Three Bianthraquinone Derivatives from the Mangrove Endophytic Fungus Alternaria sp. ZJ9-6B from the South China Sea

Abstract
:1. Introduction
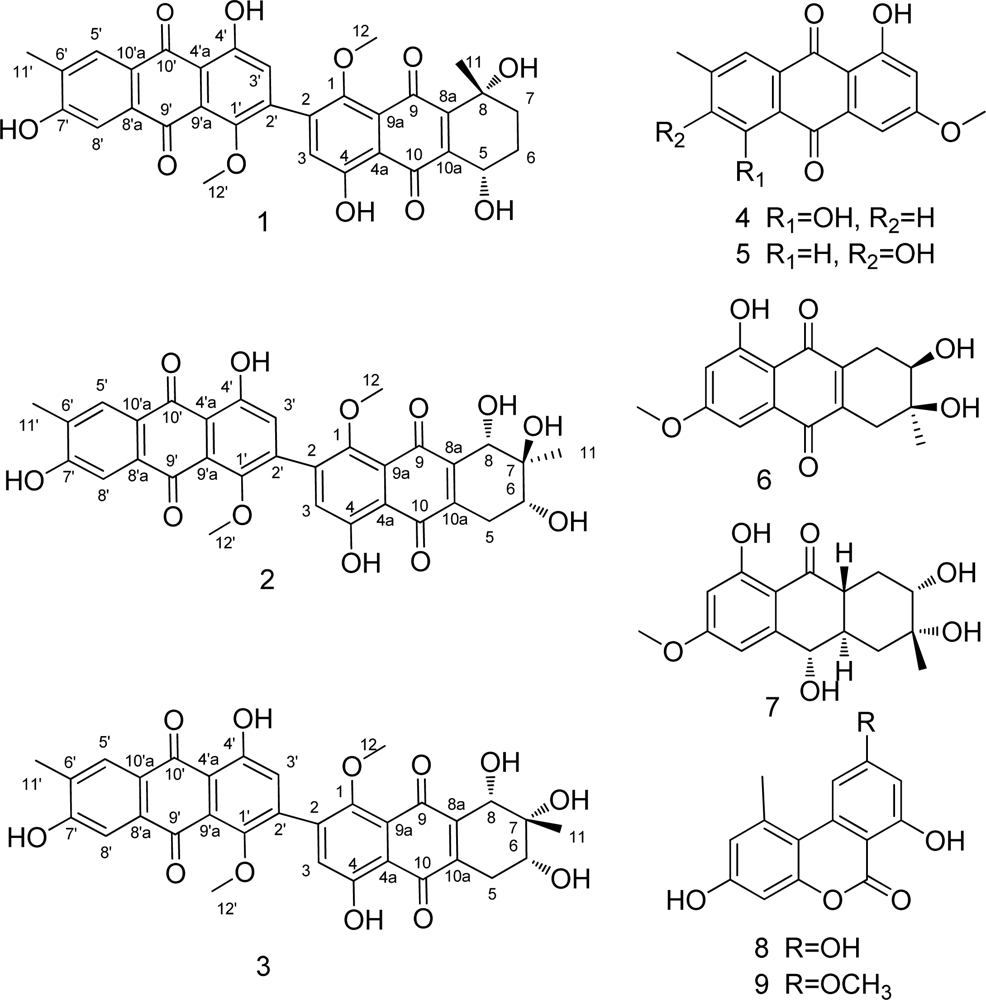
