Production of Chitooligosaccharides and Their Potential Applications in Medicine

Abstract
:1. Introduction to Chitin, Chitosans and Chitooligosaccharides (CHOS)
2. Chitosan, the Starting Material for CHOS Production
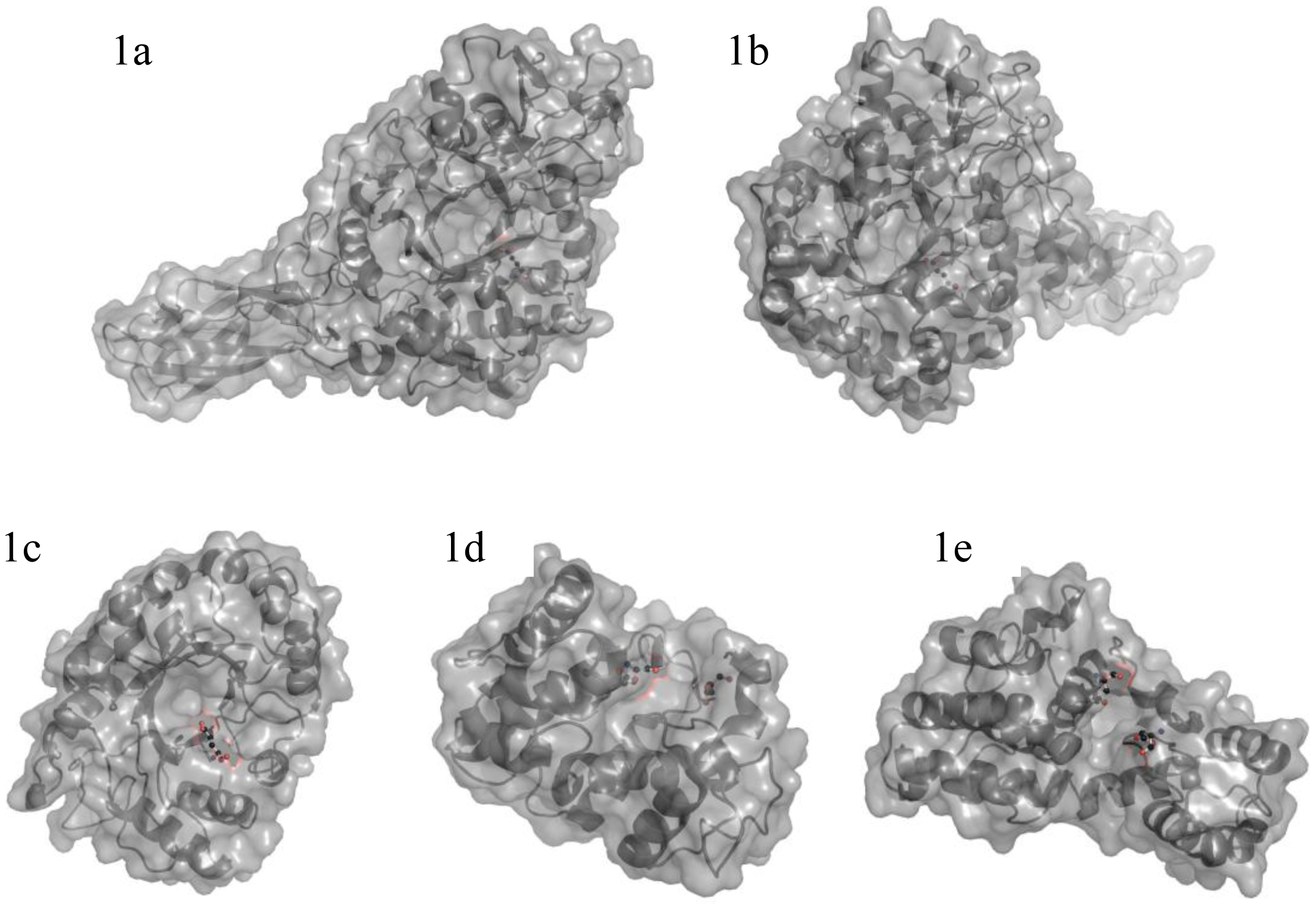
3. Enzymes Acting on Chitin and Chitosan
