Application of Spectroscopic Methods for Structural Analysis of Chitin and Chitosan

 and
and
Abstract
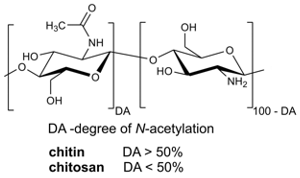
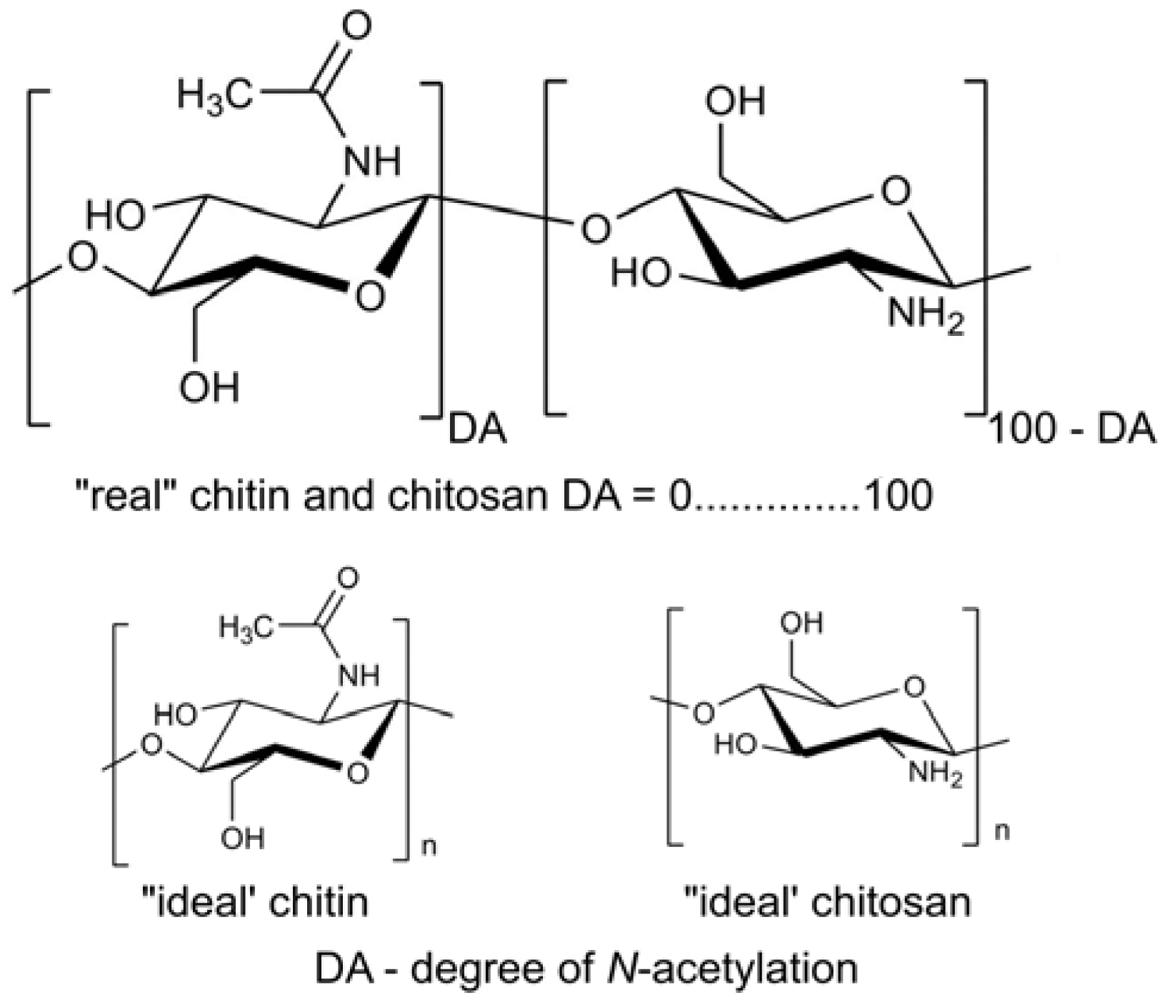
:
1. Introduction
2. Application of Spectroscopic Methods for Analyzing the Structure and Determining the Physicochemical Properties of Chitin, Chitosan and Their Derivatives
2.1. X-ray spectroscopy
2.1.1. Typical conditions of X-ray measurements
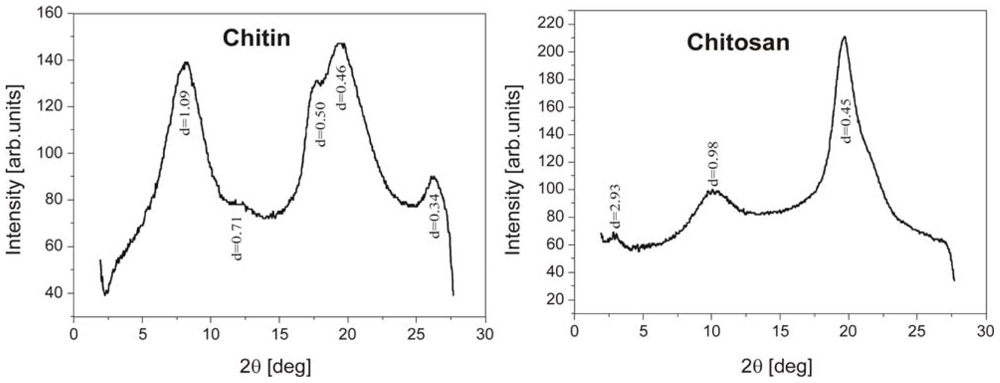
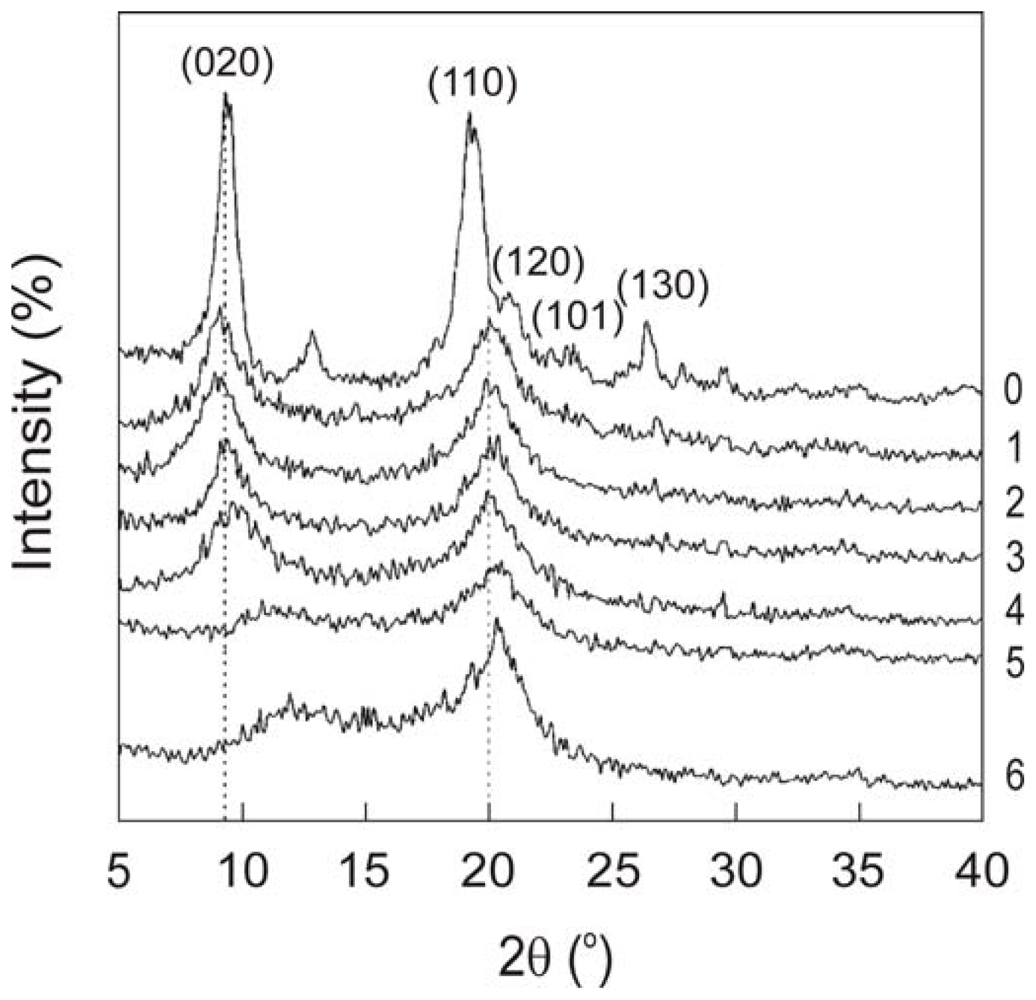
2.1.2. X-ray spectra of chitin and chitosan
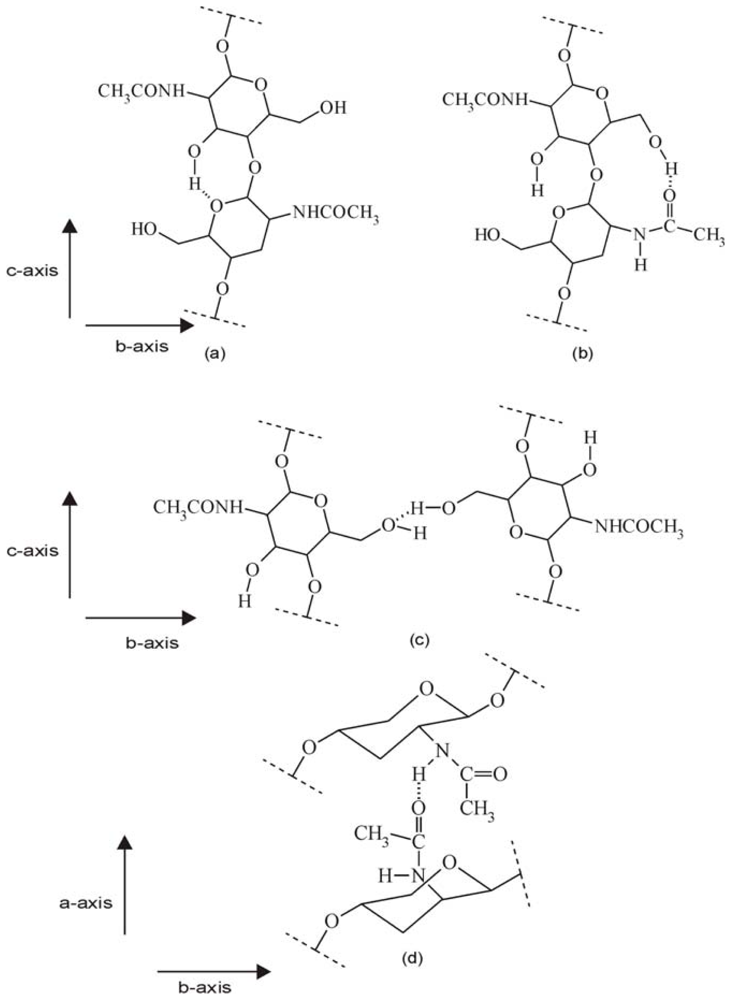
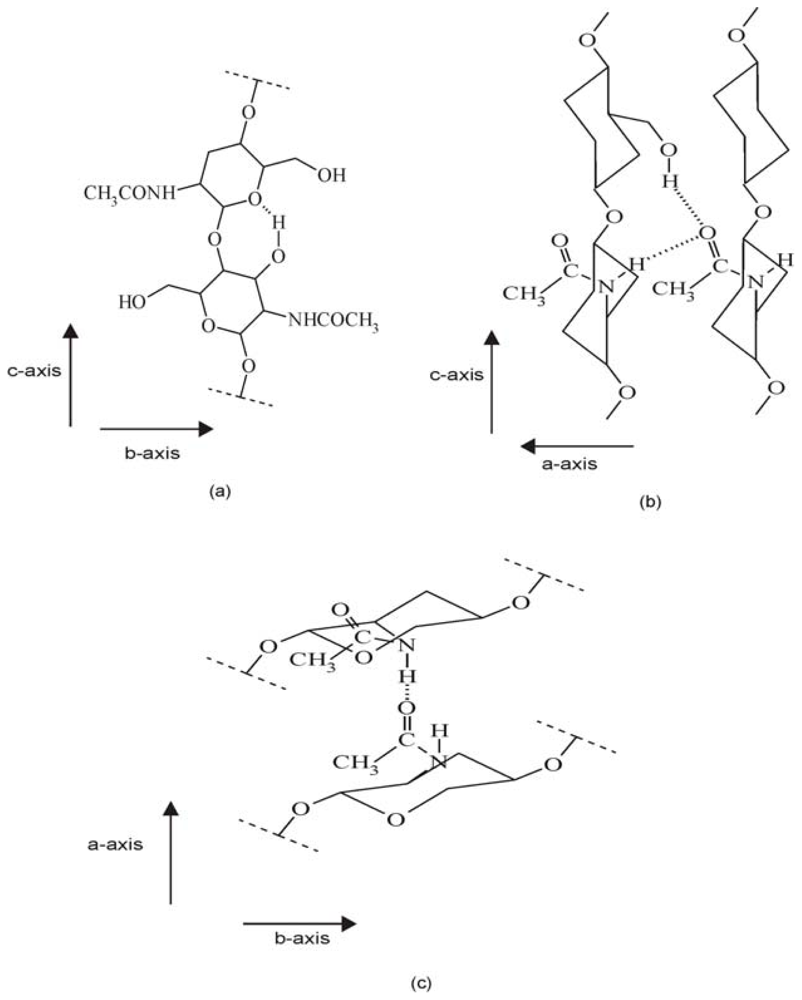
2.1.3. X-ray analysis of chitin and chitosan polymorphs
2.1.4. Physicochemical characterization of chitin and chitosan using X-ray diffraction
2.1.5. X-ray analysis of chitosan salts
2.1.6. X-ray analysis of chitosan derivatives
2.1.7. Other X-ray techniques used in chitin and chitosan analysis
2.2. Infrared spectroscopy
2.2.1. Typical conditions for the FTIR spectroscopic analysis of chitin, chitosan and their derivatives
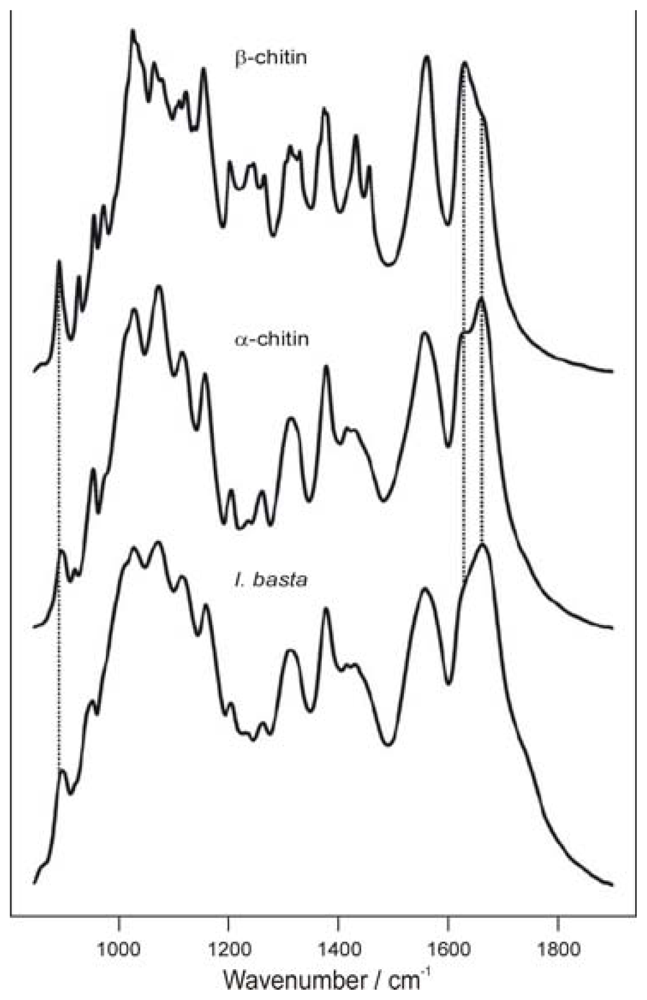
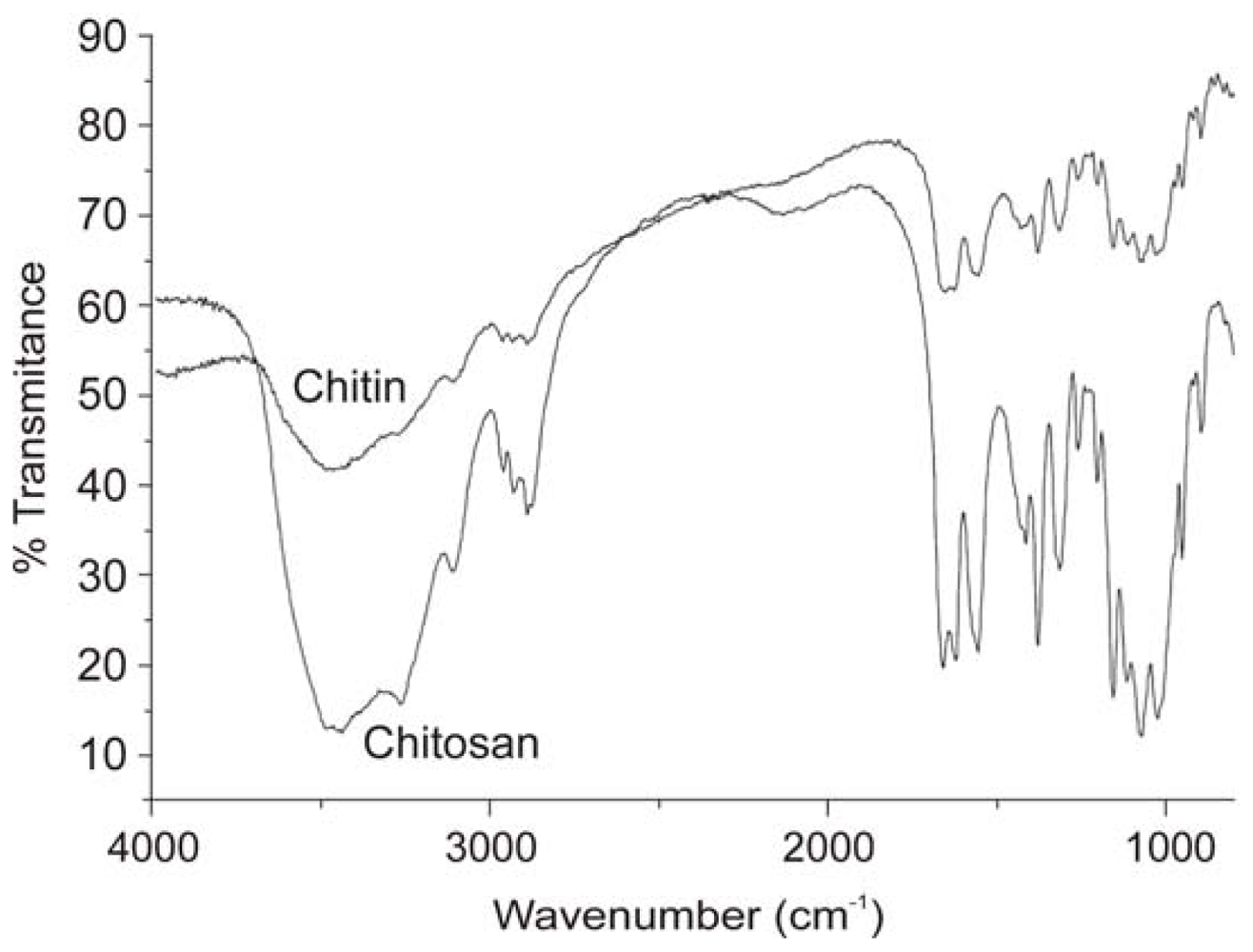
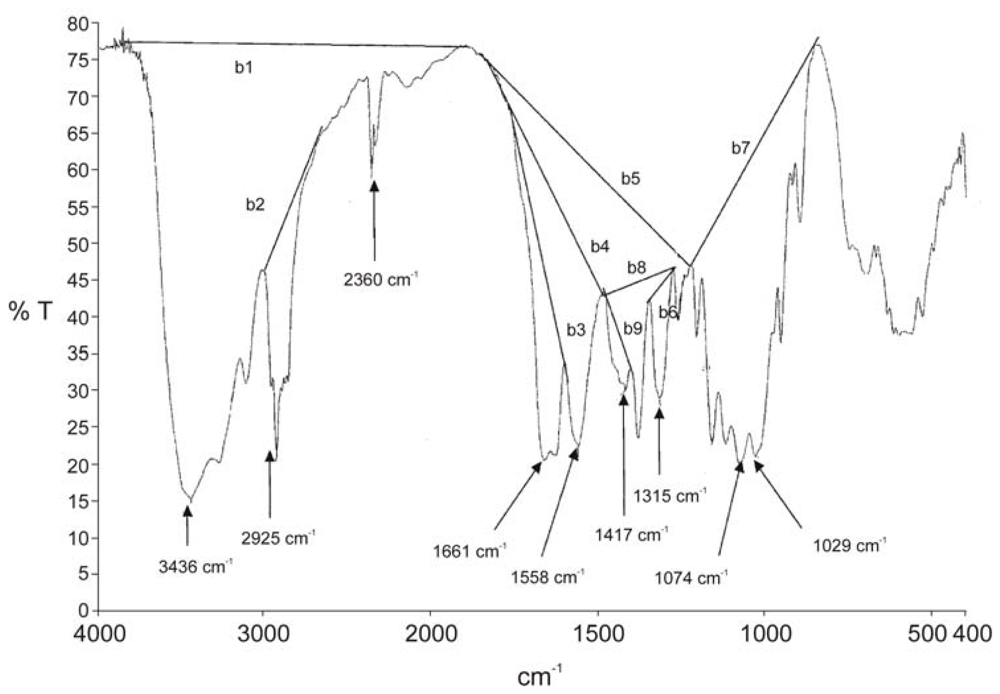
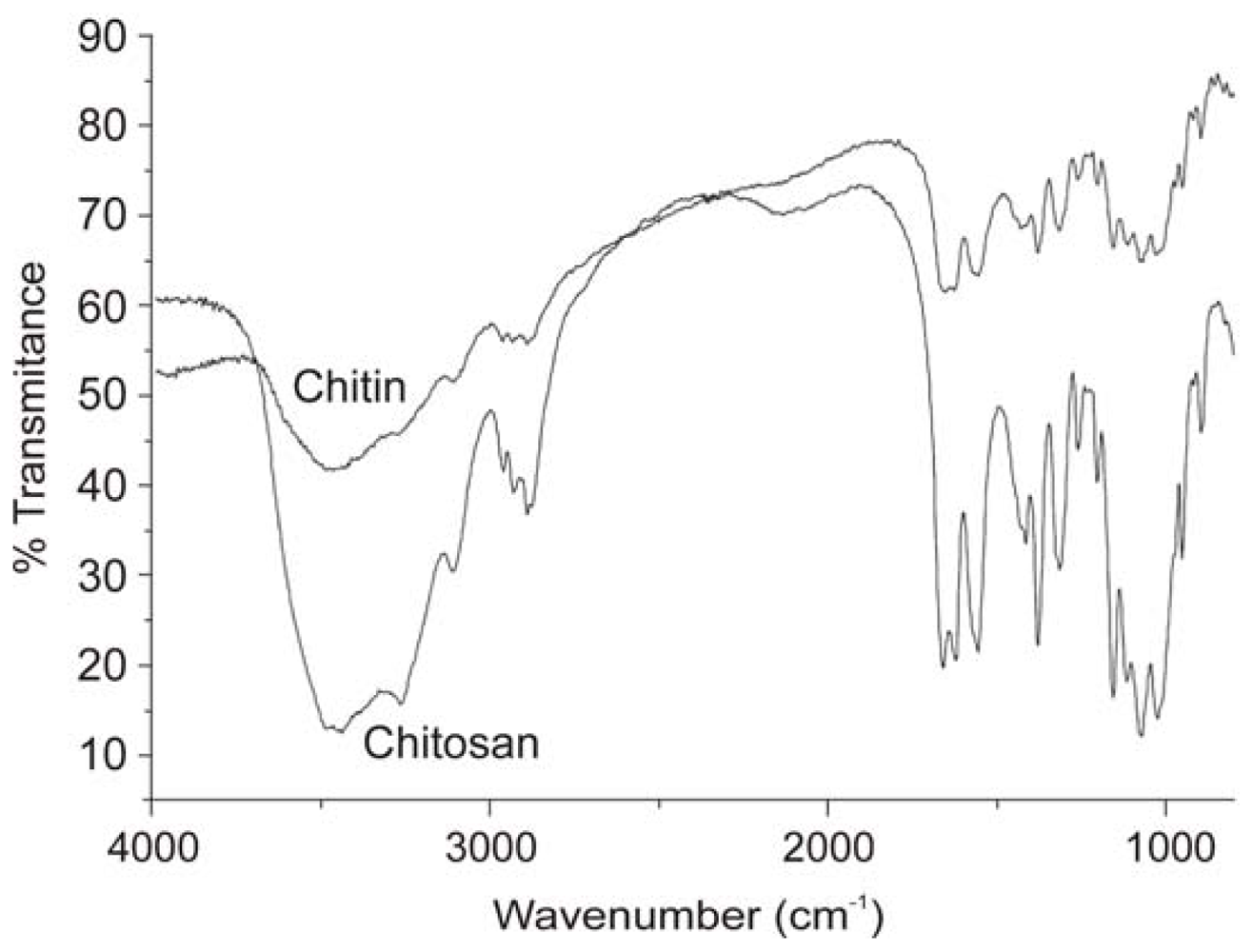
2.2.2. Physicochemical characterization of chitin and chitosan using infrared spectroscopy
2.2.3. Determination of the degree of N-acetylation of chitin and chitosan using infrared spectroscopy
- Determination of the AM/AR ratio, where AM is the intensity of the characteristic band of N-acetylation, which is a measure of the N-acetyl or amine content, and AR is the intensity of a reference band that does not change with different DA values. The DA parameter of unknown samples can be established by comparing the determined AM/AR values with similar ratios of a few reference samples of known DA.
- Drawing a calibration curve by plotting the absorption ratio of chitin/chitosan samples of known DA versus their DA as established by IR or a reference method such as 1H NMR spectroscopy. The DA values of unknown samples can then be estimated from the calibration curve.
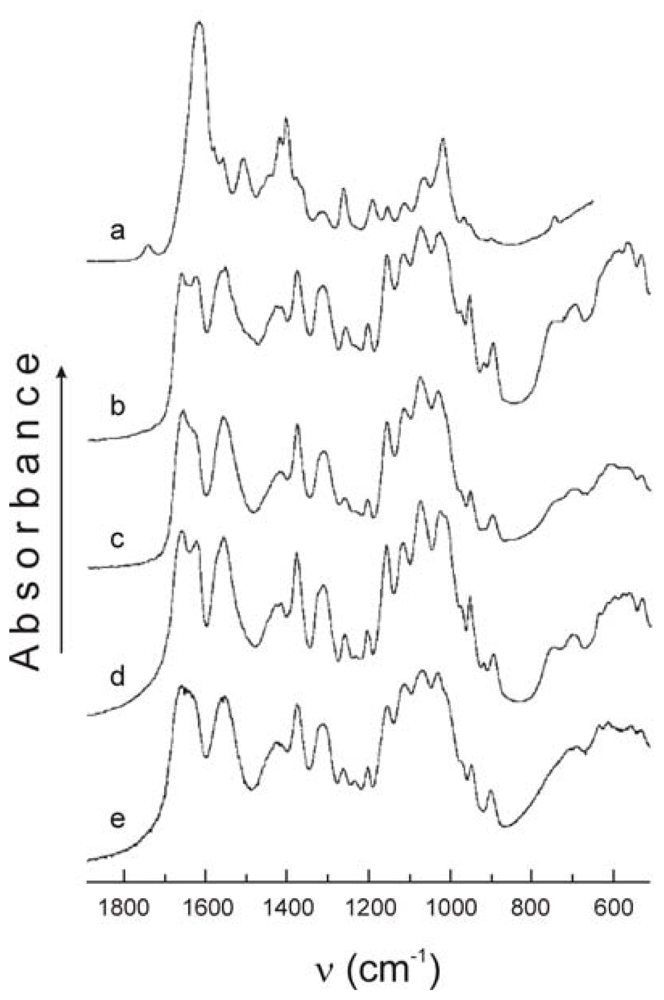
2.2.4. FTIR analysis of chitin and chitosan derivatives
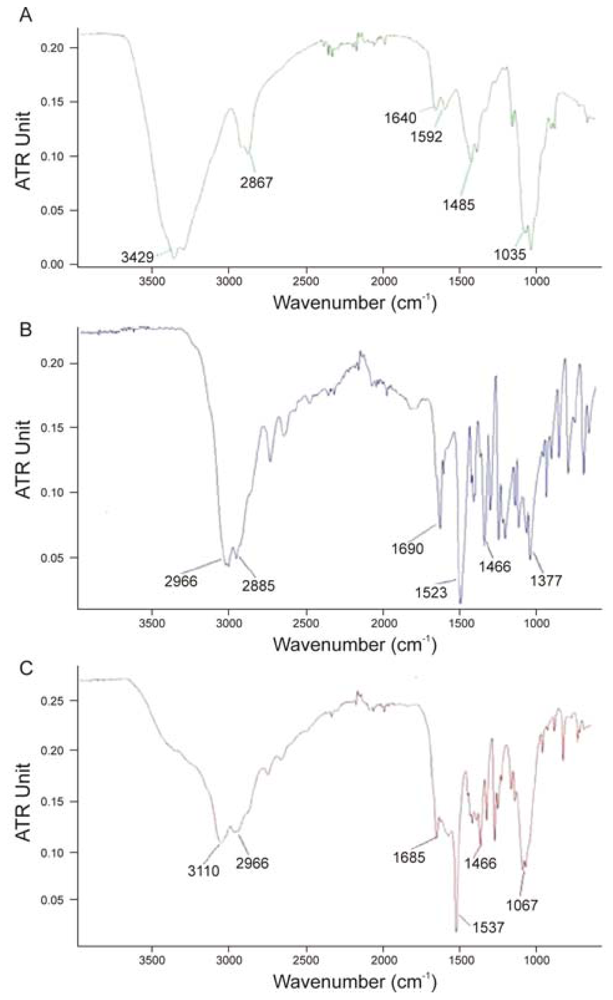
- ▪ in the IR spectra of chitosan (Figure 10A): 3429 cm−1 (O-H stretching overlapping the N-H stretching), 2921 and 2867 cm−1 (C-H stretching), 1640 cm−1 (amide II band, C-O stretching of the acetyl group), 1592 cm−1 (amide II band, N-H stretching) 1485–1380 cm−1 (asymmetrical C-H bending of the CH2 group) and 1035 cm−1 (O bridge stretching) of the glucosamine residue.
- ▪ in the IR spectra of l-GA (Figure 10B): 2966 cm−1 (O-H stretching), 2855 cm−1 for (C-H stretching), 1690 cm−1 (C=O group) and 1523 cm−1 (N-H stretching of the amino group),
- ▪ in the IR spectra of Cl-GA derivative (Figure 10C): 3110 and 2966 cm−1 (axial OH group of chitosan and glutamic acid), 1685 cm−1 (amide linkage), 1556 cm−1 (N-H bending and stretching) and 1067 cm−1 (C-O-C bridge stretching) of the chitosan residue, 1466 cm−1 (the asymmetrical deformation of CH2).
2.3. UV-Vis spectroscopy
2.3.1. Typical conditions of UV-Vis measurement
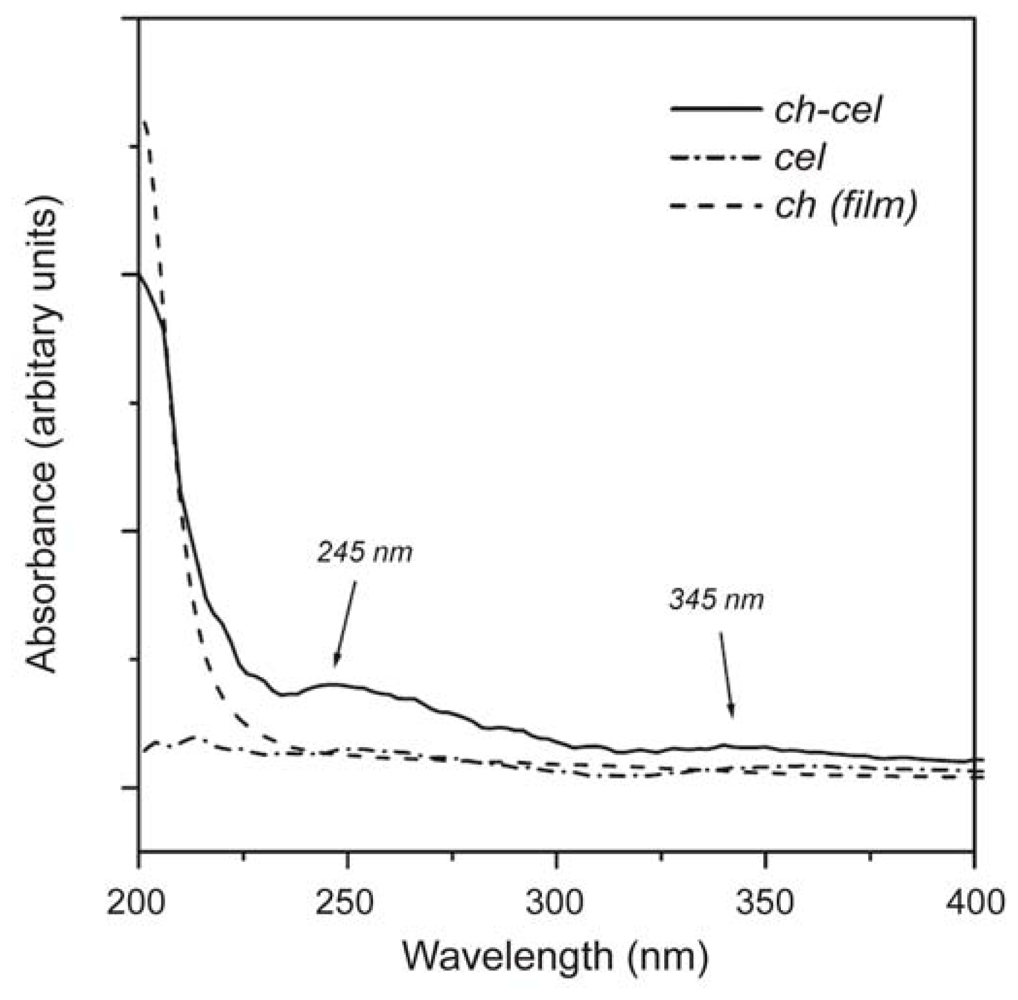
2.3.2. UV-Vis spectra of chitin and chitosan
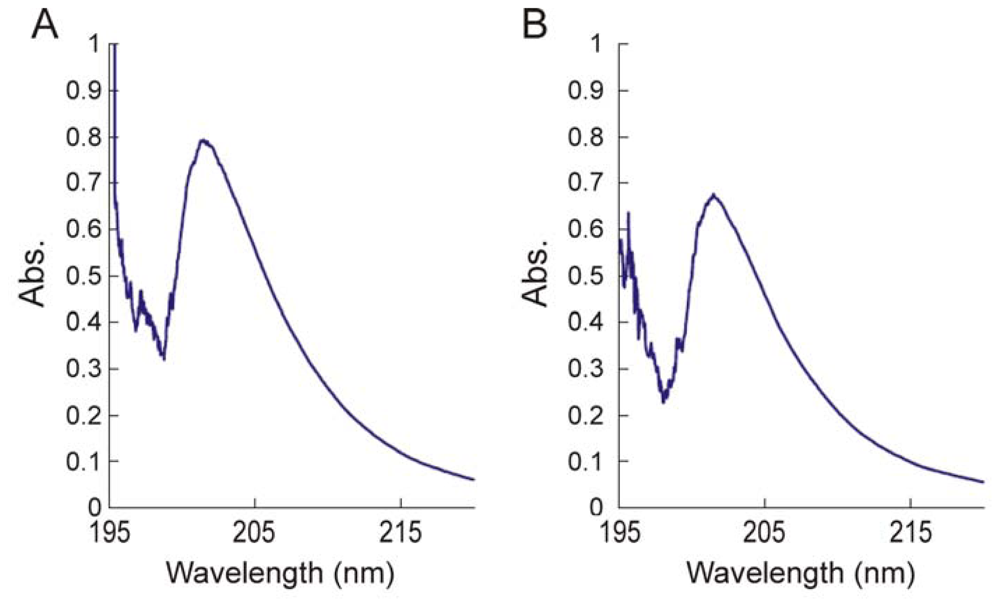
2.3.3. Determination of the degree of N-acetylation of chitin and chitosan using UV-Vis spectroscopy
2.3.4. Application of UV-Vis spectroscopy to the analysis of chitin/chitosan based compounds
2.4. Mass spectrometry
2.4.1. Typical conditions of mass spectrometric analysis of chitin/chitosan and their derivatives
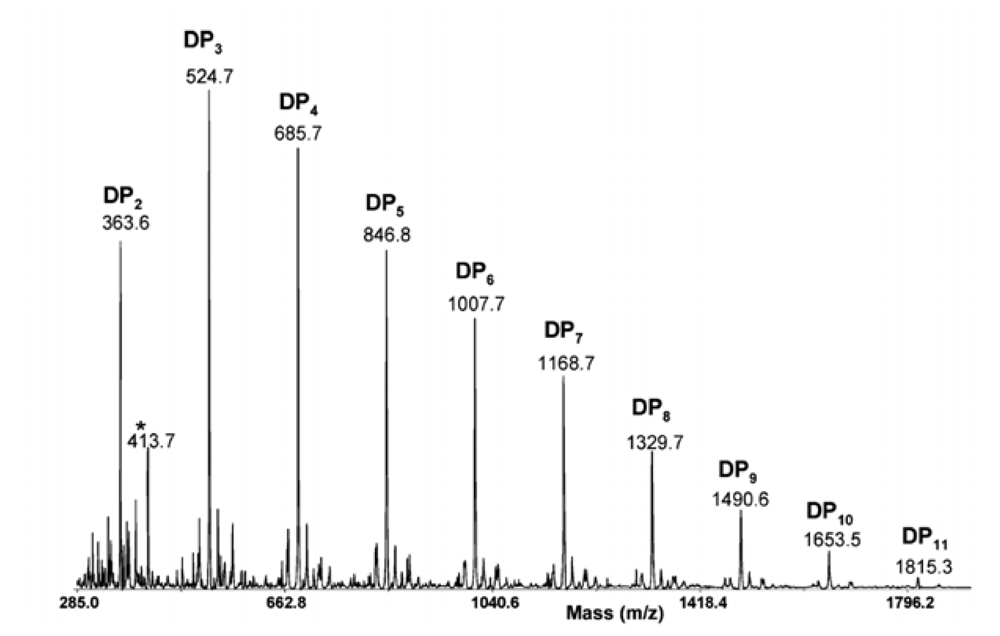
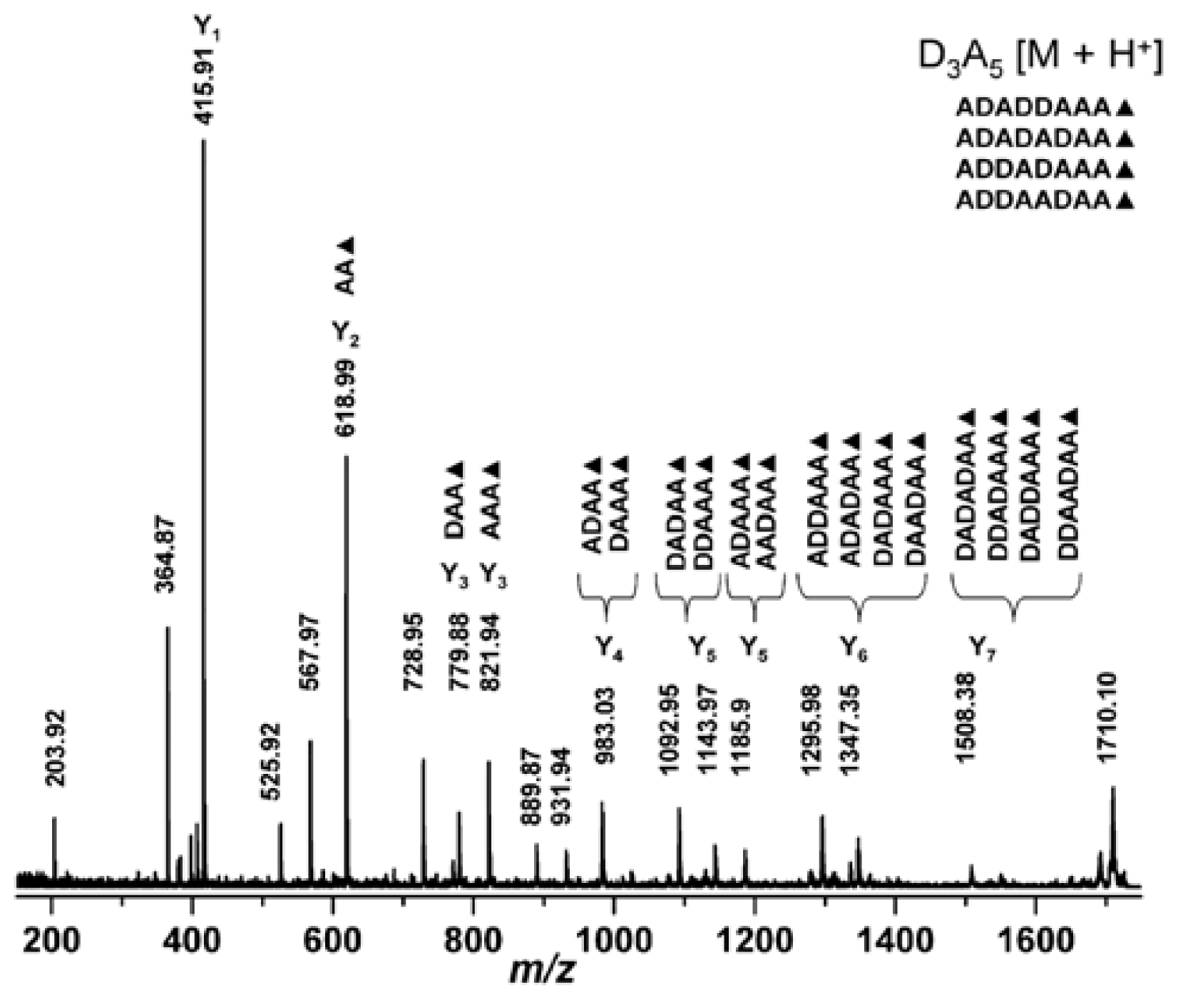
2.4.2. Mass spectrometric determination of the degree of polymerization of chitin and chitosan
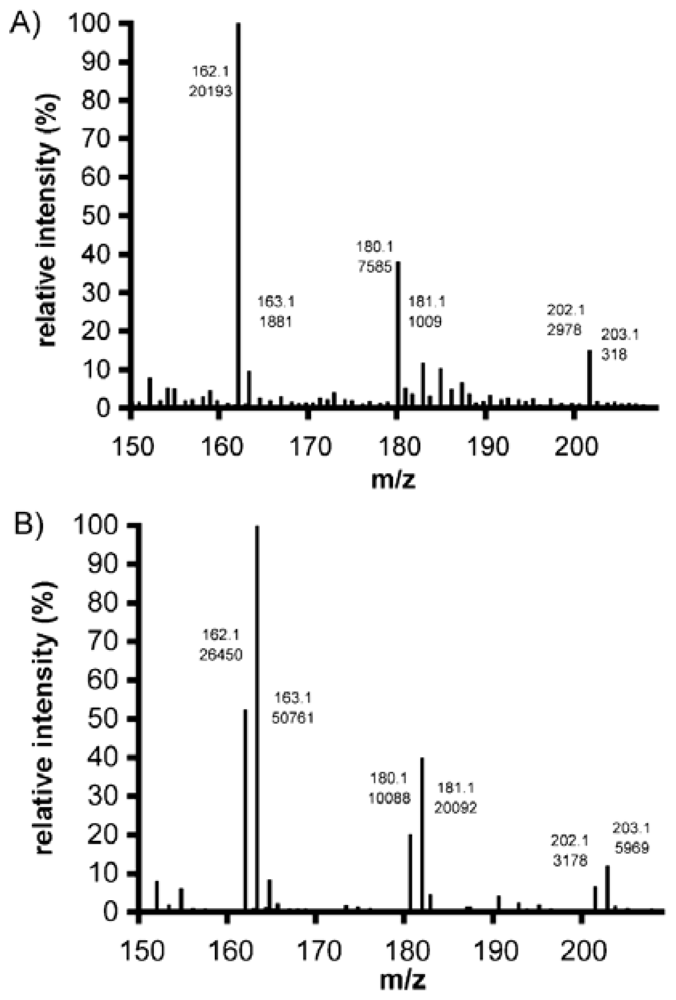
2.4.3. Determination of the degree of N-acetylation of chitin and chitosan using mass spectrometry
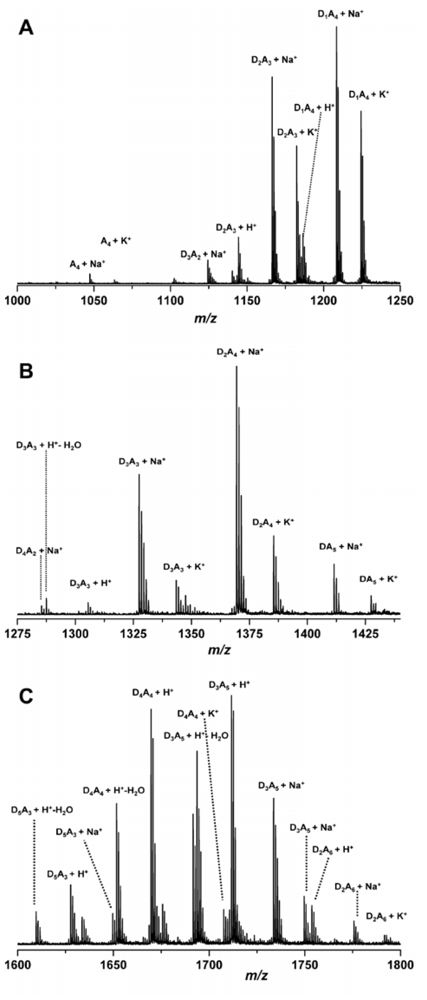
2.4.4. Analysis of chitin and chitosan derivatives using mass spectrometry
2.4.5. Application of chitosan in mass spectrometric analysis
- ▪ Cross-linked chitosan with N-2-hydroxypropyl iminodiacetic acid groups (CCS-HP/IDA), and cross-linked chitosan with N,N- iminodiacetic acid groups (CCS-IDA) [270];
- ▪ Cross-linked chitosan modified with catechol and salicylic acid [271];
- ▪ Chitosan resin derivatized with 3,4-dihydroxybenzoic acid (CCTS-DHBA) [272];
- ▪ Chitosan resin with amino acids [273].
2.5. NMR spectroscopy
2.5.1. Description of NMR techniques
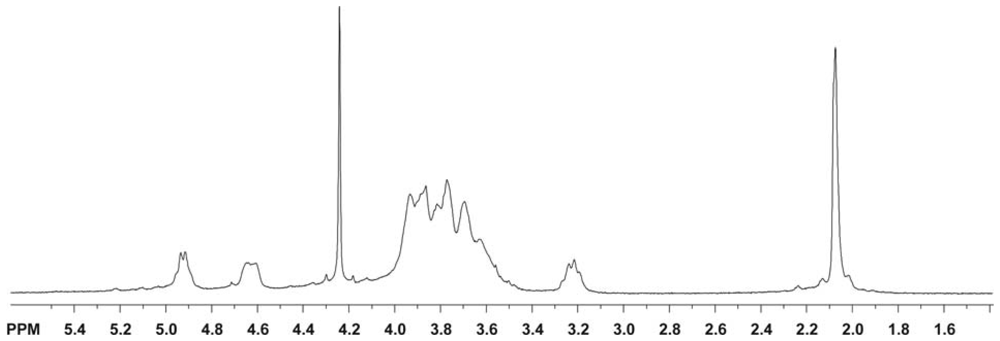
2.5.1.1. 1H NMR spectroscopy
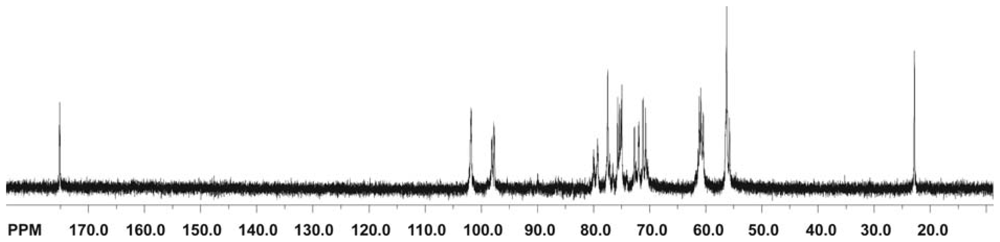
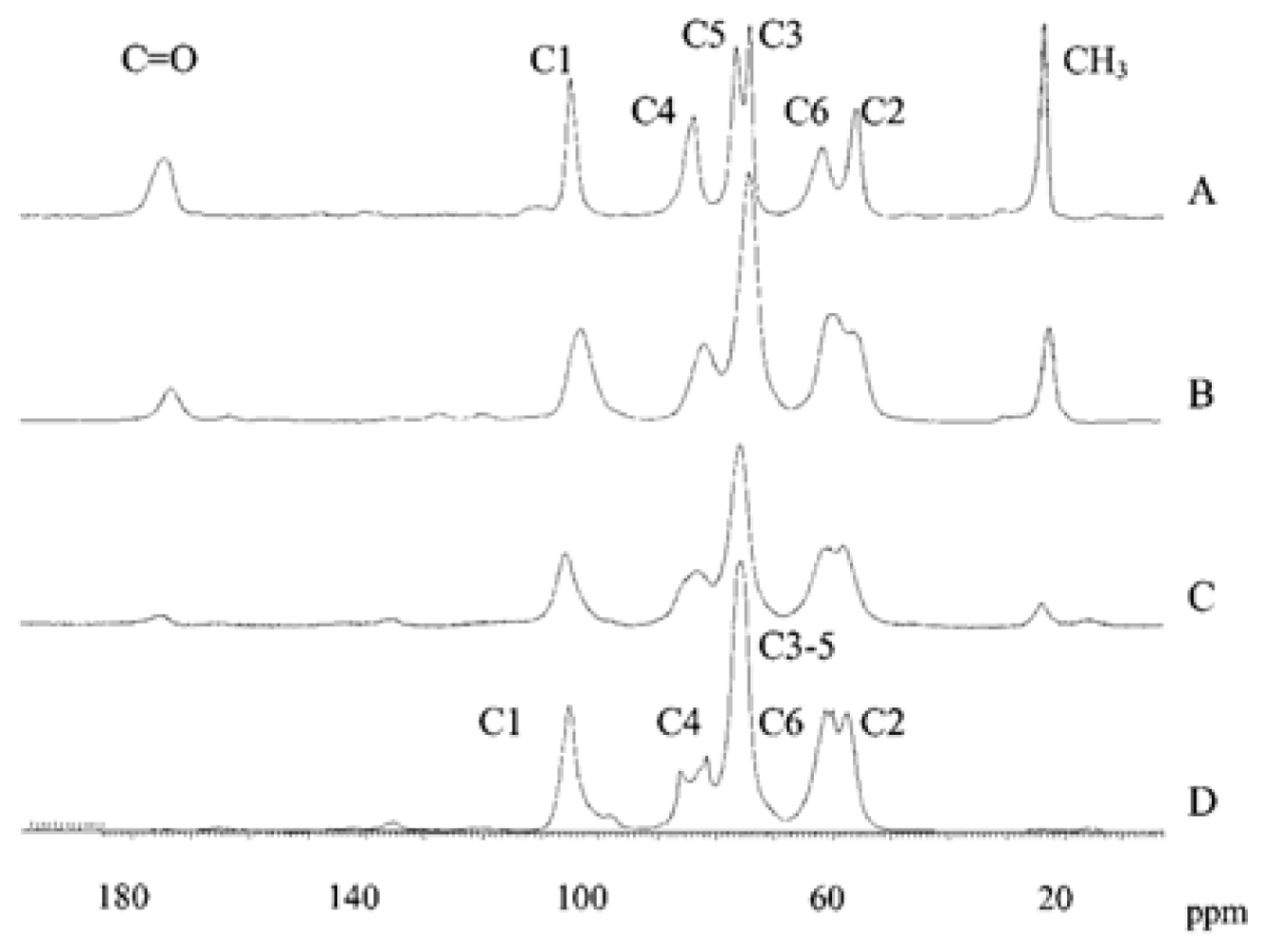
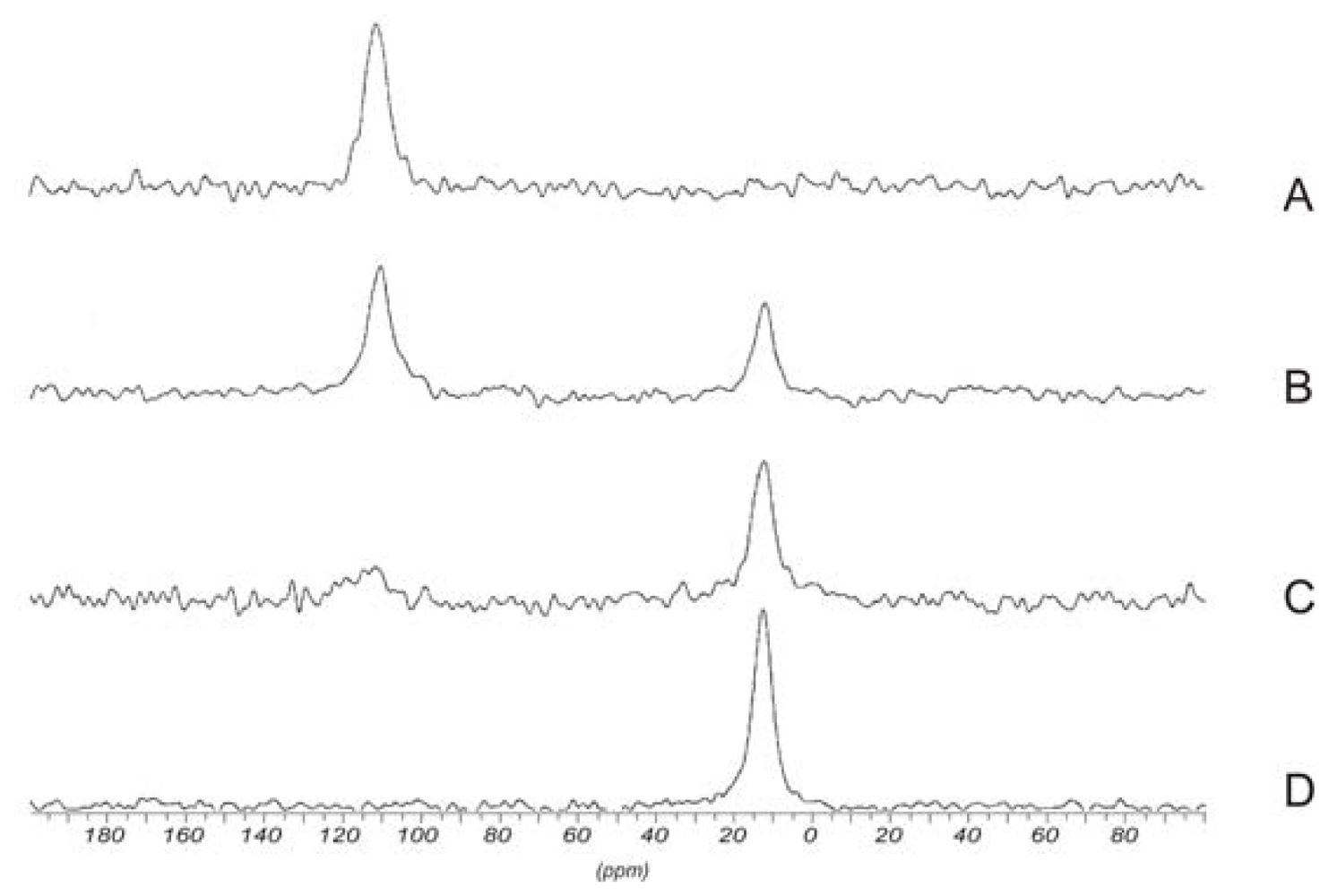
2.5.1.2. 13C NMR spectroscopy
2.5.1.3. 15N NMR spectroscopy
2.5.1.4. 31P NMR spectroscopy
2.5.1.5. Two-dimensional (2D) NMR spectroscopy
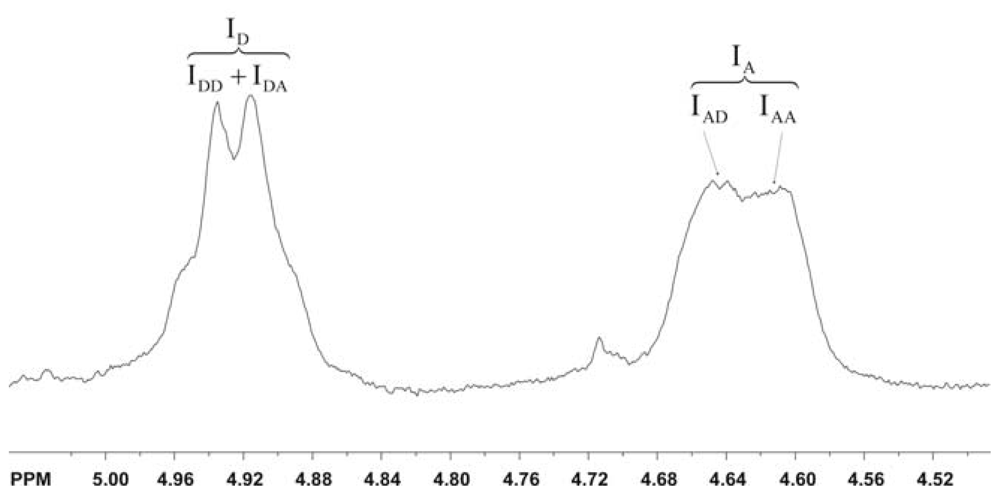
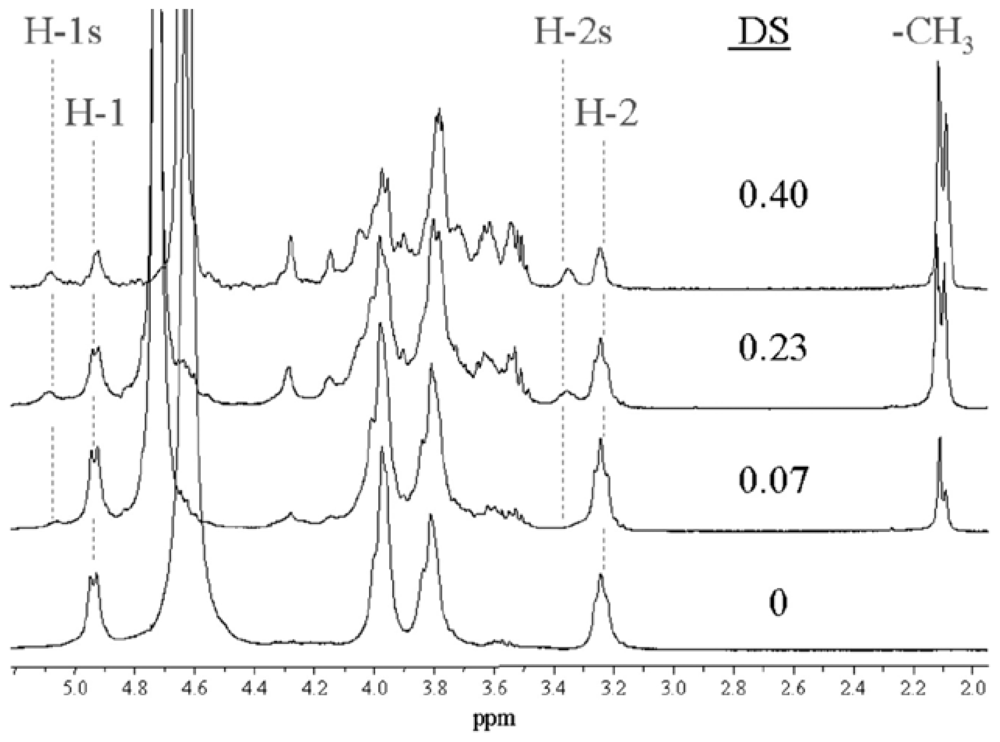
2.5.2. Determination of the degree of acetylation (DA)
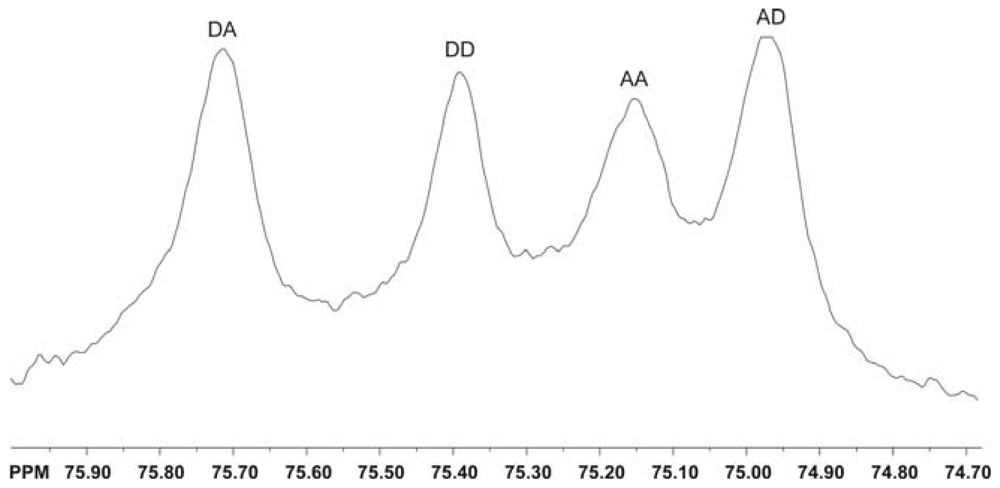
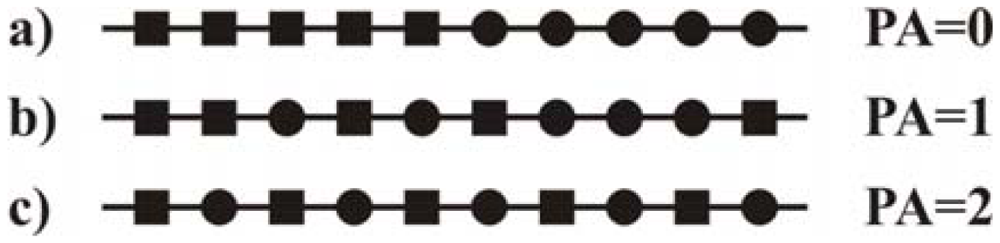
2.5.3. Determination of the pattern of N-acetylation
2.5.4. Study of chitin and chitosan derivatives
2.5.5. Physicochemical characterization of chitin and chitosan
2.5.6. Other applications of NMR techniques
2.6. Other spectroscopic methods
3. Conclusions
Acknowledgements
References and Notes
- Muzzarelli, RAA; Jeuniaux, C; Gooday, GW. Chitin in Nature and Technology; Plenum Publishing Corporation: New York, NY, USA, 1986. [Google Scholar]
- Rinaudo, M. Chitin and chitosan: Properties and application. Prog Polym Sci 2006, 31, 603–632. [Google Scholar]
- Roberts, GAF. Chitin Chemistry, 1st ed; MacMillan: London, UK, 1992. [Google Scholar]
- Austin, PR. Chitin solutions and purification of chitin. Methods Enzymol 1988, 161, 403–407. [Google Scholar]
- Kurita, K. Controlled functionalization of the polysaccharide chitin. Progr Polym Sci 2001, 26, 1921–1971. [Google Scholar]
- Muzzarelli, RAA (Ed.) Natural Chelating Polymers; Pergamon Press: New York, NY, USA, 1973; p. 83.
- Pillai, CKS; Paul, W; Sharma, CP. Chitin and chitosan polymers: Chemistry, solubility and fiber formation. Progr Polym Sci 2009, 34, 641–678. [Google Scholar]
- Ravi Kumar, MNV. A review of chitin and chitosan applications. React Funct Polym 2000, 46, 1–27. [Google Scholar]
- Roberts, GAF. The Road is long... Adv Chitin Sci 2007, 10, 3–10. [Google Scholar]
- Tajik, H; Moradi;, M; Rohani, SMR; Erfani, AM; Jalali, FSS. Preparation of Chitosan from Brine Shrimp (Artemia urmiana) Cyst Shells and Effects of Different Chemical Processing Sequences on the Physicochemical and Functional Properties of the Product. Molecules 2008, 13, 1263–1274. [Google Scholar]
- Muzzarelli, RAA; Muzzarelli, C. Chitosan chemistry: Relevance to the biomedical sciences. Adv Polym Sci 2005, 186, 151–209. [Google Scholar]
- Arai, K; Kinumaki, T; Fujita, T. Toxicity of chitosan. Bull Tokai Reg Fish Lab 1968, 43, 89–94. [Google Scholar]
- Di Martino, A; Sittinger, M; Risbud, MV. Chitosan: A versatile biopolymer for orthopaedic tissue-engineering. Biomaterials 2005, 26, 5983–5990. [Google Scholar]
- Crini, G; Badot, P-M. Application of chitosan, a natural aminopolysaccharide, for dye removal from aqueous solutions by adsorption processes using batch studies: A review of recent literature. Prog Polym Sci 2008, 33, 399–447. [Google Scholar]
- Shahidi, F; Arachchi, JKV; Jeon, Y-J. Food applications of chitin and chitosans. Trends Food Sci Technol 1999, 10, 37–51. [Google Scholar]
- Dutta, PK; Tripathi, S; Mehrotra, GK; Dutta, J. Perspectives for chitosan based antimicrobial films in food applications. Food Chem 2009, 114, 1173–1182. [Google Scholar]
- Thacharodi, D; Panduranga Rao, K. Development and in vitro evaluation of chitosan based transdermal drug delivery systems for controlled delivery of propranolol hydrochloride. Biomaterials 1995, 16, 145–148. [Google Scholar]
- Cho, Y-W; Jang, J; Park, CR; Ko, S-W. Preparation and solubility in acid and water of partially deacetylated chitins. Biomacromolecules 2000, 1, 609–614. [Google Scholar]
- Kim, I-Y; Seo, S-J; Moon, H-S; Yoo, M-K; Park, I-Y; Kim, B-C; Cho, C-S. Chitosan and its derivatives for tissue engineering applications. Biotechnol Adv 2008, 26, 1–21. [Google Scholar]
- Van der Lubben, IM; Verhoef, JC; Borchard, G; Junginger, HE. Chitosan and its derivatives in mucosal drug and vaccine delivery. Eur J Pharm Sci 2001, 14, 201–207. [Google Scholar]
- Alves, NM; Mano, JF. Chitosan derivatives obtained by chemical modifications for biomedical and environmental applications. Int J Biol Macromol 2008, 43, 401–414. [Google Scholar]
- Clasen, C; Wilhelms, T; Kulicke, W-M. Formation and Characterization of Chitosan Membranes. Biomacromolecules 2006, 7, 3210–3222. [Google Scholar]
- Sajomsang, W; Tantayanon, S; Tangpasuthadol, V; Thatte, M; Daly, WH. Synthesis and characterization of N-aryl chitosan derivatives. Int J Biol Macromol 2008, 43, 79–87. [Google Scholar]
- Silva, SS; Menezes, SMC; Garcia, RB. Synthesis and characterization of polyurethane-g-chitosan. Eur Polym J 2003, 39, 1515–1519. [Google Scholar]
- Zhao, Q; Qian, J; An, Q; Gao, C; Gui, Z; Jin, H. Synthesis and characterization of soluble chitosan/sodium carboxymethyl cellulose polyelectrolyte complexes and the pervaporation dehydration of their homogeneous membranes. J Membr Sci 2009, 333, 68–78. [Google Scholar]
- Al Sagheer, FA; Al-Sughayer, MA; Muslim, S; Elsabee, MZ. Extraction and characterization of chitin and chitosan from marine sources in Arabian Gulf. Carbohydr Polym 2009, 77, 410–419. [Google Scholar]