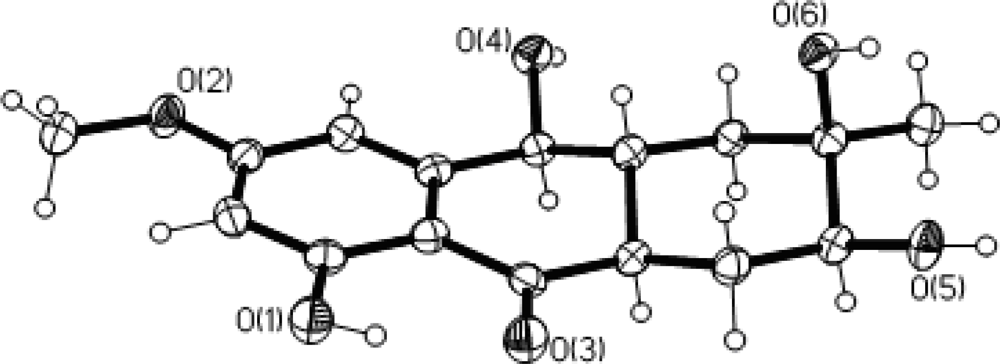
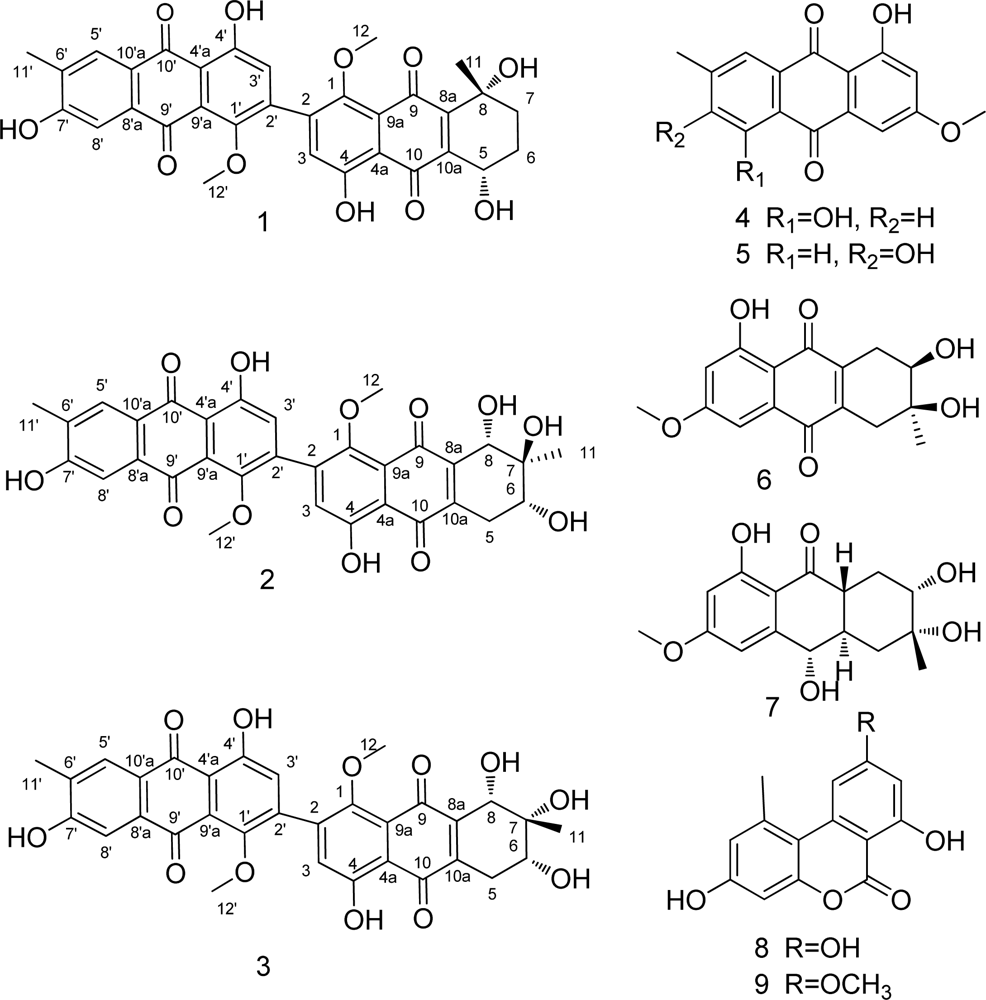
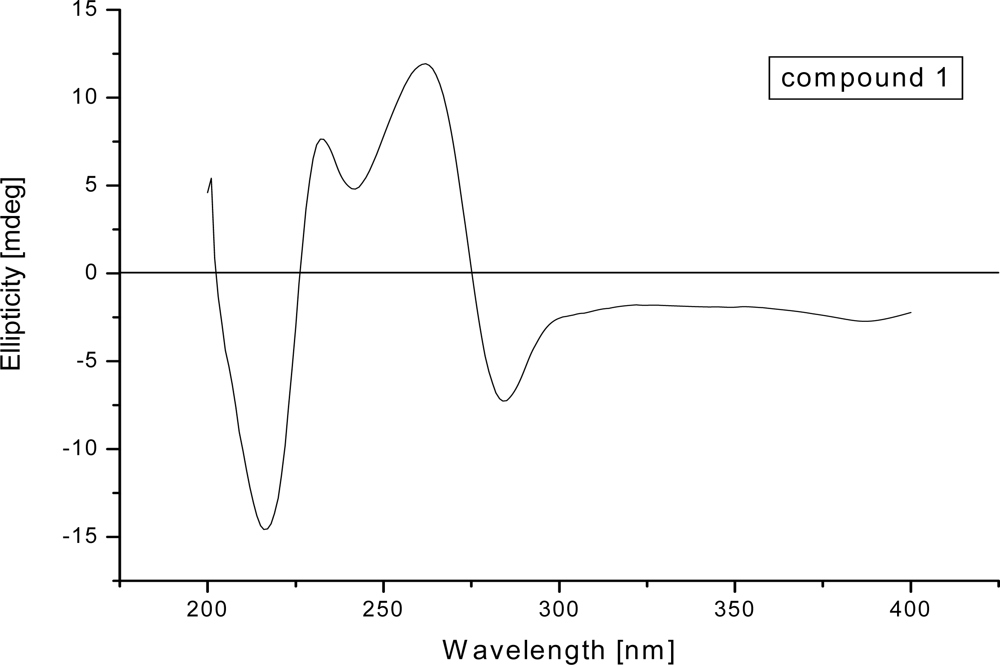
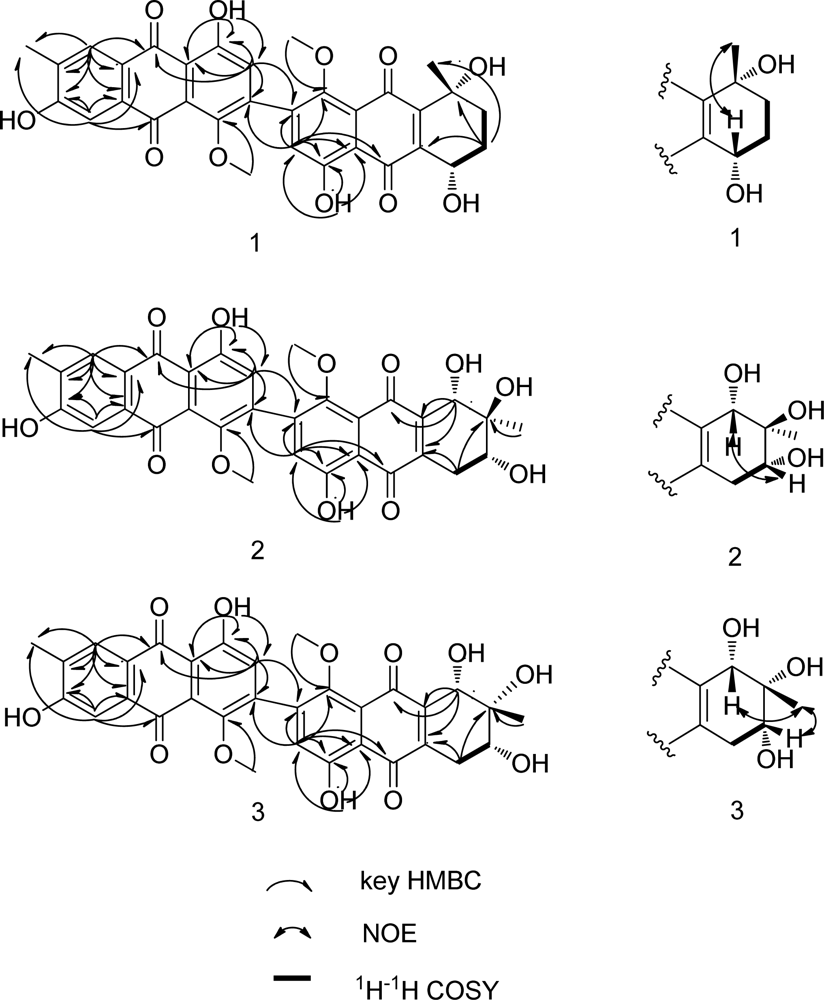
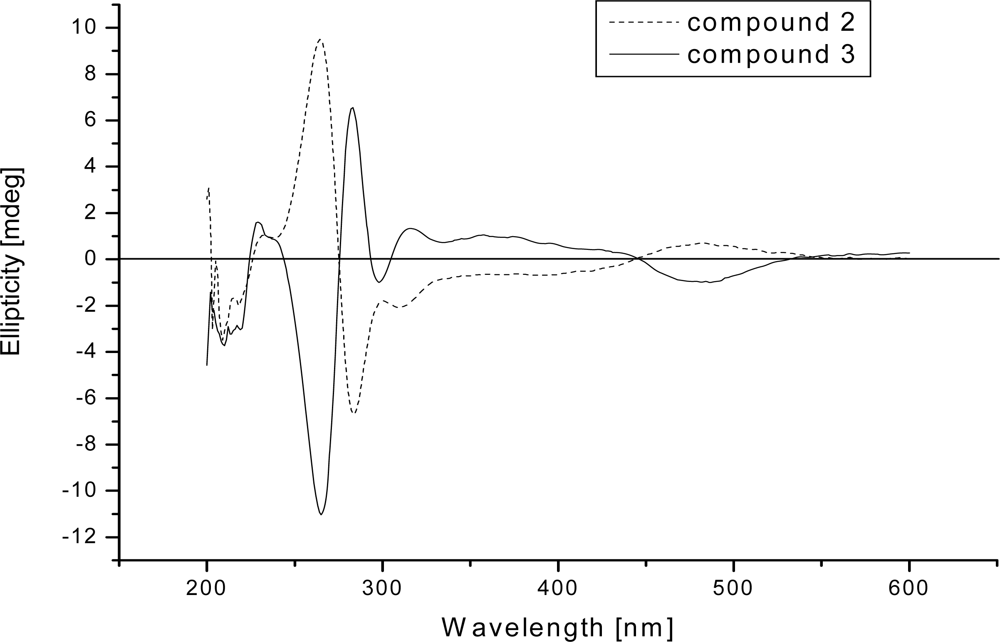