3.1. Chitinases and chitosanases
3.2. Catalytic mechanism
3.3. Human chitinases
3.4. Inhibition of family 18 chitinases with CHOS
3.5. Lysozyme
4. CHOS Production—Enzymatic Methods
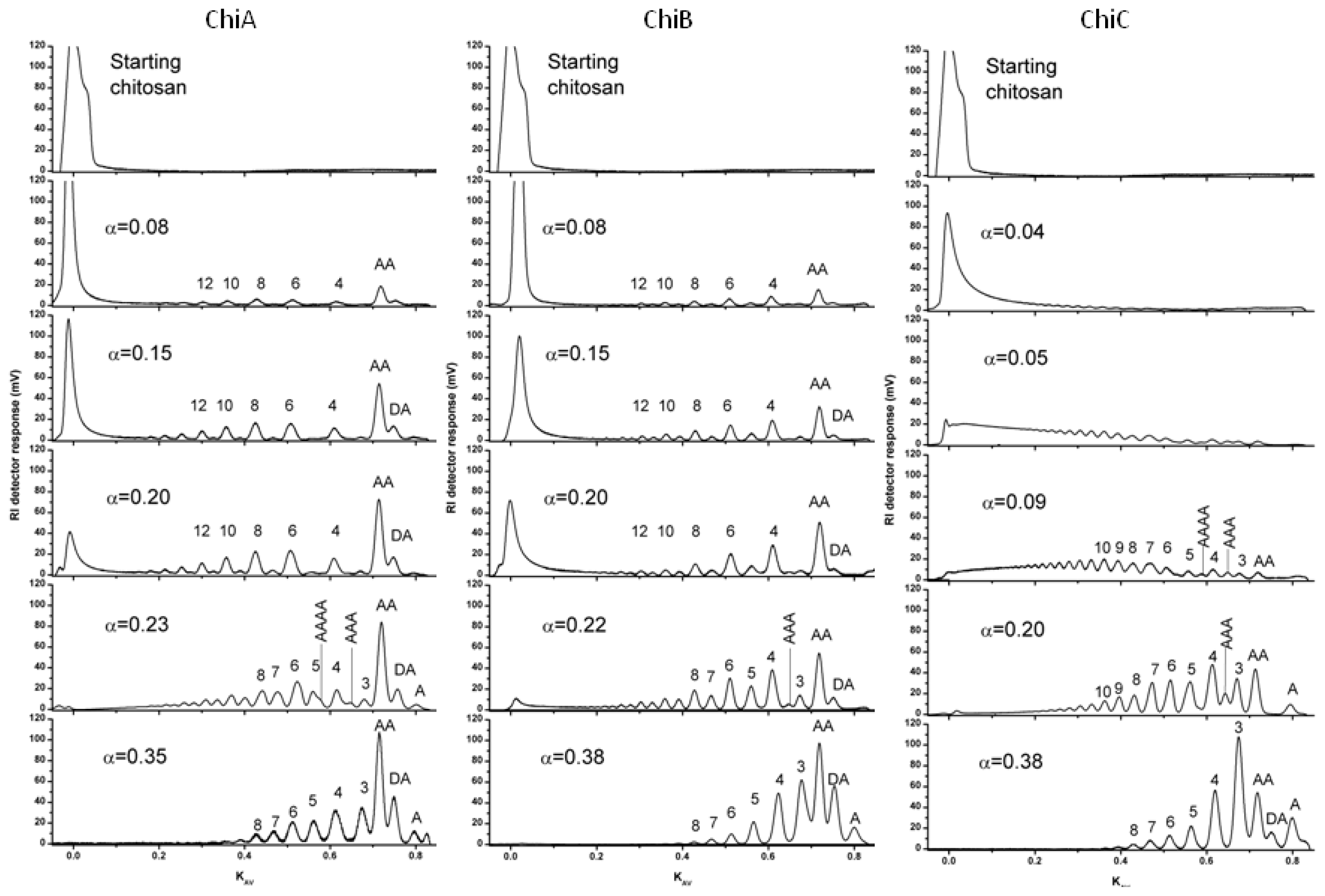
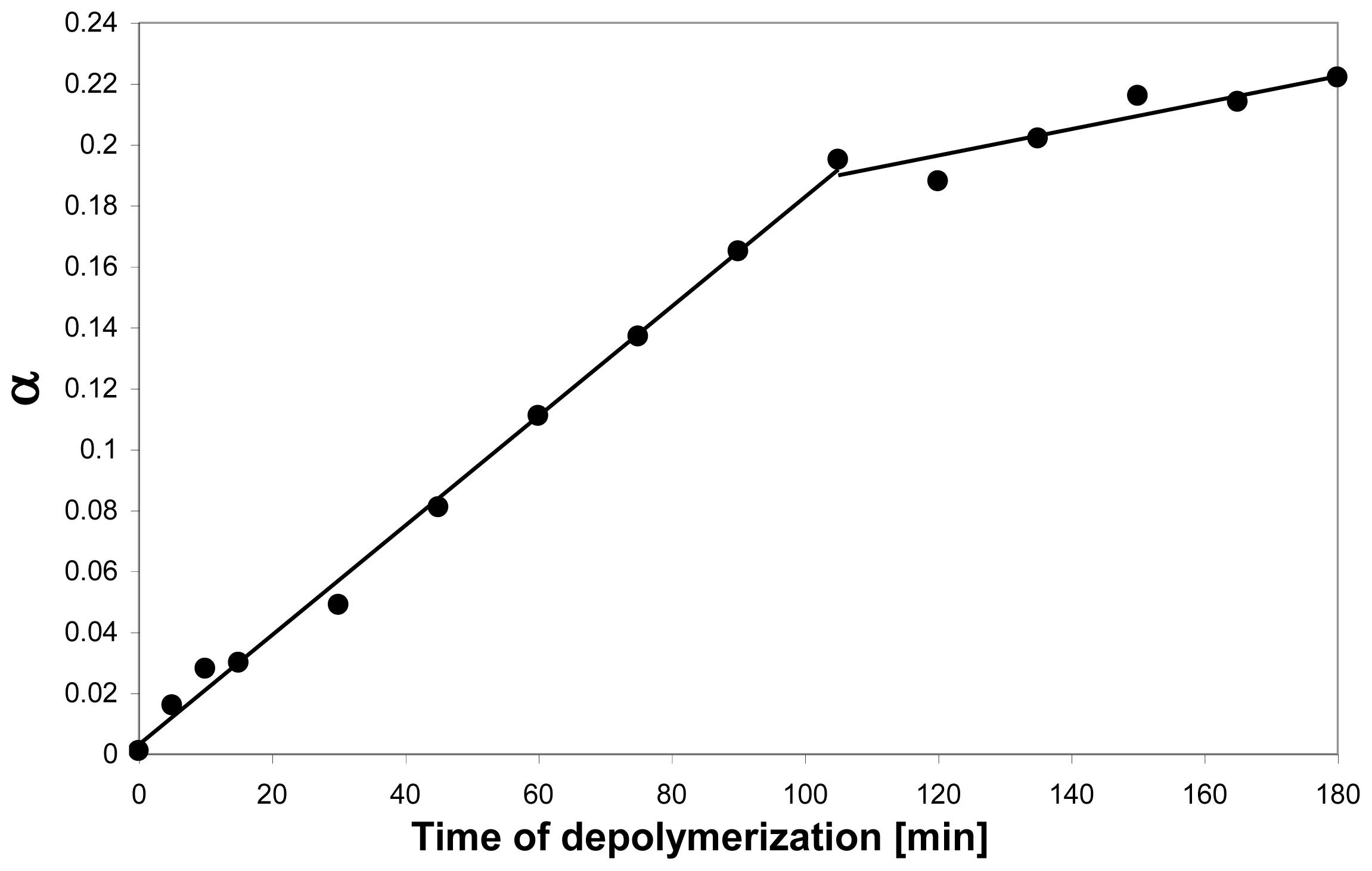
