Ercole, AJ. Insulin-like growth factor-I ameliorates demyelination induced by tumor necrosis factor-alpha in transgenic mice. J. Neurosci. Res 2007, 85, 712–722. [Google Scholar]
- Wands, JR; Mohr, L; Banerjee, K; Ganju, N; Tanaka, S; de la Monte, SM. Ethanol and IRS-1 protein in the liver and brain. In Proceedings on Signal Transduction and Alcohol; Lund: Sweden, 2001. [Google Scholar]
- Yeon, JE; Califano, S; Xu, J; Wands, JR; De La Monte, SM. Potential role of PTEN phosphatase in ethanol-impaired survival signaling in the liver. Hepatology 2003, 38, 703–714. [Google Scholar]
- Ronis, MJ; Butura, A; Korourian, S; Shankar, K; Simpson, P; Badeaux, J; Albano, E; Ingelman-Sundberg, M; Badger, TM. Cytokine and chemokine expression associated with steatohepatitis and hepatocyte proliferation in rats fed ethanol via total enteral nutrition. Exp. Biol. Med. (Maywood) 2008, 233, 344–355. [Google Scholar]
- de la Monte, SM; Wands, JR. Chronic gestational exposure to ethanol impairs insulin-stimulated survival and mitochondrial function in cerebellar neurons. CMLS, Cell Mol. Life Sci 2002, 59, 882–893. [Google Scholar]
- de la Monte, SM; Xu, XJ; Wands, JR. Ethanol inhibits insulin expression and actions in the developing brain. Cell Mol. Life Sci 2005, 62, 1131–1145. [Google Scholar]
- de la Monte, SM; Tong, M; Cohen, AC; Sheedy, D; Harper, C; Wands, JR. Insulin and insulin-like growth factor resistance in alcoholic neurodegeneration. Alcohol Clin. Exp. Res 2008, 32, 1630–1644. [Google Scholar]
- Soscia, SJ; Tong, M; Xu, XJ; Cohen, AC; Chu, J; Wands, JR; de la Monte, SM. Chronic gestational exposure to ethanol causes insulin and IGF resistance and impairs acetylcholine homeostasis in the brain. Cell Mol. Life Sci 2006, 63, 2039–2056. [Google Scholar]
- Ronis, MJ; Wands, JR; Badger, TM; de la Monte, SM; Lang, CH; Calissendorff, J. Alcohol-induced disruption of endocrine signaling. Alcohol Clin. Exp. Res 2007, 31, 1269–1285. [Google Scholar]
- Enomoto, N; Takei, Y; Hirose, M; Konno, A; Shibuya, T; Matsuyama, S; Suzuki, S; Kitamura, KI; Sato, N. Prevention of ethanol-induced liver injury in rats by an agonist of peroxisome proliferator-activated receptor-gamma, pioglitazone. J. Pharmacol. Exp. Ther 2003, 306, 846–854. [Google Scholar]
- Onishi, Y; Honda, M; Ogihara, T; Sakoda, H; Anai, M; Fujishiro, M; Ono, H; Shojima, N; Fukushima, Y; Inukai, K; Katagiri, H; Kikuchi, M; Oka, Y; Asano, T. Ethanol feeding induces insulin resistance with enhanced PI 3-kinase activation. Biochem. Biophys. Res. Commun 2003, 303, 788–794. [Google Scholar]
- Patel, BC; D’Arville, C; Iwahashi, M; Simon, FR. Impairment of hepatic insulin receptors during chronic ethanol administration. Am. J. Physiol 1991, 261, G199–205. [Google Scholar]
- Sadri, P; Legare, DJ; Takayama, S; Lautt, WW. Increased incidence of hepatic insulin-sensitizing substance (HISS)-dependent insulin resistance in female rats prenatally exposed to ethanol. Can. J. Physiol. Pharmacol 2005, 83, 383–387. [Google Scholar]
- Tomita, K; Azuma, T; Kitamura, N; Nishida, J; Tamiya, G; Oka, A; Inokuchi, S; Nishimura, T; Suematsu, M; Ishii, H. Pioglitazone prevents alcohol-induced fatty liver in rats through up-regulation of c-Met. Gastroenterology 2004, 126, 873–885. [Google Scholar]
