Preclinical Pharmacology of BA-TPQ, a Novel Synthetic Iminoquinone Anticancer Agent


Abstract
:1. Introduction
2. Results and Discussion
2.1. In vitro stability of BA-TPQ in mouse plasma
2.2. In vitro protein binding of BA-TPQ
2.3. S9 metabolism
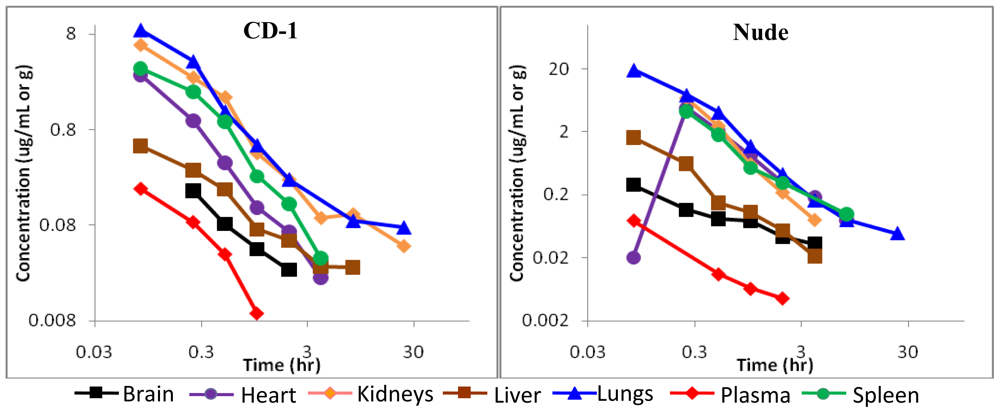
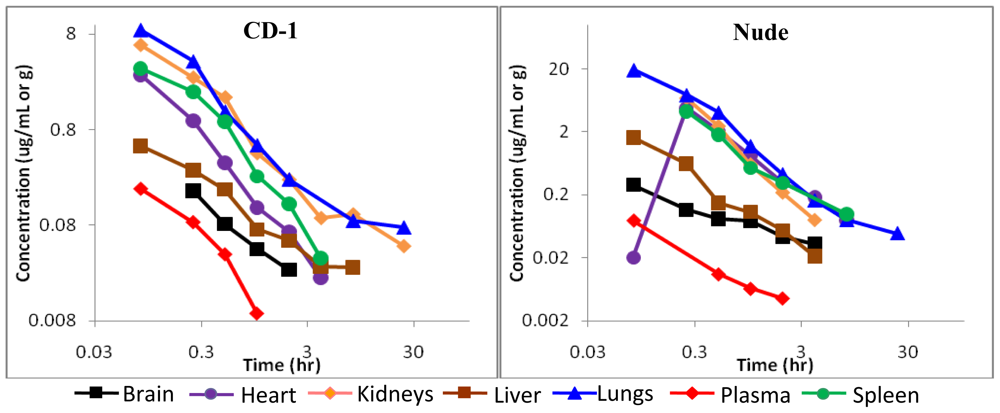
2.4. Pharmacokinetics of BA-TPQ in mice
2.5. Discussion
3. Experimental
3.1. Test compound, chemicals and reagents
3.2. Instrumentation and chromatographic conditions
3.3. Stability in mouse plasma
3.4. Binding to mouse plasma proteins
3.5. S9 metabolism
3.6. Pharmacokinetic studies
3.6.1. Animals
3.6.2. Animal dosing and sampling
3.6.3. Sample preparation
3.6.4. Pharmacokinetic analysis
4. Conclusions
Acknowledgements
- Samples Availability: Available from the authors.
