
Nickel-Refining Fumes Induced DNA Damage and Apoptosis of NIH/3T3 Cells via Oxidative Stress

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Media
2.2. Cell Culture
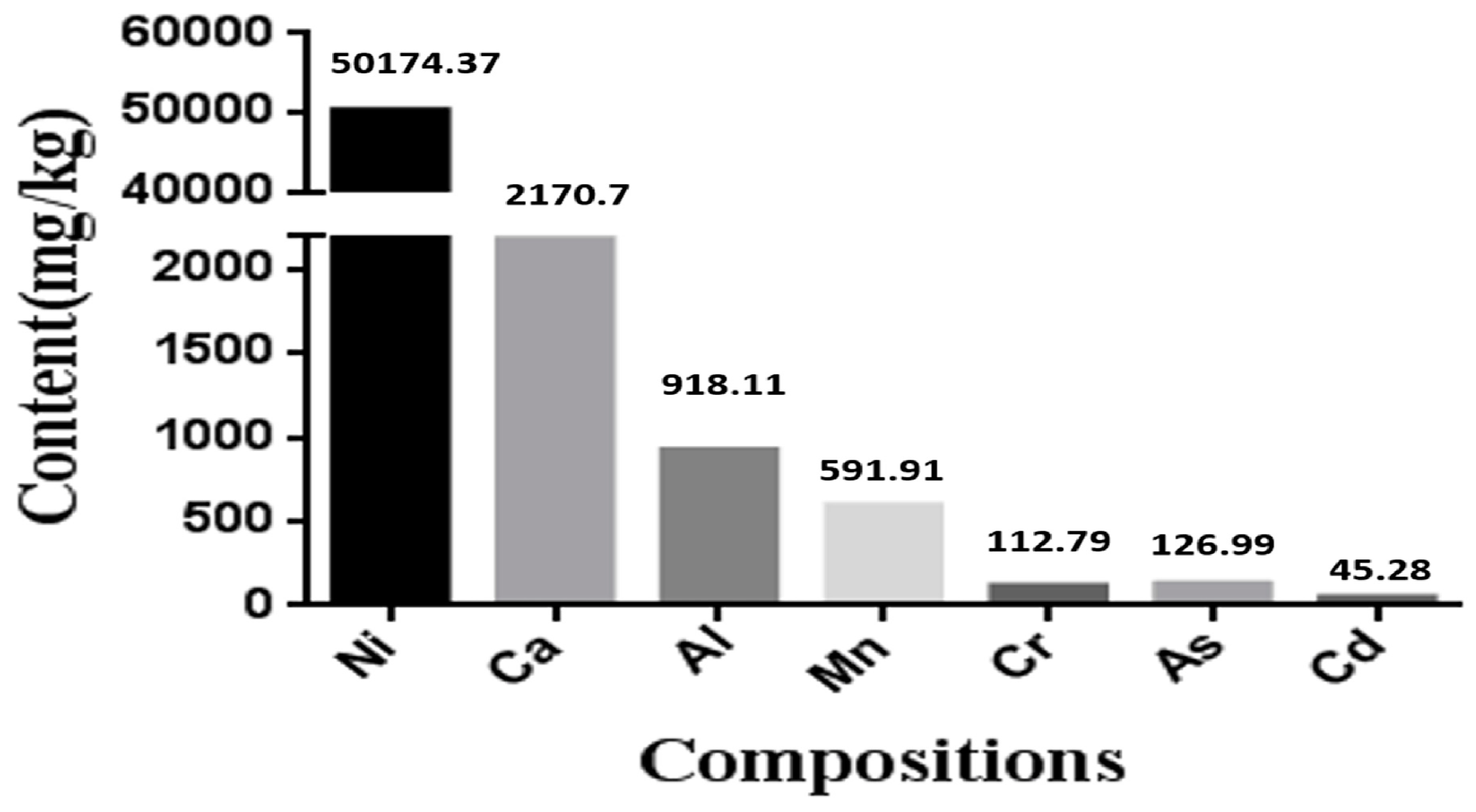
2.3. Component Analysis of Nickel-Refining Fumes
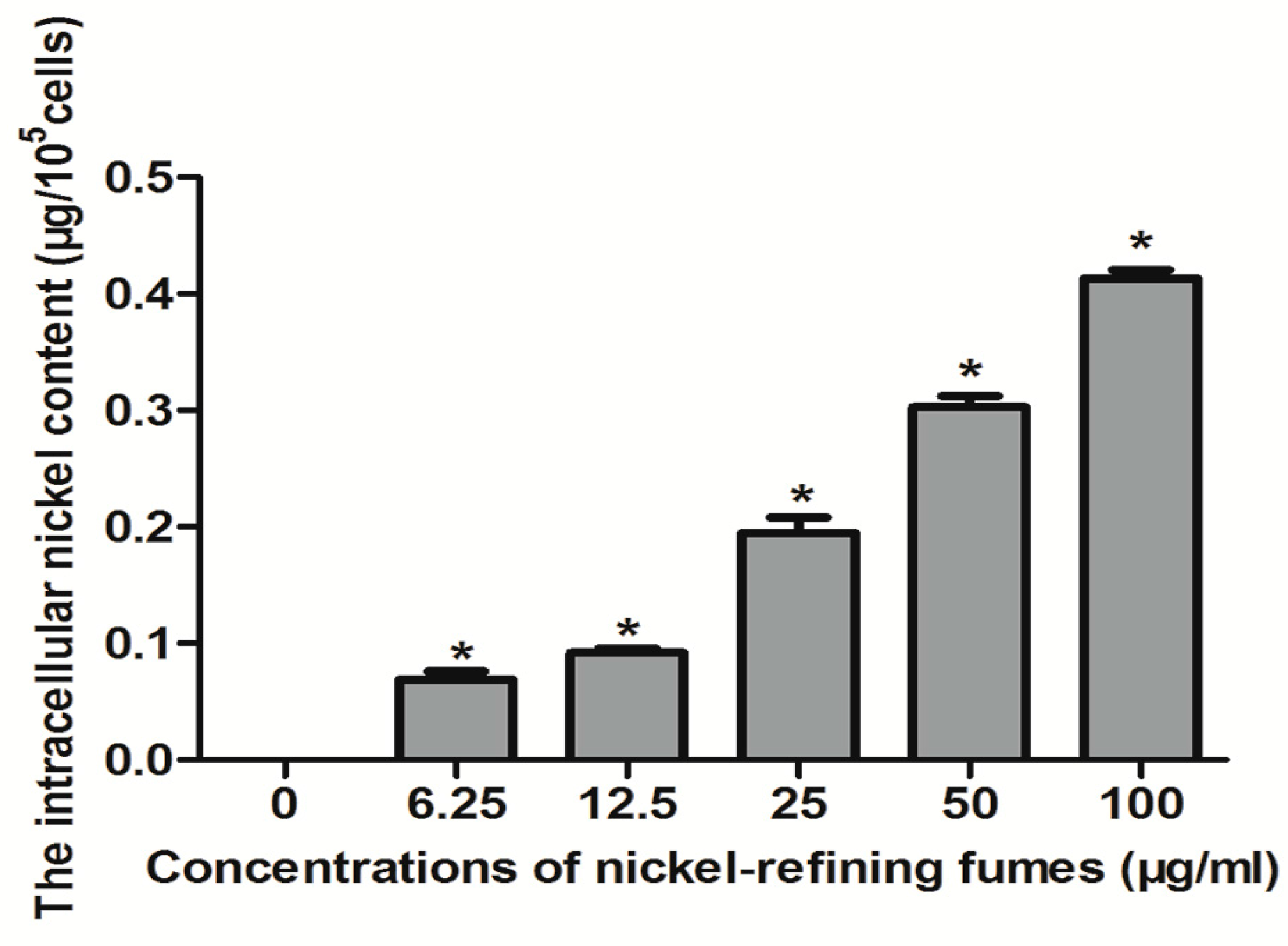
2.4. Detection of Nickel Content in Cells
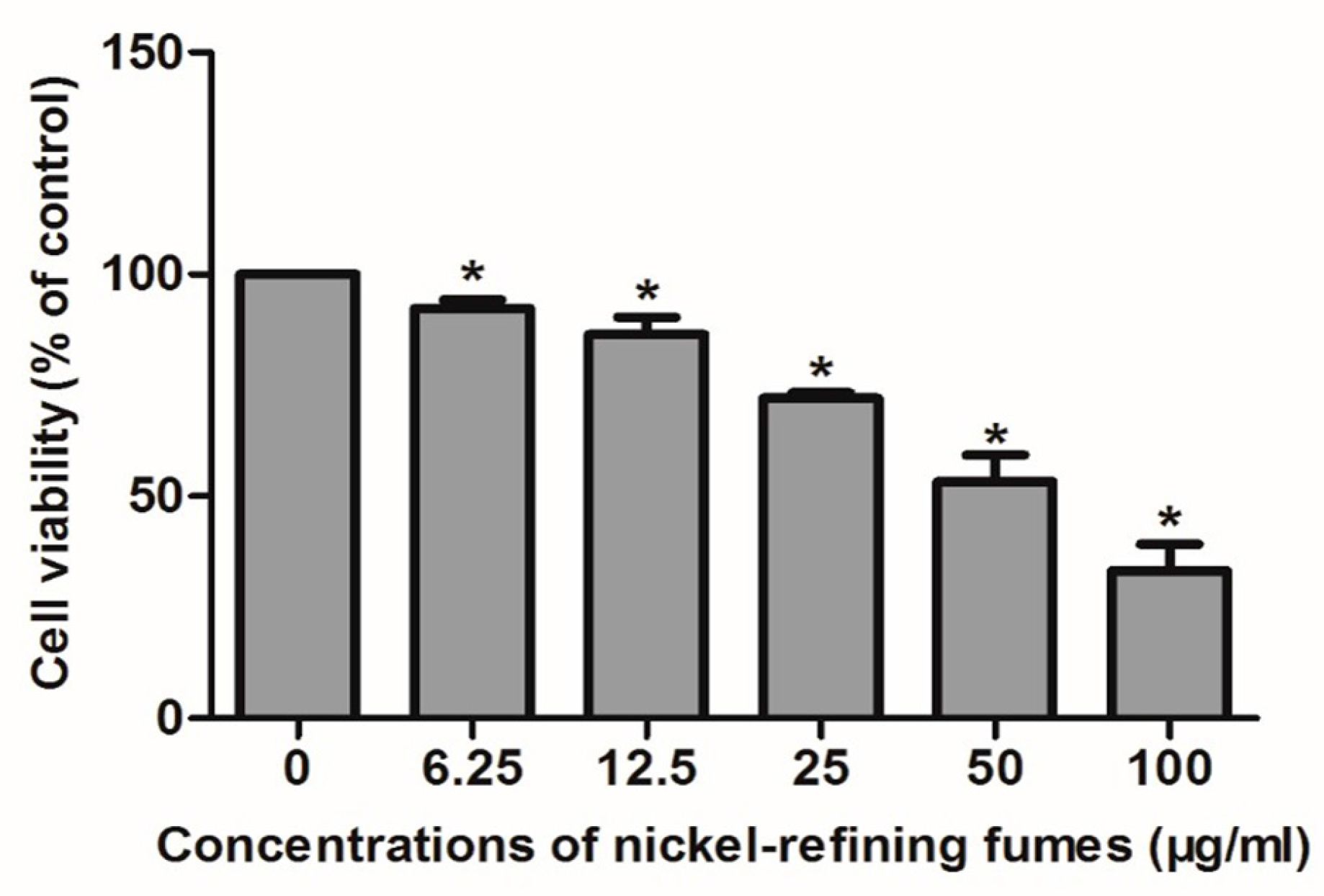
2.5. Cellular Viability
2.5.1. MTT Assay
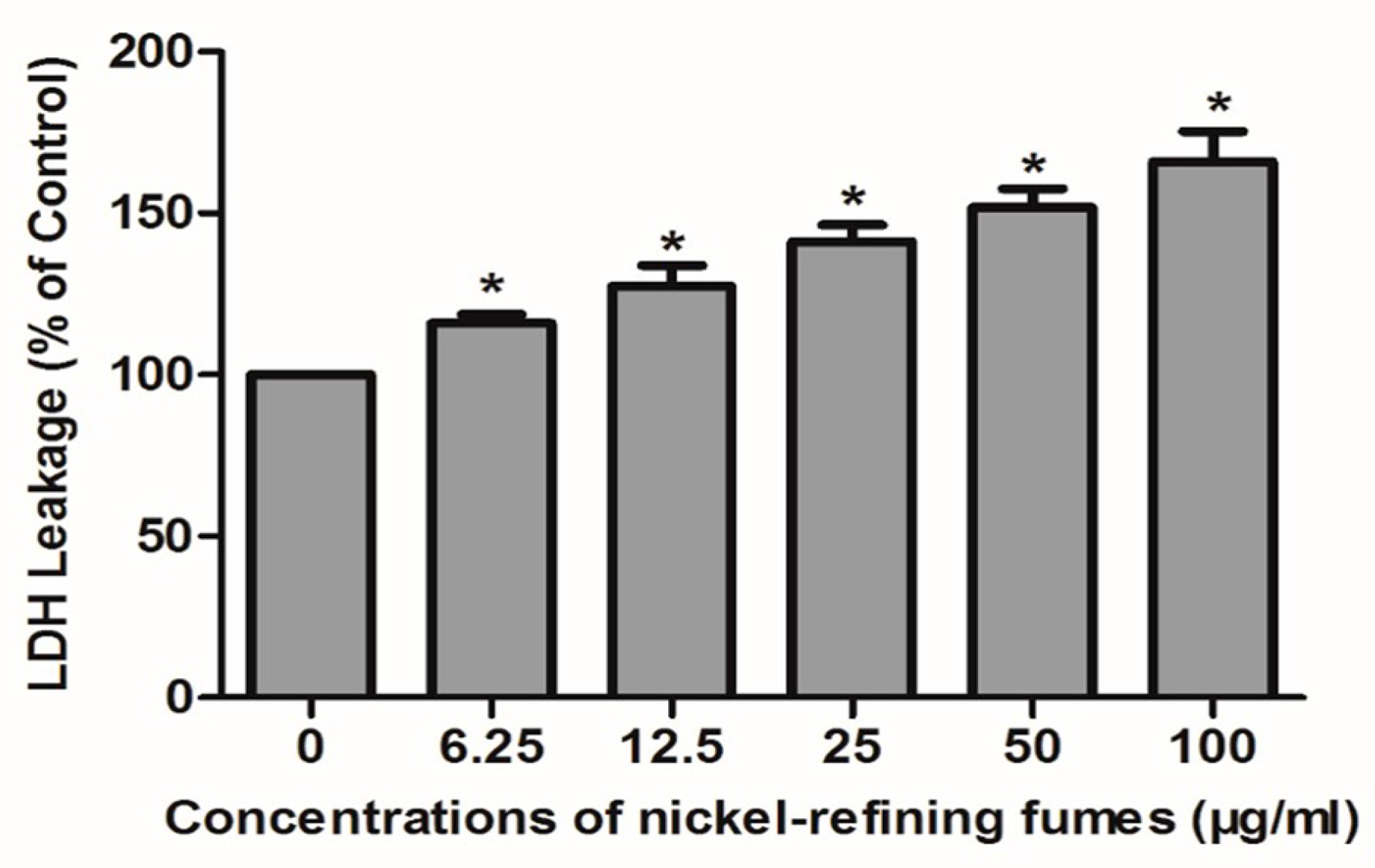
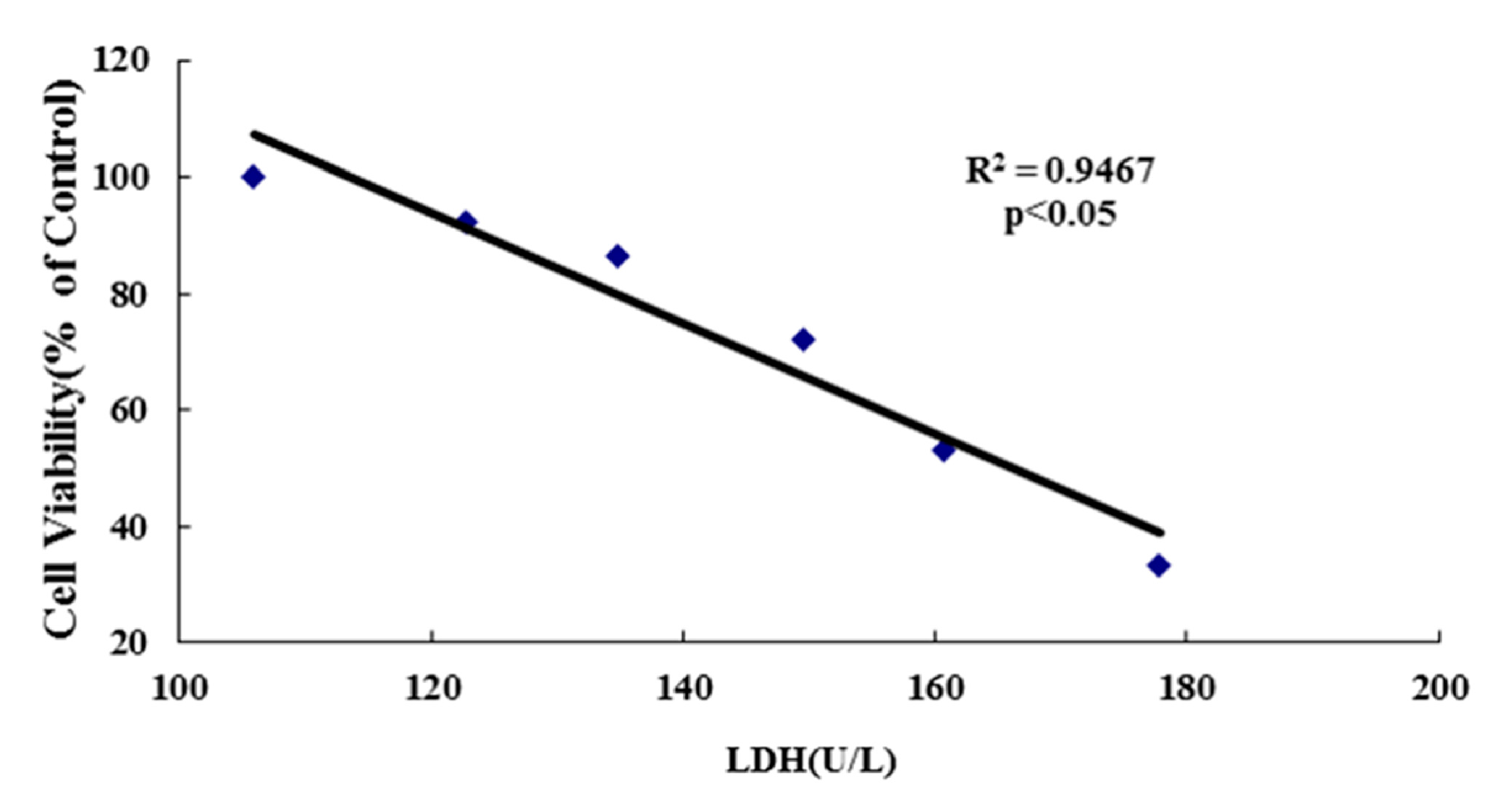
2.5.2. LDH Leakage Assay
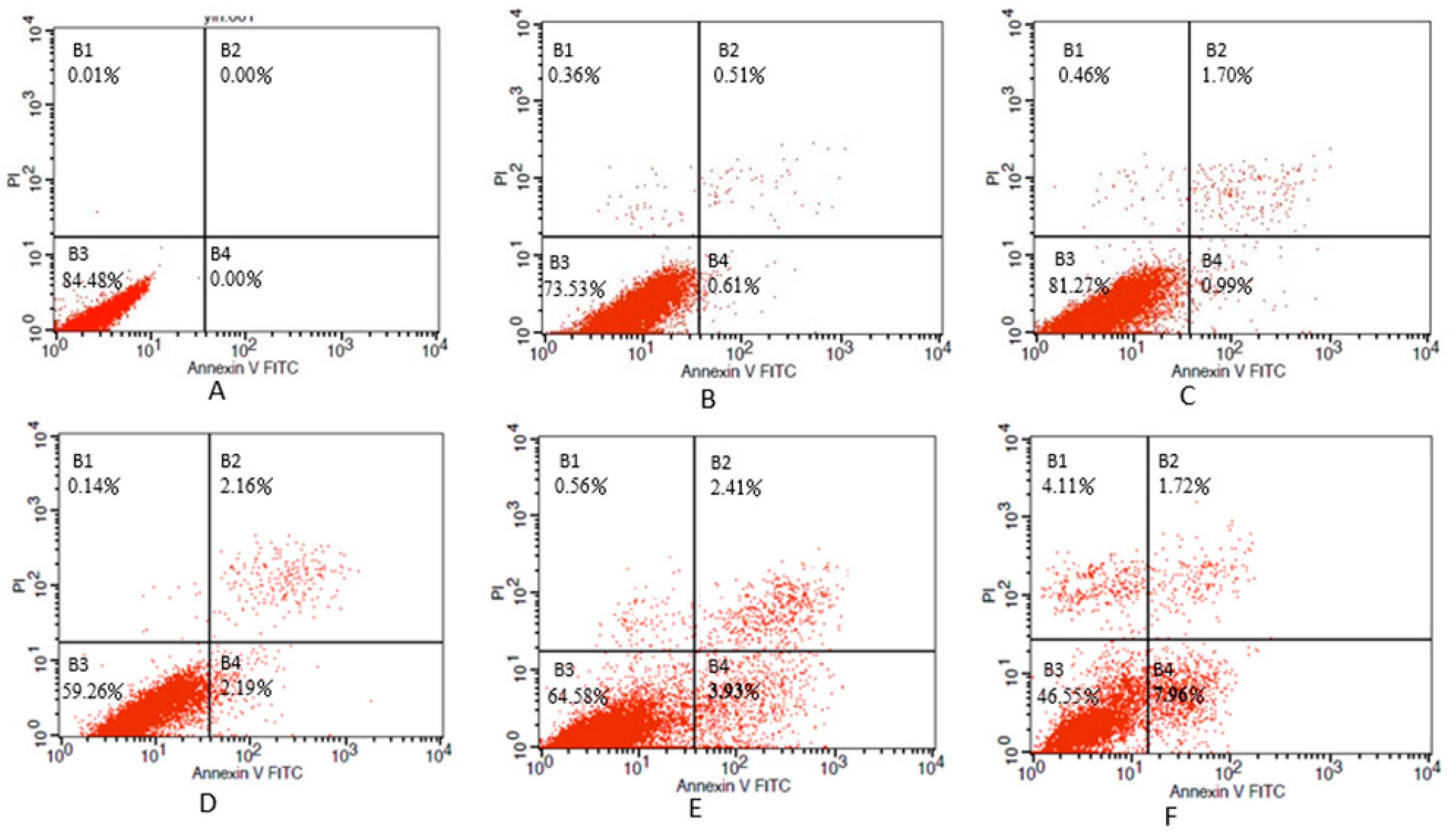
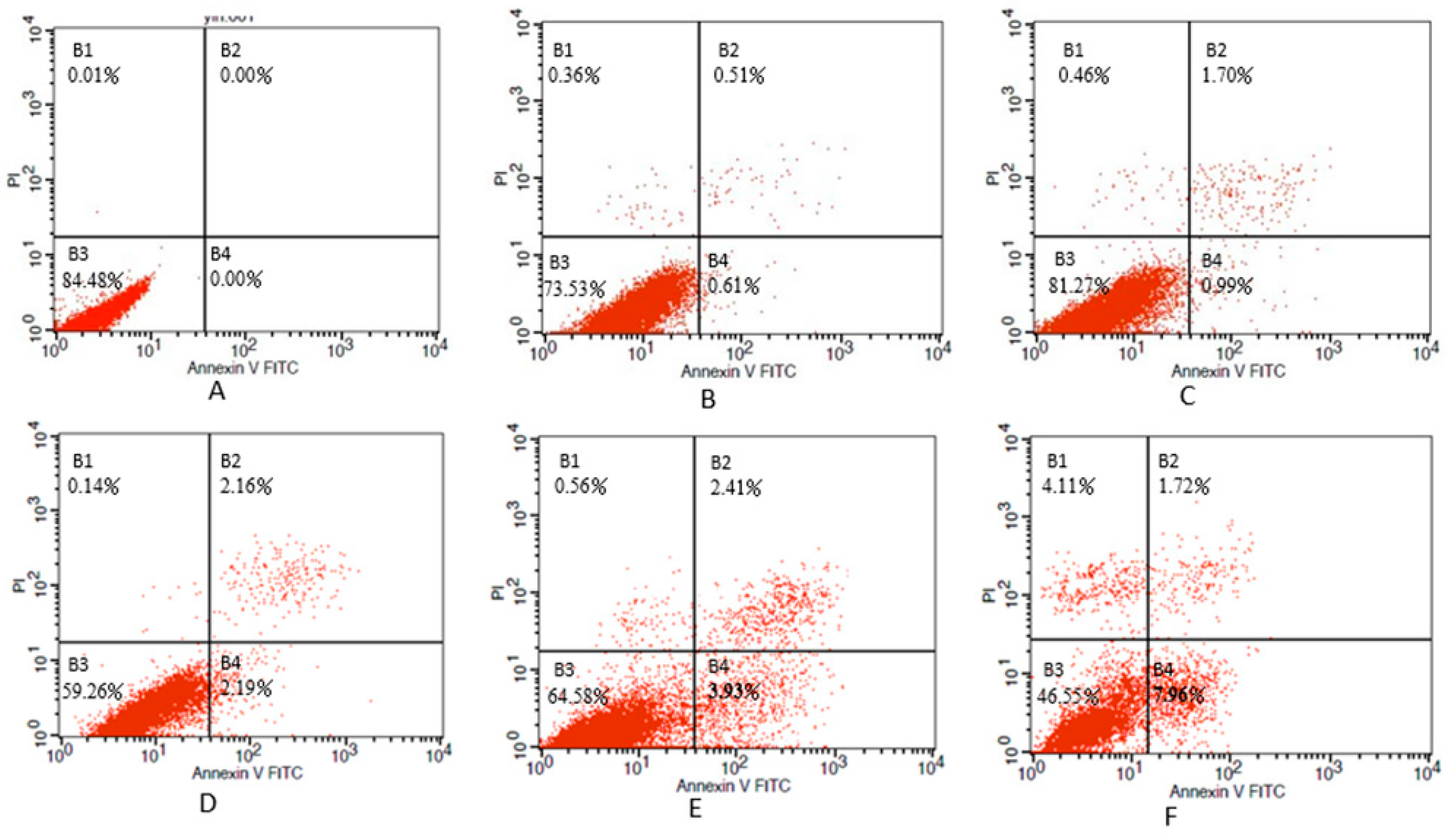
2.6. Determination of Apoptosis
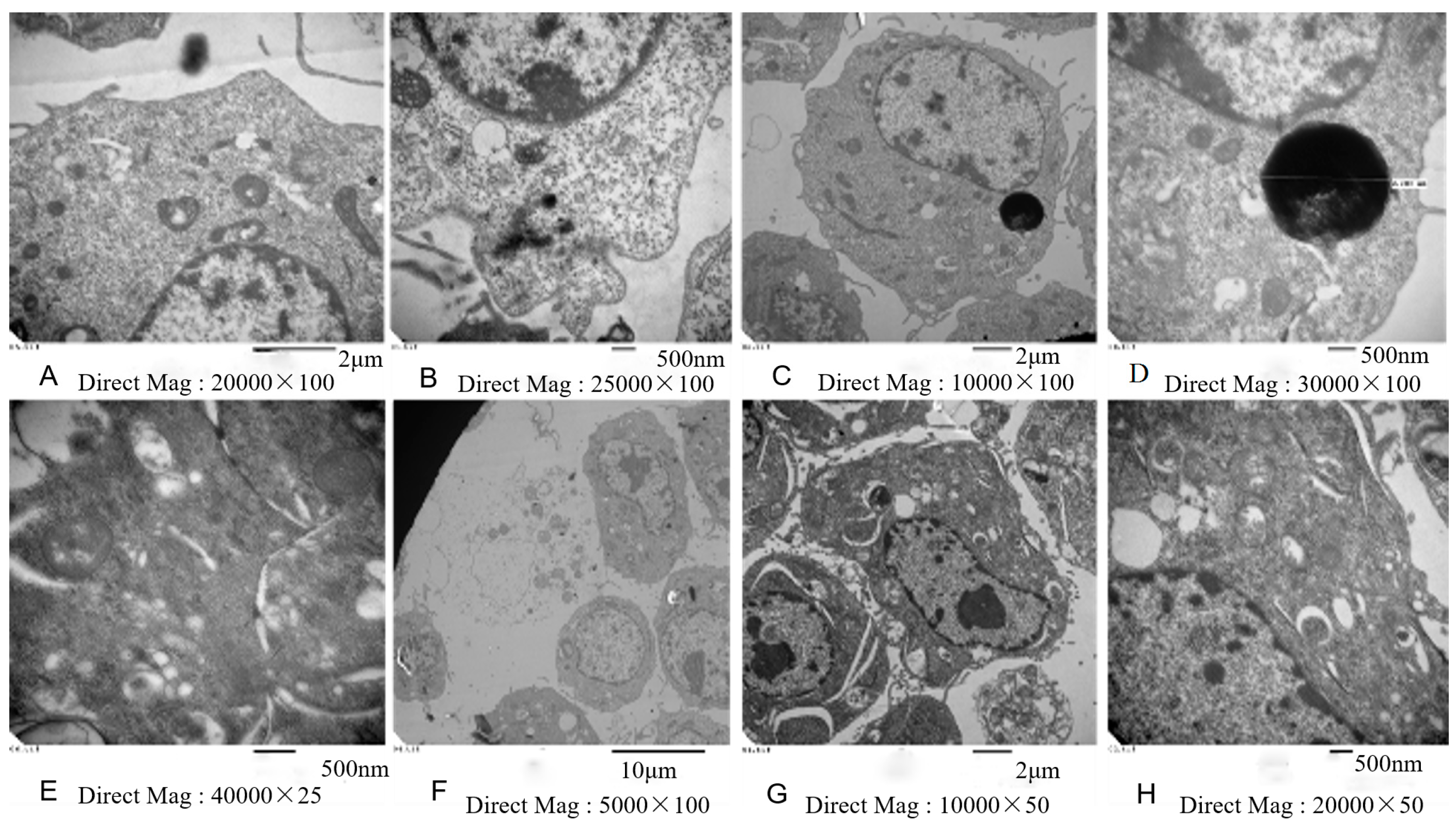
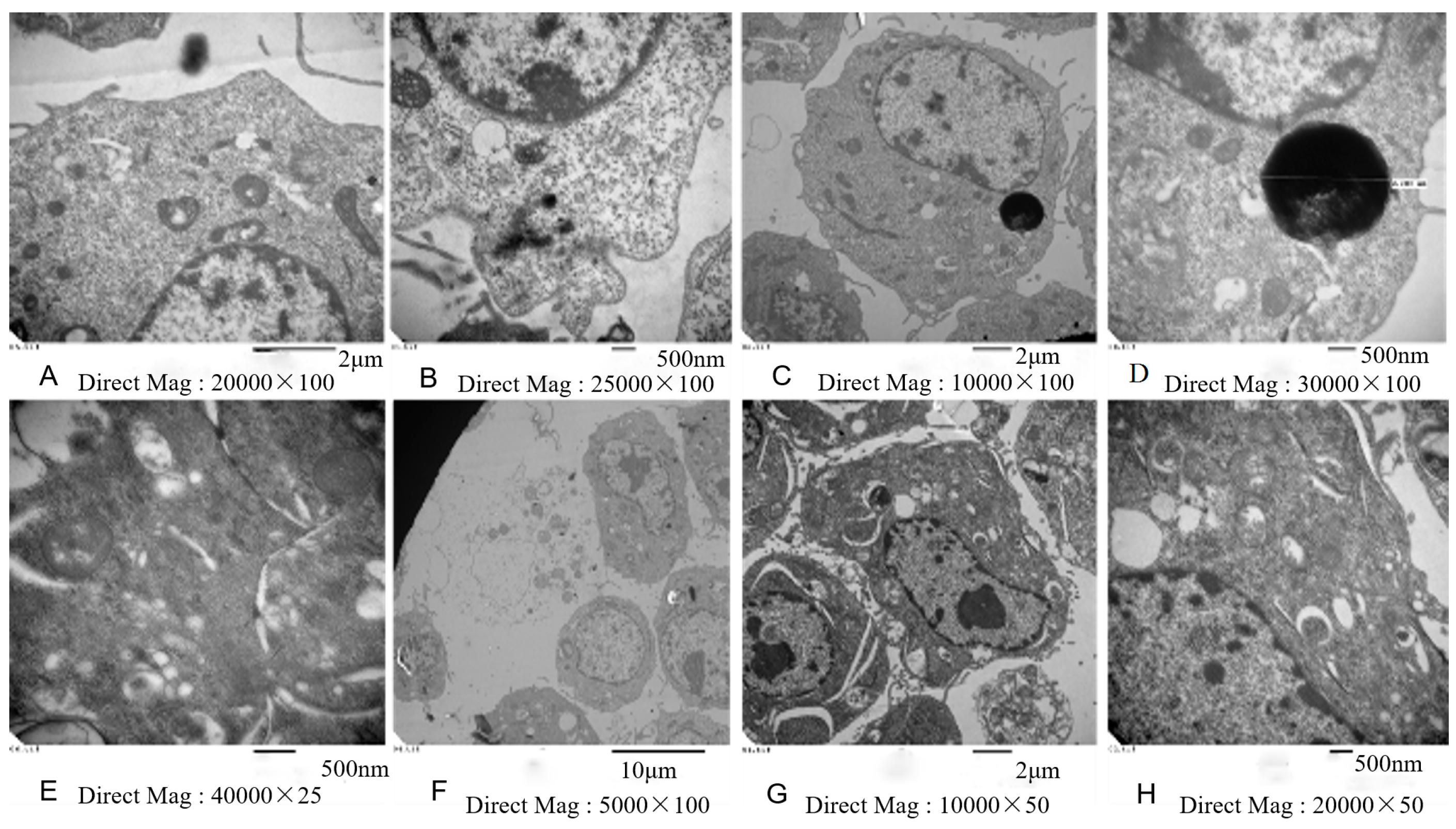
2.7. Electron Microscopy
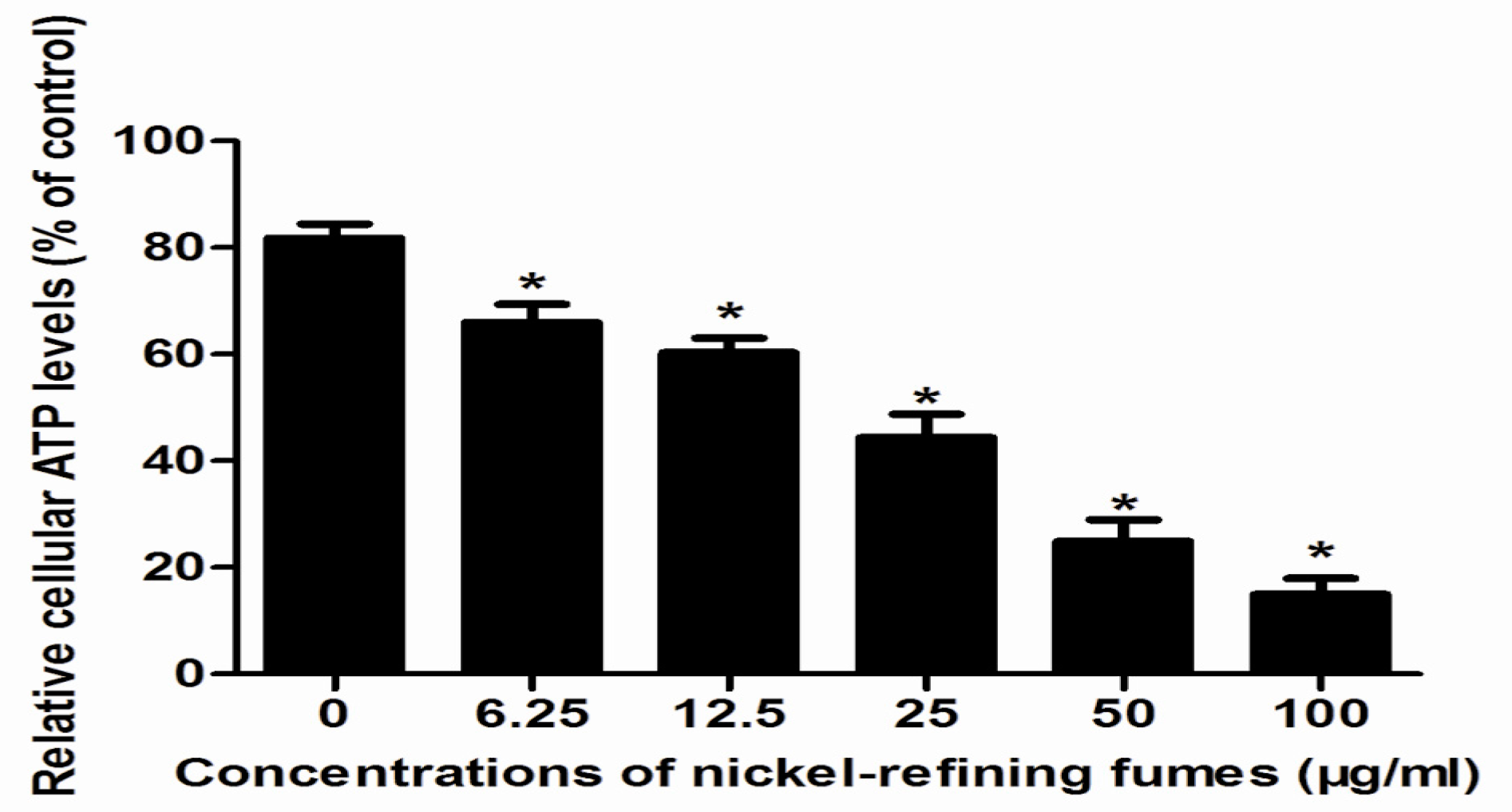
2.8. Mitochondrial ATP Measurement
2.9. Detection of ROS
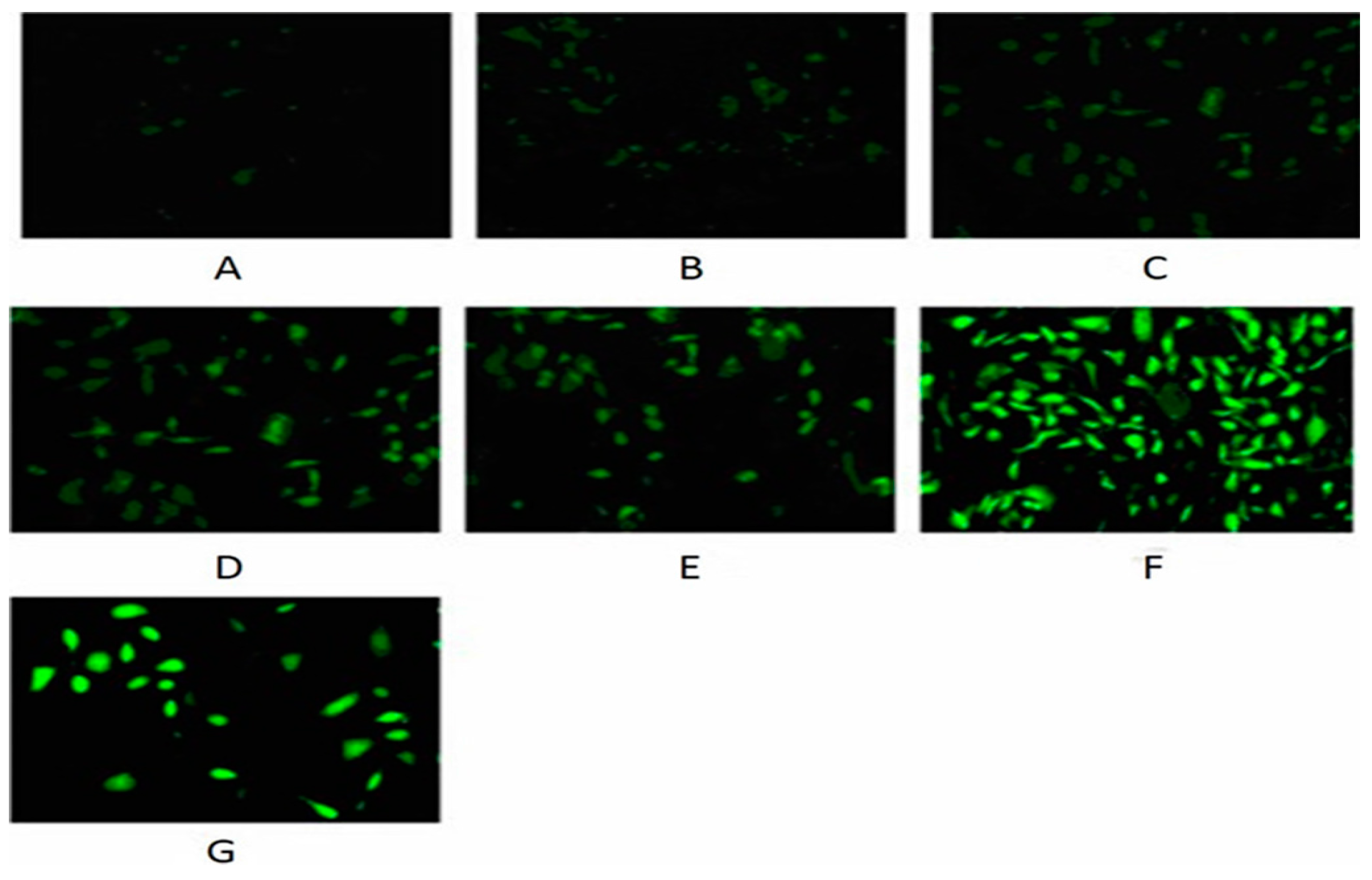
2.10. Evaluation of DNA Damage
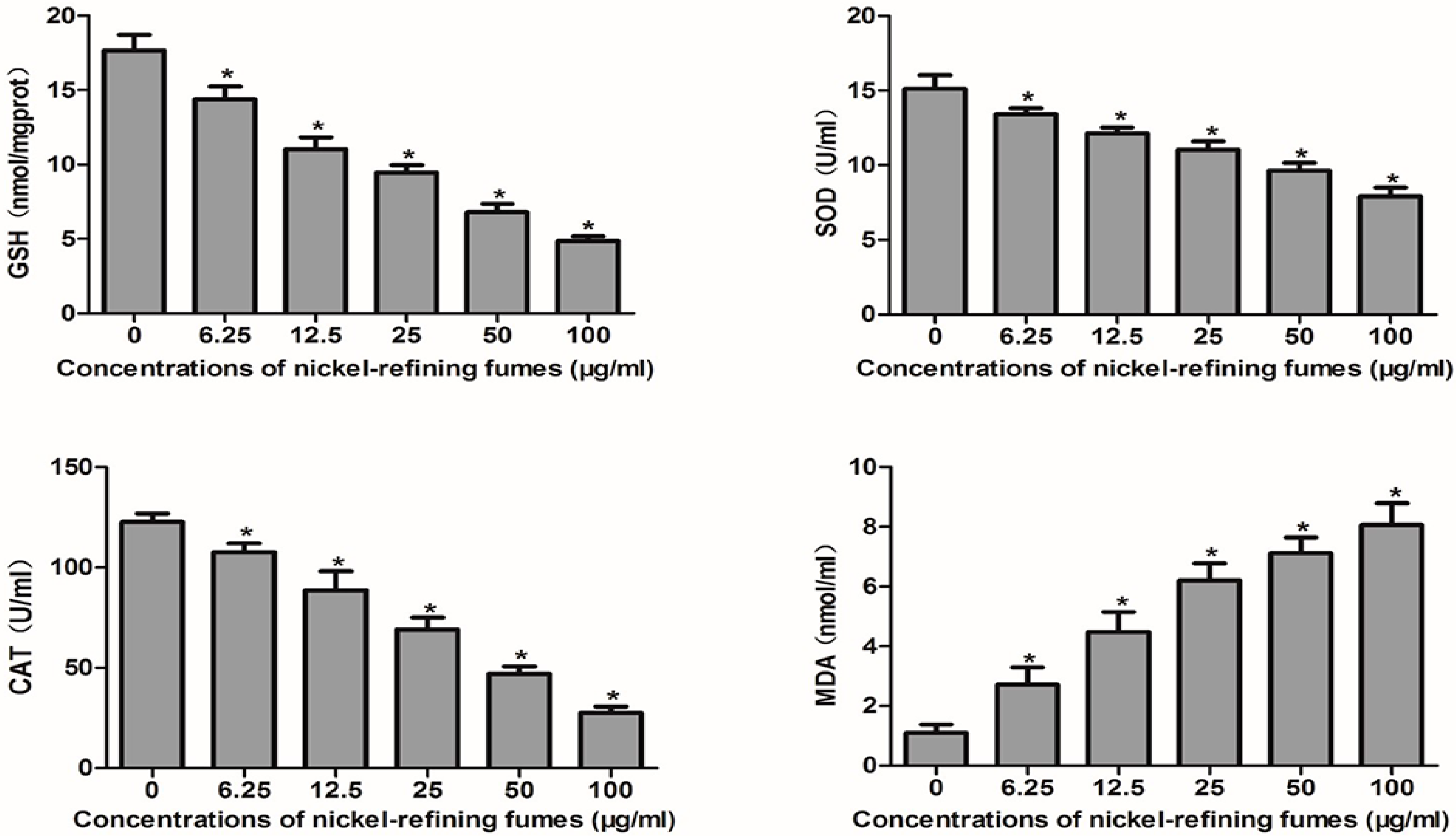
2.11. Membrane Lipid Peroxidation Assay
2.12. Detection of SOD
2.13. Detection of GSH
2.14. Measurement of CAT Level
2.15. Statistical Analysis
3. Results and Discussion
3.1. The Contents of Various Elements in Nickel-Refining Fumes
3.2. Analysis of Nickel Content in Cells
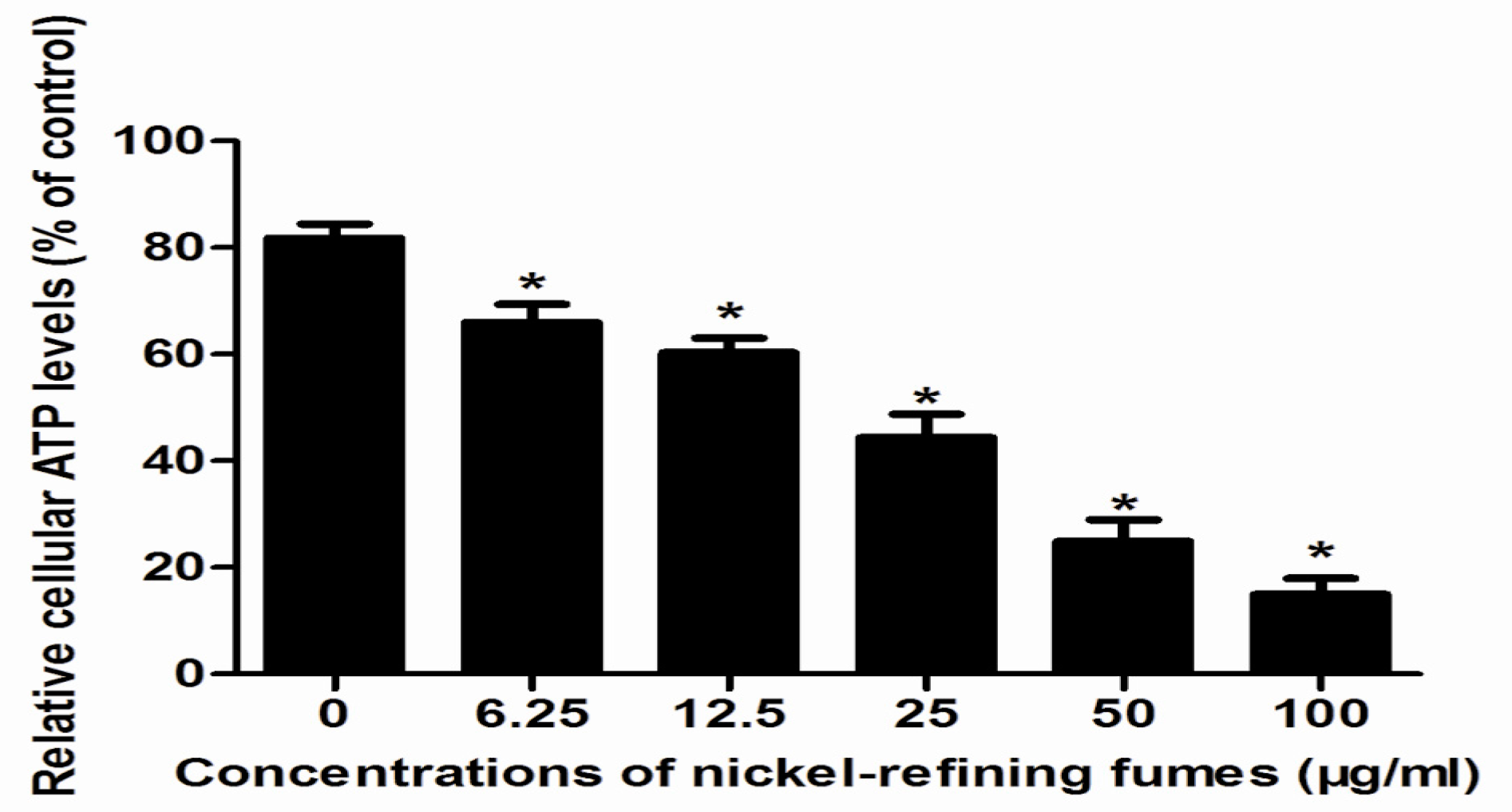
3.3. Inhibition of Cell Viability, Damage of Cell Membrane, Depletion of Mitochondrial ATP, Induction of Apoptosis by the Treatment of Nickel-Refining Fumes in NIH/3T3 Cells
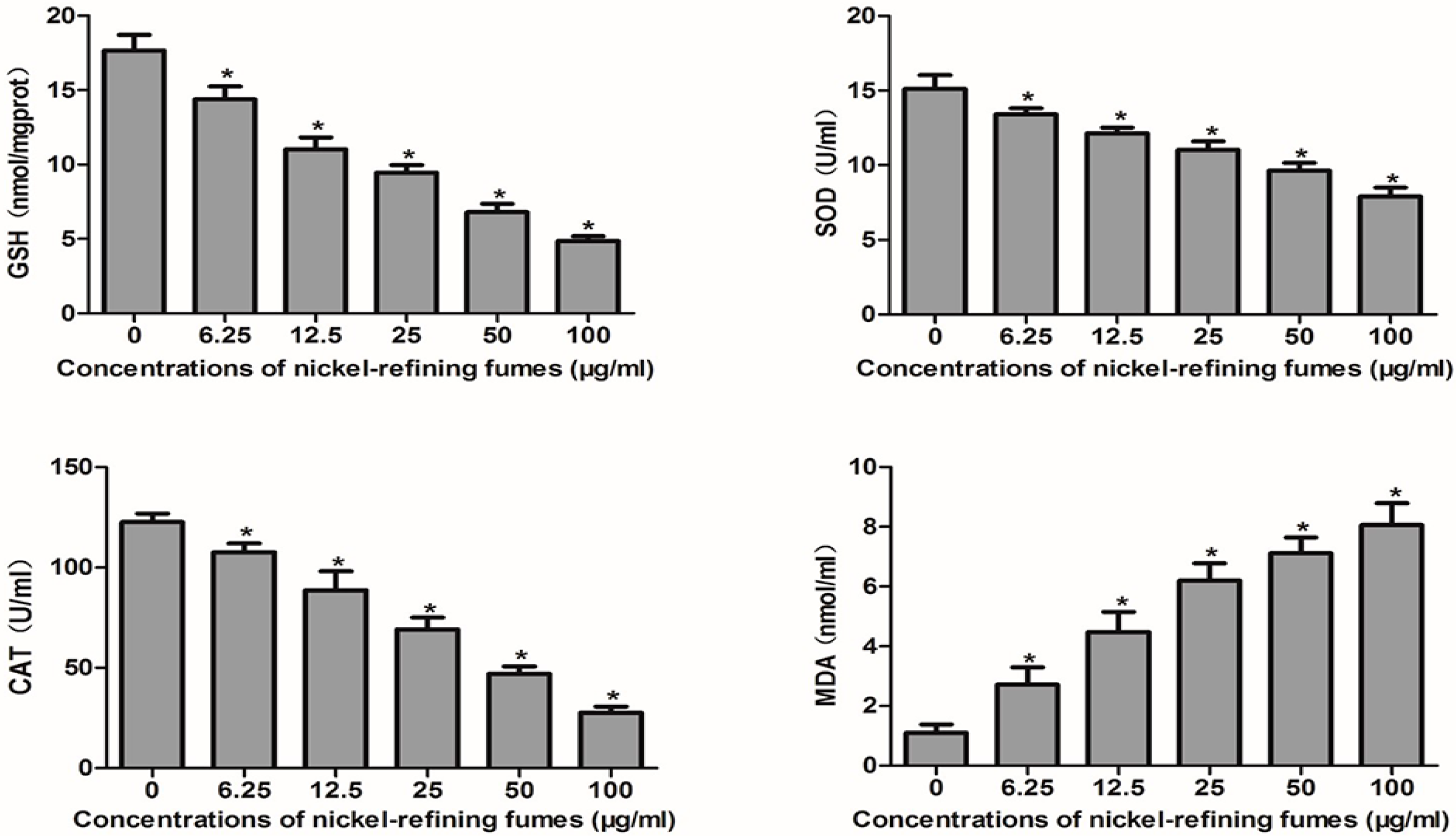
3.4. Nickel-Refining Fumes-Induced Apoptosis Is Associated with the Generation of ROS, the Disorders in the Oxidative System, Antioxidative System and DNA Damage
3.5. Nickel-Refining Fumes-Induced DNA Damage
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhao, J.; Shi, X.; Castranova, V.; Ding, M. Occupational toxicology of nickel and nickel compounds. J. Environ. Patholtoxicoloncol. 2009, 28, 177–208. [Google Scholar] [CrossRef]
- Costa, M.; Davidson, T.L.; Chen, H.; Ke, Q.; Zhang, P.; Yan, Y.; Huang, C.; Kluz, T. Nickel carcinogenesis: Epigenetics and hypoxia signaling. Mutat. Res. 2005, 592, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Krockova, J.Z.; Massanyi, P.; Sirotkin, A.V.; Pivko, J.; Makarevich, A.V.; Lukac, N.; Capcarova, M.; Toman, R.; Polakova, Z. Nickel induced structural and functional alterations in mouse Leydig cells in vitro. J. Trace Elem. Med. Biol. 2011, 25, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Wang, Y.F.; Huang, W.R.; Huang, Y.T. Nickel induces oxidative stress and genotoxicity in human lymphocytes. Toxicol. Appl. Pharmacol. 2003, 189, 153–159. [Google Scholar] [CrossRef]