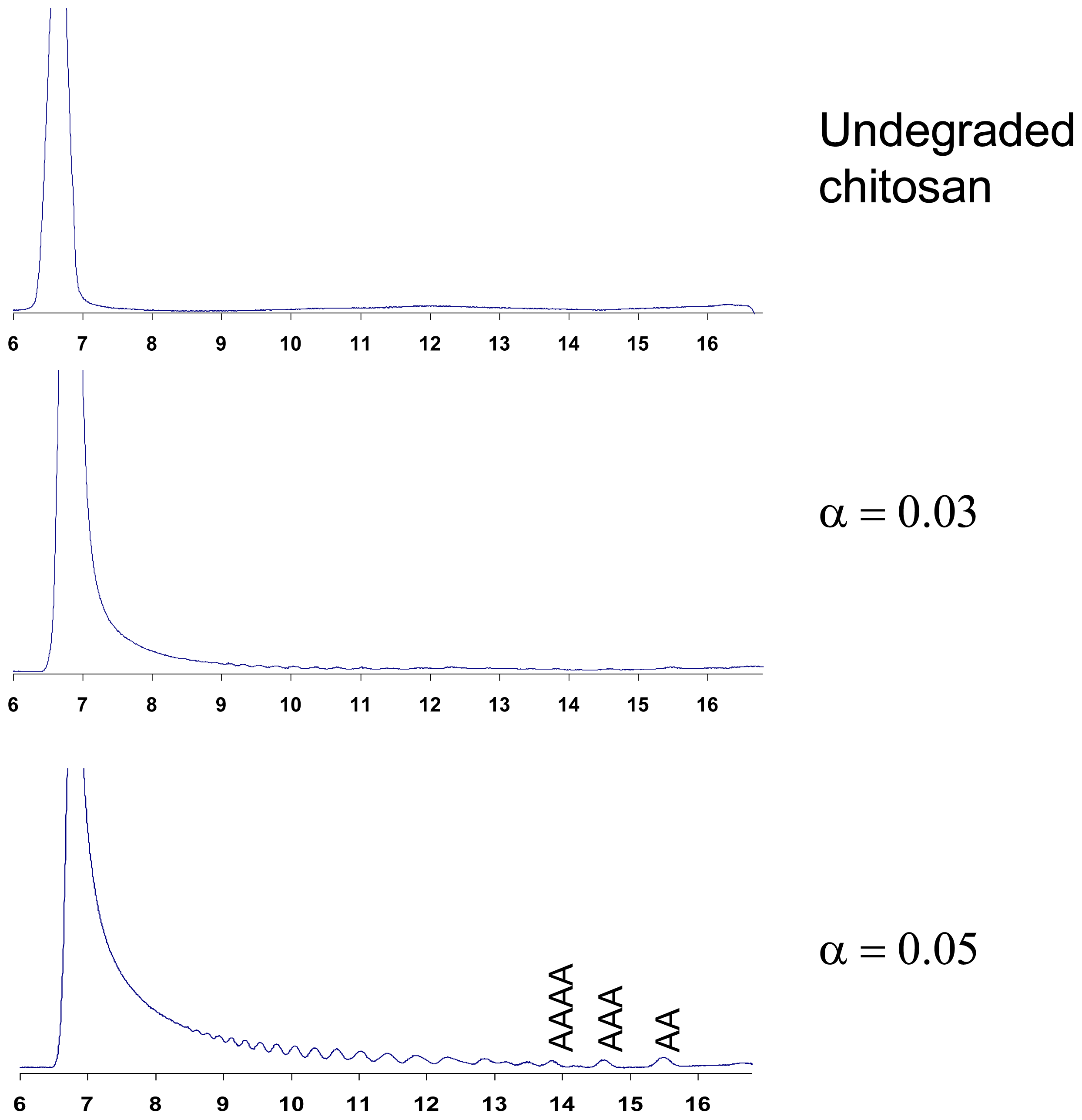
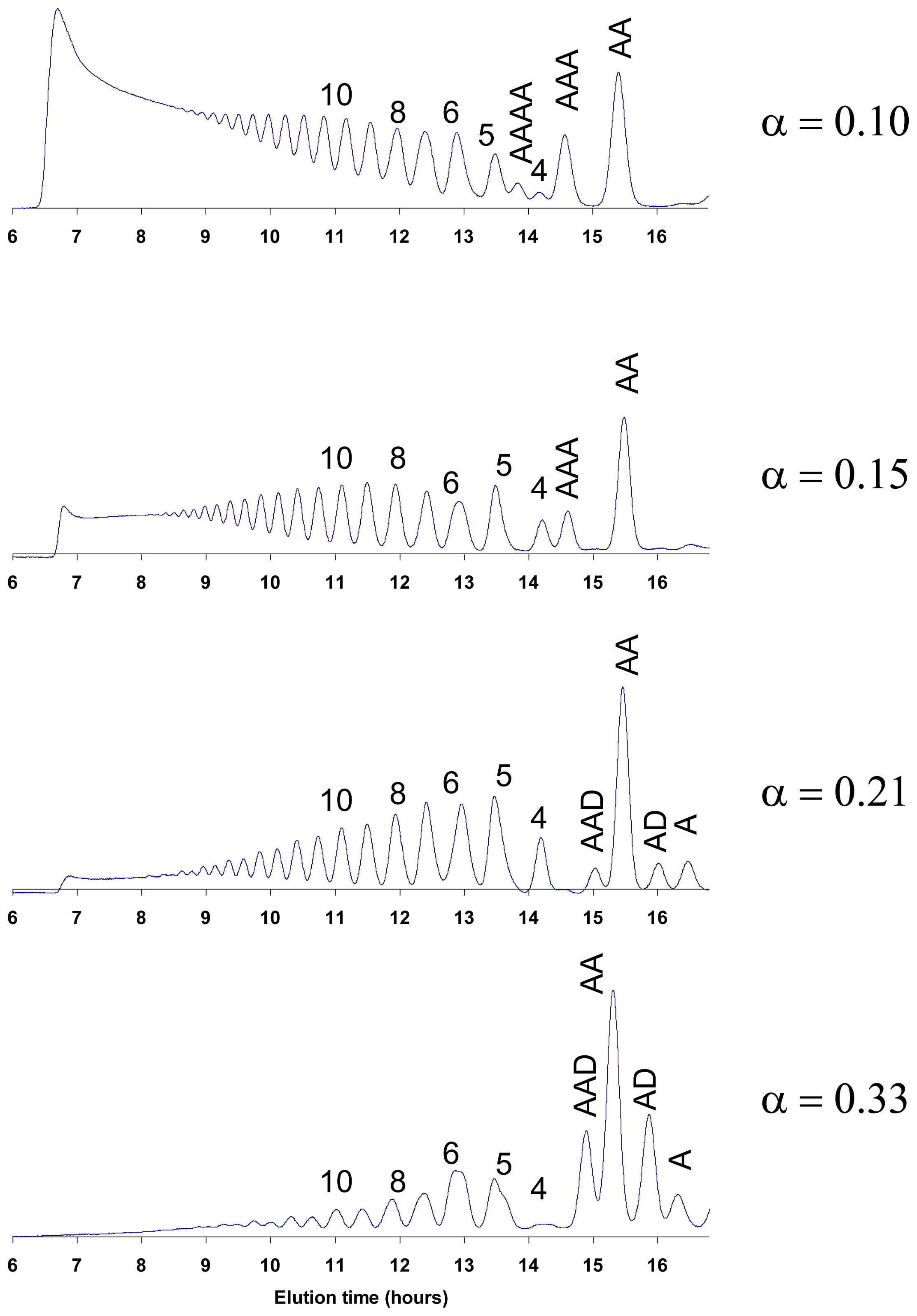
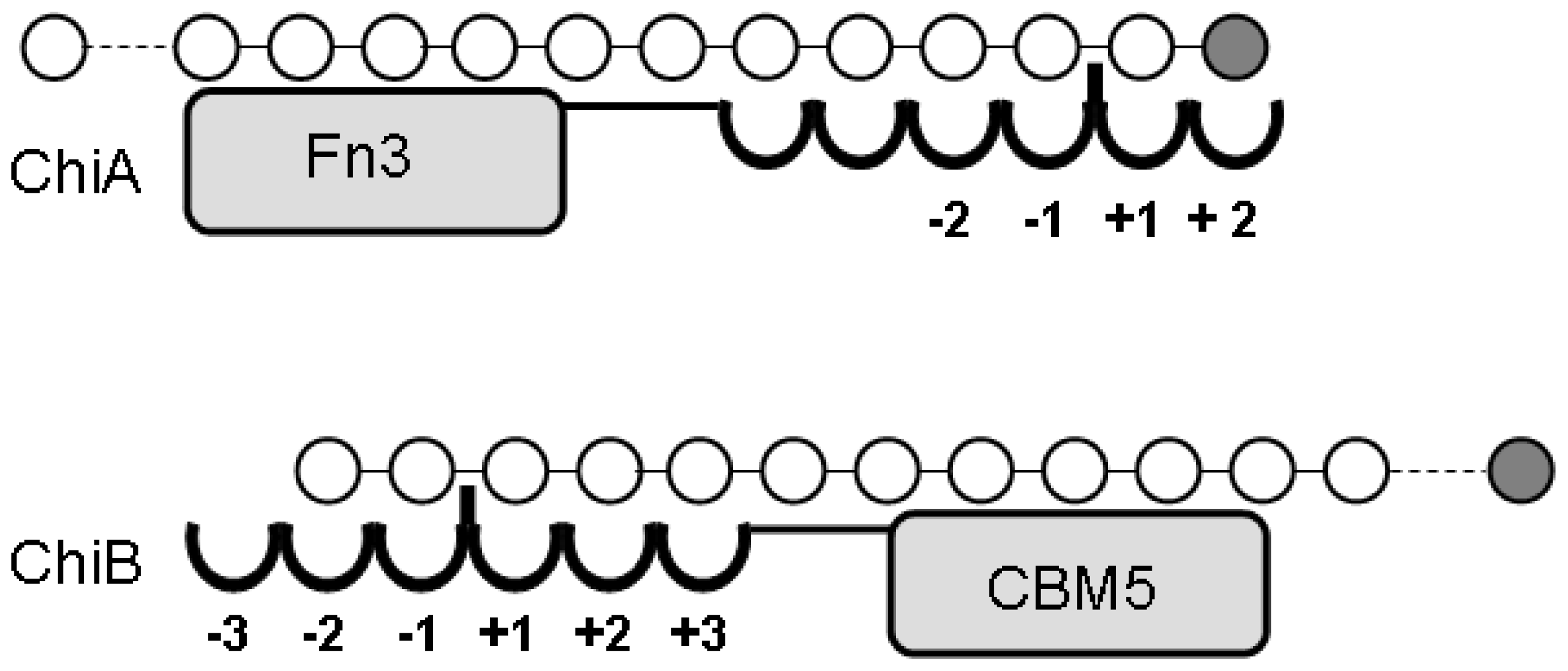
