New Tetromycin Derivatives with Anti-Trypanosomal and Protease Inhibitory Activities †

Abstract
:1. Introduction
2. Results and Discussion
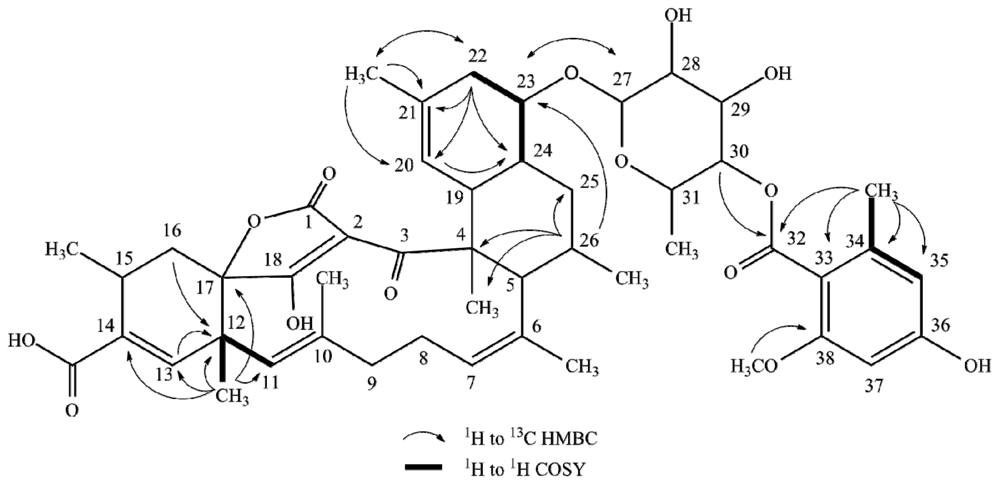
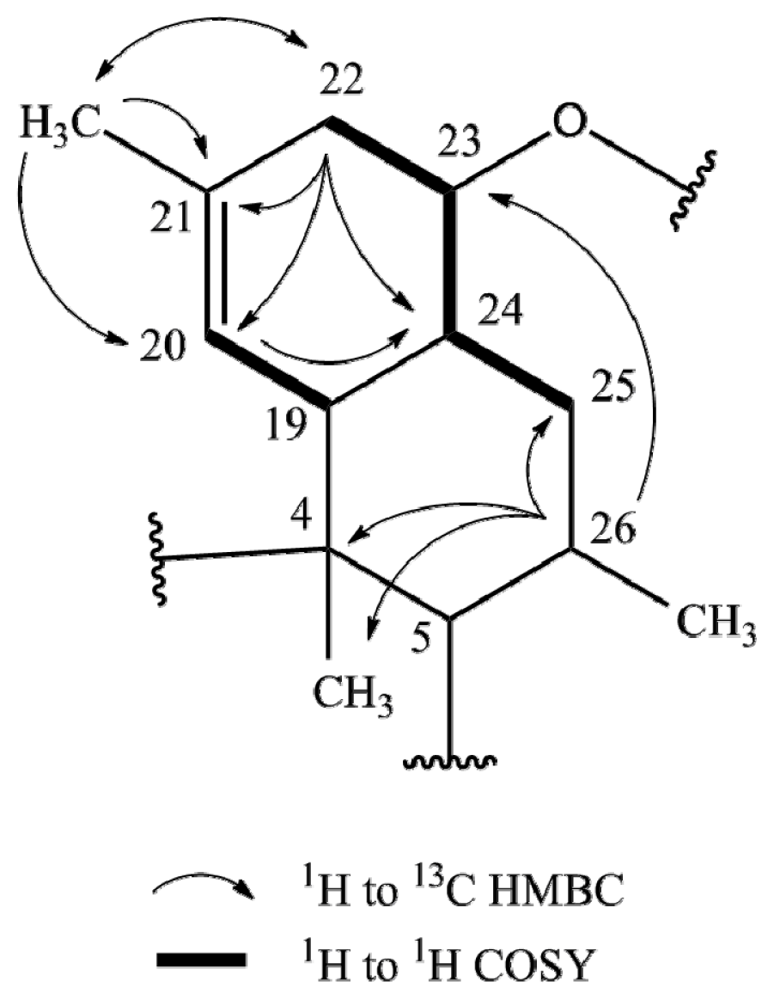
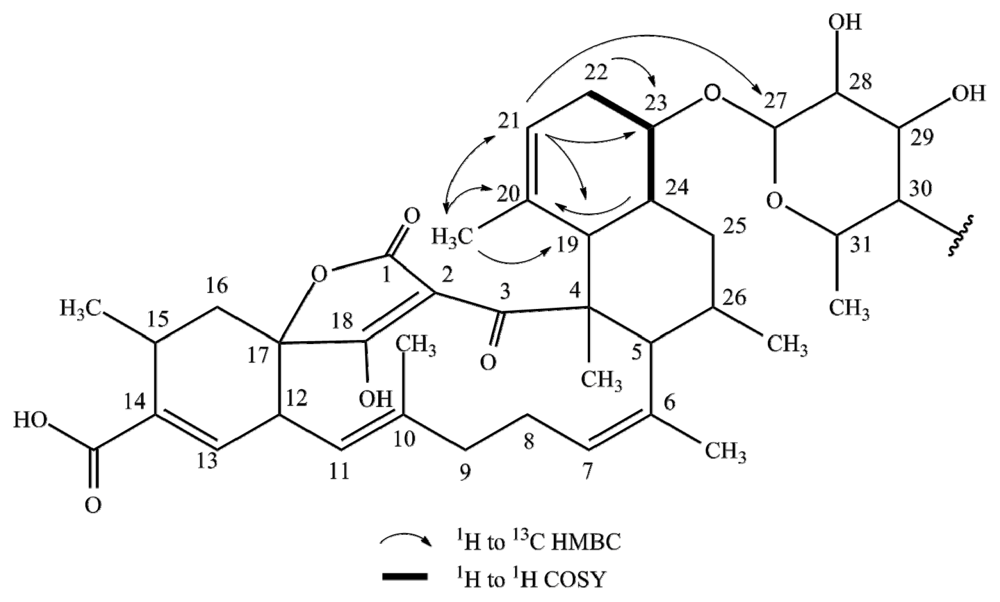
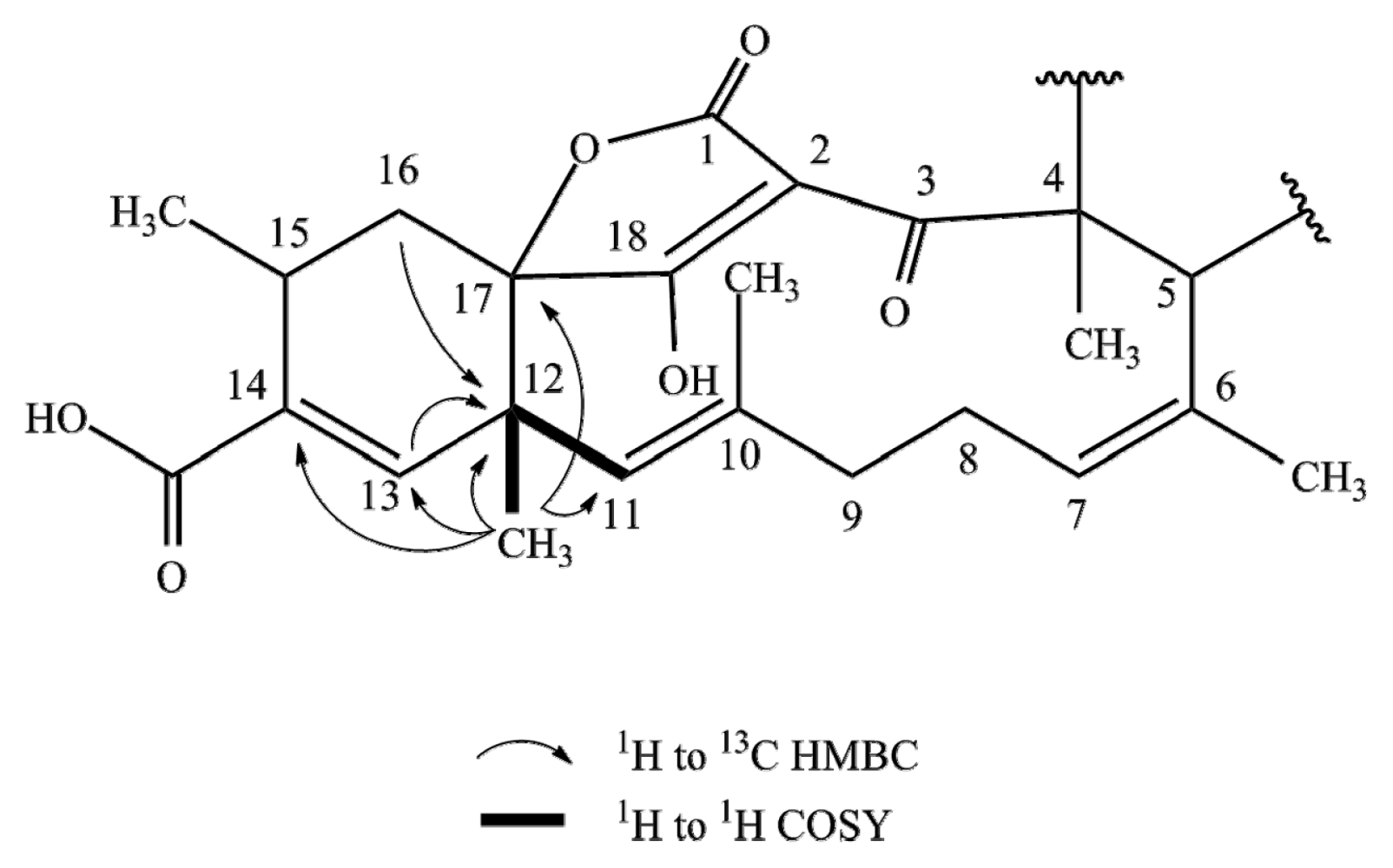
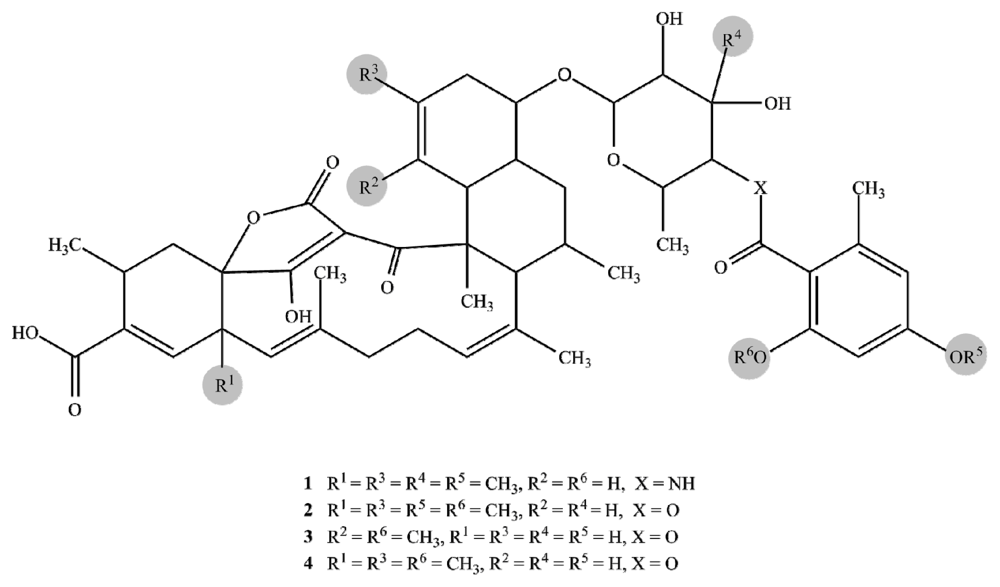
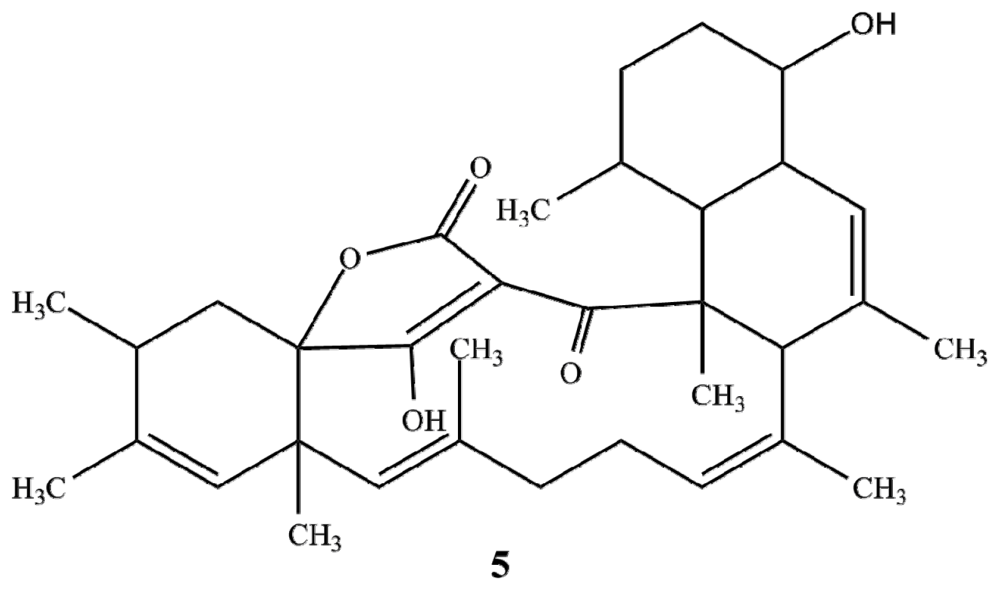
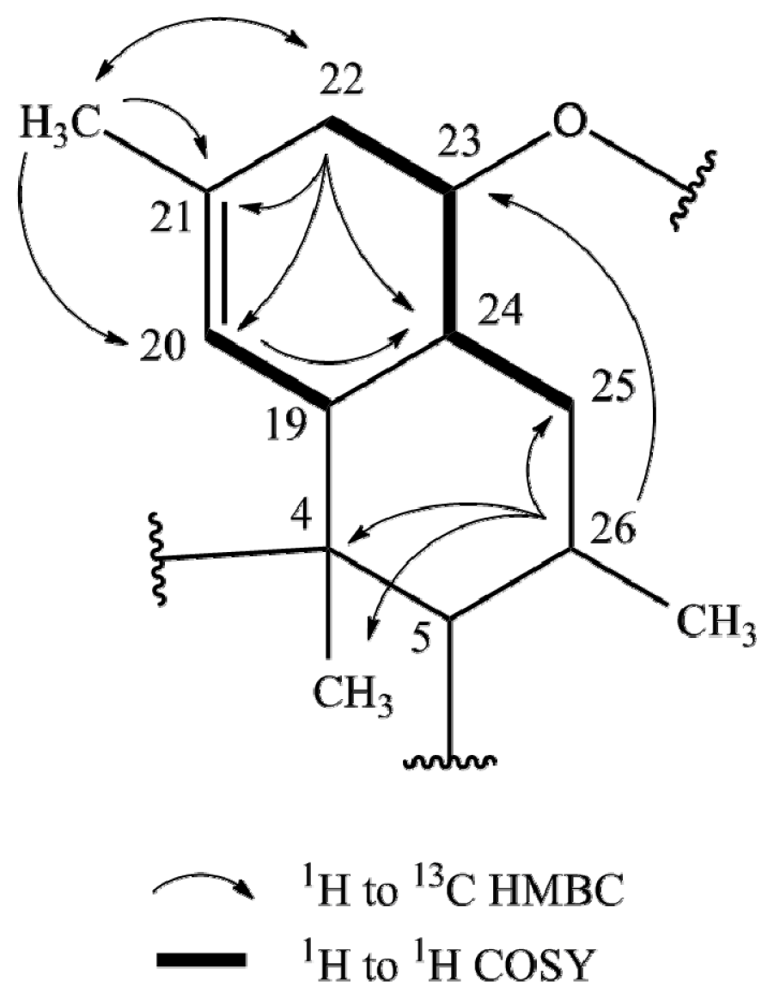
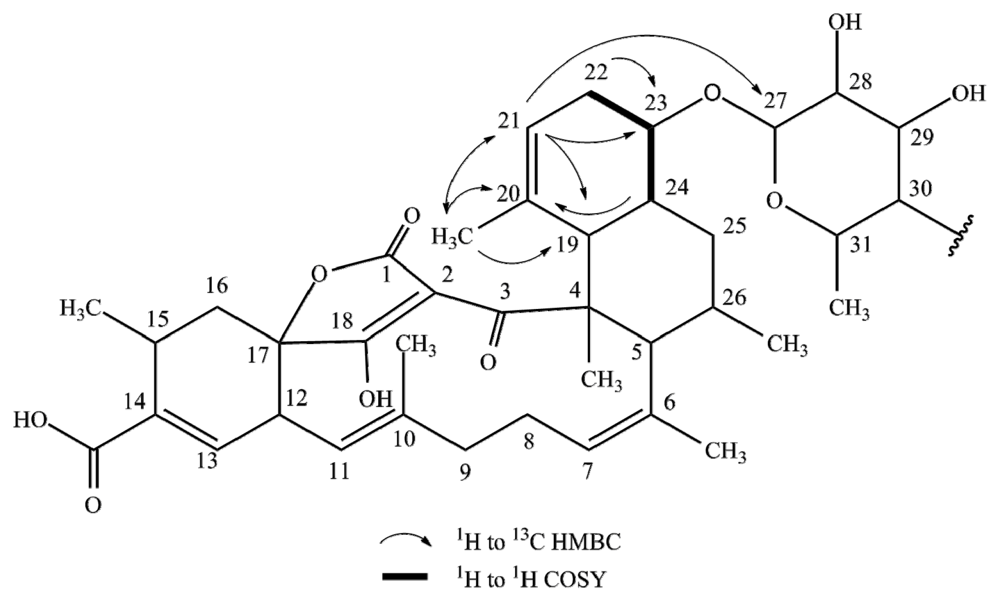
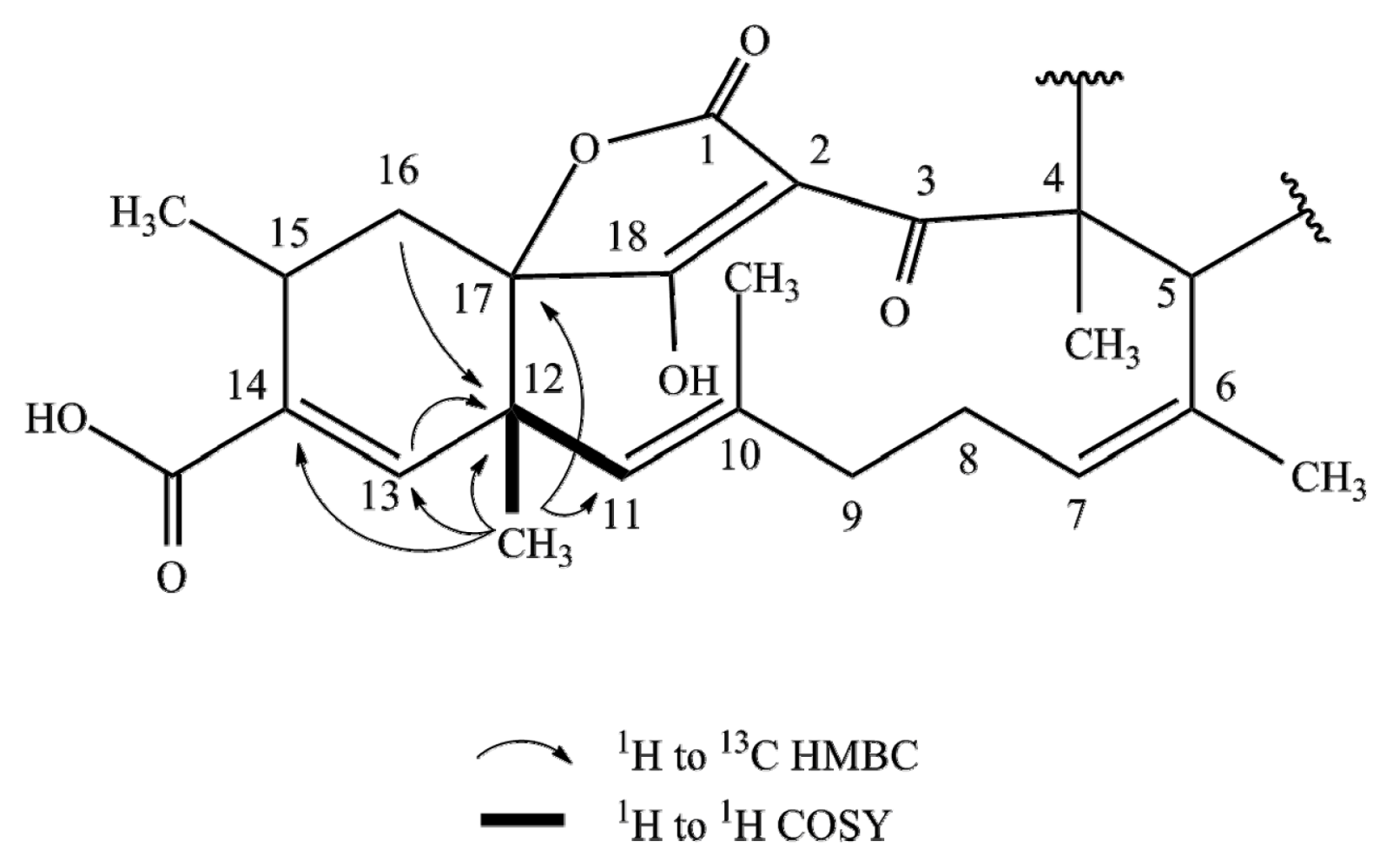
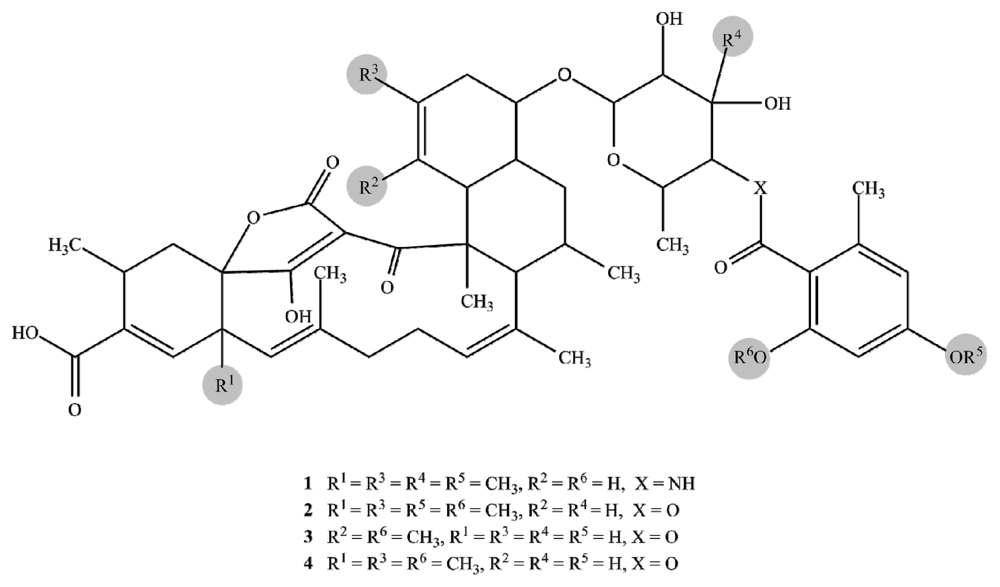
2.1. Structure Elucidation
2.2. Antiparasitic Activities
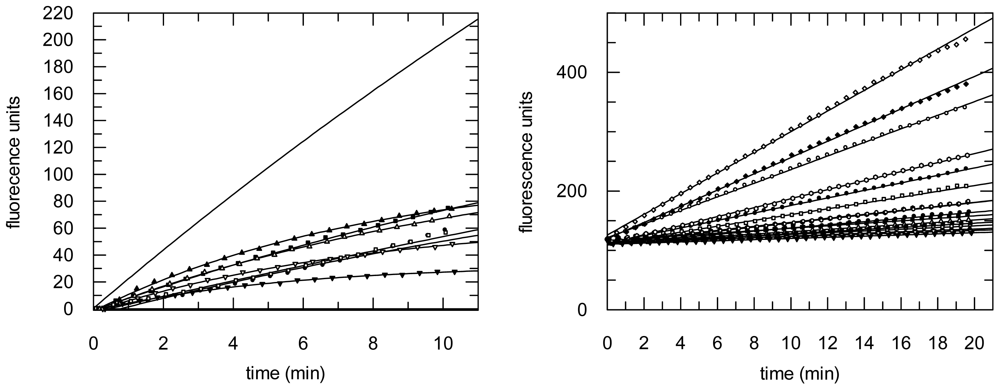
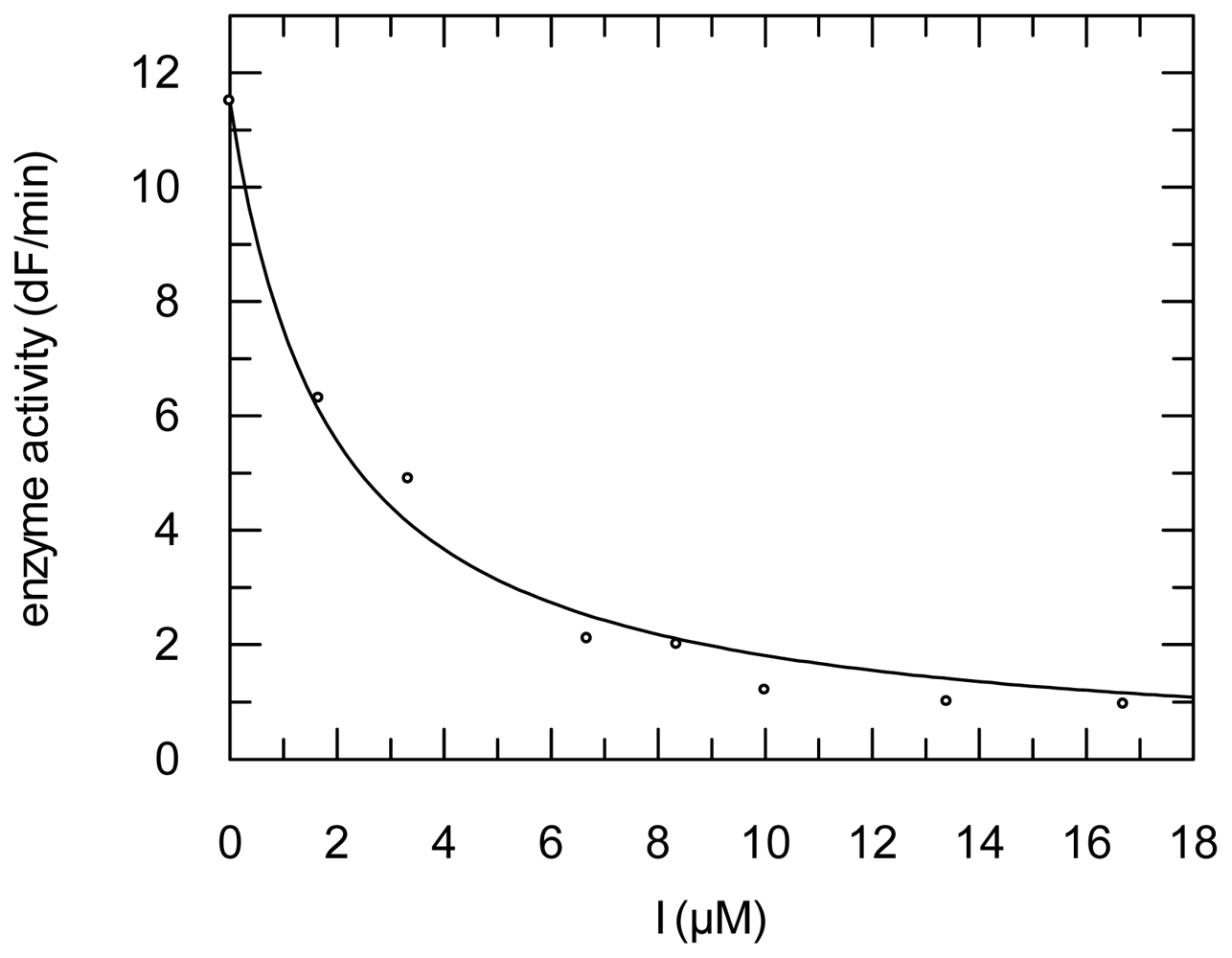
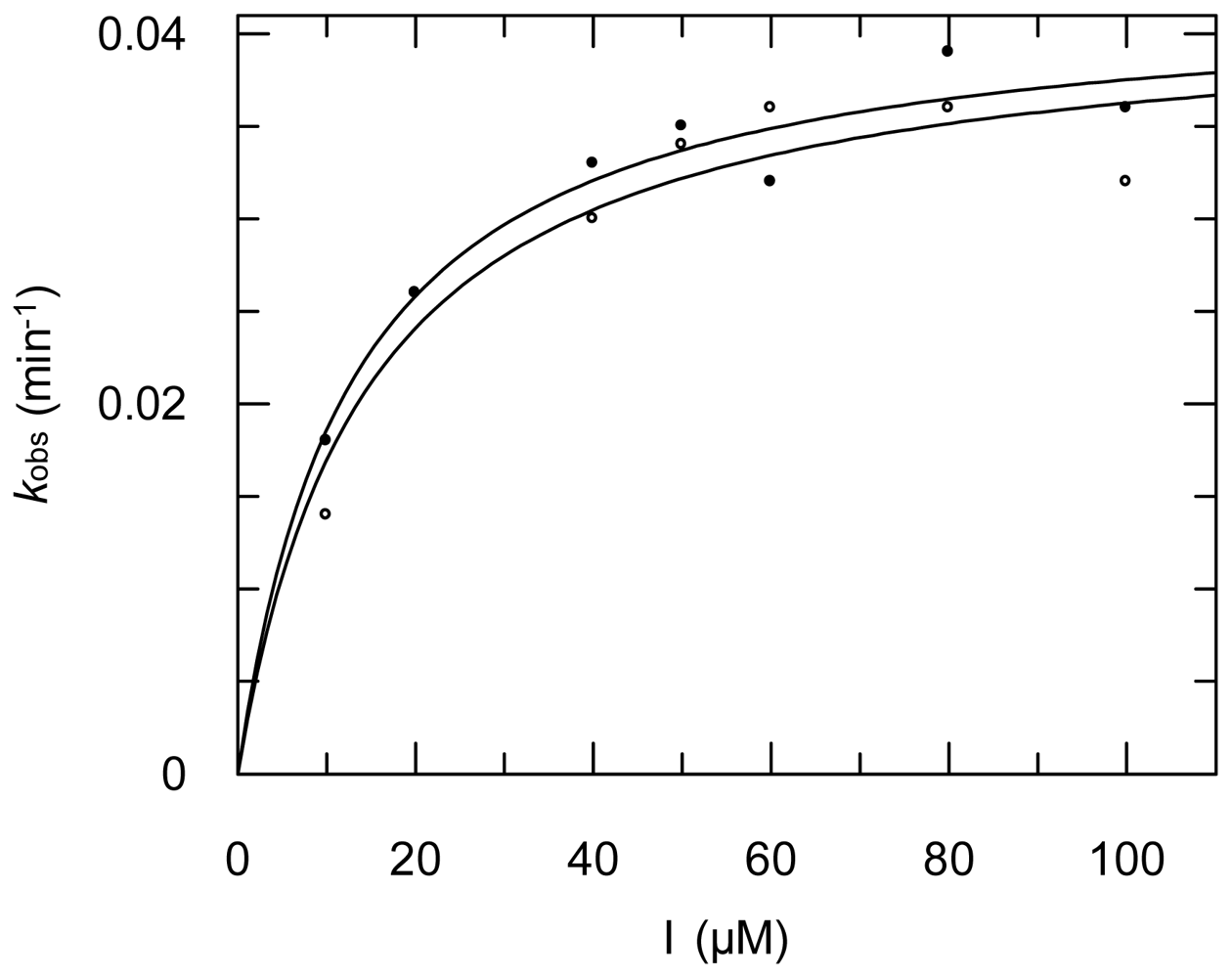
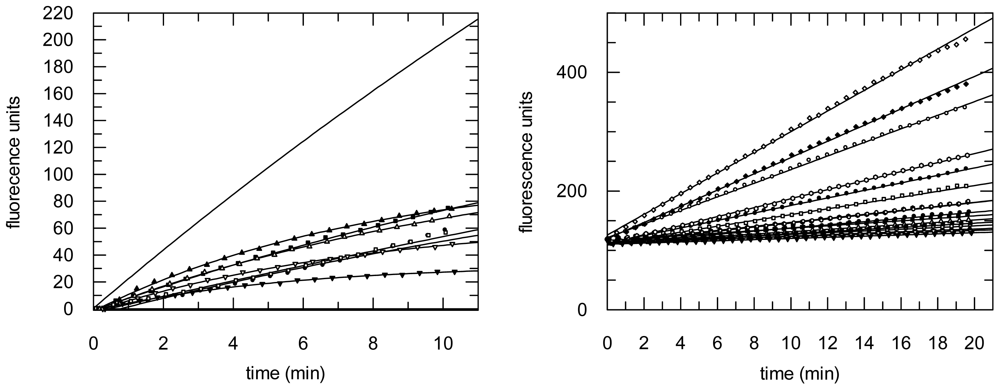
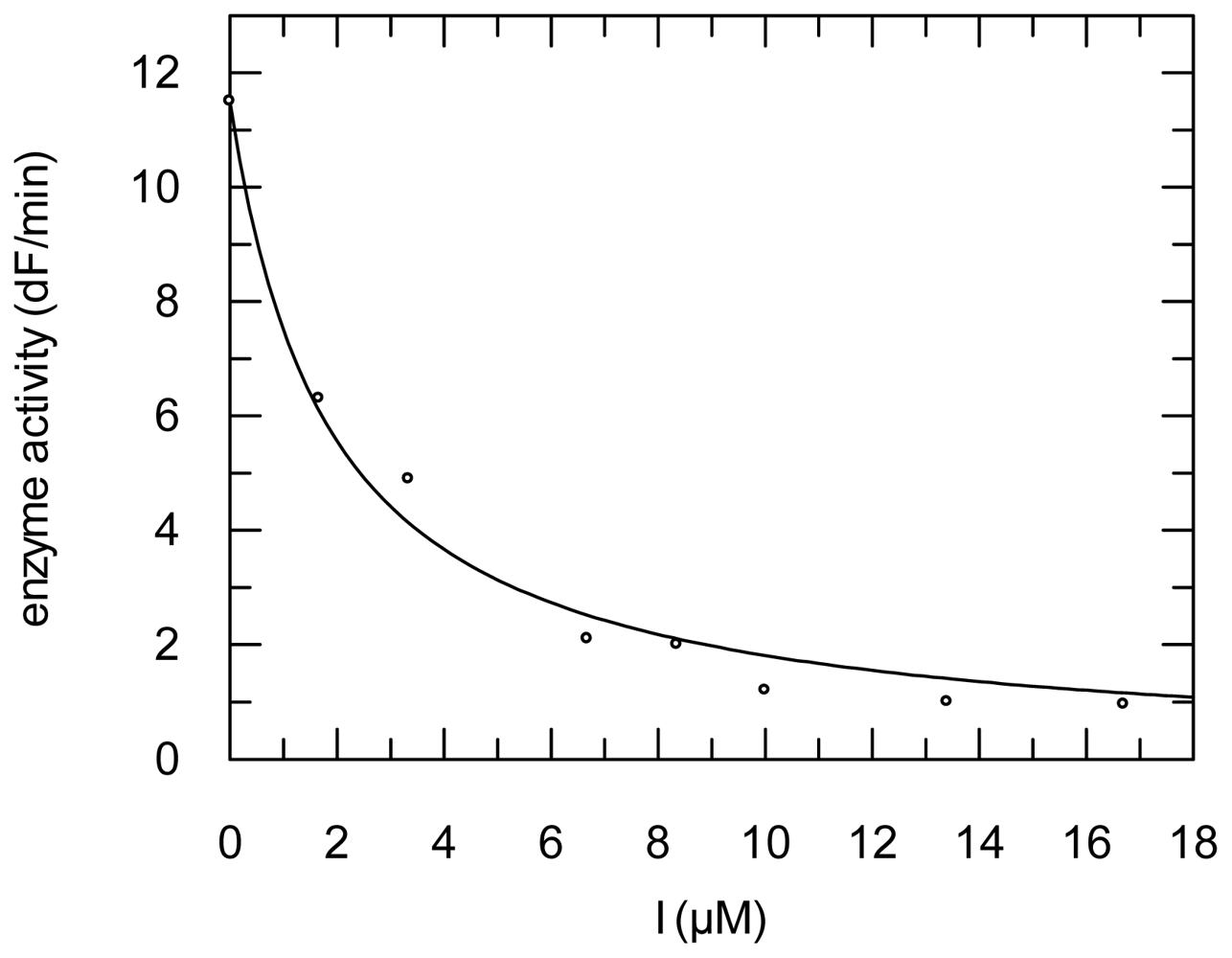
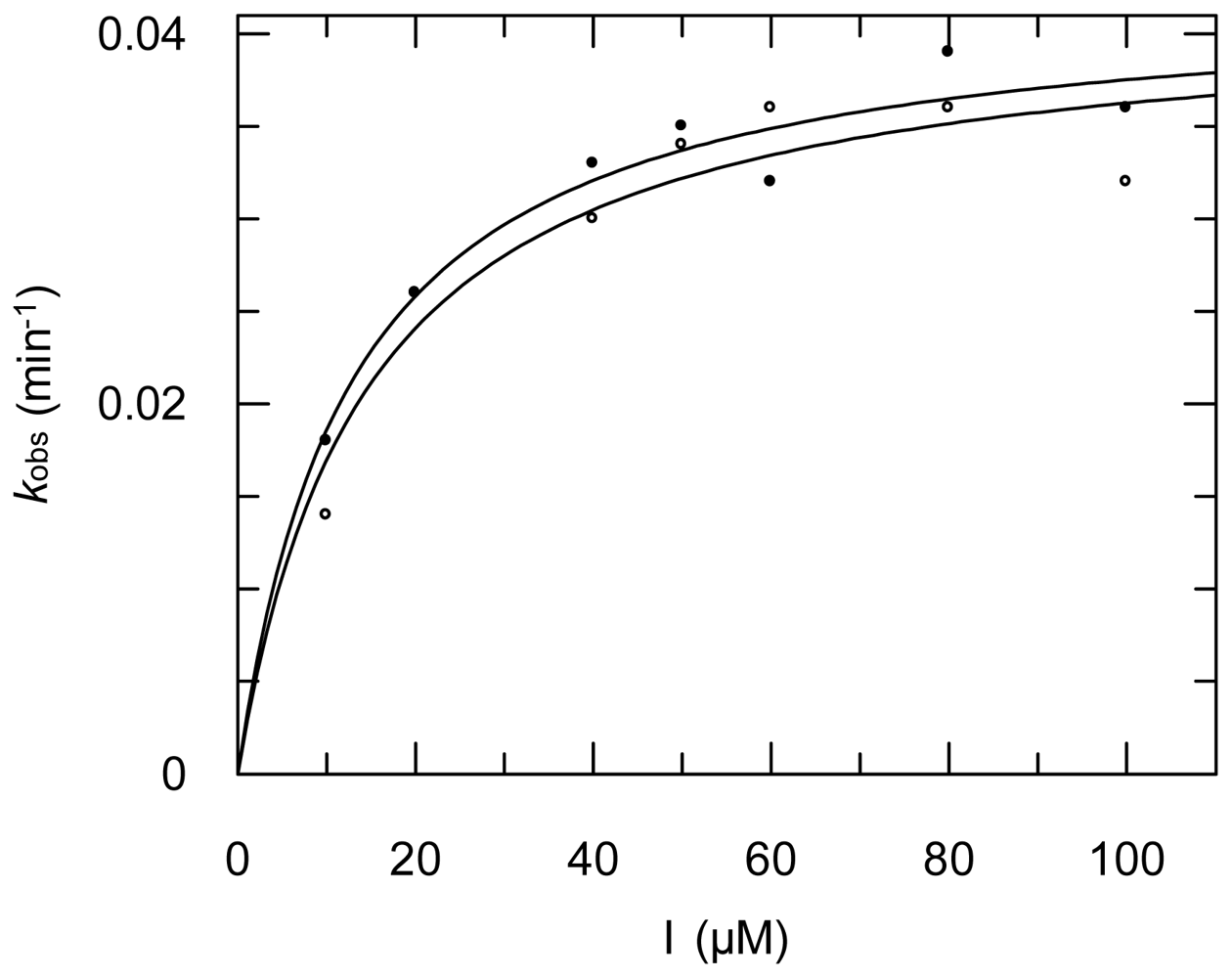
2.3. Protease Inhibition
3. Experimental Section
3.1. General Experimental Procedures
3.2. Biological Material
3.3. Extraction and Isolation
3.4. Antiparasitic Activity Assays
3.5. Cytotoxicity Assays
3.6. Protease Inhibition Assays
4. Conclusions
Acknowledgements
- †This work is dedicated to Johannes F. Imhoff on the occasion of his 60th birthday.
- Samples Availability: Available from the authors.
References
- Peiris, JS; Yuen, KY; Osterhaus, AD; Stohr, K. The severe acute respiratory syndrome. N. Engl. J. Med 2003, 349, 243–2441. [Google Scholar]
- Fear, G; Komarnytsky, S; Raskin, I. Protease inhibitors and their peptidomimetic derivatives as potential drugs. Pharmacol. Ther 2007, 113, 354–368. [Google Scholar]