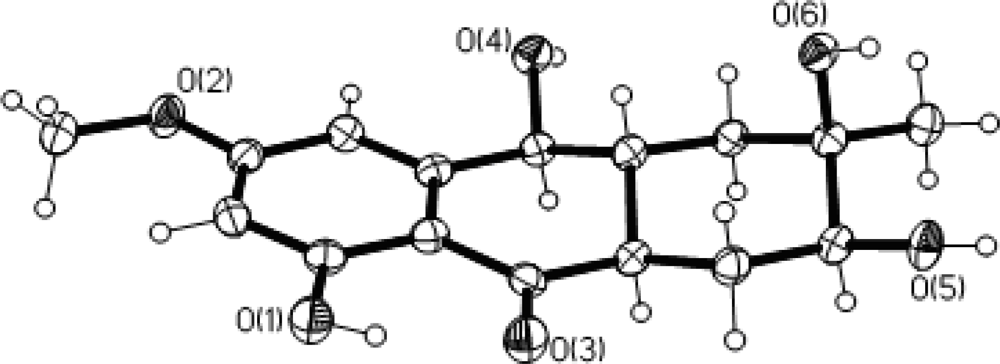
2. Results and Discussion
3. Experimental Section
3.1. General
3.2. Strain Isolation, Taxonomic Classification and Endophyte Fermentation
3.3. Extraction and Separation of Metabolites
3.4. Cytotoxic Assays
4. Conclusions
Supplementary Data
marinedrugs-09-00832-s001.pdfAcknowledgments
- Samples Availability: Available from the authors.