- Campana-Filho, SP; De Britto, D; Curti, E; Abreu, FR; Cardoso, MB; Battisti, MV; Sim, PC; Lavall, RL. Extraction, structures and properties of α- and β-chitin. Quim Nova 2007, 30, 644–650. [Google Scholar]
- Noishiki, Y; Takami, H; Nishiyama, Y; Wada, M; Okada, S; Kuga, S. Alkali-induced conversion of β-chitin to α-chitin. Biomacromolecules 2003, 4, 896–899. [Google Scholar]
- Synowiecki, J; Al-Khateeb, NA. Production, Properties, and Some New Applications of Chitin and Its Derivatives. Crit Rev Food Sci Nutr 2003, 43, 145–171. [Google Scholar]
- Hayes, M; Carney, B; Slater, J; Brück, W. Mining marine shellfish wastes for bioactive molecules: Chitin and chitosan -Part A: Extraction methods. Biotechnol J 2008, 3, 871–877. [Google Scholar]
- Struszczyk, MH. Chitin and Chitosan: Part I. Properties and production. Polimery 2002, 47, 316–325. [Google Scholar]
- Aranaz, I; Mengíbar, M; Harris, R; Paños, I; Miralles, B; Acosta, N; Galed, G; Heras, Á. Functional Characterization of Chitin and Chitosan. Curr Chem Biol 2009, 3, 203–230. [Google Scholar]
- Shirai, K; Palella, D; Castro, Y; Guerrero-Legarreta, I; Saucedo-Castaneda, G; Huerta-Ochoa, S; Hall, GM. Chen, RH, Chen, HC, Eds.; Characterization of chitins from lactic acid fermentation of prawn wastes. In Advances in Chitin Science; Elsevier: Taiwan, 1998; Volume III, pp. 103–110. [Google Scholar]
- Kurita, K; Tomita, K; Ishi, S; Nishimura, S-I; Shimoda, K. β-Chitin as a convenient starting material for acetolysis for efficient preparation of N-acetylchitooligosaccharides. J Polym Sci Part A Polym Chem 1993, 31, 2393–2395. [Google Scholar]
- Aiba, S. Studies on chitosan: 3. Evidence for the presence of random and block copolymer structures in partially N-acetylated chitosans. Int J Biol Macromol 1991, 13, 40–44. [Google Scholar]
- Weinhold, MX; Sauvageau, JCM; Keddig, N; Matzke, M; Tartsch, B; Grunwald, I; Kübel, C; Jastorff, B; Thöming, J. Strategy to improve the characterization of chitosan for sustainable biomedical applications: SAR guided multi-dimensional analysis. Green Chem 2009, 11, 498–509. [Google Scholar]
- Struszczyk, MH; Struszczyk, KJ. Medical Application of Chitin and Its Derivatives; Polish Chitin Society: Lotz, Poland, 2007; Monograph XII; pp. 139–147. [Google Scholar]
- Guo, T. More power to X-rays: New developments in X-ray spectroscopy. Laser Photon 2009, 6, 591–622. [Google Scholar]
- Broglie, M. Sur une nouveau procédé permettant d’obtenir la photographie des spectres de raies des rayons Röntgen. Comptes Rendus 1913, 157, 924–926. [Google Scholar]
- Siegbahn, K. Electron Spectroscopy for Chemical Analysis (ESCA). Philos Trans R Soc Lond A Math Phys Sci 1970, 268, 33–57. [Google Scholar]
- Cullity, BD. Elements of X-ray Diffraction; Addison-Wesley: Reading, MA, USA, 1978. [Google Scholar]
- Clark, GL; Smith, AF. X-ray diffraction studies of chitin, chitosan and derivatives. J Phys Chem 1937, 40, 863–879. [Google Scholar]
- Ogawa, K; Yui, T; Miya, M. Dependence on the preparation procedure of the polymorphism and crystallinity of chitosan membranes. Biosci Biotech Biochem 1992, 56, 858–862. [Google Scholar]
- Varma, AJ; Deshpande, SV; Kennedy, JF. Metal complexation by chitosan and its derivatives: a review. Carbohydr Polym 2004, 55, 77–93. [Google Scholar]
- Minke, R; Blackwell, J. The structure of α-chitin. J Mol Biol 1978, 120, 167. [Google Scholar]
- Matienzo, LJ; Winnacker, SK. Dry Processes for Surface Modification of a Biopolymer: Chitosan. Macromol Mater Eng 2002, 287, 871–880. [Google Scholar]
- Veleshko, AN; Rumyantseva, EV; Veleshko, IE; Teterin, AYu; Maslakov, KI; Teterin, YuA; Kulyukhin, SA; Vikhoreva, GA. X-ray Photoelectron Study of Complexation between Uranyl Group and Chitosan. Radiochemistry 2008, 5, 523–529. [Google Scholar]
- Dambies, L; Guimon, C; Yiacoumi, S; Guibal, E. Characterization of metal ion interactions with chitosan by X-ray photoelectron spectroscopy. Colloids Surf A 2001, 177, 203–214. [Google Scholar]
- Xiaoqi, S; Bo, P; Yang, J; Ji, C; Deqian, L. Chitosan (Chitin)/Cellulose Composite Biosorbents Prepared Using Ionic Liquid for Heavy Metal Ions Adsorption. Am Inst Chem Eng 2009, 55, 2062–2069. [Google Scholar]
- Kurmaev, EZ; Shin, S; Watanabe, M; Eguchi, R; Ishiwata, Y; Takeuchi, T; Moewesd, A; Ederere, DL; Gaof, Y; Iwamig, M; Yanagiharah, M. Probing oxygen and nitrogen bonding sites in chitosan by X-ray emission. J Electron Spectrosc Relat Phenom 2002, 125, 133–138. [Google Scholar]
- Klepka, M; Lawniczak-Jablonska, K; Demchenko, IN; Nedelko, N; Ślawska-Waniewska, A; Rodrigues, CA; Bordini, C. X-ray Absorption Spectroscopy investigation of Fe in metal-chitosan complexes. J Synchr Rad Nat Sci 2006, 5, 220–223. [Google Scholar]
- Jang, MK; Kong, BG; Jeong, YI; Lee, CH; Nah, JW. Physicochemical characterization of α-chitin, β-chitin and γ-chitin separated from natural resources. J Polym Sci Part A Polym Chem 2004, 42, 3423–3432. [Google Scholar]
- Cárdenas, G; Carbrera, G; Taboada, E; Miranda, SP. Chitin characterization by SEM, FTIR, XRD, and 13C cross polarization/mass angle spinning NMR. J Appl Polym Sci 2004, 93, 1876–1885. [Google Scholar]
- Kim, SS; Kim, SH; Lee, YM. Preparation, characterization, and properties of β-chitin and N-acetylated β-chitin. J Polym Sci Part B: Polym Phys 1996, 34, 2367–2374. [Google Scholar]
- Yen, MT; Mau, JL. Selected physical properties of chitin prepared from shiitake stipes. LWT– Food Sci Technol 2007, 40, 558–563. [Google Scholar]
- Muzzarelli, C; Francescanheli, O; Tosi, G; Muzzarelli, RAA. Susceptibility of dibutyryl chitin and regenerated chitin fibres to deacylation and depolymerization by lipases. Carbohydr Polym 2004, 56, 137–146. [Google Scholar]
- Wada, M; Saito, Y. Lateral thermal expansion of chitin crystals. J Polym Sci Part B Polym Phys 2001, 39(1), 168–174. [Google Scholar]
- Feng, F; Liu, Y; Hu, K. Influence of alkali-freezing treatment on the solid state structure of chitin. Carbohydr Res 2004, 339, 2321–2324. [Google Scholar]
- Muzzarelli, RAA; Morganti, P; Morganti, G; Palombo, P; Palombo, M; Biagini, G; Mattioli Belmonte, M; Giantomassi, F; Orlandi, F; Muzzarelli, C. Chitin nanofibrils/chitosan composites as wound medicaments. Carbohydr Polym 2007, 70, 274–284. [Google Scholar]
- Abdou, ES; Nagy, KSA; Elsabee, MZ. Extraction and characterization of chitin and chitosan from local sources. Bioresour Technol 2008, 99, 1359–1367. [Google Scholar]
- Samuels, RJ. Solid state characterization of the structure of chitosan films. J Polym Sci Part B: Polym Phys 1981, 19, 1081–1105. [Google Scholar]
- Sakurai, M; Takagi, M; Takahashi, T. Crystal structure of chitosan. I. Unit cell parameters. Seni Gakkaishi 1984, 40, T-246. [Google Scholar]
- Ogawa, K; Hirano, S; Miyanishi, T; Yui, T; Watanabe, T. A new polymorph of chitosan. Macromolecules 1984, 17, 973–975. [Google Scholar]
- Ogawa, K; Inukai, S. X-Ray diffraction study of sulfuric, nitric, and halogen acid salts of chitosan. Carbohydr Res 1987, 160, 425–433. [Google Scholar]
- Cairns, P; Miles, MJ; Morris, VJ; Ridout, MJ; Brownsey, GJ; Winter, WT. X-ray fibre diffraction studies of chitosan and chitosan gels. Carbohydr Res 1992, 235, 23–28. [Google Scholar]
- Saito, H; Tabeta, R. High-resolution solid-state 13C NMR study of chitosan and its salts with acids: Conformational characterization of polymorphs and helical structures as viewed from the conformation dependent 13C chemical shifts. Macromolecules 1987, 20, 2424–2430. [Google Scholar]
- Yui, T; Imada, K; Okuyama, K; Obata, Y; Suzuki, K; Ogawa, K. Molecular and Crystal Structure of the Anhydrous Form of Chitosan. Macromolecules 1994, 27, 7601–7605. [Google Scholar]
- Mazeau, K; William, WT; Chanzy, H. Molecular and Crystal Structure of a High-Temperature Polymorph of Chitosan from Electron Diffraction Data. Macromolecules 1994, 27, 7606–7612. [Google Scholar]
- Okuyama, K; Noguchi, K; Miyazawa, T; Yui, T; Ogawa, K. Molecular and Crystal Structure of Hydrated Chitosan. Macromolecules 1997, 30, 5849–5855. [Google Scholar]
- Ogawa, K; Oka, K; Miyanishi, T; Hirano, S. Chitin, Chitosan and Related Enzymes; Academic Press: Orlando, FL, USA, 1984; pp. 327–345. [Google Scholar]
- Ogawa, K. Effect of Heating an Aqueous Suspension of Chitosan on the Crystallinity and Polymorphs. Agric Biol Chem 1991, 55, 2375–2379. [Google Scholar]
- Kawada, J; Abe, Y; Yui, T; Okuyama, K; Ogawa, K. Crystalline transformation of chitosan from hydrated to anhydrous polymorph via chitosan monocarboxylic acid salts. J Carbohydr Chem 1999, 18, 559–571. [Google Scholar]
- Lima, IS; Airoldi, C. A thermodynamic investigation on chitosan–divalent cation interactions. Thermochim Acta 2004, 421, 133–139. [Google Scholar]
- Zhang, Y; Xue, C; Xue, Y; Gao, R; Zhang, X. Determination of the degree of deacetylation of chitin and chitosan by X-ray powder diffraction. Carbohydr Res 2005, 340, 1914–1917. [Google Scholar]
- Focher, B; Beltranme, PL; Naggi, A; Torri, G. Alkaline Ndeacetylation of chitin enhanced by flash treatments: reaction kinetics and structure modifications. Carbohydr Polym 1990, 12, 405–418. [Google Scholar]
- Struszczyk, H. Microcrystalline Chitosan. I Preparation and Properties of Microcrystalline Chitosan. J Appl Polym Sci 1987, 33, 177–189. [Google Scholar]
- Schiffman, JD; Stulga, LA; Schauer, CL. Chitin and Chitosan: Transformations Due to the Electrospinning Process. Polym Eng Sci 2009, 49, 1918–1928. [Google Scholar]
- Zhang, Y; Xue, C; Li, Z; Zhang, Y; Fu, X. Preparation of half-deacetylated chitosan by forced penetration and its properties. Carbohydr Polym 2006, 65, 229–234. [Google Scholar]