- Anand, K; Ziebuhr, J; Wadhwani, P; Mesters, JR; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar]
- Laport, MS; Santos, OS; Muricy, G. Marine sponges: Potential sources of new antimicrobial drugs. Curr. Pharm. Biotechnol 2009, 10, 86–105. [Google Scholar]
- Scheuermayer, M; Pimentel-Elardo, S; Fieseler, L; Grozdanov, L; Hentschel, U. Microorganisms of Sponges: Phylogenetic Diversity and Biotechnological Potential. In Frontiers in Marine Biotechnology; Proksch, P, Müller, WEG, Eds.; Horizon Bioscience: Norwich, UK, 2006; pp. 289–312. [Google Scholar]
- Thomas, TRA; Kavlekar, DP; LokaBharathi, A. Marine drugs from sponge-microbe association—A review. Mar. Drugs 2010, 8, 1417–1468. [Google Scholar]
- Fenical, W. Marine pharmaceuticals: Past, present and future. Oceanography 2006, 19, 110–119. [Google Scholar]
- Lam, KS. Discovery of novel metabolites from marine actinomycetes. Curr. Opin. Microbiol 2006, 9, 245–251. [Google Scholar]
- Bull, AT; Stach, JE. Marine actinobacteria: New opportunities for natural product search and discovery. Trends Microbiol 2007, 15, 491–499. [Google Scholar]
- Pimentel-Elardo, SM; Scheuermayer, M; Kozytska, S; Hentschel, U. Streptomyces axinellae sp. nov., isolated from the Mediterranean sponge Axinella polypoides (Porifera). Int. J. Syst. Evol. Microbiol 2009, 59, 1433–1437. [Google Scholar]
- Hobbs, G; Frazer, CM; Gardner, DCJ; Cullum, JA; Oliver, SG. Dispersed growth of Streptomyces in liquid culture. Appl. Microbiol. Biotechnol 1989, 31, 272–277. [Google Scholar]