References
- Ji, XZ; Leslie, G. Microbial transformation of amino- and hydroxyanthraquinones by Beauveria bassiana ATCC 7159. J. Nat. Prod 2006, 69, 1525–1527. [Google Scholar]
- Gizachew, A; Berhanu, A. Bianthraquinones from the seeds of Senna multiglandulosa. Phytochemistry 1996, 41, 919–921. [Google Scholar]
- Zhang, JY; Tao, LY; Liang, YJ; Chen, LM; Mi, YJ; Zheng, LS; Wang, F; She, ZG; Lin, YC; Kenneth, KWT; Fu, LW. Anthracenedione derivatives as anticancer agents isolated from secondary metabolites of the mangrove endophytic fungi. Mar Drugs 2010, 8, 1469–1481. [Google Scholar]
- Robert, HC; Zhang, YJ; Navindra, PS; Muraleedharan, GN. Inhibition of human tumor cell proliferation by novel anthraquinones from daylilies. Life Sci 2004, 74, 1791–1799. [Google Scholar]
- Debbab, A; Aly, AH; Edrada-Ebel, R; Wray, V; Müller, WEG; Totzke, F; Zirrgiebel, U; Schächtele, C; Kubbutat, MHG; Lin, WH; et al. Bioactive metabolites from the endophytic fungus Stemphylium globuliferum isolated from Mentha pulegium. J Nat Prod 2009, 72, 626–631. [Google Scholar]
- Haraguchi, H; Abo, T; Fukuda, A; Okamura, N; Yagi, A. Mode of phytotoxic action of altersolanols. Phytochemistry 1996, 43, 989–992. [Google Scholar]
- Alison, DP; Su, BN; William, JK; Douglas, AK. An anthraquinone with potent quinone reductase-inducing activity and other constituents of the fruits of Morinda citrifolia (Noni). J Nat Prod 2005, 68, 1720–1722. [Google Scholar]
- Sergio, ML; Araceli, PV; Rachel, M. Natural products with calmodulin inhibitor properties. Phytochemistry 2007, 68, 1882–1903. [Google Scholar]
- Muhammad, S; Mamona, N; Muhammad, S; Hidayat, H; Yong, S; Naheed, R; Abdul, J. Antimicrobial natural products: An update on future antibiotic drug candidates. Nat Prod Rep 2010, 27, 238–254. [Google Scholar]
- Suemitsu, R; Sano, T; Yamamoto, M; Arimoto, Y; Morimatsu, F; Nabeshima, T. Structural elucidation of alterporriol B, a novel metabolic pigment produced by Alternaria porri (Ellis) ciferri. Agric Biol Chem 1984, 48, 2611–2613. [Google Scholar]
- Suemitsu, R; Ueshima, T; Yamamoto, T; Yanagawase, S. Alterporriol C: A modified bianthraquinone from alternaria porri. Phytochemistry 1988, 27, 3251–3254. [Google Scholar]