References
- Newman, DJ; Cragg, GM. Marine natural products and related compounds in clinical and advanced preclinical trials. J. Nat. Prod 2004, 8, 1216–1238. [Google Scholar]
- Guzmán, E; Johnson, J; Carrier, M; Meyer, C; Pitts, T; Gunasekera, S; Wright, A. Selective cytotoxic activity of the marine-derived batzelline compounds against pancreatic cancer cell lines. Anticancer Drugs 2009, 20, 149–155. [Google Scholar]
- Hoang, H; LaBarbera, D; Mohammed, K; Ireland, C; Skibo, E. Synthesis and biological evaluation of imidazoquinoxalinones, imidazole analogues of pyrroloiminoquinone marine natural products. J. Med. Chem 2007, 50, 4561–4571. [Google Scholar]
- Byler, K; Wang, C; Setzer, W. Quinoline alkaloids as intercalative topoisomerase inhibitors. J. Mol. Model 2009, 15, 1417–1426. [Google Scholar]
- Delfourne, E. Analogues of marine pyrroloiminoquinone alkaloids: synthesis and antitumor properties. Anticancer Agents Med. Chem 2008, 8, 910–916. [Google Scholar]
- Marshall, K; Andjelic, C; Tasdemir, D; Concepción, G; Ireland, C; Barrows, L. Deoxyamphimedine, a pyridoacridine alkaloid, damages DNA via the production of reactive oxygen species. Mar. Drugs 2009, 7, 196–209. [Google Scholar]
- Barrows, LR; Radisky, DC; Copp, BR; Swaffar, DS; Kramer, RA; Warters, RL; Ireland, CM. Makaluvamines, marine natural products, are active anti-cancer agents and DNA topo II inhibitors. Anticancer Drug Des 1993, 8, 333–347. [Google Scholar]
- Ding, Q; Chichak, K; Lown, JW. Pyrroloquinoline and pyridoacridine alkaloids from marine sources. Curr. Med. Chem 1999, 6, 1–27. [Google Scholar]
- Shinkre, BA; Raisch, KP; Fan, L; Velu, SE. Analogs of the marine alkaloid makaluvamines: synthesis, topoisomerase II inhibition, and anticancer activity. Bioorg. Med. Chem. Lett 2007, 10, 2890–2893. [Google Scholar]
- Shinkre, BA; Raisch, KP; Fan, L; Velu, SE. Synthesis and antiproliferative activity of benzyl and phenethyl analogs of makaluvamines. Bioorg. Med. Chem 2008, 5, 2541–2549. [Google Scholar]