- Fernandez Cerveraa, M; Heinamaki, J; Rasanenc, M; Maunuc, SL; Karjalainenb, M; Nieto Acostaa, OM; Iraizoz Colartea, A; Yliruusib, J. Solid-state characterization of chitosans derived from lobster chitin. Carbohydr Polym 2004, 58, 401–408. [Google Scholar]
- Toffey, A; Glasser, WG. Chitin Derivatives. II. Time–Temperature–Transformation Cure Diagrams of the Chitosan Amidization Process. J Appl Polym Sci 1999, 73, 1879–1889. [Google Scholar]
- Ogawa, K; Yui, T. Crystalllinity of partialy N-acetylated chitosan. Biosci Biotech Biochem 1993, 57, 1466–1469. [Google Scholar]
- Seoudi, R; Nada, AMA. Molecular structure and dielectric properties studies of chitin and its treated by acid, base and hypochlorite. Carbohydr Polym 2007, 68, 728–733. [Google Scholar]
- Nishino, T; Matsui, R; Nakamae, K. Elastic Modulus of the Crystalline Regions of Chitin and Chitosan. J Polym Sci Part B Polym Phys 1999, 37, 1191–1196. [Google Scholar]
- Ogawa, K. X-ray study of Chitosan L- and D- Ascorbates. Chem Mater 1996, 8, 2349–2351. [Google Scholar]
- Muzzarelli, RAA. Chitin; Pergamon Press: Oxford, UK, 1977. [Google Scholar]
- Ogawa, K; Oka, K. X-ray Study of Chitosan-Transition Metal Complexes. Chem Mater 1993, 5, 726–728. [Google Scholar]
- Schlick, S. Binding Sites of Cu2+ in Chitin and Chitosan. An Electron Spin Resonance Study. Macromolecules 1986, 19, 192–195. [Google Scholar]
- El-Sherbiny, IM. Synthesis, characterization and metal uptake capacity of a new carboxymethyl chitosan derivative. Eur Polym J 2009, 45, 199–210. [Google Scholar]
- Hai-Bing, Li; Yuan-Yin, C; Shi-Lan, L. Synthesis, Characterization, and Metal Ions Adsorption Properties of Chitosan–Calixarenes (I). J Appl Polym Sci 2003, 89, 1139–1144. [Google Scholar]
- Trimukhe, KD; Varma, AJ. A morphological study of heavy metal complexes of chitosan and crosslinked chitosans by SEM and WAXRD. Carbohydr Polym 2008, 71, 698–702. [Google Scholar]
- Jobish, J; Vijayalakshmi, R. Thermal stability, morphology, and X-ray diffraction studies of dynamically vulcanized natural rubber/chitosan blends. J Mater Sci 2009, 44, 4087–4094. [Google Scholar]
- Sahoo, S; Sasmal, A; Nanda, R; Phani, AR; Nayak, PL. Synthesis of chitosan– polycaprolactone blend for control delivery of ofloxacin drug. Carbohydr Polym 2010, 79, 106–113. [Google Scholar]
- Zuńiga, A; Debbaudt, A; Albertengo, L; Rodríguez, MS. Synthesis and characterization of N-propyl-N-methylene phosphonic chitosan derivative. Carbohydr Polym 2010, 79, 475–480. [Google Scholar]
- Mohamed, KR; Mostafa, AA. Preparation and bioactivity evaluation of hydroxyapatite-titania/chitosangelatin polymeric biocomposites. Mater Sci Eng A 2008, 28, 1087–1099. [Google Scholar]
- Nagahama, H; Maeda, H; Kashiki, T; Jayakumar, R; Furuike, T; Tamura, H. Preparation and characterization of novel chitosan/gelatin membranes using chitosan hydrogel. Carbohydr Polym 2009, 76, 255–260. [Google Scholar]
- Tretenichenko, EM; Datsun, VM; Ignatyuk, LN; Nud’ga, LA. Preparation and Properties of Chitin and Chitosan from a Hydroid Polyp. Russ J Appl Chem 2006, 79, 1341–1346. [Google Scholar]
- Aji, PM; Laborie, MPG; Oksman, K. Cross-Linked Chitosan/Chitin Crystal Nanocomposites with Improved Permeation Selectivity and pH Stability. Biomacromolecules 2009, 10, 1627–1632. [Google Scholar]
- Kurmaev, EZ; Shin, S; Watanabe, M; Eguchi, R; Ishiwata, Y; Takeuchi, T; Moewesd, A; Ederere, DL; Gaof, Y; Iwamig, M; Yanagiharah, M. Probing oxygen and nitrogen bonding sites in chitosan by X-ray emission. J Electron Spectrosc Relat Phenom 2002, 125, 133–138. [Google Scholar]
- Stuart, BH. Infrared Spectroscopy: Fundamentals and Applications (Analytical Techniques in the Sciences (AnTs) *); John Wiley & Sons Ltd: Chichester, UK, 2004. [Google Scholar]
- Smith, BC. Fundamentals of Fourier Transform Infrared Spectroscopy; CRC Press: Boca Raton, FL, USA, 1996. [Google Scholar]
- Günzler, H; Gremlich, HU. IR Spectroscopy: An Introduction; Wiley-VCH: Weinherm, Germany, 2002. [Google Scholar]
- Griffiths, P; De Haseth, JA. Fourier Transform Infrared Spectrometry, 2nd ed; Wiley- Interscience: New York, NY, USA, 2007. [Google Scholar]
- Thanpitcha, T; Sirivat, A; Jamieson, AM; Rujiravanit, R. Dendritic polyaniline nanoparticles synthesized by carboxymethyl chitin templating. Eur Polym J 2008, 44, 3423–3429. [Google Scholar]
- Bourtoom, T; Chinnan, MS. Preparation and properties of rice starch-chitosan blend biodegradable film. LWT-Food Sci Technol 2008, 41, 1633–1641. [Google Scholar]
- Yang, J; Yao, Z; Tang, C; Darvell, BW; Zhang, H; Pan, L; Liu, J; Chen, Z. Growth of apatite on chitosan-multiwall carbon nanotube composite membranes. Appl Surf Sci 2009, 255, 8551–8555. [Google Scholar]
- Sajomsang, W; Ruktanonchai, UR; Gonil, P; Nuchuchua, O. Mucoadhesive property and biocompatibility of methylated N-aryl chitosan derivatives. Carbohydr Polym 2009, 78, 945–952. [Google Scholar]
- Urreaga, JM; de la Orden, MU. Chemical interactions and yellowing in chitosan-treated cellulose. Eur Polym J 2006, 42, 2606–2616. [Google Scholar]
- Darmon, SE; Rudall, KM. Infra-red and X-ray studies of chitin. Disc Faraday Soc 1950, 9, 251–260. [Google Scholar]
- Pearson, FG; Marchessault, RH; Liang, CY. Infrared spectra of crystalline polysaccharides. V. Chitin. J Polym Sci 1960, 13, 101–116. [Google Scholar]
- Brunner, E; Ehrlich, H; Schupp, P; Hedrich, R; Hunoldt, S; Kammer, M; Machill, S; Paasch, S; Bazhenov, VV; Kurek, DV; Arnold, T; Brockmann, S; Ruhnow, M; Born, R. Chitin-based scaffolds are an integral part of the skeleton of the marine demosponge Ianthella basta. J Struct Biol 2009, 168, 539–547. [Google Scholar]
- Focher, B; Naggi, A; Torri, G; Cosani, A; Terbojevich, M. Structural differences between chitin polymorphs and their precipitates from solutions-evidence from CP-MAS 13CNMR, FT-IR and FT-Raman spectroscopy. Carbohydr Polym 1992, 17, 97–102. [Google Scholar]
- Juárez-de la Rosa, BA; Ardisson, P-L; Azamar-Barrios, JA; Quintana, P; Alvarado-Gil, JJ. Optical, thermal, and structural characterization of the sclerotized skeleton of two antipatharian coral species. Mater Sci Eng C 2007, 27, 880–885. [Google Scholar]
- Furuhashi, T; Schwarzinger, C; Miksik, I; Smrz, M; Beran, A. Molluscan shell evolution with review of shell calcification hypothesis. Comp Biochem Physiol B Biochem Mol Biol 2009, 154, 351–371. [Google Scholar]
- Paulino, AT; Simionato, JI; Garcia, JC; Nozaki, J. Characterization of chitosan and chitin produced from silkworm chrysalides. Carbohydr Polym 2006, 64, 98–103. [Google Scholar]
- Lertwattanaseri, T; Ichikawa, N; Mizoguchi, T; Tanaka, Y; Chirachanchai, S. Microwave technique for efficient deacetylation of chitin nanowhiskers to a chitosan nanoscaffold. Carbohydr Res 2009, 344, 331–335. [Google Scholar]
- Pranshanth, KVH; Kittur, FS; Tharanathan, RN. Solid state structure of chitosan prepared under different N-deacetylating conditions. Carbohydr Polym 2002, 50, 27–33. [Google Scholar]
- Wu, T; Zivanovic, S; Draughon, FA; Conoway, WS; Sams, CE. Physicochemical Properties and Bioactivity of Fungal Chitin and Chitosan. J Agric Food Chem 2005, 53, 3888–3894. [Google Scholar]
- Prashanth, KVH; Tharanathan, RN. Crosslinked chitosan-preparation and characterization. Carbohydr Res 2006, 341, 169–173. [Google Scholar]
- Nah, J-W; Jang, M-K. Spectroscopic Characterization and Preparation of Low Molecular, Water-Soluble Chitosan with Free-Amine Group by Novel Method. J Polym Sci Part A Polym Chem 2002, 40, 3796–3803. [Google Scholar]
- Dong, Y; Wang, H; Zheng, W; Zhao, Y; Bi, D; Zhao, L; Li, X. Liquid crystalline behaviour of chitooligosaccharides. Carbohydr Polym 2004, 57, 235–240. [Google Scholar]
- Li, QD; Dunn, ET; Grandmaison, EW; Goosen, MFA. Applications and Properties of Chitosan. J Bioact Compat Polym 1992, 7, 370–397. [Google Scholar]
- Kasaai, MR. Various Methods for Determination of the Degree of N-Acetylation of Chitin and Chitosan: A Review. J Agric Food Chem 2009, 57, 1667–1676. [Google Scholar]
- Sabnis, S; Block, LH. Improved infrared spectroscopic method for the analysis of degree of N-deacetylation. Polym Bull 1997, 39, 67–71. [Google Scholar]
- Muzzarelli, RAA; Tanfani, F; Scarpini, G; Laterza, G. The degree of acetylation of chitins by gas chromatography and infrared spectroscopy. J Biochem Bioph Methods 1980, 2, 299–306. [Google Scholar]
- Domszy, JG; Roberts, GAF. Evaluation of infrared spectroscopic techniques for analysing chitosan. Die Makromolekulare Chemie 1985, 186, 1671–1677. [Google Scholar]
- Baxter, A; Dillon, M; Taylor, KD; Roberts, GAF. Improved method for IR determination of the degree of N-acetylation of chitosan. Inter J Biolog Macromol 1992, 14, 166–169. [Google Scholar]
- Shigemasa, Y; Matsuura, H; Sashiwa, H; Saimato, H. Domard, A, Jeuniaux, C, Muzzarelli, RAA, Roberts, GAF, Eds.; An improved IR spectroscopic determination of degree of deacetylation of chitin. In Advances in Chitin Science; André Publisher: Lyon, France, 1996; Volume 1, pp. 204–209. [Google Scholar]
- Shigemasa, Y; Matsuura, H; Sashiwa, H; Saimato, H. Evaluation of different absorbance ratios from infrared spectroscopy for analyzing the degree of deacetylation in chitin. Int J Biol Macromol 1996, 18, 237–242. [Google Scholar]
- Brugnerotto, J; Lizardi, J; Goyoolea, FM; Argülles-Monal, W; Desbrières, J; Rinaudo, M. An infrared investigation in relation with chitin and chitosan characterization. Polymer 2001, 42, 3569–3580. [Google Scholar]
- Duarte, ML; Ferreira, MC; Marvão, MR; Rocha, J. An optimized method to determine the degree of acetylation of chitin and chitosan by FTIR spectroscopy. Int J Biol Macromol 2002, 31, 1–8. [Google Scholar]
- Kasaai, MR. A review of several reported procedures to determine the degree of N-acetylation for chitin and chitosan using infrared spectroscopy. Carbohydr Polym 2008, 71, 497–508. [Google Scholar]