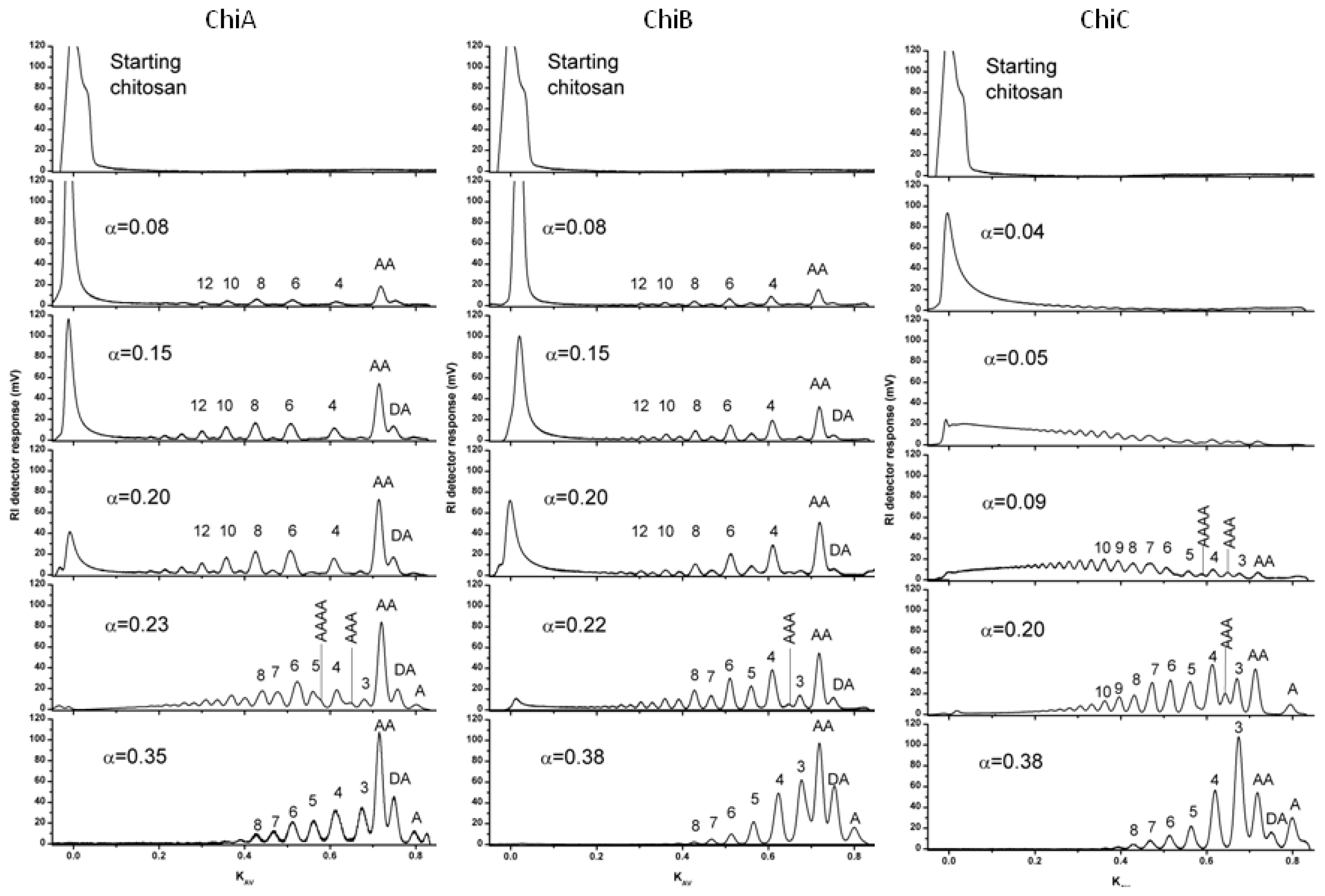
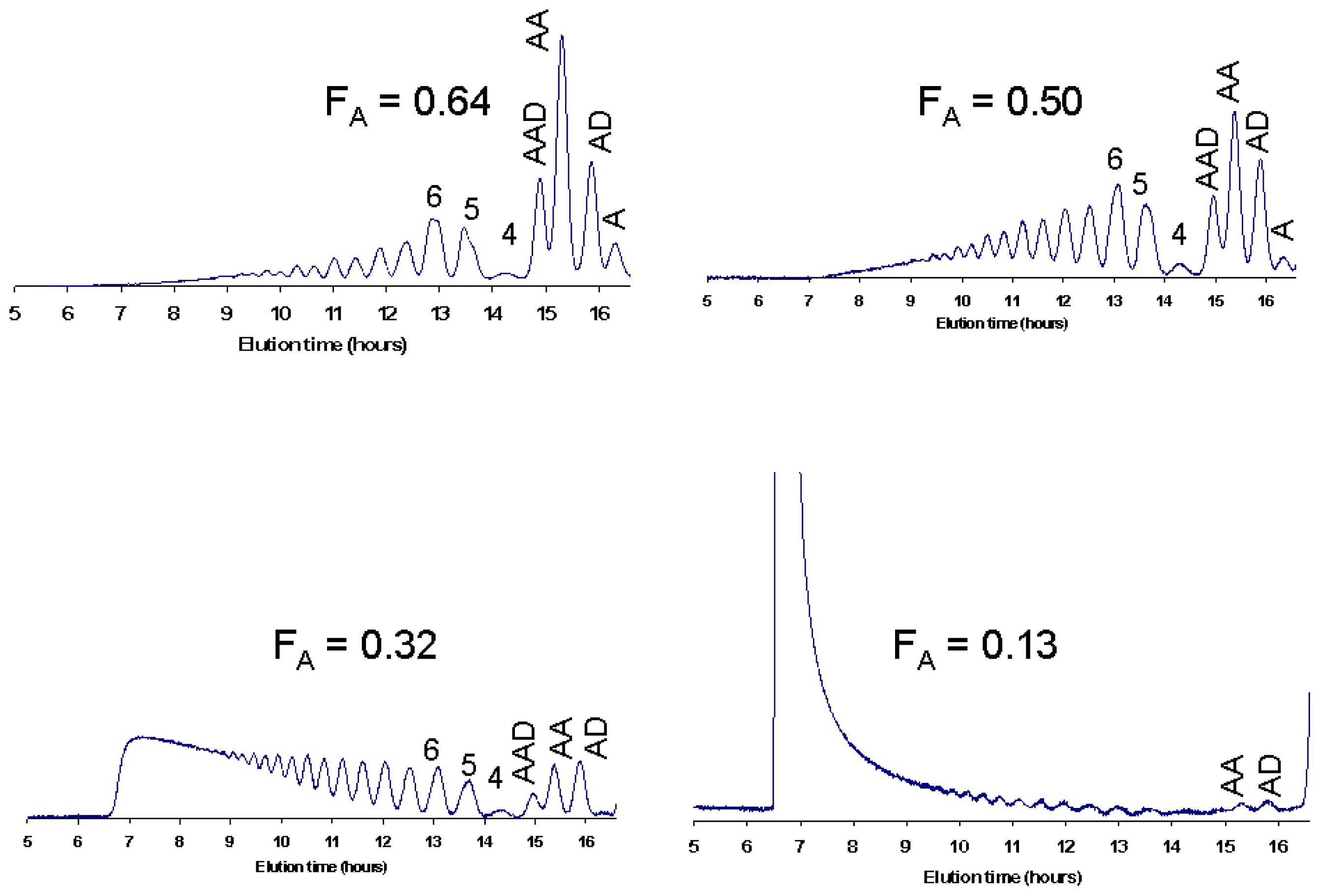
4.1. Degradation of chitosan by family 18 chitinases