- Wang, F; Ezell, SJ; Zhang, Y; Wang, W; Rayburn, ER; Nadkarni, DH; Murugesan, S; Velu, SE; Zhang, R. FBA-TPQ, a novel marine-derived compound as experimental therapy for prostate cancer. Invest. New Drugs 2010, 28, 234–241. [Google Scholar]
- Nadkarni, DH; Wang, F; Wang, W; Rayburn, ER; Ezell, SJ; Murugesan, S; Velu, SE; Zhang, R. Synthesis and in vitro anti-lung cancer activity of novel 1,3,4,8-tetrahydropyrrolo [4,3,2-de]quinolin-8(1H)-one alkaloid analogs. Med. Chem 2009, 3, 227–236. [Google Scholar]
- Wang, W; Rayburn, ER; Velu, SE; Chen, D; Nadkarni, DH; Murugesan, S; Chen, D; Zhang, R. A novel synthetic iminoquinone, BA-TPQ, as an anti-breast cancer agent: in vitro and in vivo activity and mechanisms of action. Breast Cancer Res. Treat 2010. [Google Scholar] [CrossRef]
- Geldof, AA; Mastbergen, SC; Henrar, RE; Faircloth, GT. Cytotoxicity and neurocytotoxicity of new marine anticancer agents evaluated using in vitro assays. Cancer Chemother. Pharmacol 1999, 4, 312–318. [Google Scholar]
- Ryan, DP; Supko, JG; Eder, JP; Seiden, MV; Demetri, G; Lynch, TJ; Fischman, AJ; Davis, J; Jimeno, J; Clark, JW. Phase I and pharmacokinetic study of ecteinascidin 743 administered as a 72-hour continuous intravenous infusion in patients with solid malignancies. Clin. Cancer Res 2001, 2, 231–242. [Google Scholar]
- Na, M; Ding, Y; Wang, B; Tekwani, BL; Schinazi, RF; Franzblau, S; Kelly, M; Stone, R; Li, XC; Ferreira, D; Hamann, MT. Anti-infective discorhabdins from a deep-water alaskan sponge of the genus Latrunculia. J. Nat. Prod 2010, 26, 383–387. [Google Scholar]
- Casapullo, A; Cutignano, A; Bruno, I; Bifulco, G; Debitus, C; Gomez-Paloma, L; Riccio, R. Makaluvamine P, a new cytotoxic pyrroloiminoquinone from Zyzzya cf. fuliginosa. J. Nat. Prod 2001, 64, 1354–1356. [Google Scholar]