- Takeuchi, T; Hamada, M; Osanawa, H; Takahashi, Y; Sawa, R. Tetromycin A and B manufacture with Streptomyces. Japanese Patent JP 08165286, April 1996. [Google Scholar]
- Takeuchi, T; Hamada, M; Nanagawa, H; Takahashi, Y; Sawa, R. Antibiotic tetromycin C1, C2, C3, C4 and C5 and their production. Japanese Patent JP 96-216484, March 1998. [Google Scholar]
- Ponte-Sucre, A; Vicik, R; Schultheis, M; Schirmeister, T; Moll, H. Aziridine-2,3-dicarboxylates, peptidomimetic cysteine protease inhibitors with antileishmanial activity. Antimicrob. Agents Chemother 2006, 50, 2439–2447. [Google Scholar]
- Huber, W; Koella, JC. A comparison of three methods of estimating EC50 in studies of drug resistance of malaria parasites. Acta Trop 1993, 55, 257–261. [Google Scholar]
- Vicik, R; Busemann, M; Gelhaus, C; Stiefl, N; Scheiber, J; Schmitz, W; Schulz, F; Mladenovic, M; Engels, B; Leippe, M; Baumann, K; Schirmeister, T. Aziridide-based inhibitors of cathepsin L: Synthesis, inhibition activity, and docking studies. Chem. Med. Chem 2006, 1, 1126–1141. [Google Scholar]
- Vicik, R; Hoerr, V; Glaser, M; Schultheis, M; Hansell, E; McKerrow, JK; Holzgrabe, U; Caffrey, CR; Ponte-Sucre, A; Moll, H; Stich, A; Schirmeister, T. Aziridine-2,3-dicarboxylate inhibitors targeting the major cysteine protease of Trypanosoma brucei as lead trypanocidal agents. Bioorg Med. Chem. Lett 2006, 16, 2753–2757. [Google Scholar]
