Proposed Toxic and Hypoxic Impairment of a Brainstem Locus in Autism

Abstract
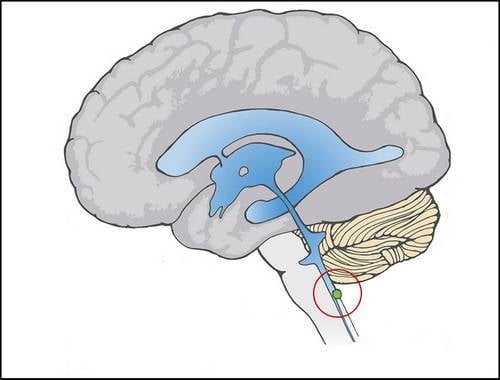
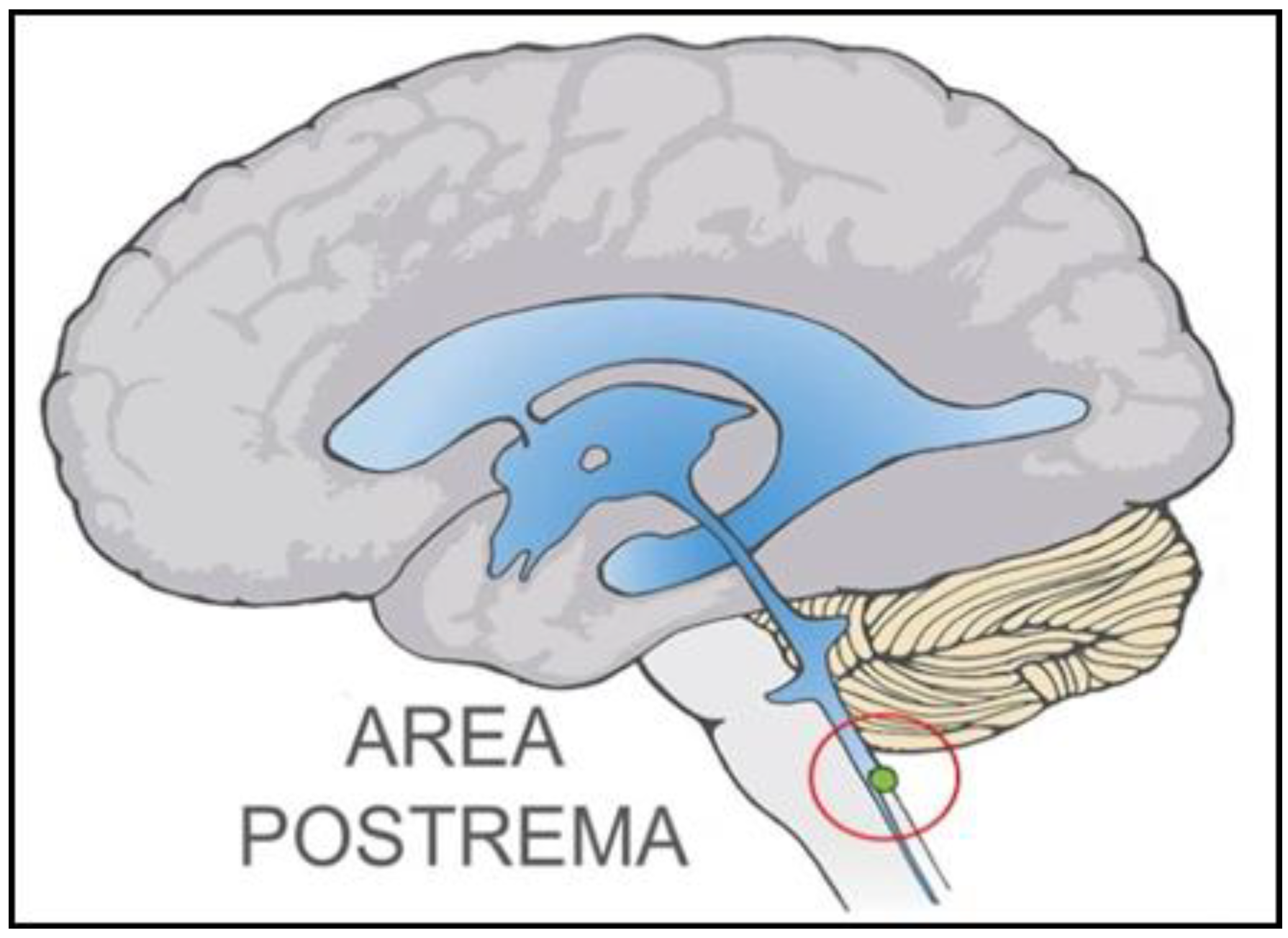
:
1. Introduction
1.1. Chelation as a Clue
2. Hypothesis
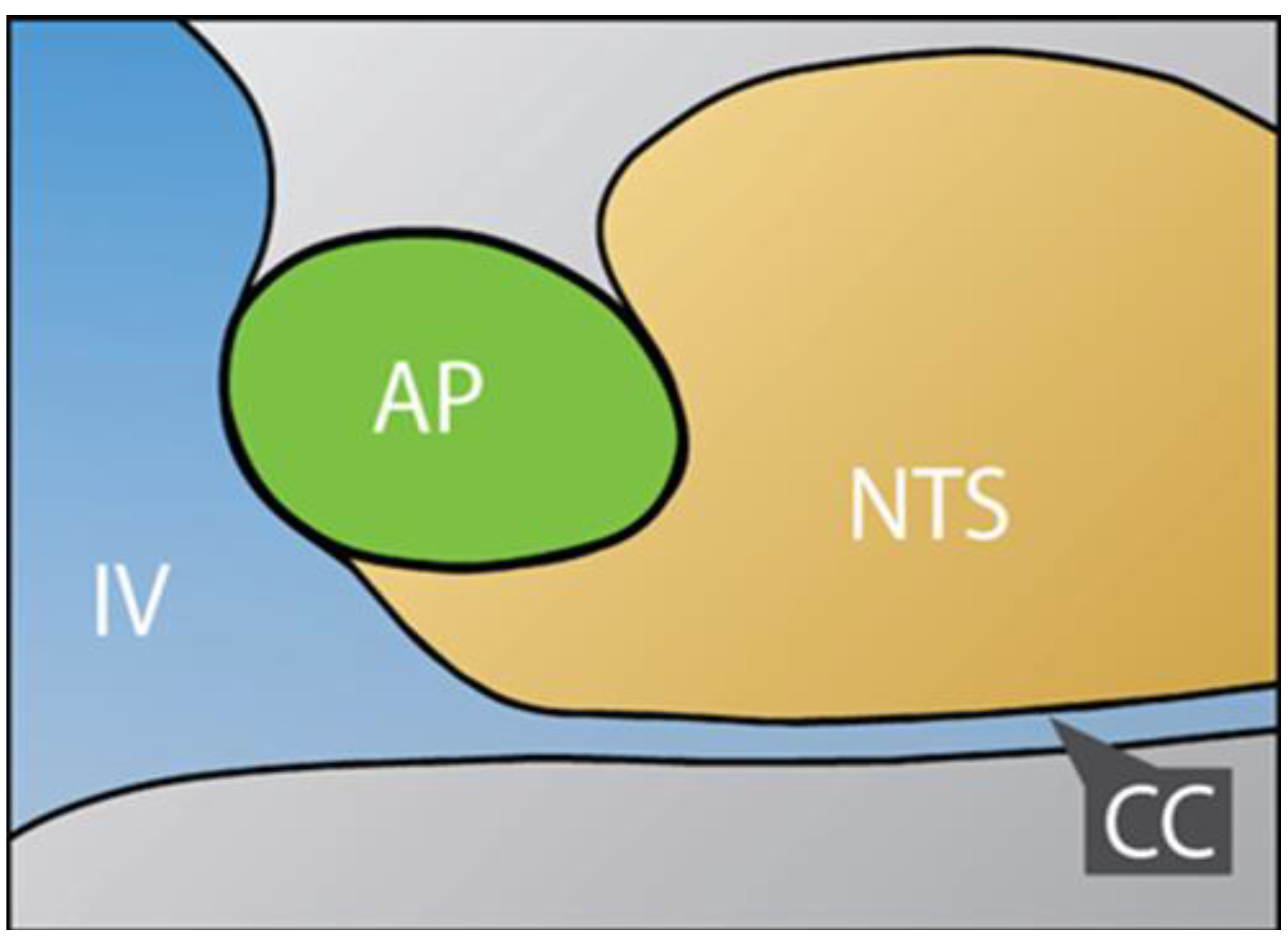
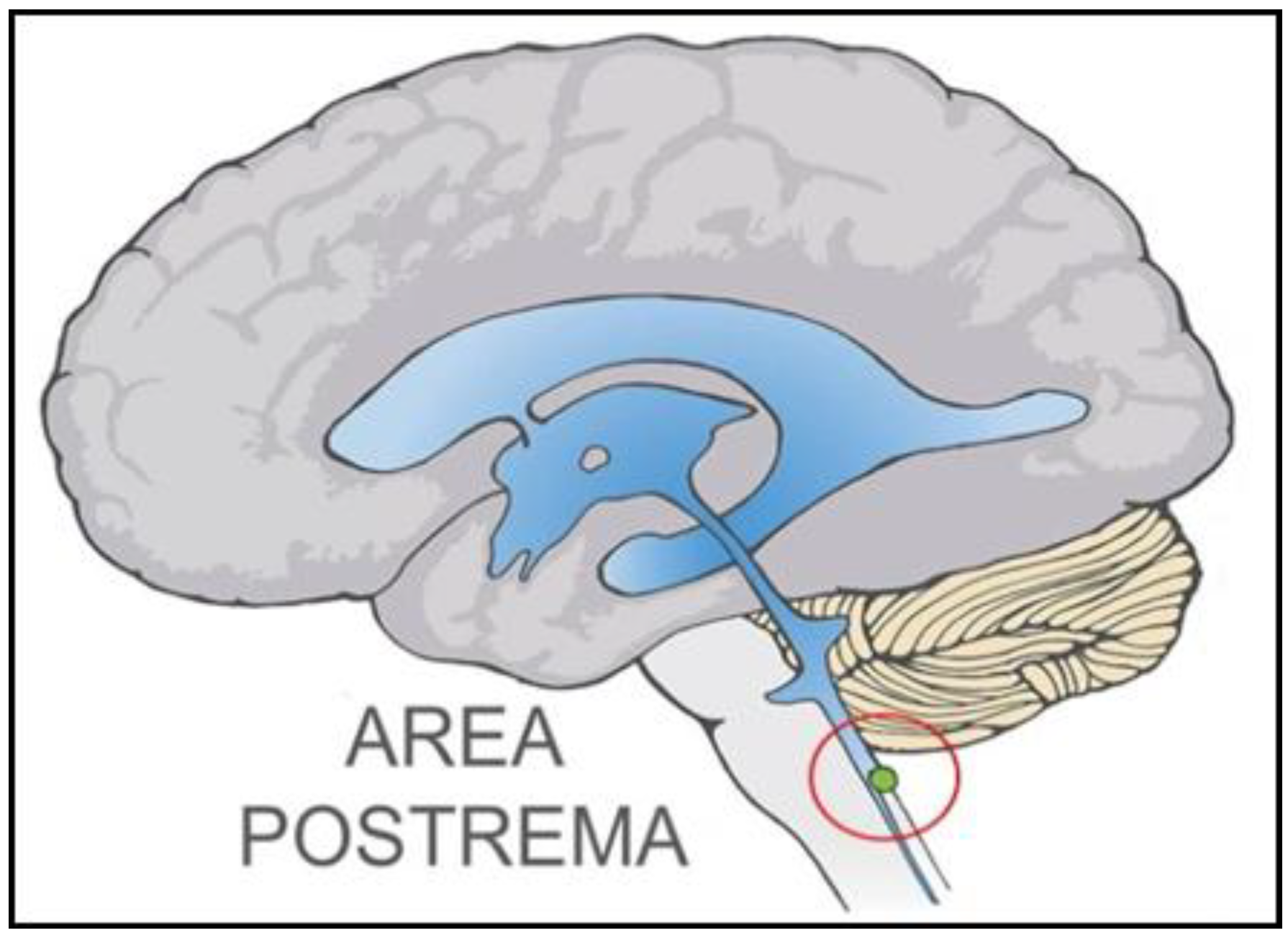
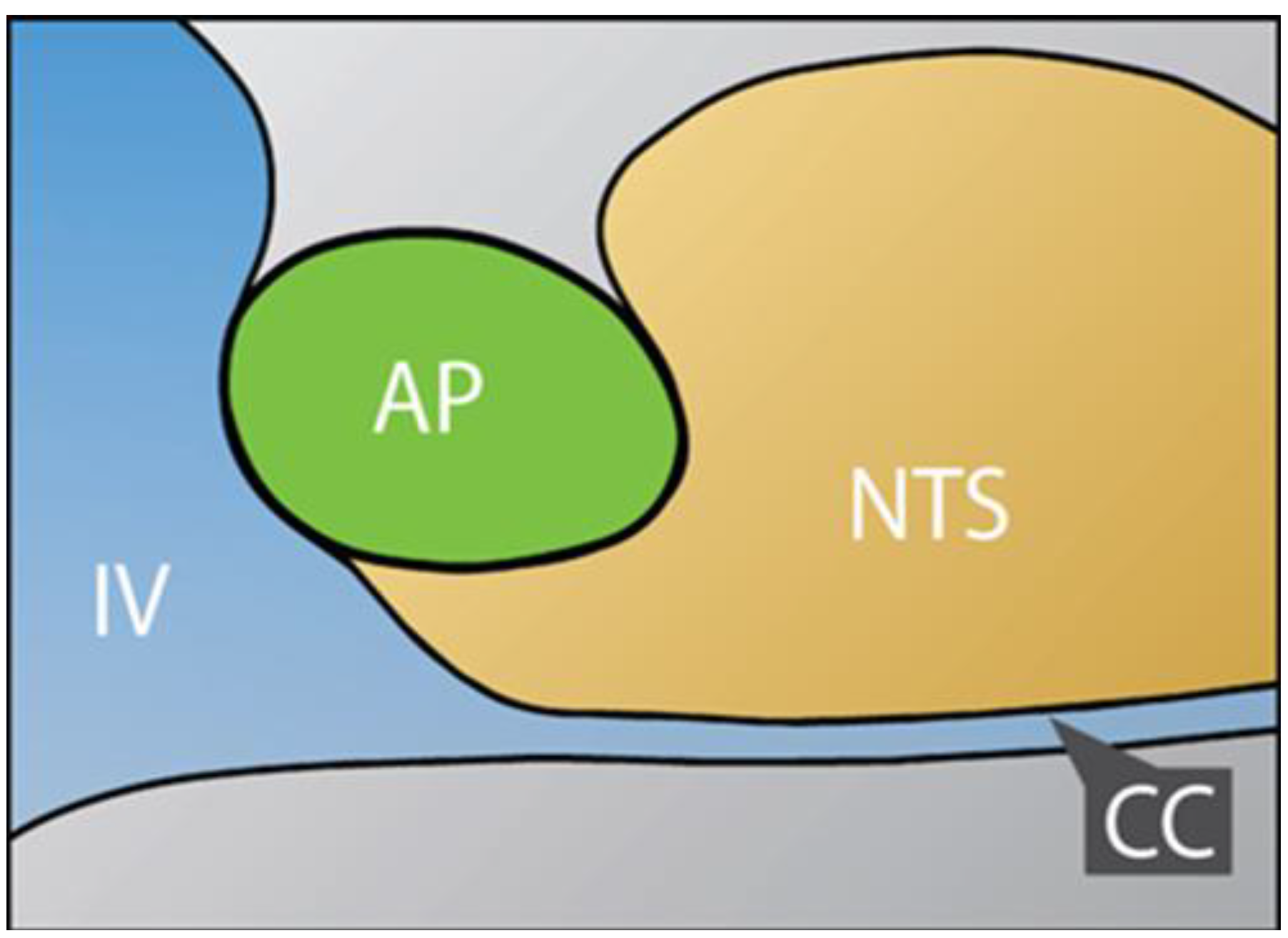
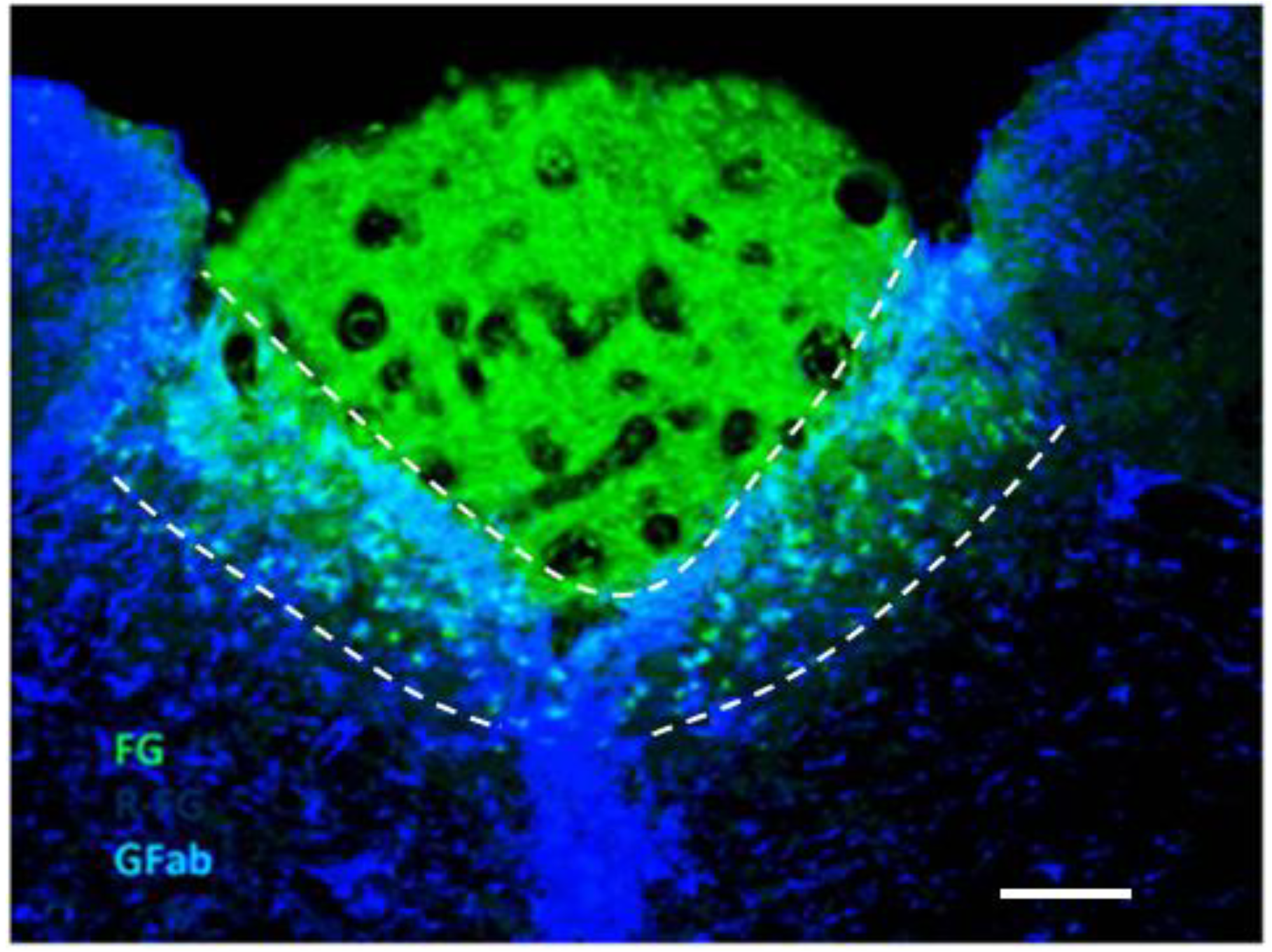
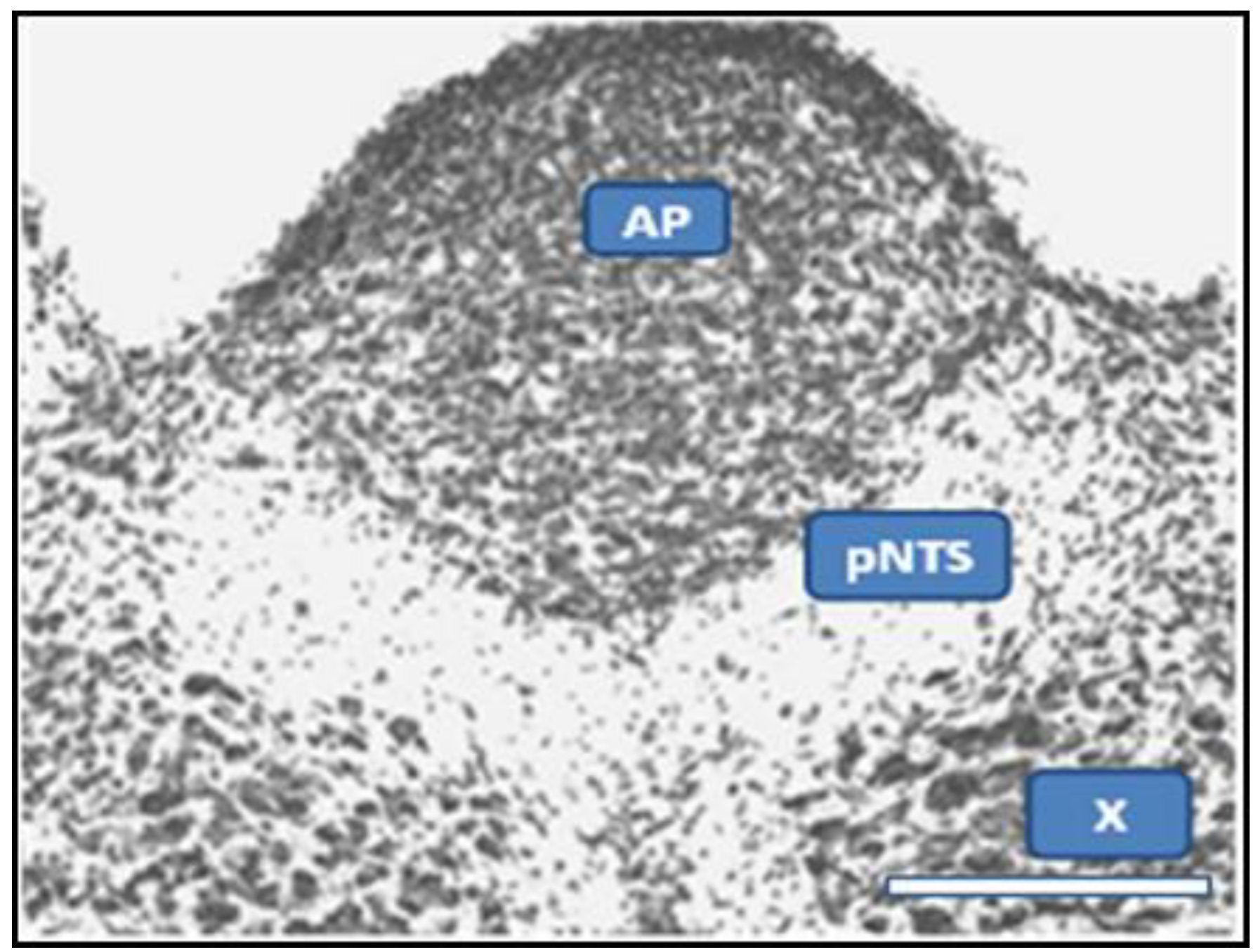
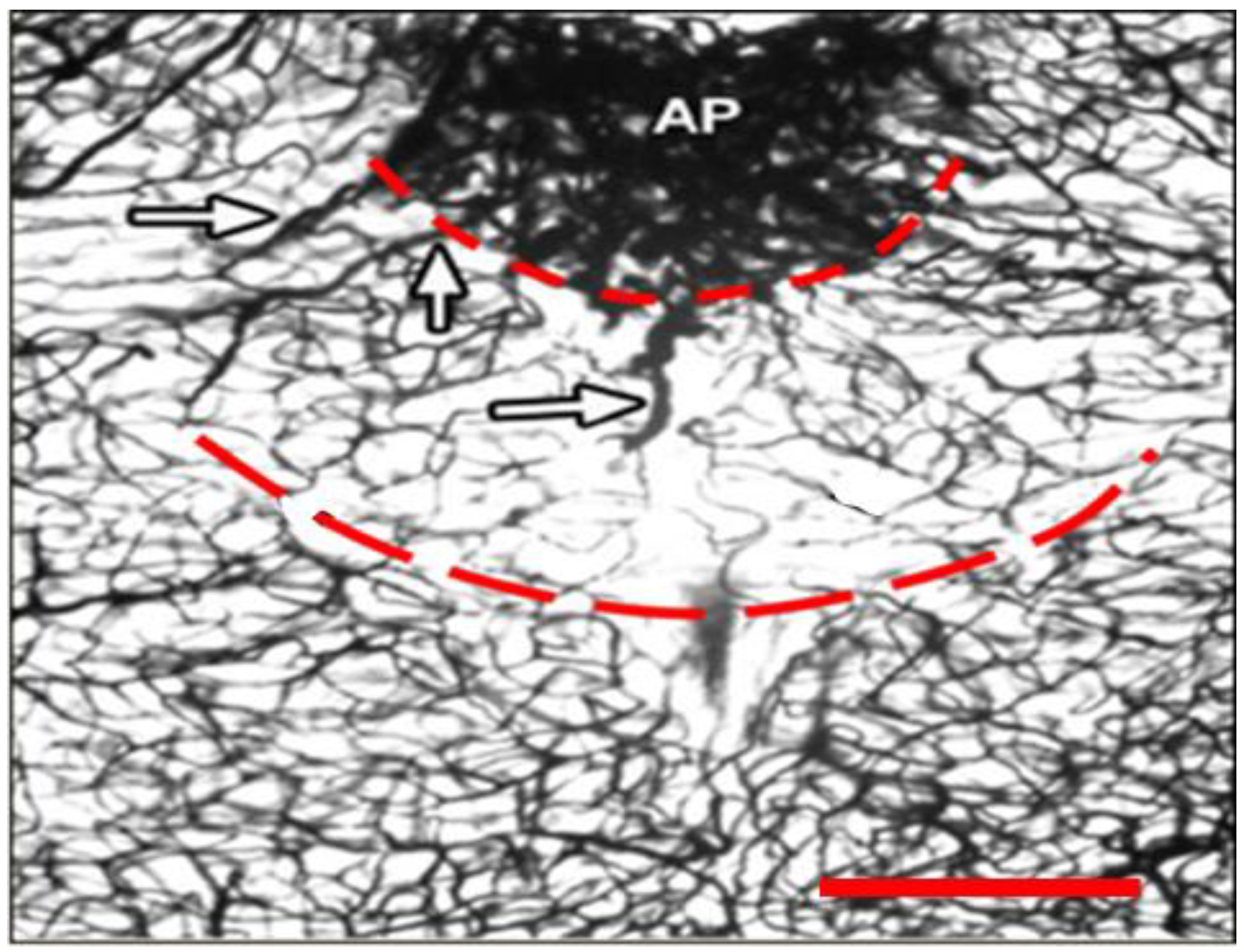
2.1. Dual Vulnerability of the NTS to Toxins and Hypoxia
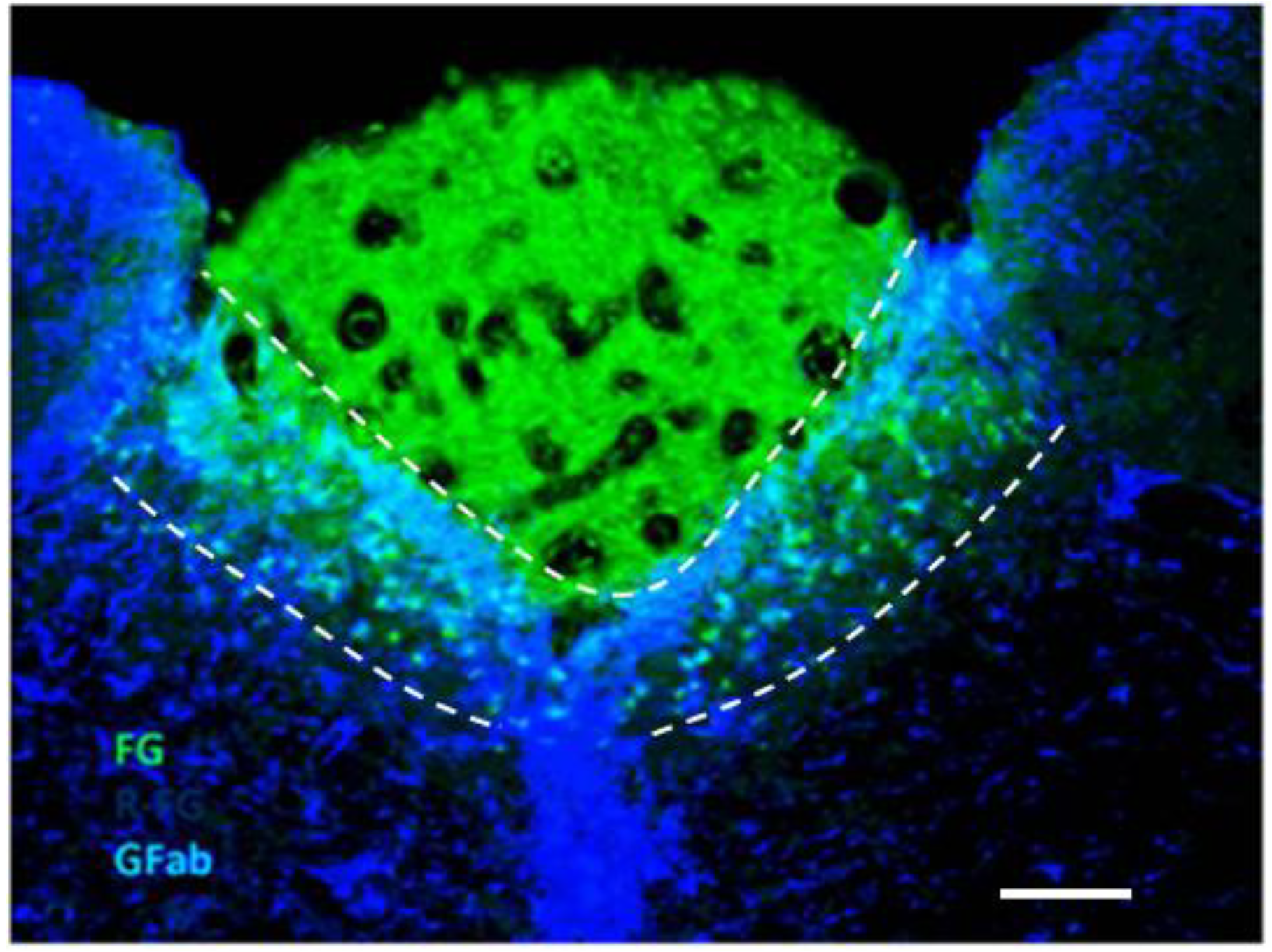
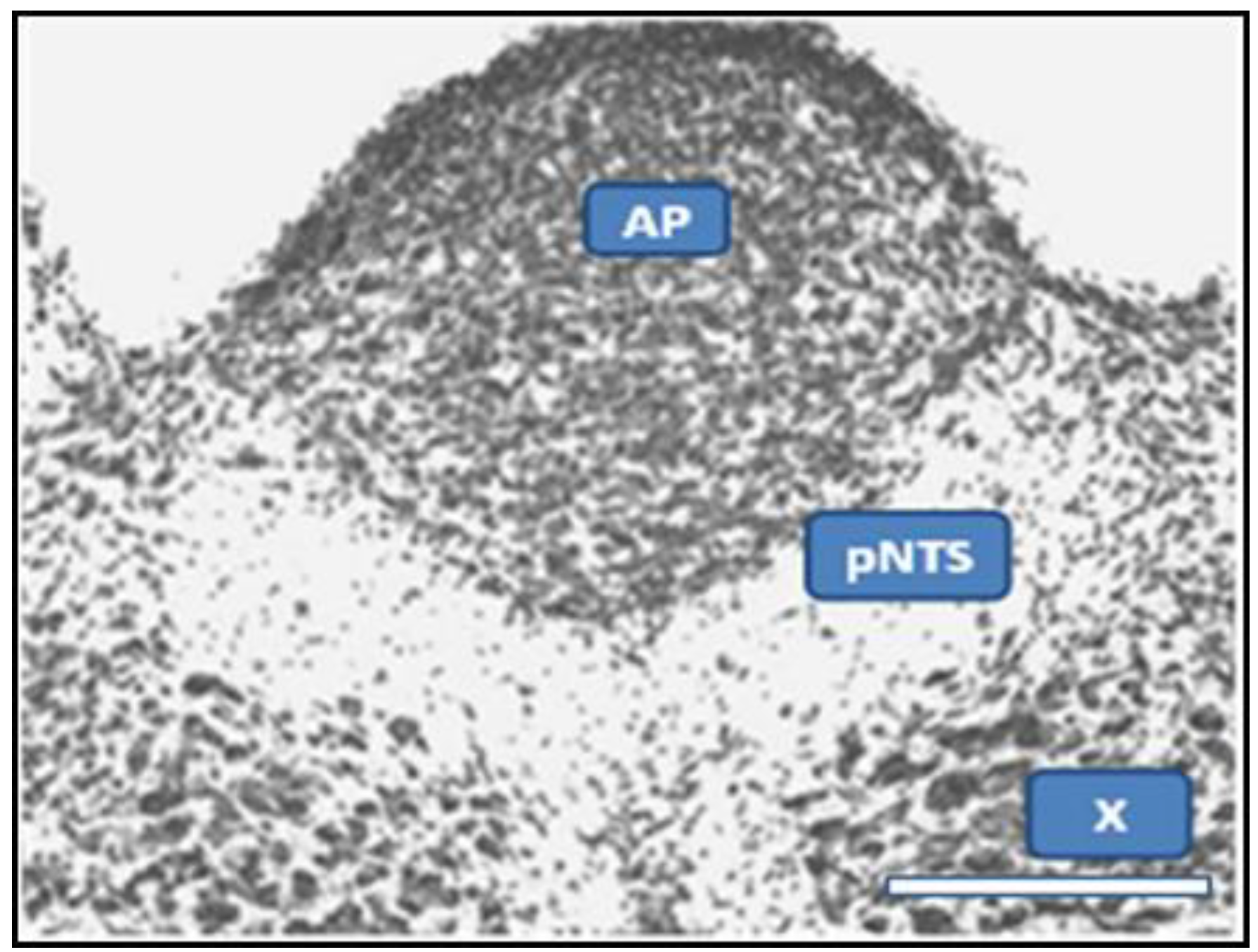
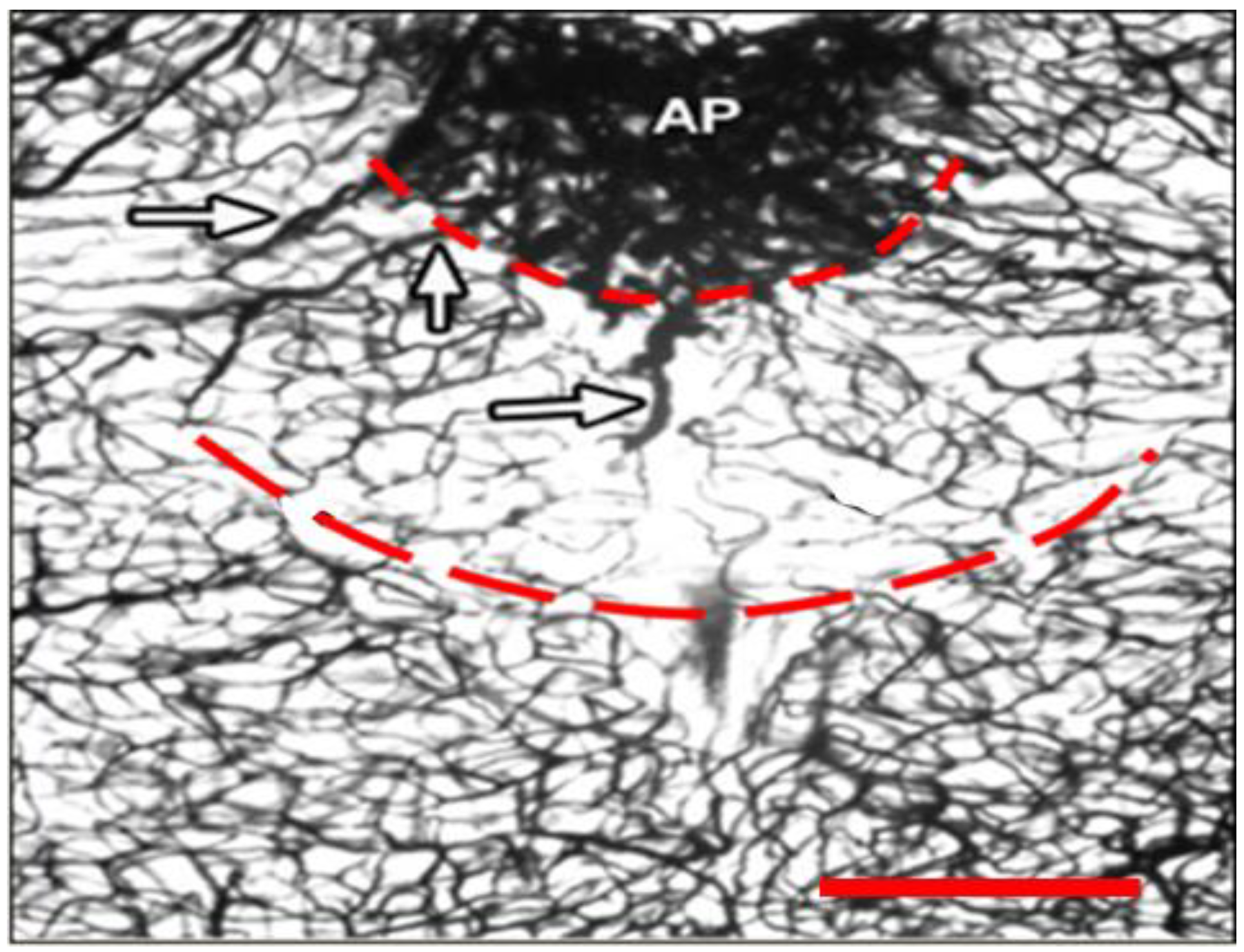
2.2. Potentiation of Hypoxia by Toxins
2.3. Abnormal Baroreflex and Associated Cardiovascular Signs in ASDs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viscera | NTS Viscerotopy | ASD Presentation |
|---|---|---|
| Baroreceptive | Dorsomedial [110,111] and Commissural [112,113] | Depressed Baroreflex [101] |
| Gastric Stretch | Dorsomedial [115] and Commissural [114] | Esophageal Reflux [120] |
| Laryngeal | “Level of AP” [116], and Commissural [117] | Altered Tone [121] and Regression-associated Whisper [10] |
| Intestinal | Commissural [114] | Retained Paneth Secretions [120] |
| Splenic | Undetermined | Inflammation [122,123,124] |
2.4. Suggestion of a Broad Role of the NTS in ASDs
3. Visceral Deafferentation Matches the ASD Phenotype
3.1. The Immune System and Deafferentation
3.2. Laryngeal Function and Deafferentation
3.3. Gastrointestinal Function and Deafferentation
| Experiment | Observation | ASD Finding |
|---|---|---|
| Electrical ablation | Baroreflex depression [157] | Depressed baroreflex [101] |
| Chemical blockade | Baroreflex depression [159] | Depressed baroreflex [101] |
| Selective A2 lesion | Blood pressure increase [160] | Higher blood pressure [101] |
| Commissural lesion | Increased water and salt intake [118] | Higher water [119] and salt [10] intake |
| Commissural stimulation | Increased cerebral blood flow [151,152] | Cerebral hypoperfusion [139,140,141] |
| Commissural lesion | Decreased autoregulation [103] | Cerebral hypoperfusion [139,140,141] |
| Commissural opioid microinjection | Blocks gastric motility and intestinal secretion [192] | Esophageal reflux and unreleased Paneth secretions [120] |
4. Cognitive Dysfunction Due to Pathology in the A2 Neurons of NTS
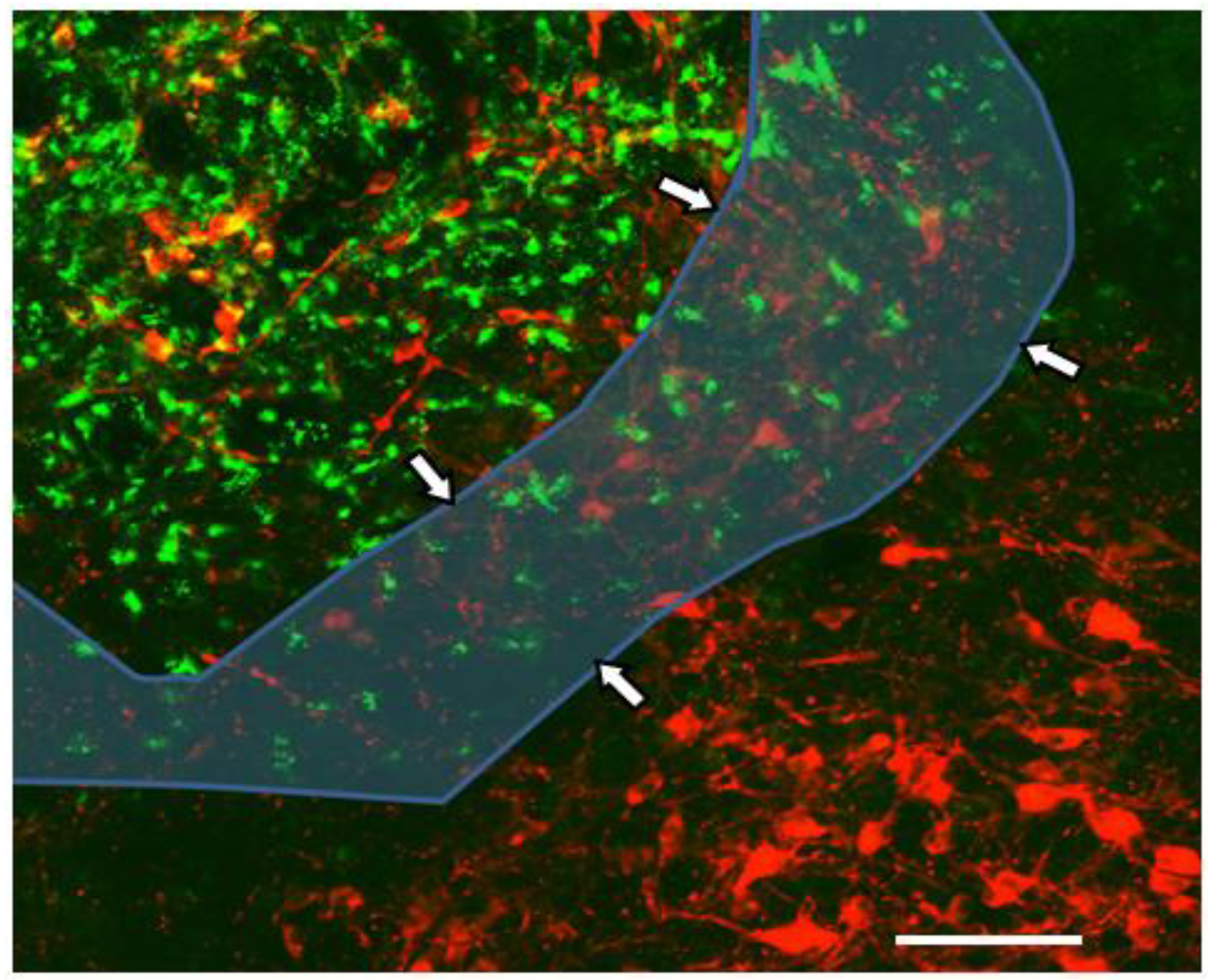
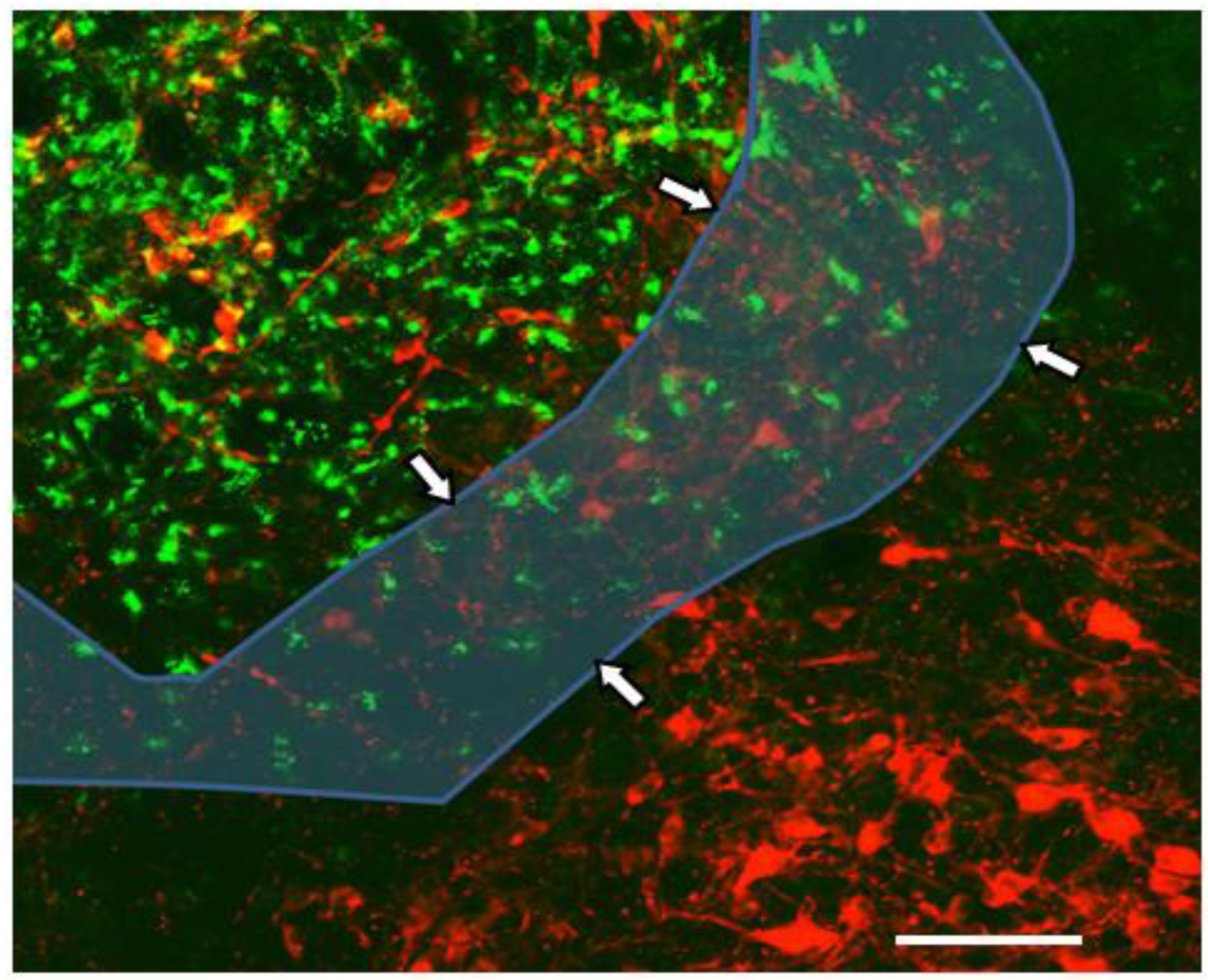
5. An Animal Model for Focal Inflammation of the NTS
6. Brain Hypoxia in ASDs
7. Research and Treatment Implications
8. Conclusions
Glossary of Abbreviations
| 5-HT | Serotonin |
| ACh | Acetylcholine |
| AChE | Acetylcholinesterase |
| ADHD | Attention Deficit Hyperactivity Disorder |
| AP | Area Postrema |
| ASD | Autism Spectrum Disorder |
| BBB | Blood-Brain Barrier |
| CBF | Cerebral Blood Flow |
| CO | Carbon Monoxide |
| CSB | Cardiac Sensitivity to Baroreflex |
| CSF | Cerebrospinal Fluid |
| CVO | Circumventricular Organ |
| CVT | Cardiac Vagal Tone |
| DBP | Diastolic Blood Pressure |
| DMSA | Dimercaptosuccinic Acid |
| DMV | Dorsal Motor Nucleus of the Vagus X |
| GCF | Glottal Closing Force |
| HBO | Hyperbaric Oxygen |
| HR | Heart Rate |
| LPS | Lipopolysaccharide/Endotoxin |
| MAP | Mean Arterial Pressure |
| MSG | Monosodium Glutamate |
| MT | Metallothionein |
| NE | Norepinephrine |
| NO | Nitric Oxide |
| NTS | Nucleus Tractus Solitarius |
| PD | Parkinson’s Disease |
| pNTS | permissive region of NTS |
| RSA | Respiratory Sinus Arrhythmia |
| SHR | Spontaneously Hypertensive Rat |
| TH | Tyrosine Hydroxylase |
| TNF | Tumor-necrosis Factor alpha |
| VNS | Vagal Nerve Stimulation |
Acknowledgements
Conflicts of Interest
References
- Windham, G.C.; Zhang, L.; Gunier, R.; Croen, L.A.; Grether, J.K. Autism spectrum disorders in relation to distribution of hazardous air pollutants in the San Francisco bay area. Environ. Health Perspect. 2006, 114, 1438–1444. [Google Scholar] [CrossRef]
- Roberts, A.L.; Lyall, K.; Hart, J.E.; Laden, F.; Just, A.C.; Bobb, J.F.; Koenen, K.C.; Ascherio, A.; Weisskopf, M.G. Perinatal air pollutant exposures and autism spectrum disorder in the children of Nurses’ Health Study II participants. Environ. Health Perspect. 2013, 121, 978–984. [Google Scholar]
- Palmer, R.F.; Blanchard, S.; Wood, R. Proximity to point sources of environmental mercury release as a predictor of autism prevalence. Health Place 2009, 15, 18–24. [Google Scholar] [CrossRef]
- Adams, J.B.; Romdalvik, J.; Ramanujam, V.M.; Legator, M.S. Mercury, lead, and zinc in baby teeth of children with autism versus controls. J. Toxicol. Environ. Health A 2007, 70, 1046–1051. [Google Scholar] [CrossRef]
- Abdullah, M.M.; Ly, A.R.; Goldberg, W.A.; Clarke-Stewart, K.A.; Dudgeon, J.V.; Mull, C.G.; Chan, T.J.; Kent, E.E.; Mason, A.Z.; Ericson, J.E. Heavy metal in children’s tooth enamel: Related to autism and disruptive behaviors? J. Autism Dev. Disord. 2012, 42, 929–936. [Google Scholar] [CrossRef]
- Farmer, J.G.; MacKenzie, A.B.; Moody, G.H. Human teeth as historical biomonitors of environmental and dietary lead: Some lessons from isotopic studies of 19th and 20th century archival material. Environ. Geochem. Health 2006, 28, 421–430. [Google Scholar] [CrossRef]
- DeSoto, M.C.; Hitlan, R.T. Blood levels of mercury are related to diagnosis of autism: A reanalysis of an important data set. J. Child Neurol. 2007, 22, 1308–1311. [Google Scholar] [CrossRef]