- Lu, H.; Shi, X.; Costa, M.; Huang, C. Carcinogenic effect of nickel compounds. Mol. Cell. Biochem. 2005, 279, 45–67. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.V. Metals and apoptosis: Recent developments. J. Trace Elem. Med. Biol. 2008, 22, 262–284. [Google Scholar] [CrossRef] [PubMed]
- Kasprzak, K.S.; Sunderman, F.W., Jr.; Salnikow, K. Nickel carcinogenesis. Mutat. Res. 2003, 533, 67–97. [Google Scholar] [CrossRef] [PubMed]
- Goodman, J.E.; Prueitt, R.L.; Dodge, D.G.; Thakali, S. Carcinogenicity assessment of water-soluble nickel compounds. Crit. Rev. Toxicol. 2009, 39, 365–417. [Google Scholar] [CrossRef] [PubMed]
- Cangul, H.; Broday, L.; Salnikow, K. Molecular mechanisms of nickel carcinogenesis. Toxicol. Lett. 2002, 127, 69–75. [Google Scholar] [CrossRef]
- Salnikow, K.; Costa, M. Epigenetic mechanisms of nickel carcinogenesis. Environ. Pathol. Toxicol. Oncol. 2000, 19, 307–318. [Google Scholar]
- Chiou, Y.H.; Liou, S.H.; Wong, R.H.; Chen, C.Y.; Lee, H. Nickel may contribute to EGFR mutation and synergistically promotes tumor invasion in EGFR-mutated lung cancer via nickel-induced microRNA-21 expression. Toxicol. Lett. 2015, 237, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.C.; Yang, C.Y.; Hung, D.Z.; Su, C.C.; Chen, K.L.; Yen, C.C.; Ho, T.J.; Su, Y.C.; Huang, C.F.; Chen, C.H.; et al. Nickel(II) induced JNK activation-regulated mitochondria-dependent apoptotic pathway leading to cultured rat pancreatic β-cell death. Toxicology 2011, 289, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Ahamed, M.; Siddiqui, M.K. Low level lead exposure and oxidative stress: Current opinions. Clin. Chim. Acta 2007, 383, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Kvansakul, M.; Hinds, M.G. Structural, biology of the Bcl-2 family and its mimicry by viral proteins. Cell Death Dis. 2013, 4, e909. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.C.; Kehrer, J.P. Acrolein-induced cell death: A caspase-influenced decision between apoptosis and oncosis/necrosis. Chem. Biol. Interact. 2002, 139, 79–95. [Google Scholar] [CrossRef]
