Lobophorin C and D, New Kijanimicin Derivatives from a Marine Sponge-Associated Actinomycetal Strain AZS17

Abstract
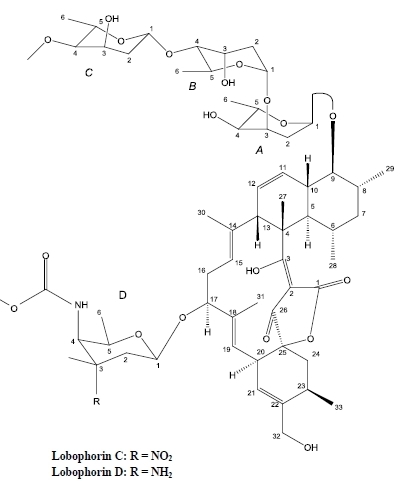
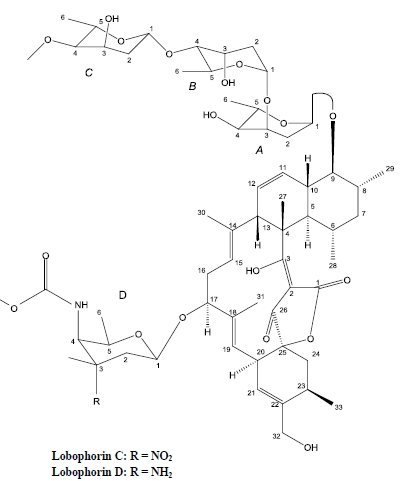
:
1. Introduction
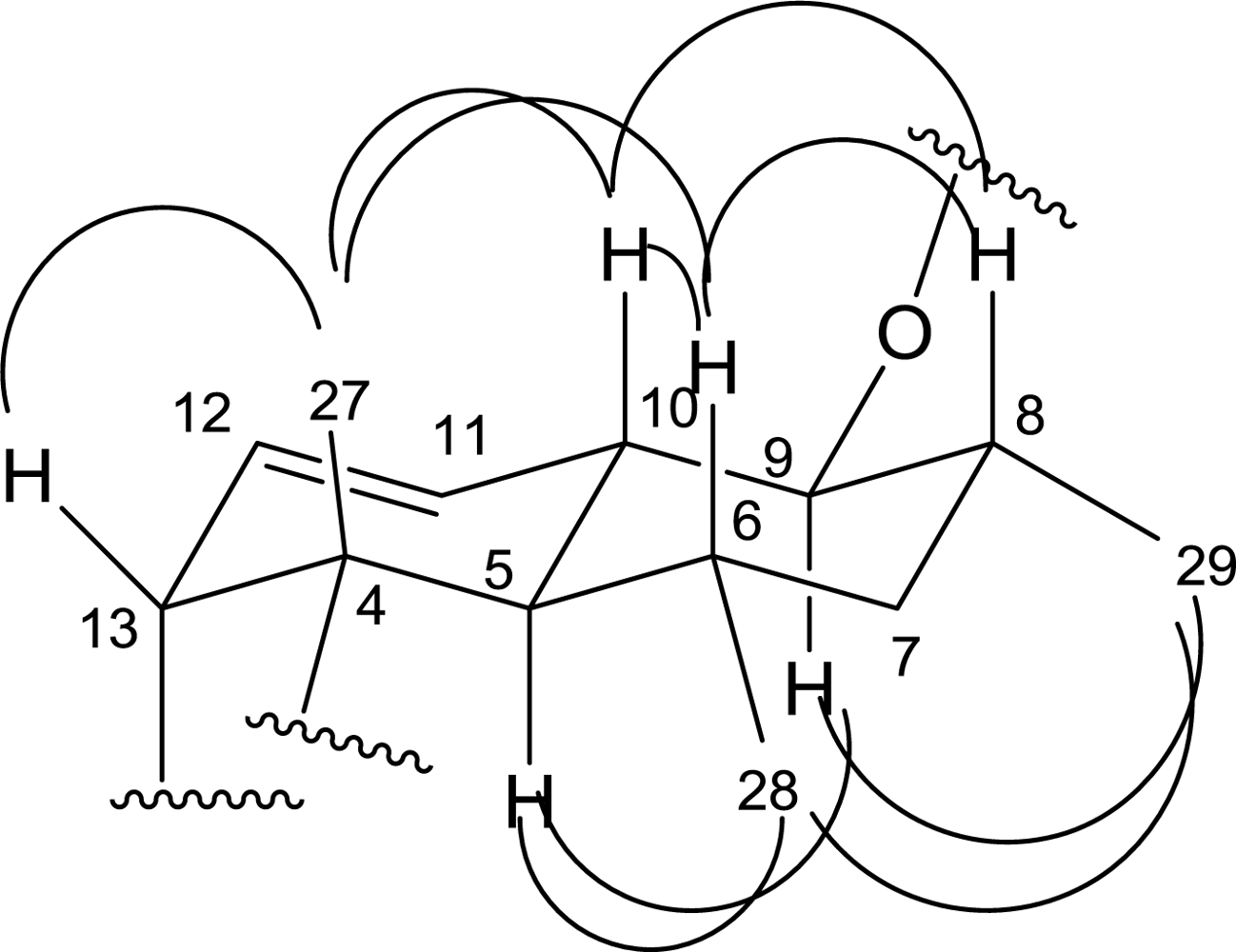
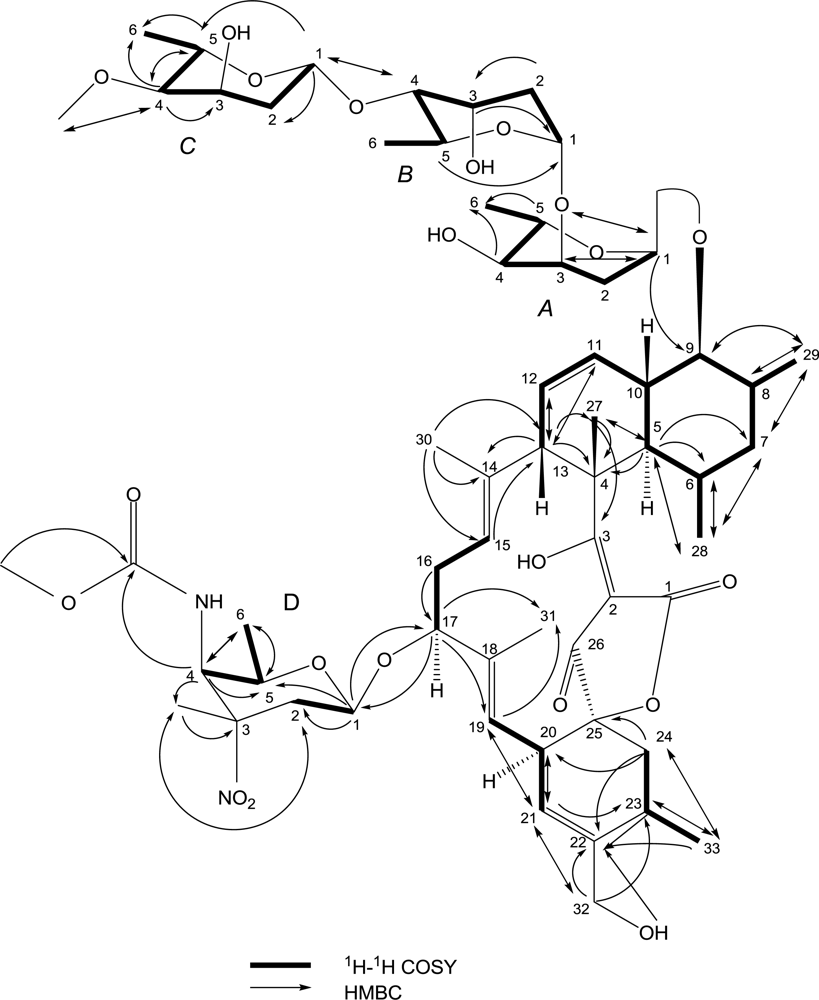
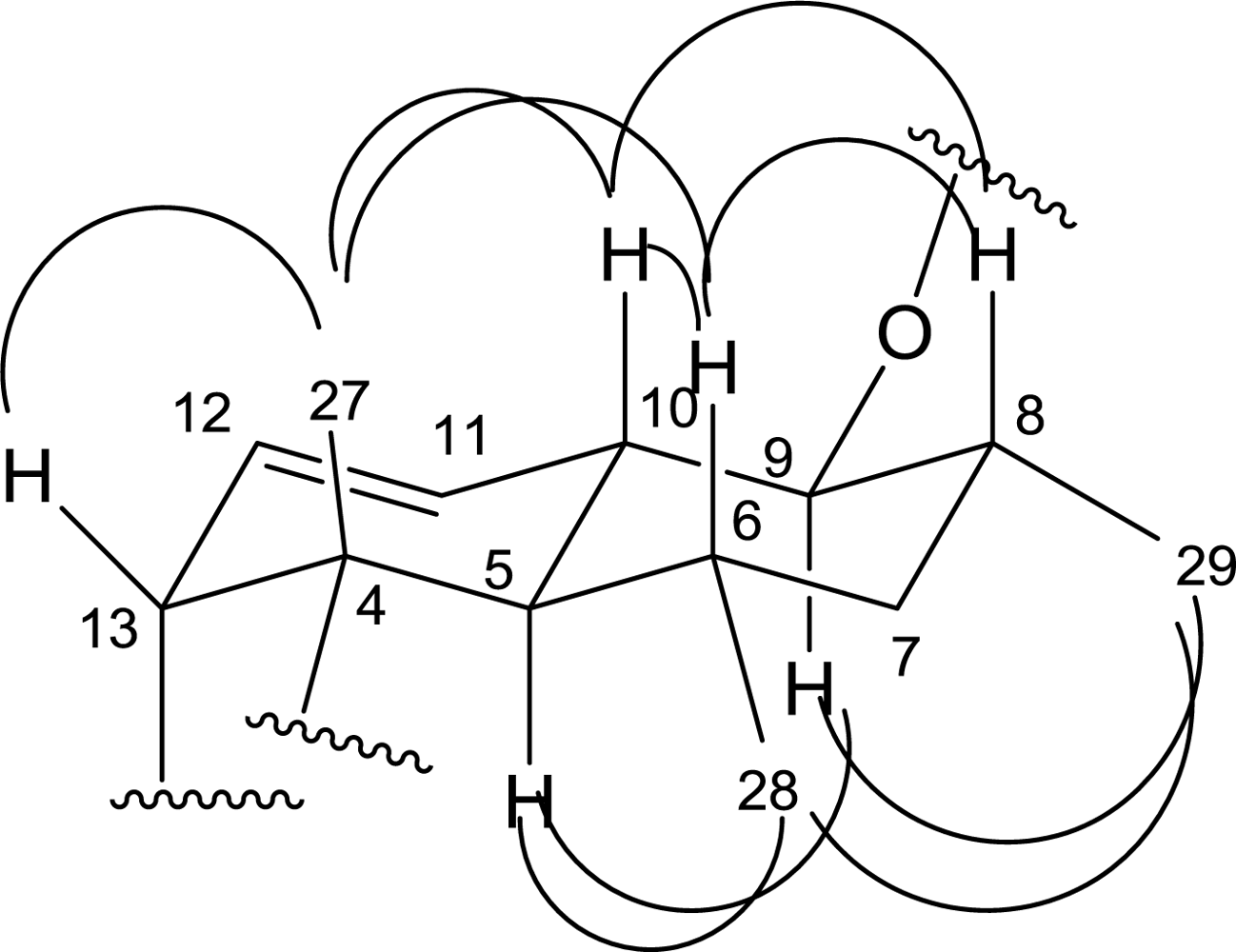