- Geier, D.A.; Audhya, T.; Kern, J.K.; Geier, M.R. Blood mercury levels in autism spectrum disorder: Is there a threshold level? Acta Neurobiol. Exp. (Wars.) 2010, 70, 177–186. [Google Scholar]
- Hertz-Picciotto, I.; Green, P.G.; Delwiche, L.; Hansen, R.; Walker, C.; Pessah, I.N. Blood mercury concentrations in CHARGE study children with and without autism. Environ. Health Perspect. 2010, 118, 161–166. [Google Scholar]
- McGinnis, W.R.; Miller, V.M.; Audhya, T. Neurotoxic Brainstem Impairment as Proposed Threshold Event in Autistic Regression. In Autism: Oxidative Stress, Inflammation and Immune Abnormalities; CRC Press: Boca Raton, FL, USA, 2010; pp. 153–176. [Google Scholar]
- Casanova, M.F. The neuropathology of autism. Brain Pathol. 2007, 17, 422–433. [Google Scholar] [CrossRef]
- Blaucok-Busch, E.; Amin, O.R.; Dessoki, H.H.; Rabah, T. Efficacy of DMSA therapy in a sample of Arab children with autistic spectrum disorder. Maedica (Buchar.) 2012, 7, 214–221. [Google Scholar]
- Adams, J.B.; Baral, M.; Geis, E.; Mitchell, J.; Ingram, J.; Hensley, A.; Zappia, I.; Newmark, S.; Gehn, E.; Rubin, R.A.; et al. Safety and efficacy of oral DMSA therapy for children with autism spectrum disorders: Part A—Medical results. BMC Clin. Pharmacol. 2009, 9, 16. [Google Scholar] [CrossRef]
- Geier, D.A.; Geier, M.R. A clinical trial of combined anti-androgen and anti-heavy metal therapy in autistic disorders. Neuroendocrinol. Lett. 2006, 27, 833–838. [Google Scholar]
- Flora, S.J.; Pachauri, V. Chelation in metal intoxication. Int. J. Environ. Res. Public Health 2010, 7, 2745–2788. [Google Scholar] [CrossRef]
- Adams, J.; Baral, M.; Geis, E.; Mitchell, J.; Ingram, J.; Hensley, A.; Zappia, I.; Newmark, S.; Gehn, E.; Rubin, R.; Mitchell, K.; et al. Safety and efficacy of oral DMSA therapy for children with autism spectrum disorders: Part B—Behavioral results. BMC Clin. Pharmacol. 2009, 9, 17. [Google Scholar] [CrossRef]
- Adams, J.B.; Audhya, T.; McDonough-Means, S.; Rubin, R.; Quig, D.; Geis, E.; Gehn, E.; Loresto, M.; Mitchell, J.; Atwood, S.; et al. Toxicological status of children with autism vs. neurotypical children and the association with autism severity. Biol. Trace Elem. Res. 2013, 151, 171–180. [Google Scholar] [CrossRef]
- Walker, E.M., Jr.; Gale, G.R.; Fody, E.P.; Atkins, L.M.; Smith, A.B.; Jones, M.M. Comparative antidotal effects of diethyldithiocarbamate, dimercaptosuccinate, and diethylenetriaminepentaacetate against cadmium-induced testicular toxicity in mice. Res. Commun. Chem. Pathol. Pharmacol. 1986, 51, 231–244. [Google Scholar]
- McGinnis, W.R. Oxidative stress in autism. Altern. Ther. Health Med. 2004, 10, 22–36. [Google Scholar]
- Russo, A.J. Anti-metallothionein IgG and levels of metallothionein in autistic children with GI disease. Drug Health Patient Saf. 2009, 1, 1–8. [Google Scholar] [CrossRef]
- Andersen, O. Oral cadmium exposure in mice: Toxicokinetics and efficiency of chelating agents. Crit. Rev. Toxicol. 1989, 20, 83–112. [Google Scholar] [CrossRef]
- Chandra, S.V.; Murthy, R.C.; Husain, T.; Bansal, S.K. Effect of interaction of heavy metals on (Na+ -K+) ATPase and the uptake of 3H-DA and 3H-NA in rat brain synaptosomes. Acta Pharmacol. Toxicol. (copenh.) 1984, 54, 210–213. [Google Scholar]
- Ali, M.M.; Murthy, R.C.; Chandra, S.V. Developmental and longterm neurobehavioral toxicity of low level in-utero cadmium exposure in rats. Neurobehav. Toxicol. Teratol. 1986, 8, 463–468. [Google Scholar]
- Nation, J.R.; Clark, D.E.; Bourgeois, A.E.; Baker, D.M. The effects of chronic cadmium exposure on schedule controlled responding and conditioned suppression in the adult rat. Neurobehav. Toxicol. Teratol. 1983, 5, 275–282. [Google Scholar]
- Murthy, R.C.; Saxena, D.K.; Sunderaraman, V.; Chandra, S.V. Cadmium induced ultrastructural changes in the cerebellum of weaned and adult rats. Ind. Health 1987, 25, 159–162. [Google Scholar] [CrossRef]
- Arvidson, B. Autoradiographic localization of cadmium in the rat brain. Neurotoxicology 1986, 7, 89–96. [Google Scholar]
- Arvidson, B. Accumulation of inorganic mercury in lower motoneurons of mice. Neurotoxicology 1992, 13, 277–280. [Google Scholar]
- Nordberg, G.F.; Serenius, F. Distribution of inorganic mercury in the guinea pig brain. Acta Pharmacol. Toxicol. (Copenh.) 1969, 27, 269–283. [Google Scholar]
- Vahter, M.; Mottet, N.K.; Friberg, L.; Lind, B.; Shen, D.D.; Burbacher, T. Speciation of mercury in the primate blood and brain following long-term exposure to methyl mercury. Toxicol. Appl. Pharm. 1994, 124, 221–229. [Google Scholar] [CrossRef]
- Waalkes, M.P.; Misra, R.R. Toxicology of Metals; Chang, L.W., Magos, L., Suzuki, T., Eds.; CRC Press, Inc.: Boca Raton, FL, USA, 2013. [Google Scholar]
- Madsen, K.M.; Vestergaard, M. MMR vaccination and autism: What is the evidence for a causal association? Drug Saf. 2004, 27, 831–840. [Google Scholar] [CrossRef]
- Allen Institute for Brain Science. Allen Brain Atlas: Mouse Brain. Available online: http://mouse.brain-map.org/ (accessed on 14 May 2013).
- Leong, S.; Ashwell, K.W. Is there a zone of vascular vulnerability in the fetal brain stem? Neurotoxicol. Teratol. 1997, 19, 265–275. [Google Scholar] [CrossRef]
- de Caro, R.; Parenti, A.; Montisci, M.; Guidolin, D.; Macchi, V. Solitary tract nuclei in acute heart failure. Stroke 2000, 31, 1187–1193. [Google Scholar] [CrossRef]
- Parenti, A.; Macchi, V.; Snenghi, R.; Porzionato, A.; Scaravilli, T.; Ferrara, S.D.; DeCaro, R. Selective stroke of the solitary tract nuclei in two cases of central sleep apnoea. Clin. Neuropathol. 2005, 24, 239–246. [Google Scholar]