- Wang, Y.F.; Shyu, H.W.; Chang, Y.C.; Tseng, W.C.; Huang, Y.L.; Lin, K.H.; Chou, M.C.; Liu, H.L.; Chen, C.Y. Nickel(II)-induced cytotoxicity and apoptosis in human proximal tubule cells through a ROS- and mitochondria-mediated pathway. Toxicol. Appl. Pharmacol. 2012, 259, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Chervona, Y.; Arita, A.; Costa, M. Carcinogenic metals and the epigenome: Understanding the effect of nickel, arsenic, and chromium. Metallomics 2012, 4, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lim, S.S.; Baek, B.J.; An, J.M.; Nam, H.S.; Woo, K.M.; Cho, M.K.; Kim, S.H.; Lee, S.H. Nickel(II)-induced nasal epithelial toxicity and oxidative mitochondrial damage. Environ. Toxicol. Pharmacol. 2016, 42, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, J.; Choi, C. Mitochondrial network determines intracellular ROS dynamics and sensitivity to oxidative stress through switching Inter-mitochondrial messengers. PLoS ONE 2011, 6, e23211. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Mihara, K. Molecular mechanisms and physiologic functions of mitochondrial dynamics. J. Biochem. 2011, 149, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, X.; Qi, H.; Li, X.; Xiao, X.; Gao, J. Ursolic acid induces apoptosis through mitochondrial intrinsic pathway and suppression of ERK1/2 MAPK in HeLa cells. J. Pharmacol. Sci. 2014, 125, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Kim, W.H.; Lim, J.H.; Song, E.H.; Song, J.; Choi, K.Y.; Jung, M.H. Mitochondrial dysfunction: Glucokinase downregulation lowers interaction of glucokinase with mitochondria, resulting in apoptosis of pancreatic β-cells. Cell Signal. 2009, 21, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.C.; Strasser, A. BH3-only proteins-essential initiators of apoptotic cell death. Cell 2000, 103, 839–842. [Google Scholar] [CrossRef]
- Gang, B.; Sun, S.Z.; Huang, A.M.; Li, S.H.; Zhang, L.F. Environmental monitoring in nickel manufacturing plants. Occup. Health Dis. 1993, 19, 323–327. [Google Scholar]