- Breuning, A; Degel, B; Schulz, F; Buchold, C; Stempka, M; Machon, U; Heppner, S; Gelhaus, C; Leippe, M; Leyh, M; et al. Michael acceptor based antiplasmodial and antitrypanosomal cysteine protease inhibitors with unusual amino acids. J. Med. Chem 2010, 53, 1951–1963. [Google Scholar]
- Ehmke, V; Heindl, C; Rottmann, M; Freymond, C; Schweizer, WB; Brun, R; Stich, A; Schirmeister, T; Diederich, F. Potent and selective inhibition of cysteine proteases from Plasmodium falciparum and Trypanosoma brucei. ChemMedChem 2011, 6, 273–278. [Google Scholar]
- Cheng, Y; Prusoff, WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor, which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [Google Scholar]
- Ludewig, S; Kossner, M; Schiller, M; Baumann, T; Schirmeister, T. Enzyme kinetics and hit validation in fluorometric protease assays. Curr. Top. Med. Chem 2010, 10, 368–382. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Posn. | 1 | 2 | 3 | 4 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δC | mult | δH | mult, (JH-H) | δC | mult | δH | mult, (JH-H) | δC | mult | δH | mult, (JH-H) | δC | mult | δH | mult, (JH-H) | |
| 1 | 167.4 | C | 168.4 | C | 174.8 | C | 169.0 | C | ||||||||
| 2 | 96.8 | C | 99.6 | C | 96.8 | C | 99.1 | C | ||||||||
| 3 | 206.3 | C | 204.5 | C | 205.4 | C | 204.6 | C | ||||||||
| 4 | 52.4 | C | 52.7 | C | 52.6 | C | 52.6 | C | ||||||||
| 4′ | 15.8 | CH3 | 1.52 | s | 16.0 | CH3 | 1.52 | s | 15.4 | CH3 | 1.54 | s | 15.6 | CH3 | 1.52 | s |
| 5 | 57.5 | CH | 3.40 | s | 57.4 | CH | 3.49 | s | 57.5 | CH | 3.35 | s | 57.0 | CH | 3.51 | s |
| 6 | 135.4 | C | 135.9 | C | 135.7 | C | 136.1 | C | ||||||||