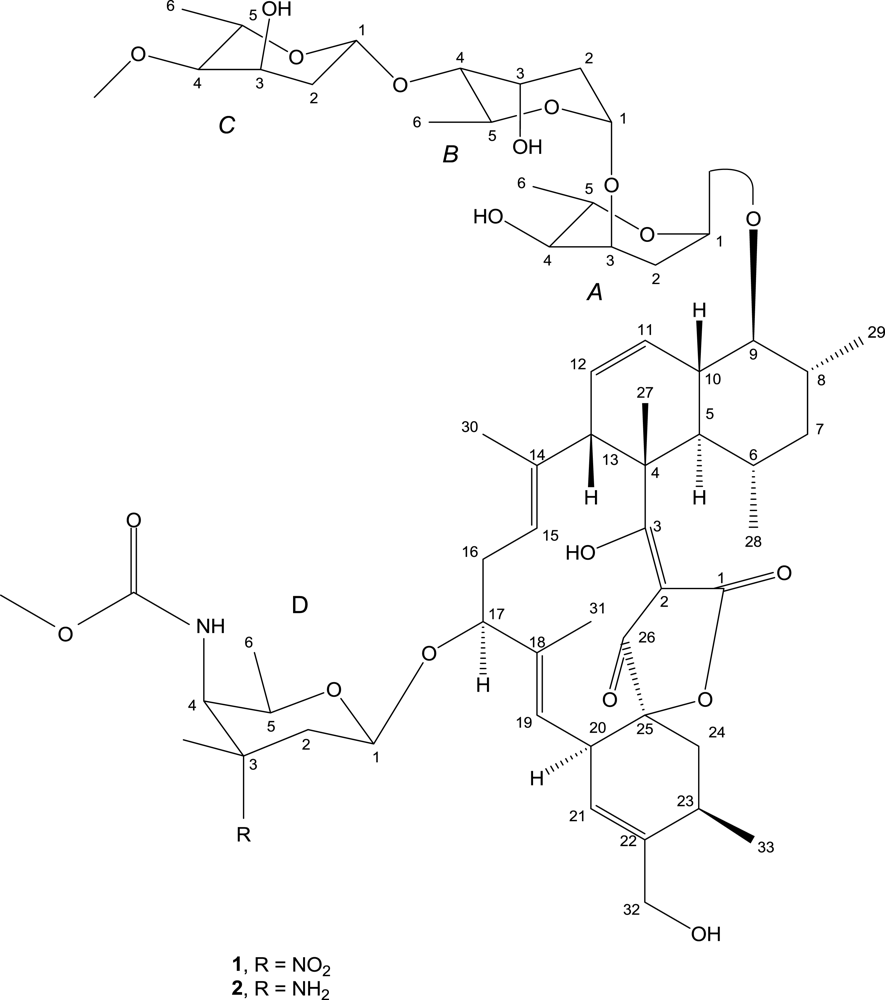
2. Results and Discussion
3. Experimental Section
3.1. General Procedures
3.2. Strain Isolation and Fermentation