- Gardener, H.; Spiegelman, D.; Buka, S.L. Prenatal risk factors for autism: Comprehensive meta-analysis. Br. J. Psychiatry 2009, 195, 7–14. [Google Scholar] [CrossRef]
- Sampson, M.B.; Mudaliar, N.A.; Lele, A.S. Fetal heart rate variability as an indicator of fetal status. Postgrad. Med. 1980, 67, 207–210, 213–215. [Google Scholar]
- Walker, C.K.; Krakowiak, P.; Baker, A.S.; Hansen, R.L.; Ozonoff, S.; Hertz-Picciotto, I. The Role of Preeclampsia in Autism Spectrum Disorders and Cognitive Function. In Proceedings of the International Meeting for Autism Research, Toronto, ON, Canada, 18 May 2012.
- Burstyn, I.; Wang, X.; Yasui, Y.; Sithole, F.; Zwaigenbaum, L. Autism spectrum disorders and fetal hypoxia in a population-based cohort: Accounting for missing exposures via estimation-maximization algorithm. BMC Med. Res. Methodol. 2011, 11, 2. [Google Scholar] [CrossRef]
- Badawi, N.; Dixon, G.; Felix, J.F.; Keogh, J.M.; Petterson, B.; Stanley, F.J.; Kurinczuk, J.J. Autism following a history of newborn encephalopathy: More than a coincidence? Dev. Med. Child Neurol. 2006, 48, 85–89. [Google Scholar] [CrossRef]
- Gillberg, I.C. Autistic syndrome with onset at age 31 years: Herpes encephalitis as a possible model for childhood autism. Dev. Med. Child Neurol. 1991, 33, 920–924. [Google Scholar] [CrossRef]
- Gillberg, C. Onset at age 14 of a typical autistic syndrome. A case report of a girl with herpes simplex encephalitis. J. Autism Dev. Disord. 1986, 16, 369–375. [Google Scholar] [CrossRef]
- Rodier, P.M. The early origins of autism. Sci. Amer. 2000, 282, 56–63. [Google Scholar] [CrossRef]
- Rodier, P.M.; Ingram, J.L.; Tisdale, B.; Croog, V.J. Linking etiologies in humans and animal models: Studies of autism. Reprod. Toxicol. 1997, 11, 417–422. [Google Scholar] [CrossRef]
- Sato, K.; Momose-Sato, Y.; Hirota, A.; Sakai, T.; Kamino, K. Optical mapping of neural responses in the embryonic rat brainstem with reference to the early functional organization of vagal nuclei. J. Neurosci. 1998, 18, 1345–1362. [Google Scholar]
- Suren, P.; Roth, C.; Bresnahan, M.; Haugen, M.; Hornig, M.; Hirtz, D.; Lie, K.K.; Lipkin, W.I.; Magnus, P.; Reichborn-Kjennerud, T.; et al. Association between maternal use of folic acid supplements and risk of autism spectrum disorders in children. JAMA 2013, 309, 570–577. [Google Scholar] [CrossRef]
- Schmidt, R.J.; Tancredi, D.J.; Ozonoff, S.; Hansen, R.L.; Hartiala, J.; Allayee, H.; Schmidt, L.C.; Tassone, F.; Hertz-Picciotto, I. Maternal periconceptional folic acid intake and risk of autism spectrum disorders and developmental delay in the CHARGE (CHildhood Autism Risks from Genetics and Environment) case-control study. Amer. J. Clin. Nutr. 2012, 96, 80–89. [Google Scholar] [CrossRef]
- Hallene, K.L.; Oby, E.; Lee, B.J.; Santaguida, S.; Bassanini, S.; Cipolla, M.; Marchi, N.; Hossain, M.; Battaglia, G.; Janigro, D. Prenatal exposure to thalidomide, altered vasculogenesis, and CNS malformations. Neuroscience 2006, 142, 267–283. [Google Scholar] [CrossRef]
- Tamilarasan, K.P.; Kolluru, G.K.; Rajaram, M.; Indhumathy, M.; Saranya, R.; Chatterjee, S. Thalidomide attenuates nitric oxide mediated angiogenesis by blocking migration of endothelial cells. BMC Cell Biol. 2006, 7, 17. [Google Scholar] [CrossRef]
- Gross, P.M.; Wall, K.M.; Pang, J.J.; Shaver, S.W.; Wainman, D.S. Microvascular specializations promoting rapid interstitial solute dispersion in nucleus tractus solitarius. Amer. J. Physiol. 1990, 259, R1131–R1138. [Google Scholar]
- Gross, P.M.; Wall, K.M.; Wainman, D.S.; Shaver, S.W. Subregional topography of capillaries in the dorsal vagal complex of rats: II. Physiological properties. J. Comp. Neurol. 1991, 306, 83–94. [Google Scholar] [CrossRef]
- Breder, C.D.; Hazuka, C.; Ghayur, T.; Klug, C.; Huginin, M.; Yasuda, K.; Teng, M.; Saper, C.B. Regional induction of tumor necrosis factor alpha expression in the mouse brain after systemic lipopolysaccharide administration. Proc. Natl. Acad. Sci. USA 1994, 91, 11393–11397. [Google Scholar] [CrossRef]
- Hyde, T.M.; Miselis, R.R. Subnuclear organization of the human caudal nucleus of the solitary tract. Brain Res. Bull. 1992, 29, 95–109. [Google Scholar] [CrossRef]
- Maolood, N.; Meister, B. Protein components of the blood-brain barrier (BBB) in the brainstem area postrema-nucleus tractus solitarius region. J. Chem. Neuroanat. 2009, 37, 182–195. [Google Scholar] [CrossRef]
- Shaver, S.W.; Pang, J.J.; Wall, K.M.; Sposito, N.M.; Gross, P.M. Subregional topography of capillaries in the dorsal vagal complex of rats: I. Morphometric properties. J. Comp. Neurol. 1991, 306, 73–82. [Google Scholar] [CrossRef]
- Fodor, M.; Palkovits, M.; Gallatz, K. Fine structure of the area subpostrema in rat. Open gate for the medullary autonomic centers. Ideggyogy. Sz. 2007, 60, 83–88. [Google Scholar]
- Broadwell, R.D.; Balin, B.J.; Salcman, M.; Kaplan, R.S. Brain-blood barrier? Yes and no. Proc. Natl. Acad. Sci. USA 1983, 80, 7352–7356. [Google Scholar] [CrossRef]
- Hu, L.; Fernstrom, J.D.; Goldsmith, P.C. Exogenous glutamate enhances glutamate receptor subunit expression during selective neuronal injury in the ventral arcuate nucleus of postnatal mice. Neuroendocrinology 1998, 68, 77–88. [Google Scholar] [CrossRef]
- Jaszai, J.; Farkas, L.M.; Gallatz, K.; Palkovits, M. Effects of glutamate-induced excitotoxicity on calretinin-expressing neuron populations in the area postrema of the rat. Cell Tissue Res. 1998, 293, 227–233. [Google Scholar] [CrossRef]