- Duarte, ML; Ferreira, MC; Marvão, MR; Rocha, J. Uragami, T, Kurita, K, Fukamizo, T, Eds.; Chitin and chitosan: An optimized methodology by FTIR spectroscopy. In Chitin and Chitosan in Life Science; Kodansha Scientific Ltd: Tokyo, Japan, 2001; pp. 86–90. [Google Scholar]
- Moore, GK; Roberts, GAF. Determination of the degree of N-acetylation of chitosan. Int J Biol Macromol 1980, 2, 115–116. [Google Scholar]
- Dong, Y; Xu, C; Wang, J; Wu, Y; Wang, M; Ruan, Y. Influence of degree of deacetylation on critical concentration of chitosan/dichlorocatic acid liquid crystalline solution. J Appl Polym Sci 2002, 83, 1204–1208. [Google Scholar]
- Qin, C; Li, H; Xiao, Q; Liu, Y; Zhu, J; Du, Y. Water-solubility of chitosan and its antimicrobial activity. Carbohydr Polym 2006, 63, 367–374. [Google Scholar]
- Miya, M; Iwamoto, R; Yoshikawa, S; Mima, S. I.R spectroscopic determination of CONH content in highly deacetylated chitosan. Int J Biol Macromol 1980, 2, 323–324. [Google Scholar]
- Qu, X; Wirsén, A; Albertsson, A-C. Effect of lactic/glycolic acid side chains on the thermal degradation kinetics of chitosan derivatives. Polymer 2000, 41, 4841–4847. [Google Scholar]
- Rathke, TD; Hudson, SM. Determination of the degree of N-acetylation in chitin and chitosan as well as their monomer sugar ratios by near infrared spectroscopy. J Polym Sci A Polym Chem 1993, 31, 749–753. [Google Scholar]
- Vårum, KM; Egelandsdal, B; Ellekjaer, MR. Characterization of partially N-acetylated chitosans by near infrared spectroscopy. Carbohydr Polym 1995, 28, 187–193. [Google Scholar]
- Baskar, D; Sampath Kumar, TS. Effect of deacetylation time on the preparation, properties and swelling behavior of chitosan films. Carbohydr Polym 2009, 78, 767–772. [Google Scholar]
- Vachoud, L; Zydowicz, N; Domarad, A. Formation and characterisation of a physical chitin gel. Carbohydr Res 1997, 302, 169–177. [Google Scholar]
- Zainol, I; Akil, HM; Mastor, A. Effect of γ-irradiation on the physical and mechanical properties of chitosan powder. Mater Sci Eng C 2009, 29, 292–297. [Google Scholar]
- Ma, G; Yang, D; Kennedy, JF; Nie, J. Synthesize and characterization of organic-soluble acylated chitosan. Carbohydr Polym 2009, 75, 390–394. [Google Scholar]
- Singh, J; Dutta, PK; Dutta, J; Hunt, AJ; Macquarrie, DJ; Clark, JH. Preparation and properties of highly soluble chitosan–l-glutamic acid aerogel derivative. Carbohydr Polym 2009, 76, 188–195. [Google Scholar]
- Xie, Y; Liu, X; Chen, Q. Synthesis and characterization of water-soluble chitosan derivate and its antibacterial activity. Carbohydr Polym 2007, 69, 142–147. [Google Scholar]
- Zhang, C; Ping, Q; Zhang, H; Shen, J. Synthesis and characterization of water-soluble O-succinyl- chitosan. Eur Polym J 2003, 39, 1629–1634. [Google Scholar]
- Ma, G; Yang, D; Zhou, Y; Xiao, M; Kennedy, JF; Nie, J. Preparation and characterization of water-soluble N-alkylated chitosan. Carbohydr Polym 2008, 74, 121–126. [Google Scholar]
- Jeong, Y-I; Kim, D-G; Jang, M-K; Nah, J-W. Preparation and spectroscopic characterization of methoxy poly(ethylene glycol)-grafted water-soluble chitosan. Carbohydr Res 2008, 343, 282–289. [Google Scholar]
- Zong, Z; Kimura, Y; Takahashi, M; Yamane, H. Characterization of chemical and solid state structures of acylated chitosans. Polymer 2000, 41, 899–906. [Google Scholar]
- Kaushik, A; Khan, R; Solanki, PR; Pandey, P; Alam, J; Ahmad, S; Malhotra, BD. Iron oxide nanoparticles–chitosan composite based glucose biosensor. Biosens Bioelectron 2008, 24, 676–683. [Google Scholar]
- Borges, O; Borchard, G; Verhoef, JC; de Sousa, A; Junginger, HE. Preparation of coated nanoparticles for a new mucosal vaccine delivery system. Int J Pharm 2005, 299, 155–166. [Google Scholar]
- Yang, D; Li, J; Jiang, Z; Lu, L; Chen, X. Chitosan/TiO2 nanocomposite pervaporation membranes for ethanol dehydration. Chem Eng Sci 2009, 64, 3130–3137. [Google Scholar]
- Tan, H; Chu, CR; Payne, KA; Marra, KG. Injectable in situ forming biodegradable chitosan–hyaluronic acid based hydrogels for cartilage tissue engineering. Biomaterials 2009, 30, 2499–2506. [Google Scholar]
- Madhumathi, K; Shalumon, KT; Divya Rani, VV; Tamura, H; Furuike, T; Selvamurugan, N; Nair, SV; Jayakumar, R. Wet chemical synthesis of chitosan hydrogel–hydroxyapatite composite membranes for tissue engineering applications. Int J Biol Macromol 2009, 45, 12–15. [Google Scholar]
- Xie, W; Xu, P; Wang, W; Liu, Q. Preparation and antibacterial activity of a water-soluble chitosan derivative. Carbohydr Polym 2002, 50, 35–40. [Google Scholar]
- Wu, C-S. A comparison of the structure, thermal properties, and biodegradability of polycaprolactone/chitosan and acrylic acid graftedpolycaprolactone/chitosan. Polymer 2005, 46, 47–155. [Google Scholar]
- Munro, NH; Hanton, LR; Moratti, SC; Robinson, BH. Synthesis and characterisation of chitosan-graft-poly(OEGMA) copolymers prepared by ATRP. Carbohydr Polym 2009, 77, 496–505. [Google Scholar]
- Wu, Z; Feng, W; Feng, Y; Liu, Q; Xu, X; Sekino, T; Fujii, A; Ozaki, M. Preparation and characterization of chitosan-grafted multiwalled carbon nanotubes and their electrochemical properties. Carbon 2007, 45, 1212–1218. [Google Scholar]
- Winie, T; Arof, AK. FT-IR studies on interactions among components in hexanoyl chitosan-based polymer electrolytes. Spectrochim Acta Part A 2006, 63, 677–684. [Google Scholar]
- Piron, E; Domarad, A. Interaction between chitosan and uranyl ions. Part 2. Mechanism of interaction. Int J Biol Macromol 1998, 22, 33–40. [Google Scholar]
- Sipos, P; Berkesi, O; Tombácz, E; StPierree, TG; Webb, J. Formation of spherical iron(III) oxyhydroxide nanoparticles sterically stabilized by chitosan in aqueous solutions. J Inorg Biochem 2003, 95, 55–63. [Google Scholar]
- Bhattarai, SR; Bahadur, KCR; Aryal, S; Khil, MS; Kim, HY. N-Acylated chitosan stabilized iron oxide nanoparticles as a novel nano-matrix and ceramic modification. Carbohydr Polym 2007, 69, 467–477. [Google Scholar]
- Yavuz, AG; Uygun, A; Bhethanabotla, VR. Substituted polyaniline/chitosan composites: Synthesis and characterization. Carbohydr Polym 2009, 75, 448–453. [Google Scholar]
- Baran, EJ. Spectroscopic investigation of the VO2+/chitosan interaction. Carbohydr Polym 2008, 74, 704–706. [Google Scholar]
- Phisalaphong, M; Jatupaiboon, N. Biosynthesis and characterization of bacteria cellulose–chitosan film. Carbohydr Polym 2008, 74, 482–488. [Google Scholar]
- Kamari, A; Wan Ngah, WS; Liew, LK. Chitosan and chemically modified chitosan beads for acid dyes sorption. J Environ Sci 2009, 21, 296–302. [Google Scholar]
- Rosa, S; Laranjeira, MCM; Riela, HG; Fávere, VT. Cross-linked quaternary chitosan as an adsorbent for the removal of the reactive dye from aqueous solutions. J Hazard Mater 2008, 155, 253–260. [Google Scholar]
- Sakkayawong, N; Thiravetyan, P; Nakbanpote, W. Adsorption mechanism of synthetic reactive dye wastewater by chitosan. J Colloid Interface Sci 2005, 286, 36–42. [Google Scholar]
- Nadavala, SK; Swayampakula, K; Boddu, VM; Abburi, K. Biosorption of phenol and o-chlorophenol from aqueous solutions on to chitosan–calcium alginate blended beads. J Hazard Mater 2009, 162, 482–489. [Google Scholar]
- Sundaram, CS; Viswanathan, N; Meenakshi, S. Fluoride sorption by nano-hydroxyapatite/ chitin composite. J Hazard Mater 2009, 172, 147–151. [Google Scholar]
- Ma, W; Ya, F-Q; Han, M; Wang, R. Characteristics of equilibrium, kinetics studies for adsorption of fluoride on magnetic-chitosan particie. J Hazard Mater 2007, 143, 296–302. [Google Scholar]
- Van de Velde, K; Kiekens, P. Structure analysis and degree of substitution of chitin, chitosan and dibutyrylchitin by FT-IR spectroscopy and solid state 13C NMR. Carbohydr Polym 2004, 58, 409–416. [Google Scholar]
- Mine, S; Izawa, H; Kaneko, Y; Kadokawa, J. Acetylation of α-chitin in ionic liquids. Carbohydr Res 2009, 344, 2263–2265. [Google Scholar]
- Hunger, M; Weitkamp, J. In situ IR, NMR, EPR, and UV/Vis spectroscopy: Tools for new insight into the mechanisms of heterogeneous catalysis. Angew Chem Int Ed 2001, 40, 2954–2971. [Google Scholar]
- Hollas, JM. Modern Spectroscopy, 3rd ed; J. Wiley & Sons: Chichester, UK, 1996. [Google Scholar]
- Ojeda, CB; Rojas, FS. Recent developments in derivative ultraviolet/visible absorption spectrophotometry. Anal Chim Acta 2004, 518, 1–24. [Google Scholar]
- Förster, H. UV/VIS Spectroscopy; Springer: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Tan, SC; Khor, E; Tan, TK; Wong, SM. The degree of deacetylation of chitosan: advocating the first derivative UV-spectrophotometry method of determination. Talanta 1998, 45, 713–719. [Google Scholar]
- De Souza, HKS; Bai, G; do Pilar Gonçalves, M; Bartos, M. Whey protein isolate–chitosan interactions: A calorimetric and spectroscopy study. Thermochim Acta 2009, 495, 108–114. [Google Scholar]
- Liu, D; Wei, Y; Yao, P; Jiang, L. Determination of the degree of acetylation of chitosan by UV spectrophotometry using dual standards. Carbohydr Res 2006, 341, 782–785. [Google Scholar]
- Fu, Z-S; Sun, B-B; Chen, J; Yuan, L. Preparation and photochromism of carboxymethyl chitin derivatives containing spirooxazine moiety. Dyes Pigm 2008, 76, 515–518. [Google Scholar]
- Sahoo, S; Sasmal, A; Nanda, R; Phani, AR; Nayak, PL. Synthesis of chitosan–polycaprolactone blend for control delivery of ofloxacin drug. Carbohydr Polym 2010, 79, 106–113. [Google Scholar]
- Yang, M; Yang, Y; Yang, H; Shen, G; Yu, R. Layer-by-layer self-assembled multilayer films of carbon nanotubes and platinum nanoparticles with polyelectrolyte for the fabrication of biosensors. Biomaterials 2006, 27, 246–255. [Google Scholar]