3.3. Extraction and Isolation
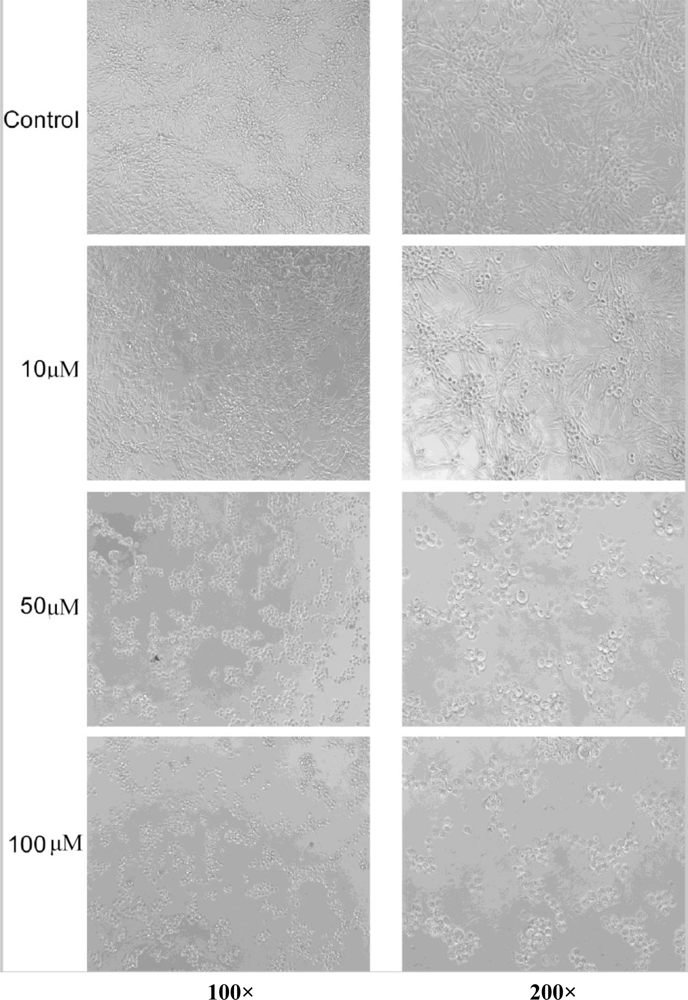
3.4. Cytotoxicity Assays and Observation
Acknowledgments
- Samples Availability: Available from the authors.