- Puchalski, TA; Ryan, DP; Garcia-Carbonero, R; Demetri, GD; Butkiewicz, L; Harmon, D; Seiden, MV; Maki, RG; Lopez-Lazaro, L; Jimeno, J; Guzman, C; Supko, JG. Pharmacokinetics of ecteinascidin 743 administered as a 24-h continuous intravenous infusion to adult patients with soft tissue sarcomas: associations with clinical characteristics, pathophysiological variables and toxicity. Cancer Chemother. Pharmacol 2002, 50, 309–319. [Google Scholar]
- Forouzesh, B; Hidalgo, M; Chu, Q; Mita, A; Mita, M; Schwartz, G; Jimeno, J; Gomez, J; Alfaro, V; Lebedinsky, C; Zintl, P; Rowinsky, EK. Phase I and pharmacokinetic study of trabectedin as a 1- or 3-hour infusion weekly in patients with advanced solid malignancies. Clin. Cancer Res 2009, 15, 3591–3599. [Google Scholar]
- Miller, MJ; Lonardo, EC; Greer, RD; Bevan, C; Edwards, DA; Smith, JH; Freeman, JJ. Variable responses of species and strains to white mineral oils and paraffin waxes. Regul. Toxicol. Pharmacol 1996, 23, 55–68. [Google Scholar]
- Diwan, BA; Rice, JM; Ward, JM. Strain-dependent effects of phenobarbital on liver tumor promotion in inbred mice. Prog. Clin. Biol. Res 1990, 331, 69–83. [Google Scholar]
- Boerrigter, ME; Wei, JY; Vijg, J. Induction and repair of benzo[a]pyrene-DNA adducts in C57BL/6 and BALB/c mice: association with aging and longevity. Mech. Ageing Dev 1995, 82, 31–50. [Google Scholar]
- Pelleitier, M; Montplaisir, S. The nude mouse: a model of deficient T-cell function. Methods Achiev. Exp. Pathol 1975, 7, 149–166. [Google Scholar]
- Giovanella, BC; Stehlin, JS, Jr; Williams, LJ, Jr; Lee, SS; Shepard, RC. Heterotransplantation of human cancers into nude mice: a model system for human cancer chemotherapy. Cancer 1978, 42, 2269–2281. [Google Scholar]
- Prakash, C; Vaz, ADN. Xie, W, Ed.; Drug Metabolism: Significance and Challenges. In Nuclear Receptors in Drug Metabolism; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp. 1–42. [Google Scholar]
- Agrawal, S; Zhang, X; Cai, Q; Zhao, H; Tan, W; Yu, D; Zhang, R. Effect of aspirin on pharmacokinetics of antisense oligonucleotides in rats. J. Drug Target 1998, 5, 303–312. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Temperature | Duration of Time | 0.5 μM | 5.0 μM | ||