- Lin, Q-K; Ren, K-F; Ji, J. Hyaluronic acid and chitosan-DNA complex multilayered thin film as surface-mediated nonviral gene delivery system. Colloids Surf B 2009, 74, 298–303. [Google Scholar]
- Ramaprasad, AT; Rao, V; Sanjeev, G; Ramananic, SP; Sabharwal, S. Grafting of polyaniline onto the radiation crosslinked chitosan. Synth Met 2009, 159, 1983–1990. [Google Scholar]
- Nosal, WH; Thompson, DW; Yan, L; Sarkar, S; Subramanian, A; Woollam, JA. UV–vis–infrared optical and AFM study of spin-cast chitosan films. Colloids Surf B 2005, 43, 131–137. [Google Scholar]
- Muzzarelli, RAA; Rocchetti, R. Determination of the degree of acetylation of chitosans by first derivative ultraviolet spectrophotometry. Carbohydr Polym 1985, 6, 461–472. [Google Scholar]
- Pedroni, VI; Gschaider, ME; Schulz, PC. UV spectrophotometry: Improvements in the study of the degree of acetylation of chitosan. Macromol Biosci 2003, 3, 531–534. [Google Scholar]
- Hsiao, HY; Tsai, CC; Chen, S; Hsieh, BC; Chen, RLC. Spectrophotometric determination of deacetylation degree of chitinous materials dissolved in phosphoric acid. Macromol Biosci 2004, 4, 919–921. [Google Scholar]
- Wu, T; Zivanovic, S. Determination of the degree of acetylation (DA) of chitin and chitosan by an improved first derivative UV method. Carbohydr Polym 2008, 73, 248–253. [Google Scholar]
- Da Silva, RMP; Mano, JF; Reis, RL. Straightforward determination of the degree of N-acetylation of chitosan by means of first-derivative UV spectrophotometry. Macromol Chem Phys 2008, 209, 1463–1473. [Google Scholar]
- Khan, TA; Peh, KK; Ch'ng, HS. Reporting degree of deacetylation values of chitosan: The influence of analytical methods. J Pharm Pharm Sci 2002, 5, 205–212. [Google Scholar]
- Liu, H; Du, Y; Wang, X; Sun, L. Chitosan kills bacteria through cell membrane damage. Int J Food Microbiol 2004, 95, 147–155. [Google Scholar]
- Tanida, F; Tojima, T; Han, S-M; Nishi, N; Tokura, S; Sakairi, N; Seino, H; Hamada, K. Novel synthesis of a water-soluble cyclodextrin-polymer having a chitosan skeleton. Polymer 1998, 39, 5261–5263. [Google Scholar]
- Mi, F-L. Synthesis and characterization of a novel chitosan-gelatin bioconjugate with fluorescence emission. Biomacromolecules 2005, 6, 975–987. [Google Scholar]
- Felinto, MCFC; Parra, DF; da Silva, CC; Angerami, J; Oliveira, MJA; Lugão, AB. The swelling behavior of chitosan hydrogels membranes obtainedby UV- and γ-radiation. Nucl Instrum Methods Phys Res Sect B 2007, 265, 418–424. [Google Scholar]
- Munro, NH; Hanton, LR; Robinson, BH; Simpson, J. Synthesis and characterisation of fluorescent chitosan derivatives containing substituted naphthalimides. React Funct Polym 2008, 68, 671–678. [Google Scholar]
- T⊘mmeraas, K; Strand, SP; Tian, W; Kenne, L; Vårum, KM. Preparation and characterisation of fluorescent chitosans using 9-anthraldehyde as fluorophore. Carbohydr Res 2001, 336, 291–296. [Google Scholar]
- Kang, B; Dai, Y; Zhang, H; Chen, D. Synergetic degradation of chitosan with gamma radiation and hydrogen peroxide. Polym Degrad Stab 2007, 92, 359–362. [Google Scholar]
- Sun, Ch; Qu, R; Chen, H; Ji, C; Wang, C; Sun, Y; Wang, B. Degradation behavior of chitosan chains in the ‘green’ synthesis of gold nanoparticles. Carbohydr Res 2008, 343, 2595–2599. [Google Scholar]
- Huang, H; Yang, X. Synthesis of chitosan-stabilized gold nanoparticles in the absence/presence of tripolyphosphate. Biomacromolecules 2004, 5, 2340–2346. [Google Scholar]
- Chen, X; Zhang, X; Yang, W; Evans, DG. Biopolymer-manganese oxide nanoflake nanocomposite films fabricated by electrostatic layer-by-layer assembly. Mater Sci Eng C 2009, 29, 284–287. [Google Scholar]
- Huang, H; Yang, X. Chitosan mediated assembly of gold nanoparticles multilayer. Colloids Surf A 2003, 226, 77–86. [Google Scholar]
- Larena, A; Cáceres, DA. Variability between chitosan membrane surface characteristics as function of its composition and environmental conditions. Appl Surf Sci 2004, 238, 273–277. [Google Scholar]
- Mao, Z; Ma, L; Yan, J; Yan, M; Gao, C; Shen, J. The gene transfection efficiency of thermoresponsive N,N,N-trimethyl chitosan chloride-g-poly(N-isopropylacrylamide) copolymer. Biomaterials 2007, 28, 4488–4500. [Google Scholar]
- Zhang, Q; Zhang, L; Li, J. Fabrication and electrochemical study of monodisperse and size controlled Prussian blue nanoparticles protected by biocompatible polymer. Electrochim Acta 2008, 53, 3050–3055. [Google Scholar]
- Johnstone, RAW; Rose, ME. Mass Spectrometry for Chemists and Biochemists, 2nd ed; Cambridge University Press: Cambridge, UK, 1996. [Google Scholar]
- Siuzdak, G. The Expanding Role of Mass Spectrometry in Biotechnology; MCC Press: San Diego, CA, USA, 2006. [Google Scholar]
- De Hoffmann, E; Stroobant, V. Mass Spectrometry: Principles and Applications, 3rd ed; J. Wiley & Sons: Chichester, UK, 2007. [Google Scholar]
- Mattai, J; Hayes, ER. Characterization of chitosan by pyrolysis—mass spectrometry. J Anal Appl Pyrolysis 1982, 3, 327–334. [Google Scholar]
- Nieto, JM; Peniche-Covas, C; Padrón, G. Characterization of chitosan by pyrolysis-mass spectrometry, thermal analysis and differential scanning calorimetry. Thermochim Acta 1991, 176, 63–68. [Google Scholar]
- Sato, H; Mitzutani, S; Tsuge, S. Determination of the degree of acetylation of chitin/chitosan by pyrolysis-gas chromatography in the presence of oxalic acid. Anal Chem 1998, 70, 7–12. [Google Scholar]
- Bierstedt, A; Stankiewicz, BA; Briggs, DEG; Evershed, RP. Quanitative and qualitative analysis of chitin in fossil arthropods using a combination of colorimetric assay and pyrolysis-gas chromatography-mass spectrometry. Analyst 1998, 123, 19–145. [Google Scholar]
- Steinbrecht, RA; Stankiewicz, BA. Molecular composition of the wall of insects alfactory sensilla-the chitin question. J Insect Physiol 1999, 45, 785–790. [Google Scholar]
- Furuhashi, T; Beran, A; Blazso, M; Czegeny, Z; Schwarzinger, C; Steiner, G. Pyrolysis GC/MS and IR spectroscopy in chitin Analysis of Molluscan shells. Biosci Biotechnol Biochem 2009, 73, 93–103. [Google Scholar]
- Lee, JW; Deng, F; Yeomans, WG; Allen, AL; Gross, RA; Kaplan, DL. Direct incorporation of glucosamine and N-acetylglucosamie into exopolymers by Gluconacetobacter xylinus (=Acetobacter xylinum) ATCC 10245: production of chitosan-cellulose and chitin-cellulose exopolymers. Appl Environ Microbiol 2001, 67, 3970–3975. [Google Scholar]
- Vesentini, D; Steward, D; Singh, AP; Ball, R; Daniel, G; Franish, R. Chitosan-mediated changes in cell wall composition, morphology and ultrastructure in two wood-inhabiting fungi. Mycol Res 2007, 111, 875–890. [Google Scholar]
- Cunha, AG; Fernandes, SCM; Freire, CSR; Silvestre, AJD; Neto, CP; Gandini, A. what is the real value of chitosan’s surface energy? Biomacromolecules 2008, 9, 610–614. [Google Scholar]
- Akiyama, K; Kawazu, K; Kobayashi, A. A novel method for chemo-enzymatic synthesis of elicitor-active chitosan oligomers and partially N-deacetylated chitin oligomers using N-acylated chitotrioses as substrates in a lysozyme-catalyzed transglycosylation reaction system. Carbohydr Res 1995, 279, 151–160. [Google Scholar]
- Zhang, H; Du, Y; Yu, X; Mitsutomi, M; Aiba, S. Preparation of chitooligosaccharides from chitosan by complex enzyme. Carbohydr Res 1999, 320, 257–260. [Google Scholar]
- Tokuyasu, K; Mitsutomi, M; Yamaguchi, I; Hayashi, K; Mori, Y. Recognition of chitooligosaccharides and their N-acetyl groups by putative subsites of chitin deacetylase from a Deuteromycete, Colletotrichum lindemuthianum. Biochemistry 2000, 39, 8837–8843. [Google Scholar]
- Vishu Kumar, AB; Varadaraj, MC; Gowda, LR; Tharanathan, RN. Characterization of chito-oligosaccharides prepared by chitosanolysis with the aid of papain and Pronase, and their bactericidal action against Bacillus cereus and Escherichia coli. Biochem J 2005, 391, 167–175. [Google Scholar]
- Makino, A; Kurosaki, K; Ohmae, M; Kobayashi, S. Chitinase-catalyzed synthesis of alternatingly N-deacetylated chitin: A chitin-chitosan hybrid polysaccharide. Biomacromolecules 2006, 7, 950–957. [Google Scholar]
- Fernandez-Megia, E; Novoa-Carballal, R; Quiñoá, E; Riguera, R. Conjugation of bioactive ligands to PEC-grafted chitosan at the distal end of PEG. Biomacromolecules 2007, 8, 833–842. [Google Scholar]
- Boesel, LF; Reis, RL; San Roman, J. Innovative Approach for producing injectable, biodegradable materials using chitooligosaccharides and Green Chemistry. Biomacromolecules 2009, 10, 465–470. [Google Scholar]
- Popa-Nita, S; Lucas, J-M; Ladavière, C; David, L; Domard, A. Mechanisms involved during the ultrasonically induced depolymerization of chitosan: characterization and control. Biomacromolecules 2009, 10, 1203–1211. [Google Scholar]
- Bahrke, S; Einarsson, JM; Gislason, J; Haebel, S; Letzel, MC; Peter-Katalinić, J; Peter, MG. Sequence analysis of chitooligosaccharides by matrix-assisted laser desorption ionization postsource decay mass spectrometry. Biomacromolecules 2002, 3, 696–704. [Google Scholar]
- Haebel, S; Bahrke, S; Peter, MG. Quantitative sequencing of complex mixtures of heterochitooligosaccharides by vMALDI-linear ion trap mass spectrometry. Anal Chem 2007, 79, 5557–5566. [Google Scholar]
- Cederkvist, FH; Parmer, MP; Vårum, KM; Eijsink, VGH; S⊘rlie, M. Inhibbition of a family 18 chitinase by chitooligosaccharides. Carbohydr Polym 2008, 74, 41–49. [Google Scholar]
- Thierry, B; Winnik, FM; Merhi, Y; Silver, J; Tabrizian, M. Bioactive coatings of endovascular stents based on polyelectrolyte multilayers. Biomacromolecules 2003, 4, 1564–1571. [Google Scholar]