| 6′ | 21.8 | CH3 | 1.29 | s | 22.0 | CH3 | 1.53 | s | 22.9 | CH3 | 1.39 | s | 25.3 | CH3 | 1.48 | s |
| 7 | 131.5 | CH | 5.09 | d (8.2) | 131.5 | CH | 5.13 | d (6.6) | 130.6 | CH | 5.00 | d (9.2) | 131.1 | CH | 5.14 | d (7.1) |
| 8 | 32.9 | CH2 | 2.27 | m | 34.2 | CH2 | 2.28 | m | 34.1 | CH2 | 2.27 | m | 33.8 | CH2 | 2.27 | m |
| 9 | 36.3 | CH2 | 2.29 | m | 35.5 | CH2 | 2.32 | m | 40.4 | CH2 | 2.23 | m | 40.6 | CH2 | 2.38 | m |
| 10 | 132.1 | C | 131.1 | C | 131.6 | C | 131.4 | C | ||||||||
| 10′ | 22.0 | CH3 | 1.31 | s | 21.8 | CH3 | 1.33 | s | 22.9 | CH3 | 1.31 | s | 21.7 | CH3 | 1.35 | s |
| 11 | 123.2 | CH | 6.17 | s | 123.1 | CH | 6.08 | s | 122.9 | CH | 6.11 | s | 122.8 | CH | 6.15 | s |
| 12 | 46.4 | C | 46.2 | C | 42.0 | C | 2.07 | br | 46.2 | C | ||||||
| 12′ | 22.8 | CH3 | 1.52 | s | 23.0 | CH3 | 1.50 | s | 22.9 | CH3 | 1.50 | s | ||||
| 13 | 145.7 | CH | 6.92 | s | 146.6 | CH | 6.92 | s | 138.6 | CH | 6.67 | s | 146.8 | CH | 6.92 | s |
| 14 | 132.1 | C | 126.0 | C | 135.1 | C | 125.7 | C | ||||||||
| 14′ | 168.1 | C | 167.7 | C | 167.5 | C | 168.5 | C | ||||||||
| 15 | 27.8 | CH | 2.97 | m | 28.0 | CH | 2.93 | m | 27.8 | CH | 2.95 | m | 28.0 | CH | 2.91 | m |
| 15′ | 13.1 | CH3 | 1.33 | m | 13.4 | CH3 | 1.34 | m | 13.8 | CH3 | 1.30 | m | 13.2 | CH3 | 1.32 | m |
| 16 | 32.9 | CH2 | 1.79 | br | 33.23 | CH2 | 1.74 | br | 27.8 | CH2 | 1.58 | br | 33.1 | CH2 | 1.69 | br |
| 2.71 | dd (14.8, 7.9) | 2.65 | dd (14.4, 7.9) | 2.23 | m | 2.65 | dd (14.6, 7.9) | |||||||||
| 17 | 85.9 | C | 85.6 | C | 82.7 | C | 85.6 | C | ||||||||