- Badding, M.A.; Fix, N.R.; Antonini, J.M.; Leonard, S.S. A comparison of cytotoxicity and oxidative stress from welding fumes generated with a new nickel-, copper-based consumable versus mild and stainless steel-based welding in RAW 264.7 mouse macrophages. PLoS ONE 2014, 9, e101310. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Gerlier, D.; Thomasset, N. Use of MTT colorimetric assay to measure cell activation. J. Immunol. Methods 1986, 94, 57–63. [Google Scholar] [CrossRef]
- Tchounwou, P.B.; Wilson, B.; Schneider, J.; Ishaque, A.; Morrow, J.D.; Roberts, L.J. Cytogenic assessment of arsenic trioxide toxicity in the Mutatox, Ames II and CAT-Tox assays. Metal Ions Biol. Med. 2000, 6, 89–91. [Google Scholar]
- Fotakis, G.; Timbrell, J.A. In vitro cytotoxicity assays: Comparison of LDH, neutral red, MTT and protein assay in hepatoma cell lines following exposure to cadmium chloride. Toxicol. Lett. 2006, 160, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wei, S.; Tang, J.M.; Guo, L.Y.; Zheng, F.; Yang, J.Y.; Kong, X.; Huang, Y.Z.; Chen, S.Y.; Wang, J.N. PEP-1-CAT protects hypoxia/reoxygenation-induced cardiomyocyte apoptosis through multiple sigaling pathways. J. Transl. Med. 2013, 11, 113. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Xue, Y.; Sun, J. Hepatotoxicity and liver injury induced by hydroxyapatite nanoparticles. J. Appl. Toxicol. 2014, 34, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Lobner, D. Comparison of the LDH and MTT assays for quantifying cell death: Validity for neuronal apoptosis. J. Neurosci. Meth. 2000, 96, 147–152. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Yedjou, C.G.; Tchounwou, P.B.; Jerkins, J.; McMurray, R. Basic mechanisms of arsenic trioxide (ATO)-induced apoptosis in human leukemia (HL-60) cells. J. Hematol. Oncol. 2010, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Yedjou, C.G.; Tchounwou, H.M.; Tchounwou, P.B. DNA Damage, cell cycle arrest, and apoptosis induction caused by lead in human leukemia cells. Int. J. Environ. Res. Public Health 2016, 13, 56. [Google Scholar] [CrossRef] [PubMed]
- Ozben, T. Oxidative stress and apoptosis: Impact on cancer therapy. J. Pharm. Sci. 2007, 96, 2181–2196. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Li, T.; Li, Y.; Cai, S.; Zhang, Z.; Zeng, Z.; Wang, X.; Gao, Y.; Li, Y.; Chen, Z. Ulinastatin inhibits oxidant-induced endothelial hyperpermeability and apoptotic signaling. Int. J. Clin. Exp. Pathol. 2014, 7, 7342–7350. [Google Scholar] [PubMed]