- Grenha, A; Seijo, B; Serra, C; Remuñán-López, C. Chitosan nanoparticle-loaded mannitol macrospheres: structure and surface characterization. Biomacromolecules 2007, 8, 2072–2079. [Google Scholar]
- Shahgholi, M; Callahan, JH; Rappoli, BJ; Rowley, DA. Investigation of copper-saccharide complexation reactions using potentiometry and electrospray mass spectrometry. J Mass Spectrom 1997, 32, 1080–1093. [Google Scholar]
- Wu, L-Q; Gadre, AP; Yi, H; Kastantin, MJ; Rubloff, GW; Bentley, WE; Payne, GF; Ghodssi, R. Voltage-dependent assembly of the polysaccharide chitosan onto an electrode surface. Langmuir 2002, 18, 8620–8625. [Google Scholar]
- Liew, S-T; Wei, A. Preparation of orthogonally protected chitosan oligosaccharides: observation of an anomalous remote substituent effect. Carbohydr Res 2002, 337, 1319–1324. [Google Scholar]
- Wu, L-Q; Embree, HD; Balgley, BM; Smith, PJ; Payne, GF. Utilizing renewable resources to create functional polymers: chitosan-based associative thickener. Environ Sci Technol 2002, 36, 3446–3454. [Google Scholar]
- Liu, Y; Gaskell, KJ; Cheng, Z; Yu, L; Payne, GF. Chitosan-coated electrodes for bimodal sensing: selective post-electrode film reaction for spectroelectrochemmical analysis. Langmuir 2008, 24, 7223–7231. [Google Scholar]
- Yao, Y-Y; Shrestha, KL; Wu, Y-J; Tasi, H-J; Chen, C-Y; Yang, J-M; Ando, A; Cheng, C-Y; Li, Y-K. Structural simulation and protein engineering to convert an endo-chitosanase to an exo-chitosanase. Protein Eng Des Sel 2008, 21, 561–566. [Google Scholar]
- Li, J; Cai, J; Fan, L. Effect of sonolysis on kinetics and physicochemical properties of treated chitosan. J Appl Polym Sci 2008, 109, 2417–2425. [Google Scholar]
- Kerwin, JL; Whitney, DL; Sheikh, A. Mass spectrometric profiling of glucosamine, glucosamine polymers and their catecholamine adducts. Model reactions and cuticular hydrolysates of Toxorhynchites amboinensis (Culicidae) pupae. Insect Biochem Mol Biol 1999, 29, 599–607. [Google Scholar]
- Trombotto, S; Ladavière, C; Delolme, F; Domard, A. Chemical preparation and structural characterization of a homogenous series of chitin/chitosan oligomers. Biomacromolecules 2008, 9, 1731–1738. [Google Scholar]
- Dennhart, N; Fukamizo, T; Brzezinski, R; Lacombe-Harvey, M-E; Letzel, T. Oligosaccharide hydrolysis by chitosanase enzymes monitored by real-time electrospray ionization-mass spectrometry. J Biotechnol 2008, 134, 253–260. [Google Scholar]
- Ishimizu, T; Mitsukami, Y; Shinkawa, T; Natsuka, S; Hase, S; Miyagi, M; Sakiyama, F; Norioka, S. Presence of asparagine-linked N-acetyloglucosamine and chitobiose in Pyrus pyrifolia S-RNases associated with gametophytic self-incompatibility. Eur J Biochem 1999, 263, 624–634. [Google Scholar]
- Li, J; Du, YM; Liang, HB; Yao, PJ; Wei, YA. Effect of immobilized neutral protease on the preparation and physicochemical properties of low molecular weight chitosan and chito-oligomers. J Appl Polym Sci 2006, 102, 4185–4193. [Google Scholar]
- Suginata, W; Pantoom, S; Prinz, H. Substrate binding modes and anomer selectivity of chitinase A from Vibrio harveyi. J Chem Biol 2009, 2, 191–202. [Google Scholar]
- Watson, HR; Apperley, DC; Dixon, DP; Edwards, R; Hodgson, DR. An efficient method for 15N-labeling of chitin in fungi. Biomacromolecules 2009, 10, 793–797. [Google Scholar]
- Kamst, E; van der Drift, KMGM; Thomas-Oates, JE; Lugtenberg, BJJ; Spaink, HP. Mass spectrometric analysis of chitin oligosaccharides produced by Rhizobium NodC Protein in Escherichia coli. J Bacteriol 1995, 177, 6282–6285. [Google Scholar]
- Van der Drift, KMGM; Spaink, HP; Bloemberg, GV; van Brussel, AAN; Lugtenberg, BJJ; Haverkamp, J; Thomas-Oates, JE. Rhizobium leguminarosum bv. trifolii produces lipo-chitin oligosaccharides with nodE-dependent highly unsaturated fatty acyl moieties. J Biol Chem 1996, 271, 22563–22569. [Google Scholar]
- Tokuyasu, K; Ono, H; Ohnishi-Kameyama, M; Hayashi, K; Mori, Y. Deacetylation of chitin oligosaccharides of dp 2–4 by chitin deacetylase from Colletotrichum lindemuthianum. Carbohydr Res 1997, 303, 353–358. [Google Scholar]
- Tokuyasu, K; Ono, H; Mitsutomi, M; Hayashi, K; Mori, Y. Synhesis of a chitosan tetramer derivative, β-D-GlcNAc-(1→4)-β-D-GlcNAc-(1→4)-β-D-GlcNAc-(1→4)-D-GlcN through a partial N-acetylation reaction by chitin deacetylase. Carbohydr Res 2000, 325, 211–215. [Google Scholar]
- Kittur, FS; Vishu Kumar, AB; Varadaraj, MC; Tharanathan, RN. Chitoologosaccharides-preparation with the aid of pectinase isozyme from Aspergillus niger and their antibacterial activity. Carbohydr Res 2005, 340, 1239–1245. [Google Scholar]
- Aronson, NN, Jr; Halloran, BA; Alexyev, MF; Amable, L; Madura, JD; Paspulati, L; Worth, C; Van Roey, P. Family 18 chitinase-oligosaccharides substrate interaction: subsite preference and anomer selectivity of S. Marcescens chitinase A. Biochem J 2003, 376, 87–95. [Google Scholar]
- Van der Drift, KMGM; Olsthoorn, MMA; Brüll, LP; Blok-Tip, L; Thomas-Oates, JE. Mass spectrometric analysis of lipo-chitin oligosaccharides-signal molecules mediating the host-specific legume-rhizobium symbiosis. Mass Spectrom Rev 1998, 17, 75–95. [Google Scholar]
- Kollár, R; Petráková, E; Ashwell, G; Robbins, PW; Cabib, E. Architecture of the yeast cell wall. J Biol Chem 1995, 270, 1170–1178. [Google Scholar]
- Lopatin, SA; Ilyn, MM; Pustobaev, VN; Bezchetnikova, ZA; Varlamov, VP; Davankov, VA. Mass-spectrometric analysis of N-acetylchitooligosaccharides prepared through enzymatic hydrolysis of chitosan. Anal Biochem 1995, 227, 285–288. [Google Scholar]
- Verbruggen, F; Heiri, O; Reichart, G-J; De Leeuw, JW; nierop, KGJ; Lotter, AF. Effects of chemical pretreatment on δ18O measurements, chemical composition, and morphology of chironomid head capsules. J Paleolimnol 2009. [Google Scholar] [CrossRef]
- Wang, YV; O’Brien, DM; Jenson, J; Francis, D; Wooler, MJ. The influence of diet and water on the stable oxygen and hydrogen isotope composition of Chironomidae (Diptere) with paleoecological implications. Oecologia 2009, 160, 225–233. [Google Scholar]
- López, FA; Mercê, ALR; Alguacil, FJ; López-Delgado, A. A kinetic study on the thermal behavior of chitosan. J Therm Anal Calorim 2008, 91, 633–639. [Google Scholar]
- Rodríguez, AT; Ramírez, MA; Cárdenas, RM; Hernández, AN; Velázquez, MG; Bautista, S. Induction of defence response of Oryza sativa L. against Pyricularia grisea (Cooke) Sacc. by treating seeds with chitosan and hydrolyzed chitosan. Pestic Biochem Physiol 2007, 89, 206–215. [Google Scholar]
- Lin, H; Wang, H; Xue, C; Ye, M. Preparation of chitosan oligomers by immobilized papain. Enzyme Microb Technol 2002, 31, 588–592. [Google Scholar]
- Lee, H-W; Park, Y-S; Jung, J-S; Shin, W-S. Chitosan oligosaccharides, dp 2–8, have prebiotic effect on the Bifidobacterium bifidium and Lactobacillus sp. Anaerobe 2002, 8, 319–324. [Google Scholar]
- Li, J; Du, Y; Yang, J; Feng, T; Li, A; Chen, P. Preparation and characterisation of low molecular weight chitosan and chito-oligomers by a commercial enzyme. Polym Degrad Stab 2005, 87, 441–448. [Google Scholar]
- Oliveira, EN, Jr; El Gueddari, NE; Moerschbacher, BM; Peter, MG; Franco, TT. Growth of phytopathogenic fungi in the presence of partially acetylated chitooligosaccharides. Mycopathologia 2008, 166, 163–174. [Google Scholar]
- Kuyama, H; Nakahara, Y; Nucada, T; Ito, J; Nakahara, Yo; Ogawa, T. Stereocontrolled synthesis of chitosan dodecamer. Carbohydr Res 1993, 243, C1–C7. [Google Scholar]
- Cabrera, JC; Messiaen, J; Cambier, P; Van Cutsem, P. Size, acetylation and concentration of chitooligosaccharide elicitors determine the switch from defence involving PAL activation to cell death and water peroxide production in Arabidopsis cell suspensions. Physiol Plant 2006, 127, 44–56. [Google Scholar]
- Lee, D-W; Baney, RH. Oligochitosan derivatives bearing electron-deficient aromatic rings for adsorption of Amitriptyline: Implications for drug detoxification. Biomacromolecules 2004, 5, 1310–1315. [Google Scholar]
- Vachoud, L; Chen, T; Payne, GF; Vazquez-Duhalt, R. Peroxidase catalyzed grafting of gallate esters onto the polysaccharide chitosan. Enzyme Microb Technol 2001, 29, 380–385. [Google Scholar]
- Fu, X; Huang, L; Zhai, M; Li, W; Liu, H. Analysis of natural carbohydrate biopolimer-high molecular chitosan and carboxymethyl chitosan by capillary zone electrophoresis. Carbohydr Polym 2007, 68, 511–516. [Google Scholar]
- Fu, X; Liu, Y; Li, W; Pang, N; Nie, H; Liu, H; Cai, Z. Analysis of aristolochic acids by CE-MS with carboxymethyl chitosan-coated capillary. Electrophoresis 2009, 30, 1783–1789. [Google Scholar]
- Katarina, RK; Takayanagi, T; Oshita, K; Oshima, M; Motomizu, S. Sample pretreatment Rusing chitosan-based chelating resin for the determination of trace metals in seawater Samales by inductively coupled plasma-mass spektrometry. Anal Sci 2008, 24, 1537–1544. [Google Scholar]
- Gao, Y; Oshita, K; Lee, K-H; Oshima, M; Motomizu, S. Development of kolumn-pretreatment chelating resins for matrix elimination/multi-element determination by inductively coupled plasma-mass spectrometry. Analyst 2002, 127, 1713–1719. [Google Scholar]
- Oshita, K; Takayanagi, T; Oshima, M; Motomizu, S. Adsorption properties of ionic species on cross-linked chitosans modified with catechol and salicylic acid moieties. Anal Sci 2008, 24, 665–668. [Google Scholar]
- Sabarudin, A; Oshima, M; Takayanagi, T; Hakim, L; Oshita, K; Yun Hua Gao; Motomizu, S. Functionalization of chitosan with 3,4-dihydroxybenzoic acid for the adsorption/collection of uranium in water samples and its determination by inductively coupled plasma-mass spectrometry. Anal Chim Acta 2007, 581, 214–220. [Google Scholar] [Green Version]