| 18 | 198.6 | C | 199.3 | C | 202.1 | C | 200.3 | C | ||||||||
| 19 | 55.7 | CH | 3.82 | br | 55.5 | CH | 3.67 | br | 42.1 | CH | 3.71 | br | 42.1 | CH | 3.60 | br |
| 20 | 124.8 | CH | 4.90 | s | 125.6 | CH | 4.93 | s | 141.5 | C | 125.8 | CH | 4.92 | s | ||
| 20′ | 14.3 | CH3 | 1.48 | s | ||||||||||||
| 21 | 140.9 | C | 140.2 | C | 120.3 | CH | 4.95 | d (2.3) | 140.4 | C | ||||||
| 21′ | 19.2 | CH3 | 1.69 | s | 19.2 | CH3 | 1.69 | s | 18.9 | CH3 | 1.69 | s | ||||
| 22 | 39.59 | CH2 | 2.29 | m | 39.9 | CH2 | 2.20 | m | 34.1 | CH2 | 2.32 | m | 39.7 | CH2 | 2.26 | m |
| 1.94 | m | 1.97 | m | 1.19 | m | 1.95 | m | |||||||||
| 23 | 84.4 | CH | 3.33 | dt (10.4, 4.8) | 85.0 | CH | 3.36 | dt (10.2, 4.8) | 84.56 | CH | 3.41 | dt (10.4, 4.8) | 84.9 | CH | 3.39 | dt (10.5, 4.8) |
| 24 | 46.2 | CH | 1.94 | m | 46.7 | CH | 1.90 | m | 46.8 | CH | 1.93 | m | 46.5 | CH | 1.90 | m |
| 25 | 35.3 | CH2 | 1.34 | br | 35.6 | CH2 | 1.31 | br | 36.4 | CH2 | 1.52 | br | 37.8 | CH2 | 1.54 | br |
| 26 | 42.6 | CH | 2.08 | br | 43.1 | CH | 2.09 | br | 37.9 | CH | 2.07 | br | 42.9 | CH | 2.09 | br |
| 26′ | 22.8 | CH3 | 0.68 | d (5.6) | 23.1 | CH3 | 0.70 | d (5.1) | 22.7 | CH3 | 0.68 | d (5.9) | 22.9 | CH3 | 0.69 | d (5.7) |
| 27 | 103.6 | CH | 4.67 | d (7.9) | 103.4 | CH | 4.77 | d (7.9) | 103.1 | CH | 4.81 | d (7.9) | 103.2 | CH | 4.81 | d (7.9) |
| 28 | 70.6 | CH | 3.79 | m | 72.4 | CH | 3.54 | dd (7.9, 2.9) | 71.9 | CH | 3.55 | dd (7.9, 3.1) | 72.0 | CH | 3.54 | dd (7.9, 3.1) |
| 29 | 73.8 | C | 70.5 | CH | 4.33 | br | 69.6 | CH | 4.38 | br | 69.7 | CH | 4.39 | br | ||