|---|---|---|---|---|---|
| Amount Remaining (%) | RSD (%) | Amount Remaining (%) | RSD (%) | ||
| 37 °C | 8 hr | 92 | 0.22 | 114 | 0.60 |
| 4 °C | 24 hr | 94 | 12 | 117 | 7 |
| −80 °C | 4 weeks | 95 | 0.36 | 100 | 0.13 |
| Tissue | Route | Dose (mg/kg) | R2* | Cmax (μg/mL) | Tmax (h) | T1/2 (h) | AUC (h·μg/mL) | Cl (mL/h/kg) |
|---|---|---|---|---|---|---|---|---|
| Plasma | IV | 5 | 1.00 | 0.36 | - | 0.14 | 0.07 | 4.92 × 105 |
| IP | 10 | 0.95 | 0.11 | 0.08 | 0.86 | 0.11 | 3.45 × 105 | |
| Heart | IV | 5 | 1.00 | 5.34 | - | 0.13 | 1.02 | 0.34 × 105 |
| IP | 10 | 0.97 | 7.24 | 0.08 | 0.71 | 4.66 | 0.08 × 105 | |
| Lungs | IV | 5 | 1.00 | 13.05 | - | 0.26 | 4.98 | 0.07 × 105 |
| IP | 10 | 0.92 | 12.38 | 0.08 | 0.50 | 9.15 | 0.04 × 105 | |
| Liver | IV | 5 | 1.00 | 0.69 | - | 0.65 | 0.65 | 0.54 × 105 |
| IP | 10 | 0.88 | 0.47 | 0.08 | 0.24 | 0.15 | 2.46 × 105 | |
| Kidneys | IV | 5 | 1.00 | 9.68 | - | 0.20 | 2.83 | 0.12 × 105 |
| IP | 10 | 0.92 | 8.36 | 0.25 | 0.48 | 8.06 | 0.04 × 105 | |
| Spleen | IV | 5 | 1.00 | 4.59 | - | 0.27 | 1.80 | 0.19 × 105 |
| IP | 10 | 0.92 | 23.00 | 0.08 | 0.41 | 11.26 | 0.03 × 105 | |
| Brain | IV | 5 | 1.00 | 0.75 | - | 0.21 | 0.22 | 1.58 × 105 |
| IP | 10 | 0.97 | 0.18 | 0.25 | 0.83 | 0.19 | 1.75 × 105 |
| Tissue | Route | Dose (mg/kg) | R2 | Cmax (μg/mL) | Tmax (h) | T1/2 (h) | AUC (h·μg/mL) | Cl (mL/h/kg) |
|---|---|---|---|---|---|---|---|---|
| Plasma | IV | 5 | 1.00 | 0.15 | - | 0.17 | 0.04 | 9.75 × 105 |
| IP | 10 | 0.95 | 0.22 | 0.25 | 1.17 | 0.15 | 2.35 × 105 | |
| Heart | IV | 5 | 1.00 | 48.80 | - | 0.09 | 6.37 | 0.05 × 105 |
| IP | 10 | 0.95 | 6.38 | 0.25 | 0.50 | 7.63 | 0.05 × 105 | |
| Lungs | IV | 5 | 0.95 | 18.38 | - | 0.35 | 9.18 | 0.04 × 105 |
| IP | 10 | 0.92 | 14.75 | 0.08 | 0.59 | 13.16 | 0.03 × 105 | |
| Liver | IV | 5 | 1.00 | 2.71 | - | 0.18 | 0.71 | 0.49 × 105 |
| IP | 10 | 0.98 | 4.56 | 0.08 | 0.53 | 2.76 | 0.13 × 105 | |
| Kidneys | IV | 5 | 1.00 | 20.76 | - | 0.18 | 5.48 | 0.06 × 105 |