References
- Hentschel, U; Hopke, J; Horn, M; Friedrich, AB; Wagner, M; Hacker, J; Moore, BS. Molecular evidence for a uniform microbial community in sponges from different oceans. Appl Environ Microb 2002, 68, 4431–4440. [Google Scholar]
- Pimentel-Elardo, SM; Kozytska, S; Bugni, TS; Ireland, CM; Moll, H; Hentschel, U. Anti-parasitic compounds from Streptomyces sp. strains isolated from Mediterranean sponges. Mar Drugs 2010, 8, 373–380. [Google Scholar]
- Liu, R; Cui, CB; Duan, L; Gu, QQ; Zhu, WM. Potent in vitro anticancer activity of metacycloprodigiosin and undecylprodigiosin from a sponge-derived actinomycete Saccharopolyspora sp. Nov Arch Pharm Res 2005, 28, 1341–1344. [Google Scholar]
- Bringmann, G; Lang, G; Steffens, S; Günther, E; Schaumann, K. Evariquinone, isoemericellin, and stromemycin from a sponge derived strain of the fungus. Emericella variecolor Phytochemistry 2003, 63, 437–443. [Google Scholar]
- Bringmann, G; Lang, G; Gulder, TAM; Tsuruta, H; Muhlbacher, J; Maksimenka, K; Steffens, S; Schaumann, K; Stohr, R; Wiese, J. The first sorbicillinoid alkaloids, the antileukemic sorbicillactones A and B, from a sponge-derived Penicillium chrysogenum strain. Tetrahedron 2005, 61, 7252–7265. [Google Scholar]
- Zheng, L; Chen, H; Han, X; Lin, W; Yan, X. Antimicrobial screening and active compound isolation from marine bacterium NJ6-3-1 associated with the sponge. Hymeniacidon perleve World J Microbiol Biotechnol 2005, 21, 201–206. [Google Scholar]
- Thomas, TR; Kavlekar, DP; LokaBharathi, PA. Marine Drugs from Sponge-Microbe Association—A Review. Mar Drugs 2010, 8, 1417–1468. [Google Scholar]
- Jiang, ZD; Jensen, PR; Fenical, W. Lobophorins A and B, new antiinflammatory macrolides produced by a tropical marine bacterium. Bioorg Med Chem Lett 1999, 9, 2003–2006. [Google Scholar]
- Mallams, AK; Puar, MS; Rossman, RR; McPhail, AT; MacFarlane, RD. Kijanimicin 2: Structure and absolute stereochemistry of kijanimicin. J Am Chem Soc 1981, 103, 3940–3943. [Google Scholar]
- Denizot, F; Lang, R. Rapid colorimetric assay for cell growth and survival: Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 1986, 89, 271–277. [Google Scholar]