- Wang, H.; Joseph, J.A. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic. Biol. Med. 1999, 27, 612–616. [Google Scholar] [CrossRef]
- Moghadamtousi, S.Z.; Kadir, H.A.; Paydar, M.; Rouhollahi, E.; Karimian, H. Annona muricata leaves induced apoptosis in A549 cells through mitochondrial-mediated pathway and involvement of NF-kappaB. BMC Complemt. Altern. Med. 2014, 14, 299. [Google Scholar] [CrossRef]
- Chang, Y.; Yang, S.T.; Liu, J.H.; Dong, E.; Wang, Y.; Cao, A.; Liu, Y.; Wang, H. In vitro toxicity evaluation of graphene oxide on A549 cells. Toxicol. Lett. 2011, 200, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.P.; Mccoy, M.T.; Tice, R.R.; Schneider, E.L. A simple technique for quantitation of low levels of DNA damage in individual cells. A simple technique for quantitation of low levels of DNA damage in in dividual cells. Exp. Cell Res. 1988, 175, 184–191. [Google Scholar] [CrossRef]
- Collins, A.R.; Ma, A.G.; Duthie, S.J. The kinetics of repair of oxidative DNA damage (strand breaks and oxidised pyrimidines) in human cells. Mutat. Res. 1995, 336, 69–77. [Google Scholar] [CrossRef]
- Collins, A.R.; Dusinská, M.; Horská, A. Detection of alkylation damage in human lymphocyte DNA with the comet assay. Acta Biochim. Pol. 2001, 48, 611–614. [Google Scholar] [PubMed]
- Yedjou, C.G.; Tchounwou, P.B. In-vitro genotoxic effect of arsenic trioxide to human leukemia (HL-60) cells using the comet assay. Mol. Cell. Biochem. 2007, 301, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Guo, W.; Ben, Y.; Jiang, J.; Tan, C.; Xu, Z.; Wang, X.; Bai, C. Preventive effects of curcumin and dexamethasone on lung transplantation-associated lung injury in rats. Crit. Care Med. 2008, 36, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Gui, D.; Huang, J.; Liu, W.; Guo, Y.; Xiao, W.; Wang, N. Astragaloside IV prevents acute kidney injury in two rodent models by inhibiting oxidative stress and apoptosis pathways. Apoptosis 2013, 18, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, N.V.; Obidin, A.B.; Marinov, B.S. Modulation of superoxide dismutase by electron donors and acceptors. FEBS Lett. 1987, 211, 211–214. [Google Scholar] [CrossRef]
- Huang, X.; Qin, J.; Lu, S. Magnesium isoglycyrrhizinate protects hepatic L02 cells from ischemia/reperfusion induced injury. Int. J. Clin. Exp. Pathol. 2014, 7, 4755–4764. [Google Scholar] [PubMed]