- Yao, XH; Chen, L; Nyomba, BL. Adult rats prenatally exposed to ethanol have increased gluconeogenesis and impaired insulin response of hepatic gluconeogenic genes. J. Appl. Physiol 2006, 100, 642–648. [Google Scholar]
- Li, XL; Man, K; Ng, KT; Sun, CK; Lo, CM; Fan, ST. The influence of phosphatidylinositol 3-kinase/Akt pathway on the ischemic injury during rat liver graft preservation. Am. J. Transplant 2005, 5, 1264–1275. [Google Scholar]
- Michl, P; Downward, J. Mechanisms of disease: PI3K/AKT signaling in gastrointestinal cancers. Z. Gastroenterol 2005, 43, 1133–1139. [Google Scholar]
- Roberts, RA; James, NH; Cosulich, SC. The role of protein kinase B and mitogen-activated protein kinase in epidermal growth factor and tumor necrosis factor alpha-mediated rat hepatocyte survival and apoptosis. Hepatology 2000, 31, 420–427. [Google Scholar]
- Rust, C; Bauchmuller, K; Fickert, P; Fuchsbichler, A; Beuers, U. Phosphatidylinositol 3-kinase-dependent signaling modulates taurochenodeoxycholic acid-induced liver injury and cholestasis in perfused rat livers. Am. J. Physiol. Gastrointest. Liver Physiol 2005, 289, G88–94. [Google Scholar]
- Valverde, AM; Fabregat, I; Burks, DJ; White, MF; Benito, M. IRS-2 mediates the antiapoptotic effect of insulin in neonatal hepatocytes. Hepatology 2004, 40, 1285–1294. [Google Scholar]
- Carmiel-Haggai, M; Cederbaum, AI; Nieto, N. Binge ethanol exposure increases liver injury in obese rats. Gastroenterology 2003, 125, 1818–1833. [Google Scholar]
- McVicker, BL; Tuma, DJ; Kubik, JL; Tuma, PL; Casey, CA. Ethanol-induced apoptosis in polarized hepatic cells possibly through regulation of the Fas pathway. Alcohol Clin. Exp. Res 2006, 30, 1906–1915. [Google Scholar]
- de la Monte, SM; Ganju, N; Banerjee, K; Brown, NV; Luong, T; Wands, JR. Partial rescue of ethanol-induced neuronal apoptosis by growth factor activation of phosphoinositol-3-kinase. Alcohol Clin. Exp. Res 2000, 24, 716–726. [Google Scholar]
- de la Monte, SM; Neely, TR; Cannon, J; Wands, JR. Ethanol impairs insulin-stimulated mitochondrial function in cerebellar granule neurons. Cell Mol. Life Sci 2001, 58, 1950–1960. [Google Scholar]
- Hallak, H; Seiler, AE; Green, JS; Henderson, A; Ross, BN; Rubin, R. Inhibition of insulin-like growth factor-I signaling by ethanol in neuronal cells. Alcohol Clin. Exp. Res 2001, 25, 1058–1064. [Google Scholar]
- Zhang, FX; Rubin, R; Rooney, TA. Ethanol induces apoptosis in cerebellar granule neurons by inhibiting insulin-like growth factor 1 signaling. J. Neurochem 1998, 71, 196–204. [Google Scholar]
- Spaissman, A; Tong, M; de la Monte, SM. Acetaldehyde-Mediated neuronal apoptosis and mitochondrial dysfunction: relevance to fetal alcohol syndrome. J Neurochem 2009. (Submitted).. [Google Scholar]
- Ramachandran, V; Perez, A; Chen, J; Senthil, D; Schenker, S; Henderson, GI. In utero ethanol exposure causes mitochondrial dysfunction, which can result in apoptotic cell death in fetal brain: a potential role for 4- hydroxynonenal. Alcohol Clin. Exp. Res 2001, 25, 862–871. [Google Scholar]
- Ikonomidou, C; Bittigau, P; Ishimaru, MJ; Wozniak, DF; Koch, C; Genz, K; Price, MT; Stefovska, V; Horster, F; Tenkova, T; Dikranian, K; Olney, JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 2000, 287, 1056–1060. [Google Scholar]