| 29′ | 19.01 | CH3 | 1.22 | s | ||||||||||||
| 30 | 58.4 | CH | 3.90 | d (10.0) | 76.1 | CH | 4.68 | dd (9.8, 2.4) | 76.8 | CH | 4.75 | dd (9.8, 2.8) | 76.8 | CH | 4.76 | dd (9.8, 2.7) |
| 31 | 75.8 | CH | 3.33 | m | 67.6 | CH | 4.03 | m | 67.3 | CH | 4.17 | m | 67.2 | CH | 4.18 | m |
| 31′ | 23.7 | CH3 | 1.32 | d (7.3) | 18.2 | CH3 | 1.31 | d (7.21) | 18.4 | CH3 | 1.24 | d (6.2) | 18.3 | CH3 | 1.25 | d (6.2) |
| 32 | 170.6 | C | 164.8 | C | 171.1 | C | 170.4 | C | ||||||||
| 33 | 113.1 | C | 108.0 | C | 106.2 | C | 106.2 | C | ||||||||
| 34 | 139.1 | C | 144.0 | C | 144.5 | C | 144.6 | C | ||||||||
| 34′ | 22.6 | CH3 | 2.5 | s | 24.6 | CH3 | 2.56 | s | 24.5 | CH3 | 2.54 | s | 24.3 | CH3 | 2.55 | s |
| 35 | 110.0 | CH | 6.33 | s | 111.8 | CH | 6.36 | br d | 111.7 | CH | 6.37 | d (2.3) | 111.7 | CH | 6.37 | d (2.5) |
| 36 | 161.9 | C | 162.6 | C | 165.1 | C | 165.8 | C | ||||||||
| 36′ | 55.9 | CH3 | 3.83 | s | ||||||||||||
| 37 | 99.9 | CH | 6.33 | s | 97.0 | CH | 6.45 | br d | 99.5 | CH | 6.34 | d (2.3) | 99.6 | CH | 6.34 | d (2.5) |
| 38 | 162.9 | C | 158.7 | C | 166.0 | C | 165.3 | C | ||||||||
| 38′ | 55.5 | CH3 | 3.78 | s | 55.9 | CH3 | 3.82 | s | 55.8 | CH3 | 3.83 | s | 55.7 | CH3 | 3.83 | s |
| Compound | L. major | T. brucei brucei (48 h) | T. brucei brucei (72 h) | 293T Kidney cells | J774.1 Macrophages |
|---|---|---|---|---|---|
| 1 | >100 | 29.30 | 31.69 | >100 | >100 |
| 2 | >100 | 45.39 | 80.27 | >100 | 50.21 |
| 3 | 36.80 | 26.90 | 30.35 | 33.38 | 25.72 |
| 4 | >100 | 35.85 | 41.61 | 58.58 | 27.54 |