- Suemitsu, R; Horiuchi, K; Kubota, M; Okamatse, T. Production of alterporriols, altersolanols and macrosporin by Alternaria porri and A Solani. Phytochemistry 1990, 29, 1509–1511. [Google Scholar]
- Pretsch, A; Proksch, P; Debbab, A. Novel Anthraquinone Derivatives. Patent WO/2010/135759, 2 December 2010. [Google Scholar]
- Wen, L; Cai, XL; Xu, F; She, ZG; Chan, WL; Vrijimoed, LLP; Jones, EBG; Lin, YC. Three metabolites from the mangrove endophytic fungus Sporothrix sp. (#4335) from the South China Sea. J Org Chem 2009, 74, 1093–1098. [Google Scholar]
- Yang, JX; Xu, F; Huang, CH; Li, J; She, ZG; Pei, Z; Lin, YC. Metabolites from the mangrove endophytic fungus Phomopsis sp. (#zsu-H76). Eur J Org Chem 2010, 19, 3692–3695. [Google Scholar]
- Chen, B; Wang, DY; Ye, Q; Li, BG; Zhang, GL. Anthranquinones from Gladiolus gandavensis. J Asian Nat Prod Res 2005, 7, 197–204. [Google Scholar]
- Bringmann, G; Kraus, J; Menche, D; Messer, K. Elucidation of the absolute configuration of knipholone and Knipholone anthrone by quantum chemical CD calculation. Tetrahedron 1999, 55, 7563–7572. [Google Scholar]
- Bringmann, G; Menche, D; Kraus, J; Mühlbacher, J; Peters, K; Peters, EM; Brun, R; Bezabih, M; Abegaz, BM. Atropo-Enantioselective total synthesis of knipholone and related antiplasmodial phenylanthrquinones. J Org Chem 2002, 67, 5595–5610. [Google Scholar]
- Bringmann, G; Mortimer, JPA; Keller, PA; Gresser, MJ; Garner, J; Breuning, M. Atroposelective synthesis of axially chiral biaryl compounds. Angew Chem Int Ed 2005, 44, 5384–5427. [Google Scholar]
- Bringmann, G; Mühlbacher, J; Reichert, M; Dreyer, M; Kolz, J; Speicher, A. Stereochemistry of isoplagiochin C, a macrocyclic bisbibenzyl from liverworts. J Am Chem Soc 2004, 126, 9283–9290. [Google Scholar]
- Gizachew, A; Berhanu, A; Snatzke, G; Duddeck, H. Bianthraquinones and a spermidine alkaloid from Cassia floribunda. Phytochemistry 1988, 27, 3255–3258. [Google Scholar]
- Suemitsu, R; Sakurai, Y; Nakachi, K; Miyoshi, I; Kubota, M; Ohnishi, K. Alterporriol D and E, modified bianthraquinones from Alternaria porri (ellis) ciferri. Agric Biol Chem 1989, 53, 1302–1304. [Google Scholar]
- Stoessl, A; Stothers, JB. Tetrahydroaltersolanol B, a hexahydroxnthronol from Alternaria solani. Can J Chem 1983, 61, 378–382. [Google Scholar]