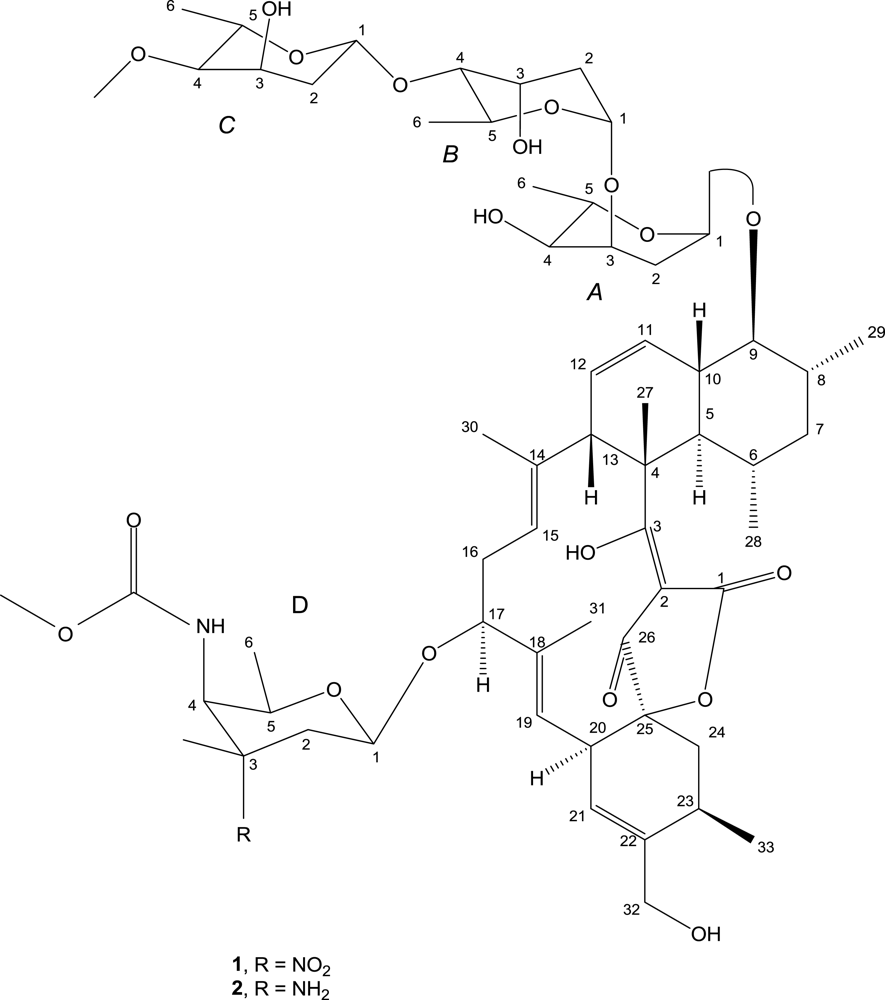


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 13C | 1H | HMBC | 1H-1H COSY | NOESY |
|---|---|---|---|---|---|
| 1 | 173.6 C | ||||
| 2 | 96.5 C | ||||
| 3 | 197.4 C | ||||
| 4 | 50.4 C | ||||
| 5 | 43.4 CH | 2.00(m,overlap *) | C-28,27,7,6,4 | H-6,10 | H-28,9 |
| 6 | 31.1 CH | 1.38(m,overlap) | C-28 | H-28 | H-27,10,8 |
| 7 | 41.7 CH2 | 1.51(m,overlap), 1.38(m,overlap) | C-29,28 | H-8,6 | |
| 8 | 34.1 CH | 2.09(m,overlap) | C-29 | H-29 | H-10,6 |
| 9 | 84.0 CH | 3.29(m,overlap) | C-29 | H-10,8 | H-29,5 |
| 10 | 38.5 CH | 1.89(m,overlap) | H-11,9,5 | H-27,8,6 | |
| 11 | 125.2 CH | 5.65(d,10.5) | C-13 | H-12 | |
| 12 | 128.0 CH | 5.28(m,overlap) | C-13 | H-13,11 | |
| 13 | 50.1 CH | 3.71(m,overlap) | C-27,14,12,11,4 | H-12 | H-27 |
| 14 | 136.0 C | ||||
| 15 | 121.4 CH | 5.06(brs) | C-13 | H-16 | |
| 16 | 35.1 CH2 | 2.06(m,overlap) | C-17 | H-17 | |
| 17 | 79.1 CH | 4.07(d,5.6) | C: 31,19, D-1 | H-16 | |
| 18 | 135.1 C | ||||
| 19 | 120.4 CH | 5.02(d,8) | C-31,21 | H-20 | |
| 20 | 39.5 CH | 3.21(d,8) | C-21 | H-21,19 | |
| 21 | 123.3 CH | 5.31(s) | C-32,23,20,19 | H-20 | |
| 22 | 140.6 C | ||||
| 23 | 27.1 CH | 2.45(m,overlap) | C-33,22 | H-33,24 | |
| 24 | 35.5 CH2 | 2.48(m,overlap) 1.43(m,overlap) | C-33,25,22,20 | H-23 | |
| 25 | 91.1 C | ||||
| 26 | 199.9 C | ||||
| 27 | 14.8 CH3 | 1.38(s) | C-5,4,3 | H-13,10,6 | |
| 28 | 22.3 CH3 | 0.55(d,4.4) | C-7,6,5 | H-6 | H-29,5 |
| 29 | 13.9 CH3 | 1.01(d,5.6) | C-9,8,7 | H-8 | H-28,9 |
| 30 | 15.0 CH3 | 1.41(s) | C-15,14,13 | ||
| 31 | 14.0 CH3 | 1.26(s) | |||
| 32 | 63.5 CH2 | 4.02(m,overlap) 3.88(m,overlap) | C-23,22,21 | OH-4.60 | |
| 33 | 19.8 CH3 | 1.16(d,5.6) | C-24,23,22 | H-23 | |
| A-2 | 29.2 CH2 | 2.21(brd,12) 1.65(brd,12) | H: A-2, A-1 | ||
| A-3 | 66.9 CH | 3.85(m,overlap) | C: A-1 | H: A-4, A-2 | |
| A-4 | 70.8 CH | 3.15(m,overlap) | C: A-6 | H: A-5, A-3, OH4.79 | |
| A-5 | 64.0 CH | 3.96(m,overlap) | C: A-6, A-1 | H: A-6, A-4 | |
| A-6 | 17.4 CH3 | 1.09(d,5.2) | H: A-5 | ||
| B-1 | 91.2 CH | 5.05(brs) | H: B-2 | ||
| B-2 | 35.2 CH2 | 1.85(m,overlap) | C: B-3 | H: B-3, B-1 | |