- Wang, J.; Sun, P.; Bao, Y.; Dou, B.; Song, D.; Li, Y. Vitamin E renders protection to PC12 cells against oxidative damage and apoptosis induced by single-walled carbon nanotubes. Toxicol. In Vitro 2012, 26, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Durak, I.; Güven, T.; Birey, M.; Oztürk, H.S.; Kurtipek, O.; Ye, M.; Dikmen, B.; Canbolat, O.; Kavutcu, M.; Kaçmaz, M. Halothane hepatotoxicity and hepatic free radical metabolism in guinea pigs; the effects of vitamin E. Can. J. Anaesth. 1996, 43, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Draper, M.H.; Duffus, J.H.; John, P.; Metcalfe, L.; Morgan, L.; Park, M.V.; Weitzner, M.I. Analysis of nickel refinery dusts. Sci. Total Environ. 1994, 148, 263–273. [Google Scholar] [CrossRef]
- Clemens, F.; Landolph, J.R. Genotoxicity of samples of nickel refinery dust. Toxicol. Sci. 2003, 73, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Pietruska, J.R.; Liu, X.; Smith, A.; McNeil, K.; Weston, P.; Zhitkovich, A.; Hurt, R.; Kane, A.B. Bioavailability, intracellular mobilization of nickel, and HIF-1α activation in human lung epithelial cells exposed to metallic nickel and nickel oxide nanoparticles. Toxicol. Sci. 2011, 124, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, A.; Costa, M. Elucidating the mechanisms of nickel compound uptake: A review of particulate and nano-nickel endocytosis and toxicity. Toxicol. Appl. Pharmacol. 2012, 260, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.C.; Yang, J.S.; Huang, A.C.; Hsia, T.C.; Chou, S.T.; Kuo, C.L.; Lu, H.F.; Lee, T.H.; Wood, W.G.; Chung, J.G. Chrysophanol induces necrosis through the production of ROS and alteration of ATP levels in J5 human liver cancer cells. Mol. Nutr. Food Res. 2010, 54, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Shui, S.; Han, X.; Guo, D.; Li, T.; Yan, L. MicroRNA-22 attenuates neuronal cell apoptosis in a cell model of traumatic brain injury. Am. J. Transl. Res. 2016, 8, 1895–1902. [Google Scholar] [PubMed]