| 5 | >100 | 30.87 | 34.22 | 71.77 | 20.20 |
| Compound | RD Ki (μM); k2nd (M−1 min−1) | FP Ki (μM); k2nd (M−1 min−1) | CL Ki (μM); k2nd (M−1 min−1) | CB Ki (μM) | SARS-CoV-PLpro Ki (μM) |
|---|---|---|---|---|---|
| 3 | 2.1 ± 0.90; 36,600 ± 1590 | 1.65 ± 0.25; 617 ± 9 | 15.0 ± 1.95; 14,977 ± 2005 | 0.57 ± 0.04 | n.d. |
| 4 | 4.00 ± 0.30; 8354 ± 1562 | 3.10 ± 0.20; 1836 ± 108 | 22.40 ± 0.80; 14,700 ± 1000 | 1.60 ± 0.10 | 40.00 ± 6.50 |
| 5 | 0.62 ± 0.03; 14,539 ± 1949 | 1.42 ± 0.01; 15,540 ± 1295 | 32.50 ± 0.05; 1576 ± 413 | 1.59 ± 0.09 | 69.60 ± 7.20 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pimentel-Elardo, S.M.; Buback, V.; Gulder, T.A.M.; Bugni, T.S.; Reppart, J.; Bringmann, G.; Ireland, C.M.; Schirmeister, T.; Hentschel, U. New Tetromycin Derivatives with Anti-Trypanosomal and Protease Inhibitory Activities. Mar. Drugs 2011, 9, 1682-1697. https://doi.org/10.3390/md9101682
Pimentel-Elardo SM, Buback V, Gulder TAM, Bugni TS, Reppart J, Bringmann G, Ireland CM, Schirmeister T, Hentschel U. New Tetromycin Derivatives with Anti-Trypanosomal and Protease Inhibitory Activities. Marine Drugs. 2011; 9(10):1682-1697. https://doi.org/10.3390/md9101682
Chicago/Turabian StylePimentel-Elardo, Sheila M., Verena Buback, Tobias A.M. Gulder, Tim S. Bugni, Jason Reppart, Gerhard Bringmann, Chris M. Ireland, Tanja Schirmeister, and Ute Hentschel. 2011. "New Tetromycin Derivatives with Anti-Trypanosomal and Protease Inhibitory Activities" Marine Drugs 9, no. 10: 1682-1697. https://doi.org/10.3390/md9101682