- Oshita, K; Takayanagi, T; Oshima, M; Motomizu, S. Adsorption behaviour of cationic and anionic species on chitosan resins possessing amino acids moieties. Anal Sci 2007, 23, 1431–1434. [Google Scholar]
- Hosoba, M; Oshita, K; Katarina, RK; Takayanagi, T; Oshima, M; Motomizu, S. Synthesis of novel chitosan resin possessing histidine moiety and its application to the determination of trace silver by ICP-AES coupled with triplet automated-pretreatment system. Anal Chim Acta 2009, 639, 51–56. [Google Scholar]
- Travan, A; Pelillo, C; Donati, I; Marsich, E; Benincasa, M; Scarpa, T; Semeraro, S; Turco, G; Gennaro, R; Paoletti, S. Non-cytotoxic silver nanoparticle-polysaccharide nanocomposites with antimicrobial activity. Biomacromolecules 2009, 10, 1429–1435. [Google Scholar]
- Kasaai, MR. Determination of the degree of N-acetylation for chitin and chitosan by various NMR spectroscopy techniques: A review. Carbohydr Polym 2010, 79, 801–810. [Google Scholar]
- Pelletier, A; Lemire, I; Sygusch, J; Chornet, E; Overend, RP. Chitin/chitosan transformation by thermo-mechano-chemical treatment including characterization by enzymatic depolymerization. Biotechnol Bioeng 1990, 36, 310. [Google Scholar]
- Raymond, L; Morin, FG; Marchessault, RH. Degree of deacetylation of chitosan using conductometric titration and solid-state NMR. Carbohydr Res 1993, 246, 331. [Google Scholar]
- Yu, G; Morin, FG; Nobes, GAR; Marchessault, RH. Degree of Acetylation of Chitin and Extent of Grafting PHB on Chitosan Determined by Solid State 15N NMR. Macromolecules 1999, 32, 518–520. [Google Scholar]
- Heux, L; Brugnerotto, J; Desbrières, J; Versali, M-F; Rinaudo, M. Solid State NMR for Determination of Degree of Acetylation of Chitin and Chitosan. Biomacromolecules 2000, 1, 746–751. [Google Scholar]
- Rinaudo, M; Le Dung, P; Gey, C; Milas, M. Substituent distribution on O,N-carboxymethylchitosans by 1H and 13C NMR. Int J Biol Macromol 1992, 14, 122–128. [Google Scholar]
- Vårum, KM; Anthonsen, MW; Grasdalen, H; Smisr⊘d, O. Determination of the degree of N-acetylation and the distribution of N-acetyl groups in partially N-deacetylated chitins (chitosans) by high-field n.m.r. spectroscopy. Carbohydr Res 1991, 211, 17–23. [Google Scholar]
- Desbrières, J; Martinez, C; Rinaudo, M. Hydrophobic derivatives of chitosan: Characterization and rheological behavior. Int J Biol Macromol 1996, 19, 21–28. [Google Scholar]
- Lebouc, F; Dez, I; Madec, P-J. NMR study of the phosphonomethylation reaction on chitosan. Polym 2005, 46, 319–325. [Google Scholar]
- Weinhold, MX; Sauvageau, JCM; Kumirska, J; Thöming, J. Studies on acetylation patterns of different chitosan preparations. Carbohydr Polym 2009, 78, 678–684. [Google Scholar]
- Yang, BY; Montgomery, R. Degree of acetylation of heteropolysaccharides. Carbohydr Res 2000, 323, 156–162. [Google Scholar]
- Duarte, ML; Ferreira, MC; Marvao, MR; Rocha, J. Determination of the degree of acetylation of chitin materials by 13C CP/MAS NMR spectroscopy. Int J Biol Macromol 2001, 28, 359–363. [Google Scholar]
- Tolaimate, A; Desbričres, J; Rhazi, M; Alagui, A; Vincendon, M; Vottero, P. On the influence of deacetylation process on the physicochemical characteristics of chitosan from squid chitin. Polymer 2000, 41, 2463–2469. [Google Scholar]
- Guinesi, LS; Cavalheiro, ETG. The use of DSC curves to determine the acetylation degree of chitin/chitosan samples. Thermochim Acta 2006, 444, 128–133. [Google Scholar]
- Rinaudo, M; Milas, M; Dung, PL. Characterization of chitosan. Influence of ionic strength and degree of acetylation on chain expansion. Int J Biol Macromol 1993, 15, 281–285. [Google Scholar]
- Hirai, A; Odani, H; Nakajima, A. Determination of degree of deacetylation of chitosan by 1H NMR spectroscopy. Polym Bullet 1991, 26, 87–94. [Google Scholar]
- Kasaai, MR; Arul, J; Chin, SL; Charlet, G. The use of intense femtosecond laser pulses for the fragmentation of chitosan. J Photochem Photobiol A 1999, 120, 201–205. [Google Scholar]
- Lavertu, M; Xia, Z; Serreqi, AN; Berrada, M; Rodrrigues, A; Wang, D; Buschmann, MD; Gupta, A. A validated H NMR method for the determination of the degree of deacetylation of chitosan. J Pharm Biomed Anal 2003, 32, 1149–1158. [Google Scholar]
- Ott⊘y, MH; Vårum, KM; Smidsr⊘d, O. Compositional heterogeneity of heterogeneously deacetylated chitosans. Carbohydr Polym 1996, 29, 17–24. [Google Scholar]
- Pelletier, A; Lemire, I; Sygusch, J; Chornet, E; Overend, RP. Chitin/chitosan transformation by thermo-mechano-chemical treatment including characterization by enzymatic depolymerization. Biotechnol Bioeng 1990, 36, 310–315. [Google Scholar]
- Fernandez-Megia, E; Novoa-Carballal, R; Quiñoá, E; Riguera, R. Optimal routine conditions for the determination of the degree of acetylation of chitosan by 1H-NMR. Carbohydr Polym 2005, 61, 155–161. [Google Scholar]
- Vårum, KM; Anthonsen, MW; Grasdalen, H; Smidsr⊘d, O. 13C NMR studies of the acetylation sequences in partially N-acetylated chitins (chitosans). Carbohydr Res 1991, 217, 19–27. [Google Scholar]
- Bovey, P; Mireau, A. NMR of Polymers; Academic Press: San Diego, CA, USA, 1996. [Google Scholar]
- Mirau, A. A Practical Guide to Understanding the NMR of Polymers; Academic Press: Hoboken, NJ, USA, 2005. [Google Scholar]
- Kumirska, J; Weinhold, MX; Steudte, S; Thöming, J; Brzozowski, K; Stepnowski, P. Determination of the pattern of acetylation of chitosan samples: Comparison of evaluation methods and some validation parameters. Int J Biol Macromol 2009, 45, 56–60. [Google Scholar]
- Kumirska, J; Weinhold, MX; Sauvageau, JCM; Thöming, J; Kaczyński, Z; Stepnowski, P. Determination of the pattern of acetylation of low molecular weight chitosan used in biomedical applications. J Pharm Biochem Anal 2009, 50, 587–590. [Google Scholar]
- Martinou, A; Bouriotis, V; Stokke, BT; Vårum, KM. Mode of action of chitin deacetylase from Mucor rouxii on partially N-acetylated chitosans. Carbohydr Res 1998, 311, 71–78. [Google Scholar]
- Tommeraas, K; Koping-Hoggard, M; Vårum, KM; Christensen, BE; Artursson, P; Smidsrod, O. Preparation and characterisation of chitosans with oligosaccharide branches. Carbohydr Res 2002, 337, 2455–2462. [Google Scholar]
- Knight, DK; Shapka, SN; Amsden, BG. Structure, depolymerization, and cytocompatibility evaluation of glycol chitosan. J Biomed Mater Res Part A 2007, 83, 787–798. [Google Scholar]
- Rabea, EI; Badawy, ME; Rogge, TM; Stevens, CV; Höfte, M; Steurbaut, W; Smagghe, G. Insecticidal and fungicidal activity of new synthesized chitosan derivatives. Pest Manag Sci 2005, 61, 951–960. [Google Scholar]
- Sun, L; Du, Y; Yang, Y; Shi, X; Li, J; Wang, X; Kennedy, JF. Conversion of crystal structure of the chitin to facilitate preparation of a 6-carboxychitin with moisture absorption–retention abilities. Carbohydr Polym 2006, 66, 168–175. [Google Scholar]
- Chen, L; Du, Y; Wu, H; Xiao, L. Relationship between Molecular Structure and Moisture- Retention Ability of Carboxymethyl Chitin and Chitosan. J Appl Polym Sci 2002, 83, 1233–1241. [Google Scholar]
- Vikhoreva, G; Bannikova, G; Stolbushkina, P; Panov, A; Drozd, N; Makarov, V; Varlamov, V; Gal’braikh, L. Preparation and anticoagulant activity of a low-molecular-weight sulfated chitosan. Carbohydr Polym 2005, 62, 327–332. [Google Scholar]
- Sieval, AB; Thanou, M; Kotzé, AF; Verhoef, JC; Brussee, J; Junginger, HE. Preparation and NMR characterization of highly substituted N-trimethyl chitosan chloride. Carbohydr Polym 1998, 36, 157–165. [Google Scholar]
- Aoi, K; Seki, T; Okada, M; Sato, H; Mizutani, S; Ohtani, H; Tsuge, S; Shiogai, Y. Synthesis of a novel N-selective ester functionalized chitin derivative and water-soluble carboxyethylchitin. Macromol Chem Phys 2000, 201, 1701–1708. [Google Scholar]
- Detchprohm, S; Aoi, K; Okada, M. Synthesis of a Novel Chitin Derivative Having Oligo(ecaprolactone) Side Chains in Aqueous Reaction Media. Macromol Chem Phys 2001, 202, 3560–3570. [Google Scholar]
- Rinaudo, M; Desbrieres, J; Le Dung, P; Thuy Binh, P; Dong, NT. NMR investigation of chitosan derivatives formed by the reaction of chitosan with levulinic acid. Carbohydr Polym 2001, 46, 339–348. [Google Scholar]
- Zhang, C; Ping, Q; Ding, Y; Cheng, Y; Shen, J. Synthesis, Characterization, and Microsphere Formation of Galactosylated Chitosan. J Appl Polym Sci 2004, 91, 659–665. [Google Scholar]
- Zou, Y; Khor, E. Preparation of sulfated-chitins under homogeneous conditions. Carbohydr Polym 2009, 77, 516–525. [Google Scholar]
- Park, IK; Park, YH. Preparation and Structural Characterization of Water-Soluble O-Hydroxypropyl Chitin Derivatives. J Appl Polym Sci 2001, 80, 2624–2632. [Google Scholar]
- Tanodekaewa, S; Prasitsilpa, M; Swasdisonb, S; Thavornyutikarna, B; Pothsreea, T; Pateepasen, R. Preparation of acrylic grafted chitin for wound dressing application. Biomaterials 2004, 25, 1453–1460. [Google Scholar]
- Changhong, P; Weijun, Y; Motang, T. Chemical Modification of Chitosan: Synthesis and Characterization of Chitosan-Crown Ethers. J Appl Polym Sci 2003, 87, 2221–2225. [Google Scholar]
- De Angelis, AA; Capitani, D; Crescenzi, V. Synthesis and 13C CP-MAS NMR Characterization of a New Chitosan-Based Polymeric Network. Macromolecules 1998, 31, 1595–1601. [Google Scholar]
- Capitani, D; De Angelis, AA; Crescenzi, V; Masci, G; Segre, AL. NMR study of a novel chitosan-based hydrogel. Carbohydr Polym 2001, 45, 245–252. [Google Scholar]
- Lebouc, F; Dez, I; Gulea, M; Madec, P-J; Jaffres, P-A. Synthesis of Phosphorus-Containing Chitosan Derivatives. Phosphorus Sulfur Silicon Relat Elem 2009, 184, 872–889. [Google Scholar]