- Karcsu, S.; Toth, L.; Jancso, G.; Poberai, M. (Na-glutamate sensitive neurons in the area postrema of the rat (author’s transl)). Acta Histochem. 1981, 68, 181–187. [Google Scholar] [CrossRef]
- Karcsu, S.; Jancso, G.; Kreutzberg, G.W.; Toth, L.; Kiraly, E.; Bacsy, E.; Laszlo, F.A. A glutamate-sensitive neuronal system originating from the area postrema terminates in and transports acetylcholinesterase to the nucleus of the solitary tract. J. Neurocytol. 1985, 14, 563–578. [Google Scholar] [CrossRef]
- Porzionato, A.; Macchi, V.; Morsut, L.; Parenti, A.; de Caro, R. Microvascular patterns in human medullary tegmentum at the level of the area postrema. J. Anat. 2005, 206, 405–410. [Google Scholar] [CrossRef]
- Choi, D.W.; Rothman, S.M. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu. Rev. Neurosci. 1990, 13, 171–182. [Google Scholar] [CrossRef]
- Aicher, S.A.; Sharma, S.; Pickel, V.M. N-methyl-d-aspartate receptors are present in vagal afferents and their dendritic targets in the nucleus tractus solitarius. Neuroscience 1999, 91, 119–132. [Google Scholar] [CrossRef]
- Sarnat, H.B. Watershed infarcts in the fetal and neonatal brainstem. An aetiology of central hypoventilation, dysphagia, Moibius syndrome and micrognathia. Eur. J. Paediatr. Neurol. 2004, 8, 71–87. [Google Scholar] [CrossRef]
- Liu, Q.; Wong-Riley, M.T. Postnatal changes in cytochrome oxidase expressions in brain stem nuclei of rats: Implications for sensitive periods. J. Appl. Physiol. 2003, 95, 2285–2291. [Google Scholar]
- Porzionato, A.; Macchi, V.; Parenti, A.; de Caro, R. The distribution of mast cells in the human area postrema. J. Anat. 2004, 204, 141–147. [Google Scholar] [CrossRef]
- Roth, G.I.; Yamamoto, W.S. The microcirculation of the area postrema in the rat. J. Comp. Neurol. 1968, 133, 329–340. [Google Scholar] [CrossRef]
- Rohrer, S.R.; Shaw, S.M.; Lamar, C.H. Cadmium induced endothelial cell alterations in the fetal brain from prenatal exposure. ACTA Neuropathol. 1978, 44, 147–149. [Google Scholar] [CrossRef]
- Webster, W.S.; Valois, A.A. The toxic effects of cadmium on the neonatal mouse CNS. J. Neuropathol. Exp. Neurol. 1981, 40, 247–257. [Google Scholar] [CrossRef]
- Lee, T.J.; Chang, H.H.; Lee, H.C.; Chen, P.Y.; Lee, Y.C.; Kuo, J.S.; Chen, M.F. Axo-axonal interaction in autonomic regulation of the cerebral circulation. ACTA Physiol. 2011, 203, 25–35. [Google Scholar] [CrossRef]
- Gwyn, D.G.; Wolstencroft, J.H. Cholinesterases in the area subpostrema. A region adjacent to the area postrema in the cat. J. Comp. Neurol. 1968, 133, 289–308. [Google Scholar] [CrossRef]
- Buller, K.M.; Wixey, J.A.; Pathipati, P.; Carty, M.; Colditz, P.B.; Williams, C.E.; Scheepens, A. Selective losses of brainstem catecholamine neurons after hypoxia-ischemia in the immature rat pup. Pediatr. Res. 2008, 63, 364–369. [Google Scholar] [CrossRef]
- Frasco, M.F.; Colletier, J.P.; Weik, M.; Carvalho, F.; Guilhermino, L.; Stojan, J.; Fournier, D. Mechanisms of cholinesterase inhibition by inorganic mercury. FEBS J. 2007, 274, 1849–1861. [Google Scholar] [CrossRef]
- Pari, L.; Murugavel, P. Diallyl tetrasulfide improves cadmium induced alterations of acetylcholinesterase, ATPases and oxidative stress in brain of rats. Toxicology 2007, 234, 44–50. [Google Scholar] [CrossRef]
- Wolf, M.B.; Baynes, J.W. Cadmium and mercury cause an oxidative stress-induced endothelial dysfunction. Biometals 2007, 20, 73–81. [Google Scholar] [CrossRef]
- Hassan, H.A.; Hafez, H.S.; Zeghebar, F.E. Garlic oil as a modulating agent for oxidative stress and neurotoxicity induced by sodium nitrite in male albino rats. Food Chem. Toxicol. 2010, 48, 1980–1985. [Google Scholar] [CrossRef]
- Gruener, N.; Shuval, H.I.; Behroozi, K.; Cohen, S.; Shechter, H. Methemoglobinemia induced by transplacental passage of nitrites in rats. Bull. Environ. Contam. Toxicol. 1973, 9, 44–48. [Google Scholar] [CrossRef]
- Slanina, L.; Slivka, P.; Struharikova, J. Transmammary transfer of nitrates and nitrites in ruminants and methemoglobin blood levels in the young and their mothers. Vet. Med. (Praha.) 1990, 35, 647–656. [Google Scholar]
- Shuval, H.I.; Gruener, N. Epidemiological and toxicological aspects of nitrates and nitrites in the environment. Amer. J. Public Health 1972, 62, 1045–1052. [Google Scholar] [CrossRef]
- Zoroglu, S.S.; Yurekli, M.; Meram, I.; Sogut, S.; Tutkun, H.; Yetkin, O.; Sivasli, E.; Savas, H.A.; Yanik, M.; Herken, H.; et al. Pathophysiological role of nitric oxide and adrenomedullin in autism. Cell Biochem. Funct. 2003, 21, 55–60. [Google Scholar] [CrossRef]
- Sweeten, T.L.; Bowyer, S.L.; Posey, D.J.; Halberstadt, G.M.; McDougle, C.J. Increased prevalence of familial autoimmunity in probands with pervasive developmental disorders. Pediatrics 2003, 112. [Google Scholar] [CrossRef]
- Sogut, S.; Zoroglu, S.S.; Ozyurt, H.; Yimaz, H.R.; Ozugurlu, F.; Sivasli, E.; Yetkin, O.; Yanik, M.; Tutkun, H.; Savas, H.A.; et al. Changes in nitric oxide levels and antioxidant enzyme activities may have a role in the pathophysiological mechanisms involved in autism. Clin. Chim. ACTA 2003, 331, 111–117. [Google Scholar] [CrossRef]
- Agency for Toxic Substances and Diseases Registry. Case Studies in Environmental Medicine: Nitrate/Nitrite Toxicity. January 2001. Available online: http://www.atsdr.cdc.gov/HEC/CSEM/nitrate/index.html (accessed 14 May 2013).