- An, YH; Zhao, TZ; Miao, J; Liu, GT; Zheng, YZ; Xu, YM; Van, E; Robert, L. Isolation, identification and mutagenicity of alternariol monomethyl ether. J Agric Food Chem 1989, 37, 1341–1343. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom | δC (ppm) | δH (ppm) (multiplicity, J (Hz)) | COSY b | HMBC b | NOESY |
|---|---|---|---|---|---|
| 1 | 164.1 | ||||
| 2 | 123.3 | ||||
| 3 | 103.7 | 6.88 (s) | C-1, 2, 2′, 4, 4a, 10 | ||
| 4 | 163.8 | ||||
| 4a | 108.6 | ||||
| 5 | 70.1 | 3.51 (ddd, 5.5, 5.5, 12.5) | H-6a, 6b, 5-OH | C-6, 7, 8a, 11 | H-11 |
| 6 | 29.1 | a: 2.53 (m) | H-5, 7a, 7b | C-5, 8, 10a | H-11 |
| b: 2.72 (m) | H-5, 6a, 7a, 7b | C-5, 8, 10a, 11 | |||
| 7 | 36.1 | a: 2.31 (m) | H-6a, 6b, 7b | C-5, 8, 8a, 11 | |
| b: 2.35 (m) | H-6a, 6b, 7a | C-5, 8, 8a, 11 | H-11 | ||
| 8 | 69.0 | ||||
| 8a | 141.6 | ||||
| 9 | 183.6 | ||||
| 9a | 128.9 | ||||
| 10 | 187.8 | ||||
| 10a | 143.2 | ||||
| 11 | 25.2 | 1.07 (s) | C-5, 7, 8 | ||
| 12 | 56.7 | 3.66 (s) | C-1, 3 | ||
| 4-OH | 13.15 (s) | C-3, 4, 4a | |||
| 5-OH | 4.69 (d, 5.5) | H-5 | C-5, 6, 8 | ||
| 8-OH | 4.30 (s) | C-5, 7, 8, 11 | |||
| 1′ | 164.7 | ||||
| 2′ | 122.5 | ||||
| 3′ | 103.8 | 6.92 (s) | C-2, 2′, 4′, 4′a, 10′ | ||
| 4′ | 165.0 | ||||
| 4′a | 110.0 | ||||
| 5′ | 110.5 | 7.55 (d, 0.8) | C-6′, 7′, 8′a, 10′, 10′a, 11′ | ||
| 6′ | 125.2 | ||||
| 7′ | 161.3 | ||||
| 8′ | 130.3 | 7.67 (d, 0.8) | C-7′, 8′a, 9′, 10′a, 11′ | ||
| 8′a | 132.4 | ||||
| 9′ | 181.1 | ||||
| 9′a | 130.7 | ||||
| 10′ | 186.7 | ||||
| 10′a | 132.2 | ||||
| 11′ | 16.1 | 2.18 (s) | C-5′, 7′, 8′ | ||
| 12′ | 56.8 | 3.69 (s) | C-1′ | ||
| 4′-OH | 13.63 (s) | C-3′, 4′, 4′a | |||
| 7′-OH | 8.11 (s) |
| Actom | 2 | 3 | ||||
|---|---|---|---|---|---|---|
| δC (ppm) | δH (ppm) (multiplicity, J (Hz)) | HMBC | δC (ppm) | δH (ppm) (multiplicity, J (Hz)) | HMBC | |
| 1 | 164.3 | 164.5 | ||||
| 2 | 123.2 | 123.5 | ||||
| 3 | 103.6 | 6.90 (s) | C-1, 2, 2′, 4, 4a, 10 | 103.4 | 6.92 (s) | C-1, 2, 2′, 4, 4a, 10 |
| 4 | 163.8 | 163.9 | ||||
| 4a | 108.8 | 108.8 | ||||
| 5 | 28.8 | a: 2.33 (dd, 19.5, 10.0) | C-6, 8a, 10a | 28.8 | a: 2.33 (dd, 19.3, 9.9) | C-6, 8a, 10a |
| b: 2.79 (dd, 19.5, 6.0) | C-6, 7, 8a, 10a | b: 2.78 (dd, 19.3, 5.9) | C-6, 7, 8a, 10, 10a | |||
| 6 | 66.7 | 3.70 c | 66.6 | 3.69 c | ||
| 7 | 71.9 | 71.9 | ||||
| 8 | 69.0 | 4.03 (d, 6.7) | C-6, 7, 9, 8a, 10a | 69.0 | 4.06 (d, 5.7) | |
| 8a | 142.6 | 142.6 | ||||