- de la Monte, SM; Wands, JR. Mitochondrial DNA damage and impaired mitochondrial function contribute to apoptosis of insulin-stimulated ethanol-exposed neuronal cells. Alcohol Clin. Exp. Res 2001, 25, 898–906. [Google Scholar]
- Xu, J; Yeon, JE; Chang, H; Tison, G; Chen, GJ; Wands, J; de la Monte, S. Ethanol impairs insulin-stimulated neuronal survival in the developing brain: role of PTEN phosphatase. J. Biol. Chem 2003, 278, 26929–26937. [Google Scholar]
- Marco, C; Ceacero, F; Gonzalez-Pacanowska, D; Garcia-Peregrin, E; Segovia, JL. Alterations induced by chronic ethanol treatment on lipid composition of microsomes, mitochondria and myelin from neonatal chick liver and brain. Biochem. Int 1986, 12, 51–60. [Google Scholar]
- Muller, G; Jung, C; Frick, W; Bandlow, W; Kramer, W. Interaction of phosphatidylinositolglycan(-peptides) with plasma membrane lipid rafts triggers insulin-mimetic signaling in rat adipocytes. Arch. Biochem. Biophys 2002, 408, 7–16. [Google Scholar]
- Lutchman, G; Promrat, K; Kleiner, DE; Heller, T; Ghany, MG; Yanovski, JA; Liang, TJ; Hoofnagle, JH. Changes in serum adipokine levels during pioglitazone treatment for nonalcoholic steatohepatitis: relationship to histological improvement. Clin. Gastroenterol. Hepatol 2006, 4, 1048–1052. [Google Scholar]
- Promrat, K; Lutchman, G; Uwaifo, GI; Freedman, RJ; Soza, A; Heller, T; Doo, E; Ghany, M; Premkumar, A; Park, Y; Liang, TJ; Yanovski, JA; Kleiner, DE; Hoofnagle, JH. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology 2004, 39, 188–196. [Google Scholar]
- Capeau, J. Insulin resistance and steatosis in humans. Diabetes Metab 2008, 34, 649–657. [Google Scholar]
- Leonard, BL; Watson, RN; Loomes, KM; Phillips, AR; Cooper, GJ. Insulin resistance in the Zucker diabetic fatty rat: a metabolic characterisation of obese and lean phenotypes. Acta Diabetol 2005, 42, 162–170. [Google Scholar]
- Kraegen, EW; Cooney, GJ. Free fatty acids and skeletal muscle insulin resistance. Curr. Opin. Lipidol 2008, 19, 235–241. [Google Scholar]
- Kao, Y; Youson, JH; Holmes, JA; Al-Mahrouki, A; Sheridan, MA. Effects of insulin on lipid metabolism of larvae and metamorphosing landlocked sea lamprey, Petromyzon marinus. Gen. Comp. Endocrinol 1999, 114, 405–414. [Google Scholar]
- Holland, WL; Summers, SA. Sphingolipids, insulin resistance, and metabolic disease: new insights from in vivo manipulation of sphingolipid metabolism. Endocr. Rev 2008, 29, 381–402. [Google Scholar]
- Langeveld, M; Aerts, JM. Glycosphingolipids and insulin resistance. Prog. Lipid. Res 2009, 48, 196–205. [Google Scholar]
- Summers, SA. Ceramides in insulin resistance and lipotoxicity. Prog. Lipid. Res 2006, 45, 42–72. [Google Scholar]
- Boden, G. Ceramide: a contributor to insulin resistance or an innocent bystander? Diabetologia 2008, 51, 1095–1096. [Google Scholar]
- Delarue, J; Magnan, C. Free fatty acids and insulin resistance. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 142–148. [Google Scholar]
- Holland, WL; Brozinick, JT; Wang, LP; Hawkins, ED; Sargent, KM; Liu, Y; Narra, K; Hoehn, KL; Knotts, TA; Siesky, A; Nelson, DH; Karathanasis, SK; Fontenot, GK; Birnbaum, MJ; Summers, SA. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 2007, 5, 167–179. [Google Scholar]