- Finegold, S.M. Desulfovibrio species are potentially important in regressive autism. Med. Hypotheses 2011, 77, 270–274. [Google Scholar] [CrossRef]
- Leopoldini, M.; Russo, N.; Toscano, M.; Dulak, M.; Wesolowski, T.A. Mechanism of nitrate reduction by Desulfovibrio desulfuricans nitrate reductase—A theoretical investigation. Chemistry 2006, 12, 2532–2541. [Google Scholar] [CrossRef]
- Volk, H.E.; Hertz-Picciotto, I.; Delwiche, L.; Lurmann, F.; McConnell, R. Residential proximity to freeways and autism in the CHARGE study. Environ. Health Perspect. 2011, 119, 873–877. [Google Scholar]
- Donnay, A. Toxicology Doctoral Program. Personal Communication, Department of Life Sciences, University of Maryland: Baltimore, MD, USA, 13 March 2012. [Google Scholar]
- Boussouar, A.; Araneda, S.; Hamelin, C.; Soulage, C.; Kitahama, K.; Dalmaz, Y. Prenatal ozone exposure abolishes stress activation of Fos and tyrosine hydroxylase in the nucleus tractus solitarius of adult rat. Neurosci. Lett. 2009, 452, 75–78. [Google Scholar] [CrossRef]
- Zhang, X.; Yao, W.; Jia, B.; Sun, D.; Ka, W.; He, D.; Wang, X.; Wen, Z. Acute dichlorvos poisoning induces hemorheological abnormalities in rabbits via oxidative stress. Clin. Hemorheol. Microcirc. 2010, 44, 207–216. [Google Scholar]
- Chung, T.W.; Ho, C.P. Changes in viscosity of low shear rates and viscoelastic properties of oxidative erythrocyte suspensions. Clin. Hemorheol. Microcirc. 1999, 21, 99–103. [Google Scholar]
- Shin, S.; Ku, Y.; Babu, N.; Singh, M. Erythrocyte deformability and its variation in diabetes mellitus. Indian J. Exp. Biol. 2007, 45, 121–128. [Google Scholar]
- Ghezzo, A.; Visconti, P.; Abruzzo, P.M.; Bolotta, A.; Ferreri, C.; Gobbi, G.; Malisardi, G.; Manfredini, S.; Marini, M.; Nanetti, L.; Pipitone, E.; Raffaelli, F.; Resca, F.; Vignini, A.; Mazzanti, L. Oxidative stress and erythrocyte membrane alterations in children with autism: correlation with clinical features. PLoS One 2013, 8. [Google Scholar] [CrossRef]
- Reneman, R.S.; Arts, T.; Hoeks, A.P. Wall shear stress—An important determinant of endothelial cell function and structure—in the arterial system in vivo. Discrepancies with theory. J. Vasc. Res. 2006, 43, 251–269. [Google Scholar] [CrossRef]
- Curran, L.K.; Newschaffer, C.J.; Lee, L.C.; Crawford, S.O.; Johnston, M.V.; Zimmerman, A.W. Behaviors associated with fever in children with autism spectrum disorders. Pediatrics 2007, 120, e1386–e1392. [Google Scholar] [CrossRef]
- Lominadze, D.; Tsakadze, N.; Sen, U.; Falcone, J.C.; D’Souza, S.E. Fibrinogen and fragment D-induced vascular constriction. Amer. J. Physiol.-Heart Circ. Phy. 2005, 288, H1257–H1264. [Google Scholar]
- Yao, Y.; Walsh, W.J.; McGinnis, W.R.; Pratico, D. Altered vascular phenotype in autism: Correlation with oxidative stress. Arch. Neurol. 2006, 63, 1161–1164. [Google Scholar] [CrossRef]
- Biaggioni, I.; Whetsell, W.O.; Jobe, J.; Nadeau, J.H. Baroreflex failure in a patient with central nervous system lesions involving the nucleus tractus solitarii. Hypertension 1994, 23, 491–495. [Google Scholar] [CrossRef]
- Germano, D.; Pochiero, M.; Romeo, G.; Nunziata, A.; Costa, G.; Caputi, A.P. Cadmium alters arterial baroreflex control of heart rate in the conscious rat. Arch. Toxicol. Suppl. 1984, 7, 374–377. [Google Scholar] [CrossRef]
- Carmignani, M.; Finelli, V.N.; Boscolo, P. Mechanisms in cardiovascular regulation following chronic exposure of male rats to inorganic mercury. Toxicol. Appl. Pharmacol. 1983, 69, 442–450. [Google Scholar] [CrossRef]
- Ming, X.; Julu, P.O.; Brimacombe, M.; Connor, S.; Daniels, M.L. Reduced cardiac parasympathetic activity in children with autism. Brain Dev. 2005, 27, 509–516. [Google Scholar] [CrossRef]
- Kasparov, S.; Paton, J.F. Differential effects of angiotensin II in the nucleus tractus solitarii of the rat—plausible neuronal mechanisms. J. Physiol. 1999, 521, 227–238. [Google Scholar] [CrossRef]
- Ishitsuka, T.; Iadecola, C.; Underwood, M.D.; Reis, D.J. Lesions of nucleus tractus solitarii globally impair cerebrovascular autoregulation. Amer. J. Physiol.-Heart Circ. Phy. 1986, 251, H269–H281. [Google Scholar]
- Talman, W.T.; Perrone, M.H.; Reis, D.J. Acute hypertension after the local injection of kainic acid into the nucleus tractus solitarii of rats. Circ. Res. 1981, 48, 292–298. [Google Scholar] [CrossRef]
- Anderson, C.J.; Colombo, J. Larger tonic pupil size in young children with autism spectrum disorder. Dev. Psychobiol. 2009, 51, 207–211. [Google Scholar] [CrossRef]
- van Steensel, F.J.; Bogels, S.M.; de Bruin, E.I. Psychiatric comorbidity in children with autism psectrum disorders: A comparison with children with ADHD. J. Child Fam. Stud. 2013, 22, 368–376. [Google Scholar] [CrossRef]
- Kushki, A.; Drumm, E.; Pla Mobarak, M.; Tanel, N.; Dupuis, A.; Chau, T.; Anagnostou, E. Investigating the autonomic nervous system response to anxiety in children with autism spectrum disorders. PLoS One 2013, 8. [Google Scholar] [CrossRef]
- Ratey, J.J.; Bemporad, J.; Sorgi, P.; Bick, P.; Polakoff, S.; O’Driscoll, G.; Mikkelsen, E. Open trial effects of beta-blockers on speech and social behaviors in 8 autistic adults. J. Autism Dev. Disord. 1987, 17, 439–446. [Google Scholar] [CrossRef]
- Beversdorf, D.Q.; Saklayen, S.; Higgins, K.F.; Bodner, K.E.; Kanne, S.M.; Christ, S.E. Effect of propranolol on word fluency in autism. Cogn. Behav. Neurol. 2011, 24, 1–17. [Google Scholar] [CrossRef]
- Andresen, M.C.; Mendelowitz, D. Sensory afferent neurotransmission in caudal nucleus tractus solitaries—common denominators. Chem. Senses 1996, 21, 387–395. [Google Scholar] [CrossRef]
- Deuchars, J.; Li, Y.W.; Kasparov, S.; Paton, J.F. Morphological and electrophysiological properties of neurones in the dorsal vagal complex of the rat activated by arterial baroreceptors. J. Comp. Neurol. 2000, 417, 233–249. [Google Scholar] [CrossRef]
- van Giersbergen, P.L.; Palkovits, M.; de Jong, W. Involvement of neurotransmitters in the nucleus tractus solitarii in cardiovascular regulation. Physiol. Rev. 1992, 72, 789–824. [Google Scholar]
- Paton, J.F.; Li, Y.W.; Deuchars, J.; Kasparov, S. Properties of solitary tract neurons receiving inputs from the sub-diaphragmatic vagus nerve. Neuroscience 1999, 95, 141–153. [Google Scholar] [CrossRef]
- Zhang, X.; Renehan, W.E.; Fogel, R. Neurons in the vagal complex of the rat respond to mechanical and chemical stimulation of the GI tract. Amer. J. Physiol.-Gastrointest. L. 1998, 274, G331–G341. [Google Scholar]
- Zheng, H.; Kelly, L.; Patterson, L.M.; Berthoud, H.R. Effect of brain stem NMDA-receptor blockade by MK-801 on behavioral and fos responses to vagal satiety signals. Am. J. Physiol. 1999, 277, R1104–R1111. [Google Scholar]
- Ambalavanar, R.; Tanaka, Y.; Selbie, W.S.; Ludlow, C.L. Neuronal activation in the medulla oblongata during selective elicitation of the laryngeal adductor response. J. Neurophysiol. 2004, 92, 2920–2932. [Google Scholar] [CrossRef]
- Jordan, D. Central nervous pathways and control of the airways. Respir. Physiol. 2001, 125, 67–81. [Google Scholar] [CrossRef]
- Ogihara, C.A.; Schoorlemmer, G.H.; Colombari, E.; Sato, M.A. Changes in sodium appetite evoked by lesions of the commissural nucleus of the tractus solitarius. Braz. J. Med. Biol. Res. 2009, 42, 561–566. [Google Scholar] [CrossRef] [Green Version]
- Terai, K.; Munesue, T.; Hiratani, M. Excessive water drinking behavior in autism. Brain Develop. 1999, 21, 103–106. [Google Scholar] [CrossRef]
- Horvath, K.; Papadimitriou, J.C.; Rabsztyn, A.; Drachenberg, C.; Tildon, J.T. Gastrointestinal abnormalities in children with autistic disorder. J. Pediatr. 1999, 135, 559–563. [Google Scholar] [CrossRef]
- Paul, R.; Augustyn, A.; Klin, A.; Volkmar, F.R. Perception and production of prosody by speakers with autism spectrum disorders. J. Autism Dev. Disord. 2005, 35, 205–220. [Google Scholar] [CrossRef]
- Gupta, S.; Aggarwal, S.; Rashanravan, B.; Lee, T. Th1- and Th2-like cytokines in CD4+ and CD8+ T cells in autism. J. Neuroimmunol. 1998, 85, 106–109. [Google Scholar] [CrossRef]
- Kern, J.K.; Geier, D.A.; Audhya, T.; King, P.G.; Sykes, L.K.; Geier, M.R. Evidence of parallels between mercury intoxication and the brain pathology in autism. ACTA Neurobiol. Exp. (Wars.) 2012, 72, 113–153. [Google Scholar]
- Ashwood, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Pessah, I.N.; van de Water, J. Altered T cell responses in children with autism. Brain Behav. Immun. 2011, 25, 840–849. [Google Scholar] [CrossRef]
- Cheshire, W.P. Highlights in clinical autonomic neuroscience: New insights into autonomic dysfunction in autism. Auton. Neurosci. 2012, 171, 4–7. [Google Scholar] [CrossRef]
- Porges, S.W. Peripheral and neurochemical parallels of psychopathology: A psychophysiological model relating autonomic imbalance to hyperactivity, psychopathy, and autism. Adv. Child Dev. Behav. 1976, 11, 35–65. [Google Scholar] [CrossRef]
- Heilman, K.J.; Bal, E.; Bazhenova, O.V.; Porges, S.W. Respiratory sinus arrhythmia and tympanic membrane compliance predict spontaneous eye gaze behaviors in young children: A pilot study. Dev. Psychobiol. 2007, 49, 531–542. [Google Scholar] [CrossRef]
- van Hecke, A.V.; Lebow, J.; Bal, E.; Lamb, D.; Harden, E.; Kramer, A.; Denver, J.; Bazhenova, O.; Porges, S.W. Electroencephalogram and heart rate regulation to familiar and unfamiliar people in children with autism spectrum disorders. Child Dev. 2009, 80, 1118–1133. [Google Scholar] [CrossRef]
- Patriquin, M.A.; Scarpa, A.; Friedman, B.H.; Porges, S.W. Respiratory sinus arrhythmia: A marker for positive social functioning and receptive language skills in children with autism spectrum disorders. Dev. Psychobiol. 2013, 55, 101–112. [Google Scholar] [CrossRef]
- Bal, E.; Harden, E.; Lamb, D.; van Hecke, A.V.; Denver, J.W.; Porges, S.W. Emotion recognition in children with autism spectrum disorders: Relations to eye gaze and autonomic state. J. Autism Dev. Disord. 2010, 40, 358–370. [Google Scholar] [CrossRef]
- Curtis, J.T.; Hood, A.N.; Chen, Y.; Cobb, G.P.; Wallace, D.R. Chronic metals ingestion by prairie voles produces sex-specific deficits in social behavior: An animal model of autism. Behav. Brain Res. 2010, 213, 42–49. [Google Scholar] [CrossRef]
- Esch, B.E.; Carr, J.E. Secretin as a treatment for autism: A review of the evidence. J. Autism Dev. Disord. 2004, 34, 543–556. [Google Scholar] [CrossRef]
- Sturmey, P. Secretin is an ineffective treatment for pervasive developmental disabilities: A review of 15 double-blind randomized controlled trials. Res. Dev. Disabil. 2005, 26, 87–97. [Google Scholar] [CrossRef]
- Nozaki, S.; Nakata, R.; Mizuma, H.; Nishimura, N.; Watanabe, Y.; Kohashi, R.; Watanabe, Y. In vitro autoradiographic localization of 125I-Secretin receptor binding sites in rat brain. Biochem. Biophys. Res. Commun. 2002, 292, 133–137. [Google Scholar] [CrossRef]
- Tay, J.; Goulet, M.; Rusche, J.; Boismenu, R. Age-related and regional differences in secretin and secretin receptor mRNA levels in the rat brain. Neurosci. Lett. 2004, 366, 176–181. [Google Scholar] [CrossRef]
- Yang, B.; Goulet, M.; Boismenu, R.; Ferguson, A.V. Secretin depolarizes nucleus tractus solitarius neurons through activation of a nonselective cationic conductance. Amer. J. Physiol. -Regul. Integr. C. 2004, 286, R927–R934. [Google Scholar] [CrossRef]
- Toda, Y.; Mori, K.; Hashimoto, T.; Miyazaki, M.; Nozaki, S.; Watanabe, Y.; Kuroda, Y.; Kagami, S. Administration of secretin for autism alters dopamine metabolism in the central nervous system. Brain Dev. 2006, 28, 99–103. [Google Scholar] [CrossRef]
- Becker, R.H.; Scholtholt, J.; Scholkens, B.A.; Jung, W.; Speth, O. A microsphere study on the effects of somatostatin and secretin on regional blood flow in anesthetized dogs. Regul. Peptides 1982, 4, 341–351. [Google Scholar] [CrossRef]
- Ohnishi, T.; Matsuda, H.; Hashimoto, T.; Kunihiro, T.; Nishikawa, M.; Uema, T.; Sasaki, M. Abnormal regional cerebral blood flow in childhood autism. Brain 2000, 123, 1838–1844. [Google Scholar] [CrossRef]
- Zilbovicius, M.; Boddaert, N.; Belin, P.; Poline, J.B.; Remy, P.; Mangin, J.F.; Thivard, L.; Barthelemy, C.; Samson, Y. Temporal lobe dysfunction in childhood autism: A PET study. Positron emission tomography. Am. J. Psychiatry 2000, 157, 1988–1993. [Google Scholar] [CrossRef]
- Yang, W.H.; Jing, J.; Xiu, L.J.; Cheng, M.H.; Wang, X.; Bao, P.; Wang, Q.X. Regional cerebral blood flow in children with autism spectrum disorders: A quantitative 99mTc-ECD brain SPECT study with statistical parametric mapping evaluation. Chin. Med. J. (Engl.) 2011, 124, 1362–1366. [Google Scholar]
- Lenz, C.; Rebel, A.; Waschke, K.F.; Koehler, R.C.; Frietsch, T. Blood viscosity modulates tissue perfusion: Sometimes and somewhere. Transfus. Altern. Transfus. Med. 2008, 9, 265–272. [Google Scholar]
- Bruneau, N.; Dourneau, M.C.; Garreau, B.; Pourcelot, L.; Lelord, G. Blood flow response to auditory stimulations in normal, mentally retarded, and autistic children: A preliminary transcranial Doppler ultrasonographic study of the middle cerebral arteries. Biol. Psychiat. 1992, 32, 691–699. [Google Scholar] [CrossRef]