| 9 | 183.3 | 183.1 | ||||
| 9a | 128.9 | 128.3 | ||||
| 10 | 188.4 | 188.3 | ||||
| 10a | 143.7 | 143.6 | ||||
| 11 | 21.8 | 1.16 (3H, s) | C-6, 7, 8 | 21.8 | 1.16 (3H, s) | C-6, 7, 8 |
| 12 | 56.7 | 3.68 (3H, s) | C-1 | 56.7 | 3.68 (3H, s) | C-1 |
| 4-OH | 13.11 (s) | C-3, 4, 4a | 13.13 (s) | C-3, 4, 4a | ||
| 6-OH | 2.17 (s) | 2.16 (s) | ||||
| 7-OH | 4.25 (s) | C-7, 8 | 4.48 (s) | |||
| 8-OH | 5.42 (d, 6.7) | C-6, 7, 8 | 5.27 (d, 5.7) | |||
| 1′ | 164.4 | 164.9 | ||||
| 2′ | 122.3 | 122.5 | ||||
| 3′ | 104.0 | 6.92(s) | C-2, 2′, 4′, 4′a, 10′ | 103.5 | 6.89 (s) | C-1′, 2, 2′, 4′, 4′a,10′ |
| 4′ | 164.9 | 165.1 | ||||
| 4′a | 110.0 | 110.0 | ||||
| 5′ | 110.5 | 7.55 (s) | C-6′, 7′, 8′a, 10′, 10′a, 11′ | 110.7 | 7.51 (s) | C-6′, 7′, 8′a, 10′, 10′a, 11′ |
| 6′ | 125.0 | 125.2 | ||||
| 7′ | 161.6 | 162.8 | ||||
| 8′ | 130.2 | 7.68 (s) | C-7′, 8′a, 9′, 10′a, 11′ | 130.1 | 7.65 (s) | C-7′, 8′a, 9′, 10′a, 11′ |
| 8′a | 132.4 | 132.4 | ||||
| 9′ | 181.1 | 180.6 | ||||
| 9′a | 131.2 | 130.7 | ||||
| 10′ | 186.8 | 186.9 | ||||
| 10′a | 132.3 | 132.4 | ||||
| 11′ | 16.1 | 2.19 (s) | C-5′, 6′, 7′, 8′, 10′a | 16.3 | 2.18 (s) | C-5′, 6′, 7′, 8′ |
| 12′ | 56.7 | 3.68 (s) | C-1′ | 56.8 | 3.71 (s) | C-1′ |
| 4′-OH | 13.61 (s) | C-3′, 4′, 9′a | 13.70 (s) | C-3′, 4′, 9′a | ||
| 7′-OH | 7.65 (s) | C-7′, 8′a, 9′, 10′, 11′ | 7.67 (s) | C-5′, 7′, 8′a, 11′ | ||
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Huang, C.-H.; Pan, J.-H.; Chen, B.; Yu, M.; Huang, H.-B.; Zhu, X.; Lu, Y.-J.; She, Z.-G.; Lin, Y.-C. Three Bianthraquinone Derivatives from the Mangrove Endophytic Fungus Alternaria sp. ZJ9-6B from the South China Sea. Mar. Drugs 2011, 9, 832-843. https://doi.org/10.3390/md9050832
Huang C-H, Pan J-H, Chen B, Yu M, Huang H-B, Zhu X, Lu Y-J, She Z-G, Lin Y-C. Three Bianthraquinone Derivatives from the Mangrove Endophytic Fungus Alternaria sp. ZJ9-6B from the South China Sea. Marine Drugs. 2011; 9(5):832-843. https://doi.org/10.3390/md9050832
Chicago/Turabian StyleHuang, Cai-Huan, Jia-Hui Pan, Bin Chen, Miao Yu, Hong-Bo Huang, Xun Zhu, Yong-Jun Lu, Zhi-Gang She, and Yong-Cheng Lin. 2011. "Three Bianthraquinone Derivatives from the Mangrove Endophytic Fungus Alternaria sp. ZJ9-6B from the South China Sea" Marine Drugs 9, no. 5: 832-843. https://doi.org/10.3390/md9050832