- Jayakumar, R; Nagahama, H; Furuike, T; Tamura, H. Synthesis of phosphorylated chitosan by novel method and its characterization. Int J Biol Macromol 2008, 42, 335–339. [Google Scholar]
- Jayakumar, R; Egawa, T; Furuike, T; Nair, SV; Tamura, H. Synthesis, Characterization, and Thermal Properties of Phosphorylated Chitin for Biomedical Applications. Polym Eng Sci 2009, 49, 844–849. [Google Scholar]
- Wang, X; Ma, J; Wang, Y; He, B. Structural characterization of phosphorylated chitosan and their applications as effective additives of calcium phosphate cements. Biomaterials 2001, 22, 2247–2255. [Google Scholar]
- Meng, S; Liu, Z; Zhong, W; Wang, Q; Du, Q. Phosphorylcholine modified chitosan: Appetent and safe material for cells. Carbohydr Polym 2007, 70, 82–88. [Google Scholar]
- Zou, Y; Khor, E. Preparation of C-6 Substituted Chitin Derivatives under Homogeneous Conditions. Biomacromolecules 2005, 6, 80–87. [Google Scholar]
- Palma, G; Casals, P; Cárdenas, G. Synthesis and Characterization of New Chitosan-O-Ethyl Phosphonate. J Chil Chem Soc 2005, 50, 597–602. [Google Scholar]
- Cárdenas, G; Cabrera, G; Taboada, E; Rinaudo, M. Synthesis and Characterization of Chitosan Alkyl phosphates. J Chil Chem Soc 2006, 51, 815–820. [Google Scholar]
- Tanner, SF; Chanzy, H; Vincendon, M; Roux, JC; Gaill, F. High resolution solid-state carbon-13 nuclear magnetic resonance study of chin. Macromolecules 1990, 23, 3576–3583. [Google Scholar]
- Kameda, T; Miyazawa, M; Ono, H; Yoshida, M. Hydrogen Bonding Structure and Stability of α-Chitin Studied by 13C Solid-State NMR. Macromol Biosci 2005, 5, 103–106. [Google Scholar]
- Cortizo, MS; Berghoff, CF; Alessandrini, JL. Characterization of chitin from Illex argentinus squid pen. Carbohydr Polym 2008, 74, 10–15. [Google Scholar]
- Manni, L; Ghorbel-Bellaaj, O; Jellouli, K; Younes, I. Nasri, Extraction and Characterization of Chitin, Chitosan, and Protein Hydrolysates Prepared from Shrimp Waste by Treatment with Crude Protease from Bacillus cereus SV1. Appl Biochem Biotechnol 2010. [Google Scholar] [CrossRef]
- Toffey, A; Samaranayake, G; Frazier, CE; Glasser, WG. Chitin derivatives. I. Kinetics of the heat-induced conversion of chitosan to chitin. J Appl Polym Sci 1996, 60, 75–85. [Google Scholar]
- Holme, HK; Foros, H; Pettersen, H; Dornish, M; Smidsrod, O. Thermal depolymerization of chitosan chloride. Carbohydr Polym 2001, 46, 287–294. [Google Scholar]
- Einbu, A; Vårum, KM. Depolymerization and de-N-acetylation of Chitin Oligomers in Hydrochloric Acid. Biomacromolecules 2007, 8, 309–314. [Google Scholar]
- Einbu, A; Vårum, KM. Characterization of Chitin and Its Hydrolysis to GlcNAc and GlcN. Biomacromolecules 2008, 9, 1870–1875. [Google Scholar]
- S⊘rbotten, A; Horn, SJ; Eijsink, VGH; Vårum, KM. Degradation of chitosans with chitinase B from Serratia marcescens. Production of chito-oligosaccharides and insight into enzyme processivity. FEBS J 2005, 272, 538–549. [Google Scholar]
- Colombo, G; Meli, M; Cañada, J; Asensio, JL; Jiménez-Barberob, J. Toward the understanding of the structure and dynamics of protein–carbohydrate interactions: molecular dynamics studies of the complexes between hevein and oligosaccharidic ligands. Carbohydr Res 2004, 339, 985–994. [Google Scholar]
- Chávez, MI; Andreu, C; Vidal, P; Aboitiz, N; Freire, F; Groves, P; Asensio, JL; Asensio, G; Muraki, M; Cañada, FJ; Jiménez-Barbero, J. On the Importance of Carbohydrate-Aromatic Interactionsfor the Molecular Recognition of Oligosaccharides by Proteins: NMR Studies of the Structure and Binding Affinity of AcAMP2-like Peptideswith Non-Natural Naphthyl and Fluoroaromatic Residues. Chem Eur J 2005, 11, 7060–7074. [Google Scholar]
- Aboitiz, N; Cañada, FJ; Hušáková, L; Kuzma, M; Křen, V; Jiménez-Barbero, J. Enzymatic synthesis of complex glycosaminotrioses and study of their molecular recognition by hevein domains. Org Biomol Chem 2004, 2, 1987–1994. [Google Scholar]
- Chan, H-Y; Chen, M-H; Yuan, G-F. Fungal chitosan. Fungal Sci 2007, 16, 39–52. [Google Scholar]
- Yen, M-T; Mau, J-L. Physico-chemical characterization of fungal chitosan from shiitake stipes. LWT–Food Sci Technol 2007, 40, 472–479. [Google Scholar]
- Yen, M-T; Yang, J-H; Mau, J-L. Physicochemical characterization of chitin and chitosan from crab shells. Carbohydr Polym 2009, 75, 15–21. [Google Scholar]
- Peter, M; Binulal, NS; Soumya, S; Nair, SV; Furuike, T; Tamura, H; Jayakumar, R. Nanocomposite scaffolds of bioactive glass ceramic nanoparticles disseminated chitosan matrix for tissue engineering applications. Carbohydr Polym 2010, 79, 284–289. [Google Scholar]
- Xi, F; Wu, J; Lin, X. Novel nylon-supported organic–inorganic hybrid membrane with hierarchical pores as a potential immobilized metal affinity adsorbent. J Chromatogr A 2006, 1125, 38–51. [Google Scholar]
- Rusua, VM; Nga, C-H; Wilkec, M; Tierscha, B; Fratzld, P; Petera, MG. Size-controlled hydroxyapatite nanoparticles as self-organized organic–inorganic composite materials. Biomaterials 2005, 26, 5414–5426. [Google Scholar]
- Maruca, R; Suder, BJ; Wightman, JP. Interaction of heavy metals with chitin and chitosan. III. Chromium. J Appl Polym Sci 1982, 27, 4827–4837. [Google Scholar]
- Gamage, A; Shahidi, F. Use of chitosan for the removal of metal ion contaminants and proteins from water. Food Chem 2007, 104, 989–996. [Google Scholar]
- Atkins, P; Paula, J. Elements of Physical Chemistry, 4th ed; Oxford University Press: New York, NY, USA, 2005. [Google Scholar]
- Kittur, FS; Vishu Kumar, AB; Tharanathan, RN. Low molecular weight chitosans-preparation by depolymerization with Aspergillus niger pectinase, and characterization. Carbohydr Res 2003, 338, 1283–1290. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | DP analysis MS method | Chitin/chitosan polymerization degree DP | Chitooligosaccharide applicability | Ref. |
|---|---|---|---|---|
| Papain | MALDI-TOF | DP 2–DP 8 | Antibacterial activity against Bacillus cereus and Escherichia coli | [222] |
| Immobilized Papain | MALDI-TOF | DP 3–DP 7 | Comparing the depolimerization efficiency between free and immobilized papain | [259] |
| Pronaze | MALDI-TOF | DP 2–DP 9 | Antibacterial activity against Bacillus cereus and Escherichia coli | [222] |
| Cellulast (Novozymes) | MALDI-TOF | DP 2–DP 8 | Induction of defence response of Oryza sativa L. against Pyricularia grisea (Cooke) Sacc. | [258] |
| Isozyme of pectinase | MALDI-TOF FAB-MS | DP 2–DP 6 | Antimicrobial activity Bacillus cereus and Escherichia coli | [250] |
| Chitinase | MALDI-TOF | Q1: DP 3–DP 8 Q2: DP 2–DP 12 Q3: DP 2–DP 10 | Affect on fungal (alternaria alternate, Rhisopus stolnifer, Botrytis cinera, Penicillinum expansum) growth rate | [262] |
| Chitosanase | MALDI-TOF | DP 2–DP 8 | Investigations of prebiotic effect on the Bifidobacterium bifidum and Lactobaccillus sp. | [260] |
| Chitinase | LC-ESI-MS | DP 2–DP 6 | Studies of mechanism of bonding COS to enzyme helpful in the drug-screening program (for drugs in allergic asthma) | [244] |
| Chitin and chitosan derivative | MS method | Chitooligosaccharide derivative applicability | Ref. |
|---|---|---|---|
| Copper-chitooligosaccharides complexes | ESI-MS, ESI-MS/MS (triple quadrupole, CAD –colision activated dissociation) | Metal-ligand associations studies | [232] |
| Lipo-chitin oligosaccharides | ESI-MS (quadrupole), FAB-MS, CID-MS/MS (QTof) | Structural studies of lipo-chitin oligosaccharides isolated from bacteria and their role as signal molecules in symbiosis | [247,252] |
| Products of electrochemical reaction between caffeic acid and glucosamine | ESI-MS | Studies of chitosan–coated electrodes for bimodal sensing | [236] |
| Methacrylated chitoligosaccharides | MALDI-TOF | Production of biodegradable biopolymers | [225] |
| Chitosan/tripolyphosphate nanoparticles | ToF-SIMS | Studies of nanoparticles as drug delivery system | [231] |
| Catechin-modified chitosan | ESI-MS | Creating polymers for technical applications | [235] |
| Benzenesulfony chitosan, Dinitrobenzenesulfonyl chitosan | MALDI-TOF | Implications for drug detoxification | [265] |
| Chitosan-g-PEG=X (where X-Man, cholesterol, coumarin, biotin) | MALDI-TOF | Producing copolymers used in active targeting and antiadhesive therapy | [224] |
| Multilayers consisting of: chitosan, hyaluronan, and poyethyleneimine | ToF-SIMS | Bioactive coating of endovascular stent | [230] |
| Dodecyl galate (DDG)-chitosan | FAB-MS, ESI-MS | Peroxidaze catalyzed production of biopolymers | [266] |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kumirska, J.; Czerwicka, M.; Kaczyński, Z.; Bychowska, A.; Brzozowski, K.; Thöming, J.; Stepnowski, P. Application of Spectroscopic Methods for Structural Analysis of Chitin and Chitosan. Mar. Drugs 2010, 8, 1567-1636. https://doi.org/10.3390/md8051567
Kumirska J, Czerwicka M, Kaczyński Z, Bychowska A, Brzozowski K, Thöming J, Stepnowski P. Application of Spectroscopic Methods for Structural Analysis of Chitin and Chitosan. Marine Drugs. 2010; 8(5):1567-1636. https://doi.org/10.3390/md8051567
Chicago/Turabian StyleKumirska, Jolanta, Małgorzata Czerwicka, Zbigniew Kaczyński, Anna Bychowska, Krzysztof Brzozowski, Jorg Thöming, and Piotr Stepnowski. 2010. "Application of Spectroscopic Methods for Structural Analysis of Chitin and Chitosan" Marine Drugs 8, no. 5: 1567-1636. https://doi.org/10.3390/md8051567
APA StyleKumirska, J., Czerwicka, M., Kaczyński, Z., Bychowska, A., Brzozowski, K., Thöming, J., & Stepnowski, P. (2010). Application of Spectroscopic Methods for Structural Analysis of Chitin and Chitosan. Marine Drugs, 8(5), 1567-1636. https://doi.org/10.3390/md8051567