- Holland, WL; Knotts, TA; Chavez, JA; Wang, LP; Hoehn, KL; Summers, SA. Lipid mediators of insulin resistance. Nutr. Rev 2007, 65, S39–46. [Google Scholar]
- Anderson, N; Borlak, J. Molecular mechanisms and therapeutic targets in steatosis and steatohepatitis. Pharmacol. Rev 2008, 60, 311–357. [Google Scholar]
- Kaplowitz, N; Than, TA; Shinohara, M; Ji, C. Endoplasmic reticulum stress and liver injury. Semin. Liver Dis 2007, 27, 367–377. [Google Scholar]
- Malhi, H; Gores, GJ. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin. Liver Dis 2008, 28, 360–369. [Google Scholar]
- Sundar Rajan, S; Srinivasan, V; Balasubramanyam, M; Tatu, U. Endoplasmic reticulum (ER) stress & diabetes. Indian J. Med. Res 2007, 125, 411–424. [Google Scholar]
- Bourbon, NA; Sandirasegarane, L; Kester, M. Ceramide-induced inhibition of Akt is mediated through protein kinase Czeta: implications for growth arrest. J. Biol. Chem 2002, 277, 3286–3292. [Google Scholar]
- Hajduch, E; Balendran, A; Batty, IH; Litherland, GJ; Blair, AS; Downes, CP; Hundal, HS. Ceramide impairs the insulin-dependent membrane recruitment of protein kinase B leading to a loss in downstream signalling in L6 skeletal muscle cells. Diabetologia 2001, 44, 173–183. [Google Scholar]
- Nogueira, TC; Anhe, GF; Carvalho, CR; Curi, R; Bordin, S; Carpinelli, AR. Involvement of phosphatidylinositol-3 kinase/AKT/PKCzeta/lambda pathway in the effect of palmitate on glucose-induced insulin secretion. Pancreas 2008, 37, 309–315. [Google Scholar]
- Powell, DJ; Hajduch, E; Kular, G; Hundal, HS. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCzeta-dependent mechanism. Mol. Cell Biol 2003, 23, 7794–7808. [Google Scholar]
- Consitt, LA; Bell, JA; Houmard, JA. Intramuscular lipid metabolism, insulin action, and obesity. IUBMB Life 2009, 61, 47–55. [Google Scholar]
- Vistisen, B; Hellgren, LI; Vadset, T; Scheede-Bergdahl, C; Helge, JW; Dela, F; Stallknecht, B. Effect of gender on lipid-induced insulin resistance in obese subjects. Eur. J. Endocrinol 2008, 158, 61–68. [Google Scholar]
- Zierath, JR. The path to insulin resistance: paved with ceramides? Cell Metab 2007, 5, 161–163. [Google Scholar]
- Adibhatla, RM; Hatcher, JF. Altered lipid metabolism in brain injury and disorders. Subcell Biochem 2008, 49, 241–268. [Google Scholar]
- Blakemore, WF; Franklin, RJ. Remyelination in experimental models of toxin-induced demyelination. Curr. Top Microbiol. Immunol 2008, 318, 193–212. [Google Scholar]
- Daigo, M; Arai, Y; Oshida, K; Kitamura, Y; Hayashi, M; Shimizu, T; Yamashiro, Y. Effect of hypoxic-ischemic injury on serine palmitoyltransferase activity in the developing rat brain. Pathobiology 2008, 75, 330–334. [Google Scholar]
- Soeda, S; Tsuji, Y; Ochiai, T; Mishima, K; Iwasaki, K; Fujiwara, M; Yokomatsu, T; Murano, T; Shibuya, S; Shimeno, H. Inhibition of sphingomyelinase activity helps to prevent neuron death caused by ischemic stress. Neurochem. Int 2004, 45, 619–626. [Google Scholar]
- Soriano, JM; Gonzalez, L; Catala, AI. Mechanism of action of sphingolipids and their metabolites in the toxicity of fumonisin B1. Prog. Lipid. Res 2005, 44, 345–356. [Google Scholar]