- Truijen, J.; van Lieshout, J.J. Parasympathetic control of blood flow to the activated human brain. Exp. Physiol. 2010, 95, 980–981. [Google Scholar] [CrossRef]
- Umemura, A.; Branston, N.M. Cerebrovascular parasympathetic innervation contributes to coupling of neuronal activation and blood flow in rat somatosensory cortex. Neurosci. Lett. 1995, 193, 193–196. [Google Scholar] [CrossRef]
- Agassandian, K.; Fazan, V.P.; Adanina, V.; Talman, W.T. Direct projections from the cardiovascular nucleus tractus solitarii to pontine preganglionic parasympathetic neurons: A link to cerebrovascular regulation. J. Comp. Neurol. 2002, 452, 242–254. [Google Scholar] [CrossRef]
- Boysen, N.C.; Dragon, D.N.; Talman, W.T. Parasympathetic tonic dilatory influences on cerebral vessels. Auton. Neurosci. 2009, 147, 101–104. [Google Scholar] [CrossRef]
- Liu, A.J.; Ling, G.; Wu, J.; Shen, F.M.; Wang, D.S.; Lin, L.L.; Liu, J.G.; Su, D.F. Arterial baroreflex function is an important determinant of acute cerebral ischemia in rats with middle cerebral artery occlusion. Life Sci. 2008, 83, 388–393. [Google Scholar] [CrossRef]
- Mihaylova, S.; Killian, A.; Mayer, K.; Pullamsetti, S.S.; Schermuly, R.; Rosengarten, B. Effects of anti-inflammatory vagus nerve stimulation on the cerebral microcirculation in endotoxinemic rats. J. Neuroinfammation 2012, 9, 183. [Google Scholar] [CrossRef]
- Walker, B.R.; Diefenbach, K.S.; Parikh, T.N. Inhibition within the nucleus tractus solitarius (NTS) ameliorates environmental exploration deficits due to cerebellum lesions in an animal model for autism. Behav. Brain Res. 2007, 176, 109–120. [Google Scholar] [CrossRef]
- Golanov, E.V.; Reis, D.J. Neurons of nucleus of the solitary tract synchronize the EEG and elevate cerebral blood flow via a novel medullary area. Brain Res. 2001, 892, 1–12. [Google Scholar] [CrossRef]
- Nakai, M.; Ogino, K. The relevance of cardio-pulmonary-vascular reflex to regulation of the brain vessels. Jpn. J. Physiol. 1984, 34, 193–197. [Google Scholar] [CrossRef]
- Fornai, F.; Ruffoli, R.; Giorgi, F.S.; Paparelli, A. The role of locus coeruleus in the antiepileptic activity induced by vagus nerve stimulation. Eur. J. Neurosci. 2011, 33, 2169–2178. [Google Scholar] [CrossRef]
- Murphy, J.V.; Wheless, J.W.; Schmoll, C.M. Left vagal nerve stimulation in six patients with hypothalamic hamartomas. Pediat. Neurol. 2000, 23, 167–168. [Google Scholar] [CrossRef]
- Danielsson, S.; Viggedal, G.; Gillberg, C.; Olsson, I. Lack of effects of vagus nerve stimulation on drug-resistant epilepsy in eight pediatric patients with autism spectrum disorders: A prospective 2-year follow-up study. Epilepsy Behav. 2008, 12, 298–304. [Google Scholar] [CrossRef]
- Park, Y.D. The effects of vagus nerve stimulation therapy on patients with intractable seizures and either Landau-Kleffner syndrome or autism. Epilepsy Behav. 2003, 4, 286–290. [Google Scholar] [CrossRef]
- Buchholz, R.A.; Nathan, M.A. Chronic lability of the arterial blood pressure produced by electrolytic lesions of the nucleus tractus solitarii in the rat. Circ. Res. 1984, 54, 227–238. [Google Scholar] [CrossRef]
- Walgenbach, S.C. Sustained interruption of the aortic baroreflex by left cervical vagotomy in dogs. Amer. J. Physiol. -Heart Circ. Phy. 1984, 246, H319–H323. [Google Scholar]
- Frigero, M.; Bonagamba, L.G.; Machado, B.H. The gain of the baroreflex bradycardia is reduced by microinjection of NMDA receptor antagonists into the nucleus tractus solitarii of awake rats. J. Autonom. Nerv. Syst. 2000, 79, 28–33. [Google Scholar] [CrossRef]
- Duale, H.; Waki, H.; Howorth, P.; Kasparov, S.; Teschemacher, A.G.; Paton, J.F. Restraining influence of A2 neurons in chronic control of arterial pressure in spontaneously hypertensive rats. Cardiovasc. Res. 2007, 76, 184–193. [Google Scholar] [CrossRef]
- Gallowitsch-Puerta, M.; Pavlov, V.A. Neuro-immune interactions via the cholinergic anti-inflammatory pathway. Life Sci. 2007, 80, 2325–2329. [Google Scholar] [CrossRef]
- Hermann, G.E.; Emch, G.S.; Tovar, C.A.; Rogers, R.C. c-Fos generation in the dorsal vagal complex after systemic endotoxin is not dependent on the vagus nerve. Amer. J. Physiol. -Regul. Integr. C. 2001, 280, R289–R299. [Google Scholar]
- Kim, W.G.; Mohney, R.P.; Wilson, B.; Jeohn, G.H.; Liu, B.; Hong, J.S. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: Role of microglia. J. Neurosci. 2000, 20, 6309–6316. [Google Scholar]
- Daulatzai, M.A. Dysfunctional nucleus tractus solitarius: Its crucial role in promoting neuropathogenetic cascade of Alzheimer’s dementia--a novel hypothesis. Neurochem. Res. 2012, 37, 846–868. [Google Scholar] [CrossRef]
- Marvel, F.A.; Chen, C.C.; Badr, N.; Gaykema, R.P.; Goehler, L.E. Reversible inactivation of the dorsal vagal complex blocks lipopolysaccharide-induced social withdrawal and c-Fos expression in central autonomic nuclei. Brain Behav. Immun. 2004, 18, 123–134. [Google Scholar] [CrossRef]
- Zitnik, R.J. Treatment of chronic inflammatory diseases with implantable medical devices. Clev. Clin. J. Med. 2011, 78, S30–S34. [Google Scholar] [CrossRef]
- van der Zanden, E.P.; Boeckxstaens, G.E.; de Jonge, W.J. The vagus nerve as a modulator of intestinal inflammation. Neurogastroenterol. Motil. 2009, 21, 6–17. [Google Scholar] [CrossRef]
- Rosas-Ballina, M.; Ochani, M.; Parrish, W.R.; Ochani, K.; Harris, Y.T.; Huston, J.M.; Chavan, S.; Tracey, K.J. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc. Natl. Acad. Sci. USA 2008, 105, 11008–11013. [Google Scholar]
- Vida, G.; Pena, G.; Kanashiro, A.; Thompson-Bonilla Mdel, R.; Palange, D.; Deitch, E.A.; Ulloa, L. Beta2-Adrenoreceptors of regulatory lymphocytes are essential for vagal neuromodulation of the innate immune system. FASEB J. 2011, 25, 4476–4485. [Google Scholar] [CrossRef]
- Huston, J.M.; Ochani, M.; Rosas-Ballina, M.; Liao, H.; Ochani, K.; Pavlov, V.A.; Gallowitsch-Puerta, M.; Ashok, M.; Czura, C.J.; Foxwell, B.; et al. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J. Exp. Med. 2006, 203, 1623–1628. [Google Scholar] [CrossRef]
- Valdes-Ferrer, S.; Rosas-Ballina, M.; Olofsson, P.; Chavan, S.; Tracey, K. Vagus nerve stimulation produces an anti-inflammatory monocyte phenotype in blood. J. Immunol. 2010, 184, 138.25. [Google Scholar]
- Chhetri, D.K.; Neubauer, J.; Berry, D.A. Graded activation of the intrinsic laryngeal muscles for vocal fold posturing. J. Acoust. Soc. Amer. 2010, 127, EL127–EL133. [Google Scholar]
- Hammer, M.J.; Barlow, S.M. Laryngeal somatosensory deficits in Parkinson’s disease: Implications for speech respiratory and phonatory control. Exp. Brain Res. 2010, 201, 401–409. [Google Scholar] [CrossRef]
- Jurgens, U.; Kirzinger, A. The laryngeal sensory pathway and its role in phonation. A brain lesioning study in the squirrel monkey. Exp. Brain Res. 1985, 59, 118–124. [Google Scholar]
- Sasaki, C.T.; Hundal, J.S.; Kim, Y.H. Protective glottic closure: Biomechanical effects of selective laryngeal denervation. Ann. Otol. Rhinol. Laryngol. 2005, 114, 271–275. [Google Scholar]
- Bauer, V.; Aleric, Z.; Jancic, E.; Miholovic, V. Voice quality in Parkinson’s disease in the Croatian language speakers. Coll. Antropol. 2011, 35, 209–212. [Google Scholar]
- Shaffer, M.J.; Jackson, C.E.; Szabo, C.A.; Simpson, C.B. Vagal nerve stimulation: Clinical and electrophysiological effects on vocal fold function. Ann. Otol. Rhinol. Laryngol. 2005, 114, 7–14. [Google Scholar]
- Braak, H.; Ghebremedhin, E.; Rub, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef]
- Sandborgh-Englund, G.; Elinder, C.G.; Langworth, S.; Schutz, A.; Ekstrand, J. Mercury in biological fluids after amalgam removal. J. Dent. Res. 1998, 77, 615–624. [Google Scholar] [CrossRef]
- Halbach, S.; Vogt, S.; Kohler, W.; Felgenhauer, N.; Welzl, G.; Kremers, L.; Zilker, T.; Melchart, D. Blood and urine mercury levels in adult amalgam patients of a randomized controlled trial: Interaction of Hg species in erythrocytes. Environ. Res. 2008, 107, 69–78. [Google Scholar] [CrossRef]
- Donnellan, A.M.; Hill, D.A.; Leary, M.R. Rethinking autism: implications of sensory and movement differences for understanding and support. Front. Integr. Neurosci. 2013, 6, 1–11. [Google Scholar]
- Thunberg, G.; Ahlsen, E.; Sandberg, A.D. Children with autistic spectrum disorders and speech-generating devices: Communication in different activities at home. Clin. Linguist. Phon. 2007, 21, 457–479. [Google Scholar] [CrossRef]
- Wadie, M.; Li, J.; Sasaki, C.T. Effect of altered core body temperature on glottal closing force. Ann. Otol. Rhinol. Laryngol. 2011, 120, 669–673. [Google Scholar]
- Ambalavanar, R.; Purcell, L.; Miranda, M.; Evans, F.; Ludlow, C.L. Selective suppression of late laryngeal adductor responses by n-methyl-d-aspartate receptor blockade in the cat. J. Neurophysiol. 2002, 87, 1252–1262. [Google Scholar]
- Fawley, J.A.; Peters, J.H.; Andresen, M.C. GABAB-mediated inhibition of multiple modes of glutamate release in the nucleus of the solitary tract. J. Neurophysiol. 2011, 106, 1833–1840. [Google Scholar] [CrossRef]
- Bailey, T.W.; Jin, Y.H.; Doyle, M.W.; Andresen, M.C. Vanilloid-sensitive afferents activate neurons with prominent A-type potassium currents in nucleus tractus solitarius. J. Neurosci. 2002, 22, 8230–8237. [Google Scholar]
- Shoudai, K.; Peters, J.H.; McDougall, S.J.; Fawley, J.A.; Andresen, M.C. Thermally active TRPV1 tonically drives central spontaneous glutamate release. J. Neurosci. 2010, 30, 14470–14475. [Google Scholar] [CrossRef]
- Laplace, J.P.; Nunes, C.S. Pancreatic size and enzyme contents after vagal deafferentation in jejunectomised pigs under free or restricted feeding. Gut 1987, 28, 169–173. [Google Scholar] [CrossRef]
- Rimland, B. Infantile Autism; Meredith Publishing Company: New York, NY, USA, 1964; p. 8. [Google Scholar]
- Beck, V.A. Confronting Autism: The Aurora on the Dark Side of Venus; New Destiny Educational Products: Bedford, NH, USA, 2013. [Google Scholar]
- Quintana, E.; Garcia-Zaragoza, E.; Martinez-Cuesta, M.A.; Calatayud, S.; Esplugues, J.V.; Barrachina, M.D. A cerebral nitrergic pathway modulates endotoxin-induced changes in gastric motility. Br. J. Pharmacol. 2001, 134, 325–332. [Google Scholar] [CrossRef]
- Glatzer, N.R.; Smith, B.N. Modulation of synaptic transmission in the rat nucleus of the solitary tract by endomorphin-1. J. Neurophysiol. 2005, 93, 2530–2540. [Google Scholar] [CrossRef]
- Afzal, N.; Murch, S.; Thirrupathy, K.; Berger, L.; Fagbemi, A.; Heuschkel, R. Constipation with acquired megarectum in children with autism. Pediatrics 2003, 112, 939–942. [Google Scholar] [CrossRef]
- Torrente, F.; Ashwood, P.; Day, R.; Machado, N.; Furlano, R.I.; Anthony, A.; Davies, S.E.; Wakefield, A.J.; Thomson, M.A.; Walker-Smith, J.A.; et al. Small intestinal enteropathy with epithelial IgG and complement deposition in children with regressive autism. Mol. Psychiat. 2002, 7, 375–382. [Google Scholar] [CrossRef]
- Sun, Z.; Cade, R. Findings in normal rats following administration of gliadorphin-7 (GD-7). Peptides 2003, 24, 321–323. [Google Scholar] [CrossRef]
- Poole, S.L.; Deuchars, J.; Lewis, D.I.; Deuchars, S.A. Subdivision-specific responses of neurons in the nucleus of the tractus solitarius to activation of mu-opioid receptors in the rat. J. Neurophysiol. 2007, 98, 3060–3071. [Google Scholar] [CrossRef]
- Cui, R.J.; Roberts, B.L.; Zhao, H.; Andresen, M.C.; Appleyard, S.M. Opioids inhibit visceral afferent activation of catecholamine neurons in the solitary tract nucleus. Neuroscience 2012, 222, 181–190. [Google Scholar] [CrossRef]
- Rhim, H.; Toth, P.T.; Miller, R.J. Mechanism of inhibition of calcium channels in rat nucleus tractus solitarius by neurotransmitters. Br. J. Pharmacol. 1996, 118, 1341–1350. [Google Scholar] [CrossRef]
- Rinaman, L. Hindbrain noradrenergic A2 neurons: Diverse roles in autonomic, endocrine, cognitive, and behavioral functions. Am. J. Physiol. -Regul. Integr. C. 2011, 300, R222–R235. [Google Scholar] [CrossRef]
- Koda, L.Y.; Bloom, F.E. Distribution of catecholamine-containing cell bodies and blood vessels in the rat nucleus tractus solitarius. Brain Res. 1983, 289, 71–78. [Google Scholar] [CrossRef]
- Viljoen, M.; Panzer, A. The central noradrenergic system: An overview. Afr. J. Psychiat. (Johannesbg.) 2007, 10, 135–141. [Google Scholar]
- Rimland, B. Infantile Autism; Meredith Publishing Company: New York, NY, USA, 1964; Chapters 5–6. [Google Scholar]
- Gaykema, R.P.; Goehler, L.E. Ascending caudal medullary catecholamine pathways drive sickness-induced deficits in exploratory behavior: Brain substrates for fatigue? Brain Behav. Immun. 2011, 25, 443–460. [Google Scholar] [CrossRef]
- Ek, M.; Kurosawa, M.; Lundeberg, T.; Ericsson, A. Activation of vagal afferents after intravenous injection of interleukin-1beta: Role of endogenous prostaglandins. J. Neurosci. 1998, 18, 9471–9479. [Google Scholar]
- Dalmaz, Y.; Lagercrantz, H.; Pequignot, J.M.; Soulier, V. Neonatal hypoxia disturbs the catecholamine turnover in the nucleus of the tractus solitarius and the peripheral chemoreceptors of the adult rat. Pediat. Pulmonol. Suppl. 1997, 16, 218–219. [Google Scholar]