- Lee, S.H.; Kim, D.K.; Seo, Y.R.; Woo, K.M.; Kim, C.S.; Cho, M.H. Nickel (II)-induced apoptosis and G2/M enrichment. Exp. Mol. Med. 1998, 30, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lee, S.H.; Seo, Y.R.; Perkins, S.N.; Kasprzak, K.S. Nickel(II)-induced apoptosis in murine T cell hybridoma cells is associated with increased fas ligand expression. Toxicol. Appl. Pharmacol. 2002, 185, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Ahamed, M.; Akhtar, M.J.; Siddiqui, M.A.; Ahmad, J.; Musarrat, J.; Al-Khedhairy, A.A.; AlSalhi, M.S.; Alrokayan, S.A. Oxidative stress mediated apoptosis induced by nickel ferrite nanoparticles in cultured A549 cells. Toxicology 2011, 283, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Lin, T.K.; Chang, Y.C.; Wang, Y.F.; Shyu, H.W.; Lin, K.H.; Chou, M.C. Nickel (II)-induced oxidative stress, apoptosis, G2/M arrest, and genotoxicity in normal rat kidney cells. J. Toxicol. Environ. Health A. 2010, 73, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Schedle, A.; Samorapoompichit, P.; Rausch-Fan, X.H.; Franz, A.; Füreder, W.; Sperr, W.R.; Sperr, W.; Ellinger, A.; Slavicek, R.; Boltz-Nitulescu, G.; et al. Response of L-929 fibroblasts, human gingival fibroblasts, and human tissue mast cells to various metal cations. J. Dent. Res. 1995, 74, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Shiao, Y.H.; Lee, S.H.; Kasprzak, K.S. Cell cycle arrest, apoptosis and p53 expression in nickel(II) acetate-treated Chinese hamster ovary cells. Carcinogenesis 1998, 19, 1203–1237. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Choi, J.G.; Cho, M.H. Apoptosis, bcl2 expression, and cell cycle analyses in nickel(II)-treated normal rat kidney cells. J. Korean Med. Sci. 2001, 16, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Mariggió, M.A.; Fulle, S.; Calissano, P.; Nicoletti, I.; Fanó, G. The brain protein S-100ab induces apoptosis in PC12 cells. Neuroscience 1994, 60, 29–35. [Google Scholar] [CrossRef]
- Pan, J.; Chang, Q.; Wang, X.; Son, Y.; Zhang, Z.; Chen, G.; Luo, J.; Bi, Y.; Chen, F.; Shi, X. Reactive oxygen species-activated Akt/ASK1/p38 signaling pathway in nickel compound-induced apoptosis in BEAS 2B cells. Chem. Res. Toxicol. 2010, 23, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Song, M.; Zhang, Y.; Yan, M.; Zhang, M.; Bi, H. Nickel nanowires induce cell cycle arrest and apoptosis by generation of reactive oxygen species in HeLa cells. Toxicol. Rep. 2014, 1, 114–121. [Google Scholar] [CrossRef]