| B-3 | 66.2 CH | 3.91(brs) | C: B-1 | H: B-2, B-4 | |
| B-4 | 81.4 CH | 3.14(m,overlap) | C: C-1 | H: B-3, B-5 | |
| B-5 | 61.9 CH | 3.96(m,overlap) | C: B-1 | H: B-4, B-6 | |
| B-6 | 17.9 CH3 | 1.10(d,5.2) | H: B-5 | ||
| C-1 | 99.1 CH | 4.82(d,7.6) | C: C-5, C-2, B-4 | H: C-2 | |
| C-2 | 37.8 CH2 | 1.54(m,overlap) | H: C-3, C-1 | ||
| C-3 | 61.9 CH | 4.15(brs) | H: C-4, C-2, OH4.60 | ||
| C-4 | 82.1 CH | 2.76(dd,9.5,2.5) | C: C-6, C-5, C-3, C-4OCH3 | H: C-5, C-3 | |
| C-5 | 67.5 CH | 3.69(m,overlap) | C: C-6, C-4 | H: C-6, C-4 | |
| C-6 | 18.2 CH3 | 1.13(d,5.2) | H: C-5 | ||
| C-4OCH3 | 55.7 CH3 | 3.26(s) | C: C-4 | ||
| D-1 | 97.6 CH | 4.36(d,7.6) | C: 17, D-5, D-2 | H: D-2 | |
| D-2 | 30.8 CH2 | 2.27(m,overlap) 2.08(m,overlap) | C: D-3CH3 | H: D-2, D-1 | |
| D-3 | 82.0 C | ||||
| D-4 | 53.1 CH | 4.24(d,8.4) | C: D-6,D-5, D-4C=O, D-3CH3 | H: D-5, NH | |
| D-5 | 68.6 CH | 3.43(m,overlap) | C: D-6 | H: D-6, D-4 | |
| D-6 | 16.8 CH3 | 1.01(d,5.2) | C: D-5, D-4 | H: D-5 | |
| D-3CH3 | 24.9 CH3 | 1.43(s) | C: D-3, D-2 | ||
| D-4C=O | 157.7 C | ||||
| D-4OCH3 | 51.9 CH3 | 3.59(s) | C: D-4 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wei, R.-B.; Xi, T.; Li, J.; Wang, P.; Li, F.-C.; Lin, Y.-C.; Qin, S. Lobophorin C and D, New Kijanimicin Derivatives from a Marine Sponge-Associated Actinomycetal Strain AZS17. Mar. Drugs 2011, 9, 359-368. https://doi.org/10.3390/md9030359
Wei R-B, Xi T, Li J, Wang P, Li F-C, Lin Y-C, Qin S. Lobophorin C and D, New Kijanimicin Derivatives from a Marine Sponge-Associated Actinomycetal Strain AZS17. Marine Drugs. 2011; 9(3):359-368. https://doi.org/10.3390/md9030359
Chicago/Turabian StyleWei, Rong-Bian, Tao Xi, Jing Li, Ping Wang, Fu-Chao Li, Yong-Cheng Lin, and Song Qin. 2011. "Lobophorin C and D, New Kijanimicin Derivatives from a Marine Sponge-Associated Actinomycetal Strain AZS17" Marine Drugs 9, no. 3: 359-368. https://doi.org/10.3390/md9030359
APA StyleWei, R.-B., Xi, T., Li, J., Wang, P., Li, F.-C., Lin, Y.-C., & Qin, S. (2011). Lobophorin C and D, New Kijanimicin Derivatives from a Marine Sponge-Associated Actinomycetal Strain AZS17. Marine Drugs, 9(3), 359-368. https://doi.org/10.3390/md9030359