- Salchner, P.; Engidawork, E.; Hoeger, H.; Lubec, B.; Singewald, N. Perinatal asphyxia exerts lifelong effects on neuronal responsiveness to stress in specific brain regions in the rat. J. Investig. Med. 2003, 51, 288–294. [Google Scholar] [CrossRef]
- Zhang, W.; Carreno, F.R.; Cunningham, J.T.; Mifflin, S.W. Chronic sustained hypoxia enhances both evoked EPSCs and norepinephrine inhibition of glutamatergic afferent inputs in the nucleus of the solitary tract. J. Neurosci. 2009, 29, 3093–3102. [Google Scholar] [CrossRef]
- Ronai, A.Z.; Kato, E.; Al-Khrasani, M.; Hajdu, M.; Mullner, K.; Elor, G.; Gyires, K.; Furst, S.; Palkovits, M. Age and monosodium glutamate treatment cause changes in the stimulation-induced[3H]-norepinephrine release from rat nucleus tractus solitarii-dorsal vagal nucleus slices. Life Sci. 2004, 74, 1573–1580. [Google Scholar] [CrossRef]
- Rosenberg, P.A.; Li, Y. Adenylyl cyclase activation underlies intracellular cyclic AMP accumulation, cyclic AMP transport, and extracellular adenosine accumulation evoked by beta-adrenergic receptor stimulation in mixed cultures of neurons and astrocytes derived from rat cerebral cortex. Brain Res. 1995, 692, 227–232. [Google Scholar] [CrossRef]
- Mehler, M.F.; Purpura, D.P. Autism, fever, epigenetics and the locus coeruleus. Brain Res. Rev. 2009, 59, 388–392. [Google Scholar] [CrossRef]
- Jaselskis, C.A.; Cook, E.H., Jr.; Fletcher, K.E.; Leventhal, B.L. Clonidine treatment of hyperactive and impulsive children with autistic disorder. J. Clin. Psychopharmacol. 1992, 12, 322–327. [Google Scholar]
- Fankhauser, M.P.; Karumanchi, V.C.; German, M.L.; Yates, A.; Karumanchi, S.D. A double-blind, placebo-controlled study of the efficacy of transdermal clonidine in autism. J. Clin. Psychiat. 1992, 53, 77–82. [Google Scholar]
- Ming, X.; Gordon, E.; Kang, N.; Wagner, G.C. Use of clonidine in children with autism spectrum disorders. Brain Dev. 2008, 30, 454–460. [Google Scholar] [CrossRef]
- Golubchik, P.; Sever, J.; Weizman, A. Reboxetine treatment for autistic spectrum disorder of pediatric patients with depressive and inattentive/hyperactive symptoms: An open-label trial. Clin. Neuropharmacol. 2013, 36, 37–41. [Google Scholar] [CrossRef]
- Anderson, C.J.; Colombo, J.; Unruh, K.E. Pupil and salivary indicators of autonomic dysfunction in autism spectrum disorder. Dev. Psychobiol. 2013, 55, 465–482. [Google Scholar] [CrossRef]
- Philbin, K.E.; Bateman, R.J.; Mendelowitz, D. Clonidine, an alpha2-receptor agonist, diminishes GABAergic neurotransmission to cardiac vagal neurons in the nucleus ambiguus. Brain Res. 2010, 1347, 65–70. [Google Scholar] [CrossRef]
- Judy, W.V.; Farrell, S.K. Arterial baroreceptor reflex control of sympathetic nerve activity in the spontaneously hypertensive rat. Hypertension 1979, 1, 605–614. [Google Scholar] [CrossRef]
- Simms, A.E.; Paton, J.F.; Pickering, A.E. Hierarchical recruitment of the sympathetic and parasympathetic limbs of the baroreflex in normotensive and spontaneously hypertensive rats. J. Physiol. 2007, 579, 473–486. [Google Scholar]
- Waki, H.; Gouraud, S.S.; Maeda, M.; Paton, J.F. Evidence of specific inflammatory condition in nucleus tractus solitarii of spontaneously hypertensive rats. Exp. Physiol. 2010, 95, 595–600. [Google Scholar] [CrossRef]
- Washington, B.; Williams, S.; Armstrong, P.; Mtshali, C.; Robinson, J.T.; Myles, E.L. Cadmium toxicity on arterioles vascular smooth muscle cells of spontaneously hypertensive rats. Int. J. Environ. Res. Public Health 2006, 3, 323–328. [Google Scholar] [CrossRef]
- Waki, H.; Liu, B.; Miyake, M.; Katahira, K.; Murphy, D.; Kasparov, S.; Paton, J.F. Junctional Adhesion Molecule-1 is upregulated in spontaneously hypertensive rats: Evidence for a prohypertensive role within the brain stem. Hypertension 2007, 49, 1321–1327. [Google Scholar] [CrossRef]
- Paton, J.F.; Waki, H. Is neurogenic hypertension related to vascular inflammation of the brainstem? Neurosci. Biobehav. R 2009, 33, 89–94. [Google Scholar] [CrossRef]
- Supowit, S.C.; Zhao, H.; DiPette, D.J. Nerve growth factor enhances calcitonin gene-related peptide expression in the spontaneously hypertensive rat. Hypertension 2001, 37, 728–732. [Google Scholar] [CrossRef]
- Zaidi, M.; Bevis, P.J. Enhanced circulating levels of neurally derived calcitonin gene related peptide in spontaneously hypertensive rats. Cardiovasc. Res. 1991, 25, 125–128. [Google Scholar] [CrossRef]
- Nelson, K.B.; Grether, J.K.; Croen, L.A.; Dambrosia, J.M.; Dickens, B.F.; Jelliffe, L.L.; Hansen, R.L.; Phillips, T.M. Neuropeptides and neurotrophins in neonatal blood of children with autism or mental retardation. Ann. Neurol. 2001, 49, 597–606. [Google Scholar] [CrossRef]
- Abdallah, M.W.; Larsen, N.; Grove, J.; Bonefeld-Jorgensen, E.C.; Norgaard-Pedersen, B.; Hougaard, D.M.; Mortensen, E.L. Neonatal chemokine levels and risk of autism spectrum disorders: Findings from a Danish historic birth cohort follow-up study. Cytokine 2013, 61, 370–376. [Google Scholar] [CrossRef]
- Bona, E.; Andersson, A.L.; Blomgren, K.; Gilland, E.; Puka-Sundvall, M.; Gustafson, K.; Hagberg, H. Chemokine and inflammatory cell response to hypoxia-ischemia in immature rats. Pediatr. Res. 1999, 45, 500–509. [Google Scholar] [CrossRef]
- Lyng, K.; Munkeby, B.H.; Scheie, D.; Mallard, C.; Hagberg, H.; Stray-Pedersen, B.; Saugstad, O.D.; Froen, J.F. Fetal brain injury in experimental intrauterine asphyxia and inflammation in Gottingen minipigs. J. Perinat. Med. 2006, 34, 226–234. [Google Scholar]
- Mangano, E.N.; Hayley, S. Inflammatory priming of the substantia nigra influences the impact of later paraquat exposure: Neuroimmune sensitization of neurodegeneration. Neurobiol. Aging 2009, 30, 1361–1378. [Google Scholar] [CrossRef]
- Charleston, J.S.; Bolender, R.P.; Mottet, N.K.; Body, R.L.; Vahter, M.E.; Burbacher, T.M. Increases in the number of reactive glia in the visual cortex of Macaca fascicularis following subclinical long-term methyl mercury exposure. Toxicol. Appl. Pharm. 1994, 129, 196–206. [Google Scholar] [CrossRef]
- Souza, V.; Escobar Md Mdel, C.; Gomez-Quiroz, L.; Bucio, L.; Hernandez, E.; Cossio, E.C.; Gutierrez-Ruiz, M. Acute cadmium exposure enhances AP-1 DNA binding and induces cytokines expression and heat shock protein 70 in HepG2 cells. Toxicology 2004, 197, 213–228. [Google Scholar] [CrossRef]
- Chaparro-Huerta, V.; Rivera-Cervantes, M.C.; Torres-Mendoza, B.M.; Beas-Zírate, C. Neuronal death and tumor necrosis factor-alpha response to glutamate-induced excitotoxicity in the cerebral cortex of neonatal rats. Neurosci. Lett. 2002, 333, 95–98. [Google Scholar] [CrossRef]
- Nadeau, S.; Rivest, S. Regulation of the gene encoding tumor necrosis factor alpha (TNF-[alpha]) in the rat brain and pituitary in response to different models of systemic immune challenge. J. Neuropathol. Exp. Neur. 1999, 58, 61–77. [Google Scholar] [CrossRef]
- Zimmerman, A.W.; Jyonouchi, H.; Comi, A.M.; Connors, S.L.; Milstien, S.; Varsou, A.; Heyes, M.P. Cerebrospinal Fluid and Serum Markers of Inflammation in Autism. Pediat. Neurol. 2005, 33, 195–201. [Google Scholar] [CrossRef]
- Chez, M.G.; Dowling, T.; Patel, P.B.; Khanna, P.; Kominsky, M. Elevation of tumor necrosis factor-alpha in cerebrospinal fluid of autistic children. Pediatr. Neurol. 2007, 36, 361–365. [Google Scholar] [CrossRef]
- Hermann, G.E.; Rogers, R.C. TNF alpha: A trigger of autonomic dysfunction. Neuroscientist 2008, 14, 53–67. [Google Scholar] [CrossRef]
- Saeed, R.W.; Varma, S.; Peng-Nemeroff, T.; Sherry, B.; Balakhaneh, D.; Huston, J.; Tracey, K.J.; Al-Abed, Y.; Metz, C.N. Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J. Exp. Med. 2005, 201, 1113–1123. [Google Scholar] [CrossRef]
- Fisher, M. Pericyte signaling in the neurovascular unit. Stroke 2009, 40, S13–S15. [Google Scholar] [CrossRef]
- Peppiatt, C.M.; Howarth, C.; Mobbs, P.; Attwell, D. Bidirectional control of CNS capillary diameter by pericytes. Nature 2006, 443, 700–704. [Google Scholar] [CrossRef]
- Nguyen, P.T.; Nakamura, T.; Hori, E.; Urakawa, S.; Uwano, T.; Zhao, J.; Li, R.; Bac, N.D.; Hamashima, T.; Ishii, Y.; et al. Cognitive and socio-emotional deficits in platelet-derived growth factor receptor-beta gene knockout mice. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Zakareia, F.A.; Al-Ayadhi, L.Y.; Al-Drees, A.A. Study of dual angiogenic/neurogenic growth factors among Saudi autistic children and their correlation with the severity of this disorder. Neuroscience (Riyadh.) 2012, 17, 213–218. [Google Scholar]
- Oishi, K.; Kamiyashiki, T.; Ito, Y. Isometric contraction of microvascular pericytes from mouse brain parenchyma. Microvasc. Res. 2007, 73, 20–28. [Google Scholar] [CrossRef]
- Sakagami, K.; Kawamura, H.; Wu, D.M.; Puro, D.G. Nitric oxide/cGMP-induced inhibition of calcium and chloride currents in retinal pericytes. Microvasc. Res. 2001, 62, 196–203. [Google Scholar] [CrossRef]
- Kim, J.; Kim, O.S.; Kim, C.S.; Kim, N.H.; Kim, J.S. Cytotoxic role of methylglyoxal in rat retinal pericytes: Involvement of a nuclear factor-kappaB and inducible nitric oxide synthase pathway. Chem-Biol. Inter. 2010, 188, 86–93. [Google Scholar] [CrossRef]
- Bisserbe, J.C.; Patel, J.; Marangos, P.J. Autoradiographic localization of adenosine uptake sites in rat brain using [3H]nitrobenzylthioinosine. J. Neurosci. 1985, 5, 544–550. [Google Scholar]
- Li, Q.; Puro, D.G. Adenosine activates ATP-sensitive K+ currents in pericytes of rat retinal microvessels: Role of A1 and A2a receptors. Brain Res. 2001, 907, 93–99. [Google Scholar] [CrossRef]
- Matsugi, T.; Chen, Q.; Anderson, D.R. Adenosine-induced relaxation of cultured bovine retinal pericytes. Invest. Ophthalmol. Vis. Sci. 1997, 38, 2695–2701. [Google Scholar]
- Toth, A.; Boczan, J.; Kedei, N.; Lizanecz, E.; Bagi, Z.; Papp, Z.; Edes, I.; Csiba, L.; Blumberg, P.M. Expression and distribution of vanilloid receptor 1 (TRPV1) in the adult rat brain. Brain Res. Mol. Brain Res. 2005, 135, 162–168. [Google Scholar] [CrossRef]
- Koide, M.; Bonev, A.D.; Nelson, M.T.; Wellman, G.C. Inversion of neurovascular coupling by subarachnoid blood depends on large-conductance Ca2+-activated K+ (BK) channels. Proc. Natl. Acad. Sci. USA 2012, 109, E1387–E1395. [Google Scholar] [CrossRef]
- Welsh, J.P.; Yuen, G.; Placantonakis, D.G.; Vu, T.Q.; Haiss, F.; O’Hearn, E.; Molliver, M.E.; Aicher, S.A. Why do Purkinje cells die so easily after global brain ischemia? Aldolase C, EAAT4, and the cerebellar contribution to posthypoxic myoclonus. Adv. Neurol. 2002, 89, 331–359. [Google Scholar]
- Cavanagh, J.B. Is Purkinje cell loss in Leigh’s disease an excitotoxic event secondary to damage to inferior olivary nuclei? Neuropathol. Appl. Neurobiol. 1994, 20, 599–603. [Google Scholar] [CrossRef]
- Franco, J.L.; Braga Hde, C.; Nunes, A.K.; Ribas, C.M.; Stringari, J.; Silva, A.P.; Pomblum, S.C.; Moro, M.; Bohrer, D.; Santos, A.R.; et al. Lactational exposure to inorganic mercury: evidence of neurotoxic effects. Neurotoxicol. Teratol. 2007, 29, 360–367. [Google Scholar] [CrossRef]
- Piven, J.; Bailey, J.; Ranson, B.J.; Arndt, S. An MRI study of the corpus callosum in autism. Amer. J. Psychiat. 1997, 154, 1051–1056. [Google Scholar]
- Hardan, A.Y.; Pabalan, M.; Gupta, N.; Bansal, R.; Melhem, N.M.; Fedorov, S.; Keshavan, M.S.; Minshew, N.J. Corpus callosum volume in children with autism. Psychiat. Res. 2009, 174, 57–61. [Google Scholar] [CrossRef]
- Kohlhauser, C.; Mosgoller, W.; Hoger, H.; Lubec, B. Myelination deficits in brain of rats following perinatal asphyxia. Life Sci. 2000, 67, 2355–2368. [Google Scholar] [CrossRef]
- Casanova, M.F.; Buxhoeveden, D.P.; Brown, C. Clinical and macroscopic correlates of minicolumnar pathology in autism. J. Child Neurol. 2002, 17, 692–695. [Google Scholar] [CrossRef]
- Herr, K.J.; Herr, D.R.; Lee, C.W.; Noguchi, K.; Chun, J. Stereotyped fetal brain disorganization is induced by hypoxia and requires lysophosphatidic acid receptor 1 (LPA1) signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 15444–15449. [Google Scholar] [CrossRef]
- Kulesza, R.J.; Lukose, R.; Stevens, L.V. Malformation of the human superior olive in autistic spectrum disorders. Brain Res. 2011, 1367, 360–371. [Google Scholar] [CrossRef]
- Windle, W.F. Brain damage by asphyxia at birth. Sci. Amer. 1969, 221, 76–84. [Google Scholar] [CrossRef]
- Myers, R.E. Two patterns of perinatal brain damage and their conditions of occurrence. Am. J. Obstet. Gynecol. 1972, 112, 246–276. [Google Scholar]
- Jiang, Z.D.; Brosi, D.M.; Wilkinson, A.R. Differences in impaired brainstem conduction between neonatal chronic lung disease and perinatal asphyxia. Clin. Neurophysiol. 2010, 121, 725–733. [Google Scholar] [CrossRef]
- Cohen, I.L.; Gardner, J.M.; Karmel, B.Z.; Phan, H.T.; Kittler, P.; Gomez, T.R.; Gonzalez, M.G.; Lennon, E.M.; Parab, S.; Barone, A. Neonatal brainstem function and 4-month arousal-modulated attention are jointly associated with autism. Autism Res. 2013, 6, 11–22. [Google Scholar] [CrossRef]