| IP | 10 | 0.89 | 10.84 | 0.5 | 0.49 | 12.62 | 0.03 × 105 | |
| Spleen | IV | 5 | 1.00 | 10.99 | - | 0.31 | 4.98 | 0.07 × 105 |
| IP | 10 | 0.90 | 31.22 | 0.25 | 1.65 | 18.29 | 0.02 × 105 | |
| Brain | IV | 5 | 1.00 | 0.62 | - | 0.38 | 0.34 | 1.04 × 105 |
| IP | 10 | 0.92 | 0.09 | 1.00 | 4.06 | 0.77 | 0.39 × 105 |
Abbreviations
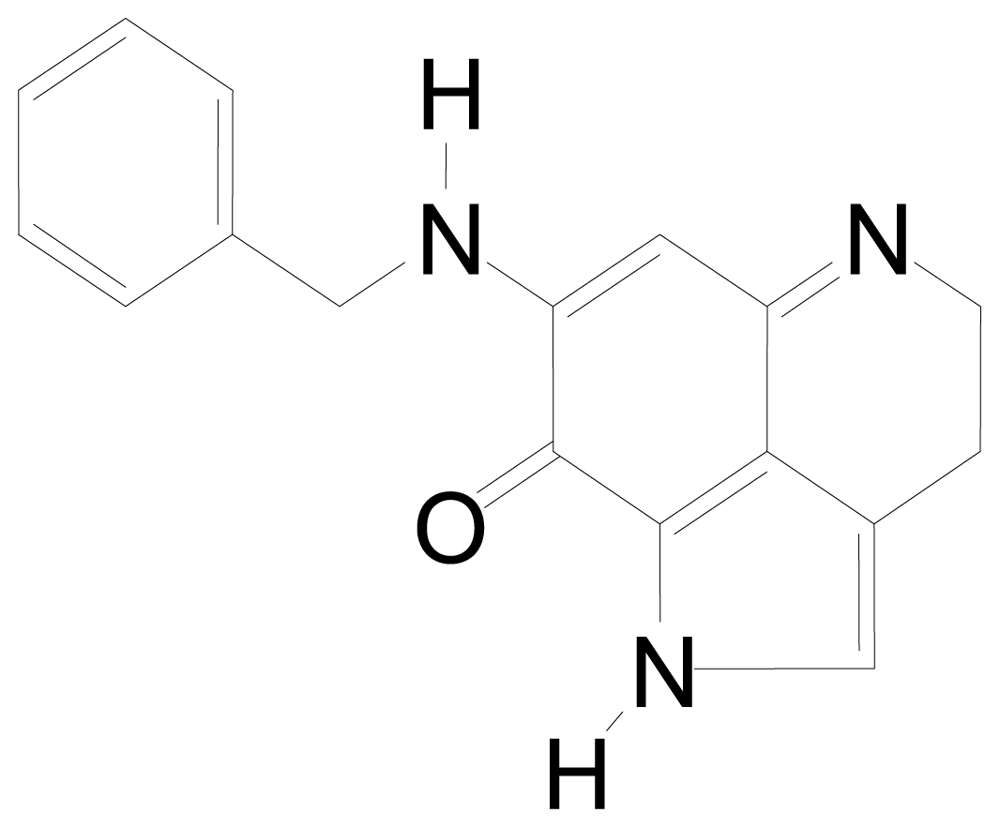
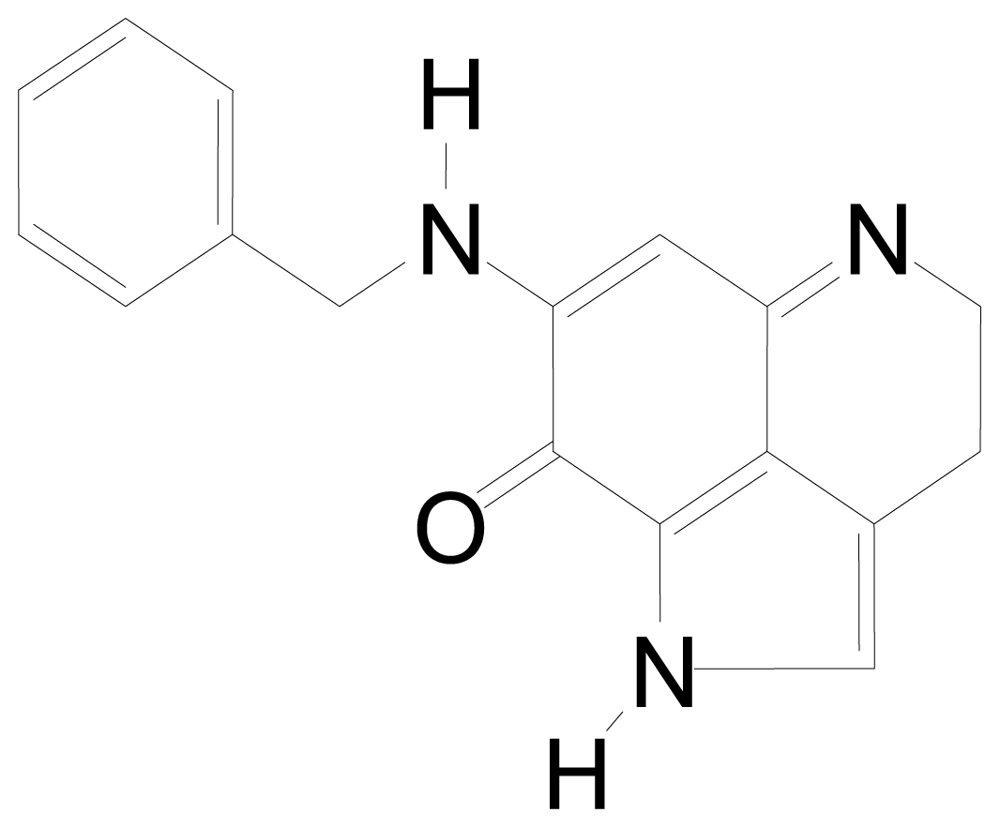
| BA-TPQ | [(11,12),7-(benzylamino)-1,3,4,8-tetrahydropyrrolo[4,3,2-de]quinolin-8(1H)-one] |
| Cmax | Maximum concentration |
| Tmax | Time at maximum concentration |
| T1/2 | Half-life |
| AUC | Area under the concentration-time curve |
| Cl | Clearance |
| ADME(T) | Absorption distribution metabolism excretion (toxicity) |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ezell, S.J.; Li, H.; Xu, H.; Zhang, X.; Gurpinar, E.; Zhang, X.; Rayburn, E.R.; Sommers, C.I.; Yang, X.; Velu, S.E.; et al. Preclinical Pharmacology of BA-TPQ, a Novel Synthetic Iminoquinone Anticancer Agent. Mar. Drugs 2010, 8, 2129-2141. https://doi.org/10.3390/md8072129
Ezell SJ, Li H, Xu H, Zhang X, Gurpinar E, Zhang X, Rayburn ER, Sommers CI, Yang X, Velu SE, et al. Preclinical Pharmacology of BA-TPQ, a Novel Synthetic Iminoquinone Anticancer Agent. Marine Drugs. 2010; 8(7):2129-2141. https://doi.org/10.3390/md8072129
Chicago/Turabian StyleEzell, Scharri J., Haibo Li, Hongxia Xu, Xiangrong Zhang, Evrim Gurpinar, Xu Zhang, Elizabeth R. Rayburn, Charnell I. Sommers, Xinyi Yang, Sadanandan E. Velu, and et al. 2010. "Preclinical Pharmacology of BA-TPQ, a Novel Synthetic Iminoquinone Anticancer Agent" Marine Drugs 8, no. 7: 2129-2141. https://doi.org/10.3390/md8072129
APA StyleEzell, S. J., Li, H., Xu, H., Zhang, X., Gurpinar, E., Zhang, X., Rayburn, E. R., Sommers, C. I., Yang, X., Velu, S. E., Wang, W., & Zhang, R. (2010). Preclinical Pharmacology of BA-TPQ, a Novel Synthetic Iminoquinone Anticancer Agent. Marine Drugs, 8(7), 2129-2141. https://doi.org/10.3390/md8072129