- Adams, JM, II; Pratipanawatr, T; Berria, R; Wang, E; DeFronzo, RA; Sullards, MC; Mandarino, LJ. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 2004, 53, 25–31. [Google Scholar]
- Aerts, JM; Ottenhoff, R; Powlson, AS; Grefhorst, A; van Eijk, M; Dubbelhuis, PF; Aten, J; Kuipers, F; Serlie, MJ; Wennekes, T; Sethi, JK; O’Rahilly, S; Overkleeft, HS. Pharmacological inhibition of glucosylceramide synthase enhances insulin sensitivity. Diabetes 2007, 56, 1341–1349. [Google Scholar]
- JeBailey, L; Wanono, O; Niu, W; Roessler, J; Rudich, A; Klip, A. Ceramide- and oxidant-induced insulin resistance involve loss of insulin-dependent Rac-activation and actin remodeling in muscle cells. Diabetes 2007, 56, 394–403. [Google Scholar]
- Schmitz-Peiffer, C; Craig, DL; Biden, TJ. Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J. Biol. Chem 1999, 274, 24202–24210. [Google Scholar]
- Teruel, T; Hernandez, R; Lorenzo, M. Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes 2001, 50, 2563–25671. [Google Scholar]
- Tong, M; de la Monte, SM. Mechanisms of ceramide-mediated neurodegeneration. J. Alzheimers Dis 2009, 16, 705–714. [Google Scholar]
- Schmidt, KS; Gallo, JL; Ferri, C; Giovannetti, T; Sestito, N; Libon, DJ; Schmidt, PS. The neuropsychological profile of alcohol-related dementia suggests cortical and subcortical pathology. Dement. Geriatr. Cogn. Disord 2005, 20, 286–291. [Google Scholar]
- Kopelman, MD; Thomson, AD; Guerrini, I; Marshall, EJ. The Korsakoff syndrome: clinical aspects, psychology and treatment. Alcohol Alcoholism 2009, 44, 148–154. [Google Scholar]
- Elwing, JE; Lustman, PJ; Wang, HL; Clouse, RE. Depression, anxiety, and nonalcoholic steatohepatitis. Psychosom. Med 2006, 68, 563–569. [Google Scholar]
- Loftis, JM; Huckans, M; Ruimy, S; Hinrichs, DJ; Hauser, P. Depressive symptoms in patients with chronic hepatitis C are correlated with elevated plasma levels of interleukin-1beta and tumor necrosis factor-alpha. Neurosci. Lett 2008, 430, 264–268. [Google Scholar]
- Perry, W; Hilsabeck, RC; Hassanein, TI. Cognitive dysfunction in chronic hepatitis C: a review. Dig. Dis. Sci 2008, 53, 307–321. [Google Scholar]
- Karaivazoglou, K; Assimakopoulos, K; Thomopoulos, K; Theocharis, G; Messinis, L; Sakellaropoulos, G; Labropoulou-Karatza, C. Neuropsychological function in Greek patients with chronic hepatitis C. Liver Int 2007, 27, 798–805. [Google Scholar]
- Weiss, JJ; Gorman, JM. Psychiatric behavioral aspects of comanagement of hepatitis C virus and HIV. Curr. HIV/AIDS Rep 2006, 3, 176–181. [Google Scholar]
- de la Monte, SM; Tong, M; Lester-Coll, N; Plater, M, Jr; Wands, JR. Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: relevance to Alzheimer’s disease. J. Alzheimers Dis 2006, 10, 89–109. [Google Scholar]
- Lester-Coll, N; Rivera, EJ; Soscia, SJ; Doiron, K; Wands, JR; de la Monte, SM. Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer’s disease. J. Alzheimers Dis 2006, 9, 13–33. [Google Scholar]
- Tong, M; Lawton, M; Neusner, A; Longato, L; Wands, JR; de la Monte, SM. Nitrosamine-mediated type 2 diabetes mellitus, hepatic steatosis, and alzheimer-type neurodegeneration: Potential role of environmental exposures in our insulin resistance diseases pandemic. J Alzheimers Dis, 2009. (In Press). [Google Scholar]