- Janusonis, S. Origin of the blood hyperserotonemia of autism. Theor. Biol. Med. Model. 2008, 5, 10. [Google Scholar] [CrossRef]
- Kemperman, R.; Muskiet, F.; Boutier, A.I.; Kema, I.P.; Muskiet, F.A. Brief report: Normal intestinal permeability at elevated platelet serotonin levels in a subgroup of children with pervasive developmental disorders in Curacao (The Netherlands Antilles). J. Autism Dev. Disord. 2008, 38, 401–406. [Google Scholar] [CrossRef]
- Li, Y.; Wu, X.Y.; Zhu, J.X.; Owyang, C. Intestinal serotonin acts as paracrine substance to mediate pancreatic secretion stimulated by luminal factors. Amer. J. Physiol. -Gastrointest. L. 2001, 281, G916–G923. [Google Scholar]
- Ohuoha, D.C.; Knable, M.B.; Wolf, S.S.; Kleinman, J.E.; Hyde, T.M. The subnuclear distribution of 5-HT3 receptors in the human nucleus of the solitary tract and other structures of the caudal medulla. Brain Res. 1994, 637, 222–226. [Google Scholar] [CrossRef]
- Raul, L. Serotonin2 receptors in the nucleus tractus solitarius: Characterization and role in the baroreceptor reflex arc. Cell Mol. Neurobiol. 2003, 23, 709–726. [Google Scholar] [CrossRef]
- Yamasaki, K.; Lico, M.C. Electroencephalographic serotonin synchronization: Area postrema and solitary tract nucleus. Am. J. Physiol. -Regul. Integr. C. 1981, 241, R158–R162. [Google Scholar]
- Raymond, G.V.; Bauman, M.L.; Kemper, T.L. Hippocampus in autism: A Golgi analysis. ACTA Neuropathol. 1996, 91, 117–119. [Google Scholar] [CrossRef]
- Evans, T.A.; Siedlak, S.L.; Lu, L.; Fu, X.; Wang, Z.; McGinnis, W.R.; Fakhoury, E.; Castellani, R.J.; Hazen, S.L.; Walsh, W.J.; et al. The autistic phenotype exhibits a remarkably localized modification of brain protein by products of free radical-induced lipid oxidation. Amer. J. Biochem. Biotechnol. 2008, 4, 61–72. [Google Scholar] [CrossRef]
- Palmieri, L.; Papaleo, V.; Porcelli, V.; Scarcia, P.; Gaita, L.; Sacco, R.; Hager, J.; Rousseau, F.; Curatolo, P.; Manzi, B.; et al. Altered calcium homeostasis in autism-spectrum disorders: Evidence from biochemical and genetic studies of the mitochondrial aspartate/glutamate carrier AGC1. Mol. Psychiat. 2010, 15, 38–52. [Google Scholar] [CrossRef]
- Gagliardi, R.J. Neuroprotection, excitotoxicicity and NMDA antagonists. Arq. Neuro-Psiquiat. 2000, 58, 583–588. [Google Scholar] [CrossRef]
- Sajdel-Sulkowska, E.M.; Lipinski, B.; Windom, H.; Audhya, T.; McGinnis, W. Oxidative stress in autism: Elevated cerebellar 3-nitrotyrosine levels. Amer. J. Biochem. Biotechnol. 2008, 4, 73–84. [Google Scholar] [CrossRef]
- Dosman, C.F.; Brian, J.A.; Drmic, I.E.; Senthilselvan, A.; Harford, M.M.; Smith, R.W.; Sharieff, W.; Zlotkin, S.H.; Moldofsky, H.; Roberts, S.W. Children with autism: Effect of iron supplementation on sleep and ferritin. Pediat. Neurol. 2007, 36, 152–158. [Google Scholar] [CrossRef]
- Dolske, M.C.; Spollen, J.; McKay, S.; Lancashire, E.; Tolbert, L. A preliminary trial of ascorbic acid as supplemental therapy for autism. Prog. Neuropsychopharmacol. Biol. Psychiat. 1993, 17, 765–774. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Bradstreet, J.J.; van Dyke, K.; Schneider, C.; Freedenfeld, S.H.; O’Hara, N.; Cave, S.; Buckley, J.A.; Mumper, E.A.; Frye, R.E. Hyperbaric oxygen treatment in autism spectrum disorders. Med. Gas Res. 2012, 2, 16. [Google Scholar] [CrossRef]
- Lin, H.C.; Wan, F.J. Hyperbaric oxygenation reduces overexpression of c-Fos and oxidative stress in the brain stem of experimental endotoxemic rats. Intensive Care Med. 2008, 34, 1122–1132. [Google Scholar] [CrossRef]
- Ames, B.N.; Elson-Schwab, I.; Silver, E.A. High-dose vitamin therapy stimulates variant enzymes with decreased coenzyme binding affinity (increased Km): Relevance to genetic disease and polymorphisms. Amer. J. Clin. Nutr. 2002, 75, 616–658. [Google Scholar]
- Rimland, B.; Callaway, E.; Dreyfus, P. The effect of high doses of vitamin B6 on autistic children: A double-blind crossover study. Amer. J. Psychiat. 1978, 135, 472–475. [Google Scholar]
- Eto, K.; Kimura, H. A novel enhancing mechanism for hydrogen sulfide-producing activity of cystathionine beta-Synthase. J. Biol. Chem. 2002, 277, 42680–42685. [Google Scholar] [CrossRef]
- Austgen, J.R.; Hermann, G.E.; Dantzler, H.A.; Rogers, R.C.; Kline, D.D. Hydrogen sulfide augments synaptic neurotransmission in the nucleus of the solitary tract. J. Neurophysiol. 2011, 106, 1822–1832. [Google Scholar] [CrossRef]
- Yoshida, E.; Toyama, T.; Shinkai, Y.; Sawa, T.; Akaike, T.; Kumagai, Y. Detoxification of methylmercury by hydrogen sulfide-producing enzyme in mammalian cells. Chem. Res. Toxicol. 2011, 24, 1633–1635. [Google Scholar] [CrossRef]
- Zhao, Y.X.; He, W.; Jing, X.H.; Liu, J.L.; Rong, P.J.; Ben, H.; Liu, K.; Zhu, B. Transcutaneous auricular vagus nerve stimulation protects endotoxemic rat from lipopolysaccharide-induced inflammation. Evid-Based Complement. Altern. 2012. [Google Scholar] [CrossRef]
- 2Cheng, C.H.; Yi, P.L.; Lin, J.G.; Chang, F.C. Endogenous opiates in the nucleus tractus solitarius mediate electroacupuncture-induced sleep activities in rats. Evid-Based Complement. Altern. 2011. [Google Scholar] [CrossRef]
- Cohen, I.L.; Liu, X.; Lewis, M.E.; Chudley, A.; Forster-Gibson, C.; Gonzalez, M.; Jenkins, E.C.; Brown, W.T.; Holden, J.J. Autism severity is associated with child and maternal MAOA genotypes. Clin. Genet. 2011, 79, 355–362. [Google Scholar] [CrossRef]
- Brielmaier, J.; Matteson, P.G.; Silverman, J.L.; Senerth, J.M.; Kelly, S.; Genestine, M.; Millonig, J.H.; Cicco-Bloom, E.; Crawley, J.N. Autism-relevant social abnormalities and cognitive deficits in engrailed-2 knockout mice. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Muscarella, L.A.; Guarnieri, V.; Sacco, R.; Curatolo, P.; Manzi, B.; Alessandrelli, R.; Giana, G.; Militerni, R.; Bravaccio, C.; Lenti, C.; et al. Candidate gene study of HOXB1 in autism spectrum disorder. Mol. Autism 2010, 1, 9. [Google Scholar] [CrossRef]
- Tan, W.H.; Baris, H.N.; Burrows, P.E.; Robson, C.D.; Alomari, A.I.; Mulliken, J.B.; Fishman, S.J.; Irons, M.B. The spectrum of vascular anomalies in patients with PTEN mutations: Implications for diagnosis and management. J. Med. Genet. 2007, 44, 594–602. [Google Scholar] [CrossRef]
- Qian, Y.; Shirasawa, S.; Chen, C.L.; Cheng, L.; Ma, Q. Proper development of relay somatic sensory neurons and D2/D4 interneurons requires homeobox genes Rnx/Tlx-3 and Tlx-1. Gene. Develop. 2002, 16, 1220–1233. [Google Scholar] [CrossRef]
- Dauger, S.; Pattyn, A.; Lofaso, F.; Gaultier, C.; Goridis, C.; Gallego, J.; Brunet, J.F. Phox2b controls the development of peripheral chemoreceptors and afferent visceral pathways. Development 2003, 130, 6635–6642. [Google Scholar] [CrossRef]
- Kim, B.N.; Kim, J.W.; Kang, H.; Cho, S.C.; Shin, M.S.; Yoo, H.J.; Hong, S.B.; Lee, D.S. Regional differences in cerebral perfusion associated with the alpha-2A-adrenergic receptor genotypes in attention deficit hyperactivity disorder. J. Psychiat. Neurosci. 2010, 35, 330–336. [Google Scholar]
- Miller, V.M.; Zhu, Y.; Bucher, C.; McGinnis, W.; Ryan, L.K.; Siegel, A.; Zalcman, S. Gestational flu exposure induces changes in neurochemicals, affiliative hormones and brainstem inflammation, in addition to autism-like behaviors in mice. Brain Behav. Immun. 2013, 33, 153–163. [Google Scholar] [CrossRef]
- Matsuda, K.; Park, C.H.; Sunden, Y.; Kimura, T.; Ochiai, K.; Kida, H.; Umemura, T. The vagus nerve is one route of transneural invasion for intranasally inoculated influenza A virus in mice. Vet. Pathol. 2004, 41, 101–107. [Google Scholar] [CrossRef]
- Shinya, K.; Shimada, A.; Ito, T.; Otsuki, K.; Morita, T.; Tanaka, H.; Takada, A.; Kida, H.; Umemura, T. Avian influenza virus intranasally inoculated infects the central nervous system of mice through the general visceral afferent nerve. Arch. Virol. 2000, 145, 187–195. [Google Scholar] [CrossRef]
- DeLong, G.R.; Bean, S.C.; Brown, F.R., 3rd. Acquired reversible autistic syndrome in acute encephalopathic illness in children. Arch. Neurol. 1981, 38, 191–194. [Google Scholar] [CrossRef]
- Friedman, D.P. Abnormalities of the posterior inferior cerebellar artery: MR imaging findings. Amer. J. Roentgenol. 1993, 160, 1257–1263. [Google Scholar] [CrossRef]
- Boris, M. Private Practice, Allergy and Immunology. Personal Communication, Woodbury, NY, USA, 20 April 2010. [Google Scholar]
- Prilipko, O.; Dehdashti, A.R.; Zaim, S.; Seeck, M. Orthostatic intolerance and syncope associated with Chiari type I malformation. J. Neurol. Neurosur. 2005, 76, 1034–1036. [Google Scholar] [CrossRef]
- Siclari, F.; Burger, I.M.; Fasel, J.H.; Gailloud, P. Developmental anatomy of the distal vertebral artery in relationship to variants of the posterior and lateral spinal arterial systems. Amer. J. Neuroradiol. 2007, 28, 1185–1190. [Google Scholar]
- Macchi, V.; Porzionato, A.; Parenti, A.; de Caro, R. The course of the posterior inferior cerebellar artery may be related to its level of origin. Surg. Radiol. Anatomy 2004, 26, 60–65. [Google Scholar] [CrossRef]
- Fein, J.M.; Frishman, W. Neurogenic hypertension related to vascular compression of the lateral medulla. Neurosurgery 1980, 6, 615–622. [Google Scholar] [CrossRef]
- Slotkin, T.A.; Seidler, F.J. Terbutaline impairs the development of peripheral noradrenergic projections: Potential implications for autism spectrum disorders and pharmacotherapy of preterm labor. Neurotoxicol. Teratol. 2013, 36, 91–96. [Google Scholar] [CrossRef]
- Saigal, S.; Usher, R.H. Sympathetic neonatal plethora. Biol. Neonate 1977, 32, 62–72. [Google Scholar]
- Rabe, H.; Diaz-Rossello, J.L.; Duley, L.; Dowswell, T. Effect of timing of umbilical cord clamping and other strategies to influence placental transfusion at preterm birth on maternal and infant outcomes. Cochrane Database Syst. Rev. 2012, 8. [Google Scholar] [CrossRef]
- Chavez-Valdez, R.; Mason, A.; Nunes, A.R.; Northington, F.J.; Tankersley, C.; Ahlawat, R.; Johnson, S.M.; Gauda, E.B. Effect of hyperoxic exposure during early development on neurotrophin expression in the carotid body and nucleus tractus solitarii. J. Appl. Physiol. 2012, 112, 1762–1772. [Google Scholar]
- Moyse, E.; Bauer, S.; Charrier, C.; Coronas, V.; Krantic, S.; Jean, A. Neurogenesis and neural stem cells in the dorsal vagal complex of adult rat brain: New vistas about autonomic regulations, a review. Auton. Neurosci. 2006, 126–127, 50–58. [Google Scholar]
- Bauer, S.; Hay, M.; Amilhon, B.; Jean, A.; Moyse, E. In vivo neurogenesis in the dorsal vagal complex of the adult rat brainstem. Neuroscience 2005, 130, 75–90. [Google Scholar] [CrossRef]
- Pecchi, E.; Dallaporta, M.; Charrier, C.; Pio, J.; Jean, A.; Moyse, E.; Troadec, J.D. Glial fibrillary acidic protein (GFAP)-positive radial-like cells are present in the vicinity of proliferative progenitors in the nucleus tractus solitarius of adult rat. J. Comp. Neurol. 2007, 501, 353–368. [Google Scholar] [CrossRef]
- Clark, C.G.; Hasser, E.M.; Kunze, D.L.; Katz, D.M.; Kline, D.D. Endogenous brain-derived neurotrophic factor in the nucleus tractus solitarius tonically regulates synaptic and autonomic function. J. Neurosci. 2011, 31, 12318–12329. [Google Scholar] [CrossRef]
- Kline, D.D.; Ogier, M.; Kunze, D.L.; Katz, D.M. Exogenous brain-derived neurotrophic factor rescues synaptic dysfunction in Mecp2-null mice. J. Neurosci. 2010, 30, 5303–5310. [Google Scholar] [CrossRef]
- Russell, J.A.; Ciucci, M.R.; Connor, N.P.; Schallert, T. Targeted exercise therapy for voice and swallow in persons with Parkinson’s disease. Brain Res. 2010, 1341, 3–11. [Google Scholar]
- Xu, D.; Gilkerson, J.; Richards, J.; Yapanel, U.; Gray, S. Child Vocalization Composition as Discriminant Information for Automatic Autism Detection. In Proceedings of the Engineering in Medicine and Biology Society Meeting, Minneapolis, MN, USA, 3 September 2009.
- Hoeve, L.J.; Goedegebure, A.; Joosten, K.F. Observations in a cohort of infants with severe laryngeal dyskinesia: Auditory brainstem response may aid in the diagnosis. Int. J. Pediat. Otorhinolaryngol. 2006, 70, 683–687. [Google Scholar] [CrossRef]
- Damasio, A.; Carvalho, G.B. The nature of feelings: Evolutionary and neurobiological origins. Nat. Rev. Neurosci. 2013, 14, 143–152. [Google Scholar]
- Mayer, E.A. Gut feelings: The emerging biology of gut-brain communication. Nat. Rev. Neurosci. 2011, 12, 453–466. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
McGinnis, W.R.; Audhya, T.; Edelson, S.M. Proposed Toxic and Hypoxic Impairment of a Brainstem Locus in Autism. Int. J. Environ. Res. Public Health 2013, 10, 6955-7000. https://doi.org/10.3390/ijerph10126955
McGinnis WR, Audhya T, Edelson SM. Proposed Toxic and Hypoxic Impairment of a Brainstem Locus in Autism. International Journal of Environmental Research and Public Health. 2013; 10(12):6955-7000. https://doi.org/10.3390/ijerph10126955
Chicago/Turabian StyleMcGinnis, Woody R., Tapan Audhya, and Stephen M. Edelson. 2013. "Proposed Toxic and Hypoxic Impairment of a Brainstem Locus in Autism" International Journal of Environmental Research and Public Health 10, no. 12: 6955-7000. https://doi.org/10.3390/ijerph10126955