- Zheng, G.H.; Liu, C.M.; Sun, J.M.; Feng, Z.J.; Cheng, C. Nickel-induced oxidative stress and apoptosis in Carassiusauratus liver by JNK pathway. Aquat. Toxicol. 2014, 147, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Ahamed, M. Toxic response of nickel nanoparticles in human lung epithelial A549 cells. Toxicol. In Vitro 2011, 25, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, A.H.; Kerr, J.F.; Currie, A.R. Cell death: The significance of apoptosis. Int. Rev. Cytol. 1980, 68, 251–306. [Google Scholar] [PubMed]
- Kasprzak, K.S.; Jaruga, P.; Zastawny, T.H.; North, S.L.; Riggs, C.W.; Olinski, R.; Dizdaroglu, M. Oxidative DNA base damage and its repair in kidneys and livers of nickel(II)-treated male F344 rats. Carcinogenesis 1997, 18, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Ahamed, M.; Siddiqui, M.A.; Akhtar, M.J.; Ahmad, I.; Pant, A.B.; Alhadlaq, H.A. Genotoxic potential of copper oxide nanoparticles in human lung epithelial cells. Biochem. Biophys. Res. Commun. 2010, 396, 578–583. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Tail DNA Contents (%) | Comet Cell Rate (%) | Tail Length(x ± s, µm) |
|---|---|---|---|
| negative control | 5.480 ± 0.138 | 3.50 | 8.721 ± 0.350 |
| 6.25 | 6.334 ± 0.247 | 6.17 | 9.041 ± 0.326 |
| 12.50 | 8.010 ± 0.316 * | 15.33 | 10.509 ± 0.698 |
| 25.00 | 10.031 ± 0.469 * | 32.83 | 15.175 ± 0.712 * |
| 50.00 | 13.883 ± 0.683 * | 51.83 | 17.624 ± 0.659 * |
| 100.0 | 16.964 ± 0.572 * | 66.50 | 24.974 ± 0.562 * |
| B[a]P | 24.232 ± 0.764 | 96.17 | 34.721 ± 0.650 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Wang, S.-Y.; Jia, L.; Zhang, L.; Ba, J.-C.; Han, D.; Yu, C.-P.; Wu, Y.-H. Nickel-Refining Fumes Induced DNA Damage and Apoptosis of NIH/3T3 Cells via Oxidative Stress. Int. J. Environ. Res. Public Health 2016, 13, 629. https://doi.org/10.3390/ijerph13070629
Wang Y, Wang S-Y, Jia L, Zhang L, Ba J-C, Han D, Yu C-P, Wu Y-H. Nickel-Refining Fumes Induced DNA Damage and Apoptosis of NIH/3T3 Cells via Oxidative Stress. International Journal of Environmental Research and Public Health. 2016; 13(7):629. https://doi.org/10.3390/ijerph13070629
Chicago/Turabian StyleWang, Yue, Sheng-Yuan Wang, Li Jia, Lin Zhang, Jing-Chong Ba, Dan Han, Cui-Ping Yu, and Yong-Hui Wu. 2016. "Nickel-Refining Fumes Induced DNA Damage and Apoptosis of NIH/3T3 Cells via Oxidative Stress" International Journal of Environmental Research and Public Health 13, no. 7: 629. https://doi.org/10.3390/ijerph13070629