- Lyn-Cook, LE; Lawton, M; Tong, M; Silbermann, E; Longato, L; Jiao, P; Mark, P; Wands, JR; Xu, H; de la Monte, SM. Hepatic ceramide mediates brain insulin resistance and neurodegeneration in obesity with type 2 diabetes mellitus and Non-alcoholoic steatohepatitis. J Alzheimers Dis, 2008; (Submitted). [Google Scholar]
- Moroz, N; Tong, M; Longato, L; Xu, H; de la Monte, SM. Limited Alzheimer-type neurodegeneration in experimental obesity and Type 2 diabetes mellitus. J. Alzheimers Dis 2008, 15, 29–44. [Google Scholar]
- Tong, M; Longato, L; de la Monte, SM. Nitrosamine exposure exacerbates high fat diet-mediated neurodegeneration. BMC Cell Biol, 2009; (Under Review). [Google Scholar]
- Tong, M; Neusner, A; Longato, L; Lawton, M; Wands, JR; de la Monte, SM. Nitrosamine Exposure Causes Insulin Resistance Diseases: Relevance to Type 2 Diabetes Mellitus, Non-Alcoholic Steatohepatitis, and Alzheimer’s Disease. J Alzheimers Dis, 2009 Jun 19; (Epub ahead of print). [Google Scholar]
- Tong, M; Neusner, A; Longato, L; Lawton, M; Wands, JR; de la Monte, SM. Nitosamine exposure causes insulin resistance diseases: relevance to type 2 diabetes mellitus, non-alcoholic steatohepatitis, and Alzheimer disease. J Alzheimers Dis, 2009. (In Press) [Google Scholar]
- Arboleda, G; Huang, TJ; Waters, C; Verkhratsky, A; Fernyhough, P; Gibson, RM. Insulin-like growth factor-1-dependent maintenance of neuronal metabolism through the phosphatidylinositol 3-kinase-Akt pathway is inhibited by C2-ceramide in CAD cells. Eur. J. Neurosci 2007, 25, 3030–3038. [Google Scholar]
- Chalfant, CE; Kishikawa, K; Mumby, MC; Kamibayashi, C; Bielawska, A; Hannun, YA. Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A. Activation is stereospecific and regulated by phosphatidic acid. J. Biol. Chem 1999, 274, 20313–20317. [Google Scholar]
- Liu, B; Obeid, LM; Hannun, YA. Sphingomyelinases in cell regulation. Semin. Cell Dev. Biol 1997, 8, 311–322. [Google Scholar]
- Bryan, L; Kordula, T; Spiegel, S; Milstien, S. Regulation and functions of sphingosine kinases in the brain. Biochim. Biophys. Acta 2008, 1781, 459–466. [Google Scholar]
- Van Brocklyn, JR. Sphingolipid signaling pathways as potential therapeutic targets in gliomas. Mini. Rev. Med. Chem 2007, 7, 984–990. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De la Monte, S.M.; Longato, L.; Tong, M.; DeNucci, S.; Wands, J.R. The Liver-Brain Axis of Alcohol-Mediated Neurodegeneration: Role of Toxic Lipids. Int. J. Environ. Res. Public Health 2009, 6, 2055-2075. https://doi.org/10.3390/ijerph6072055
De la Monte SM, Longato L, Tong M, DeNucci S, Wands JR. The Liver-Brain Axis of Alcohol-Mediated Neurodegeneration: Role of Toxic Lipids. International Journal of Environmental Research and Public Health. 2009; 6(7):2055-2075. https://doi.org/10.3390/ijerph6072055
Chicago/Turabian StyleDe la Monte, Suzanne M., Lisa Longato, Ming Tong, Sarah DeNucci, and Jack R. Wands. 2009. "The Liver-Brain Axis of Alcohol-Mediated Neurodegeneration: Role of Toxic Lipids" International Journal of Environmental Research and Public Health 6, no. 7: 2055-2075. https://doi.org/10.3390/ijerph6072055