4.2. Degradation of chitosan by family 19 chitinases
4.3. Degradation of chitosan by family 46 chitosanases
4.4. Degradation of chitosan by unspecific enzymes
5. CHOS Production—Chemical Methods
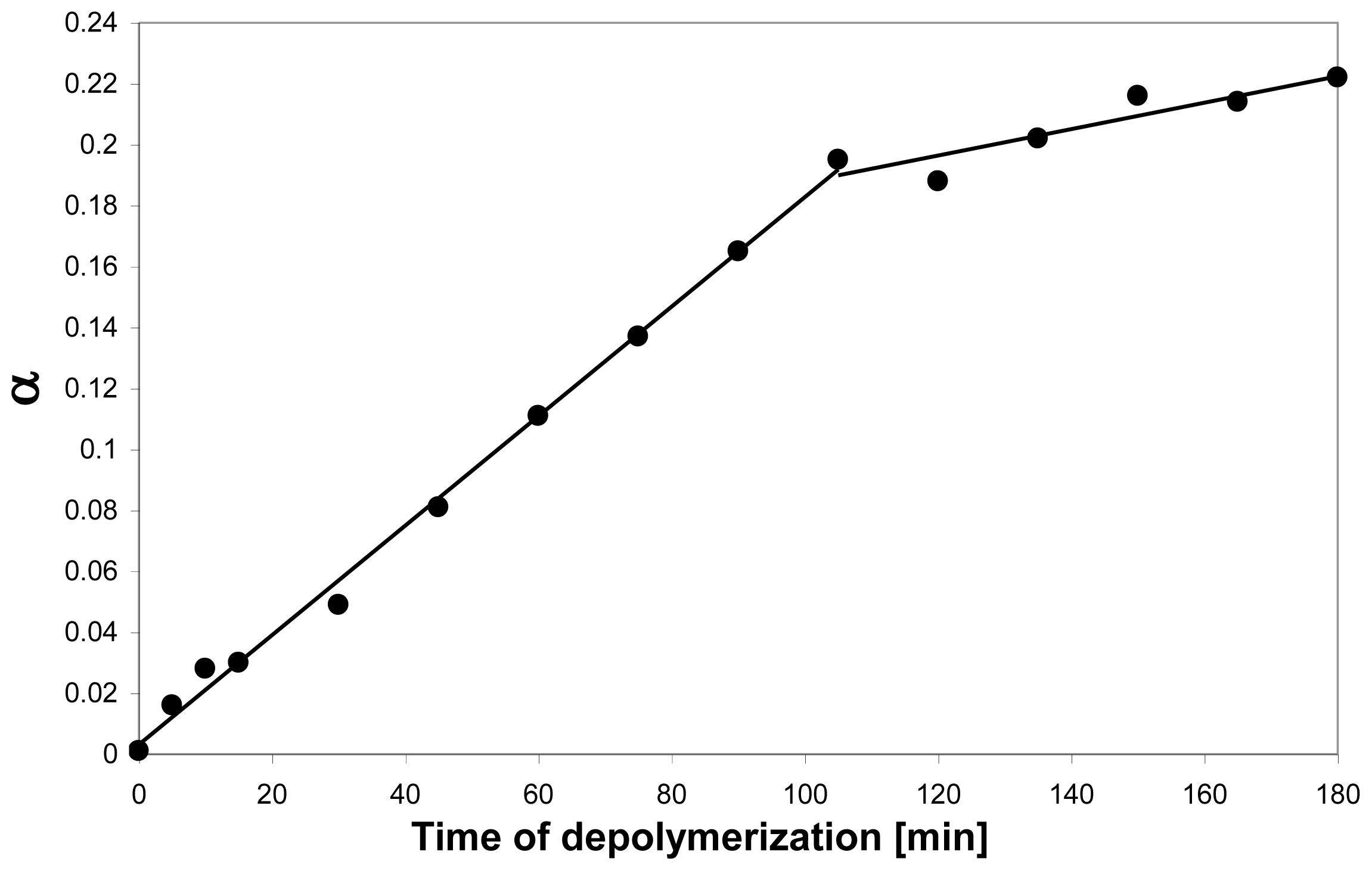
5.1. Acid hydrolysis of chitosan
5.2. Chemical synthesis of CHOS
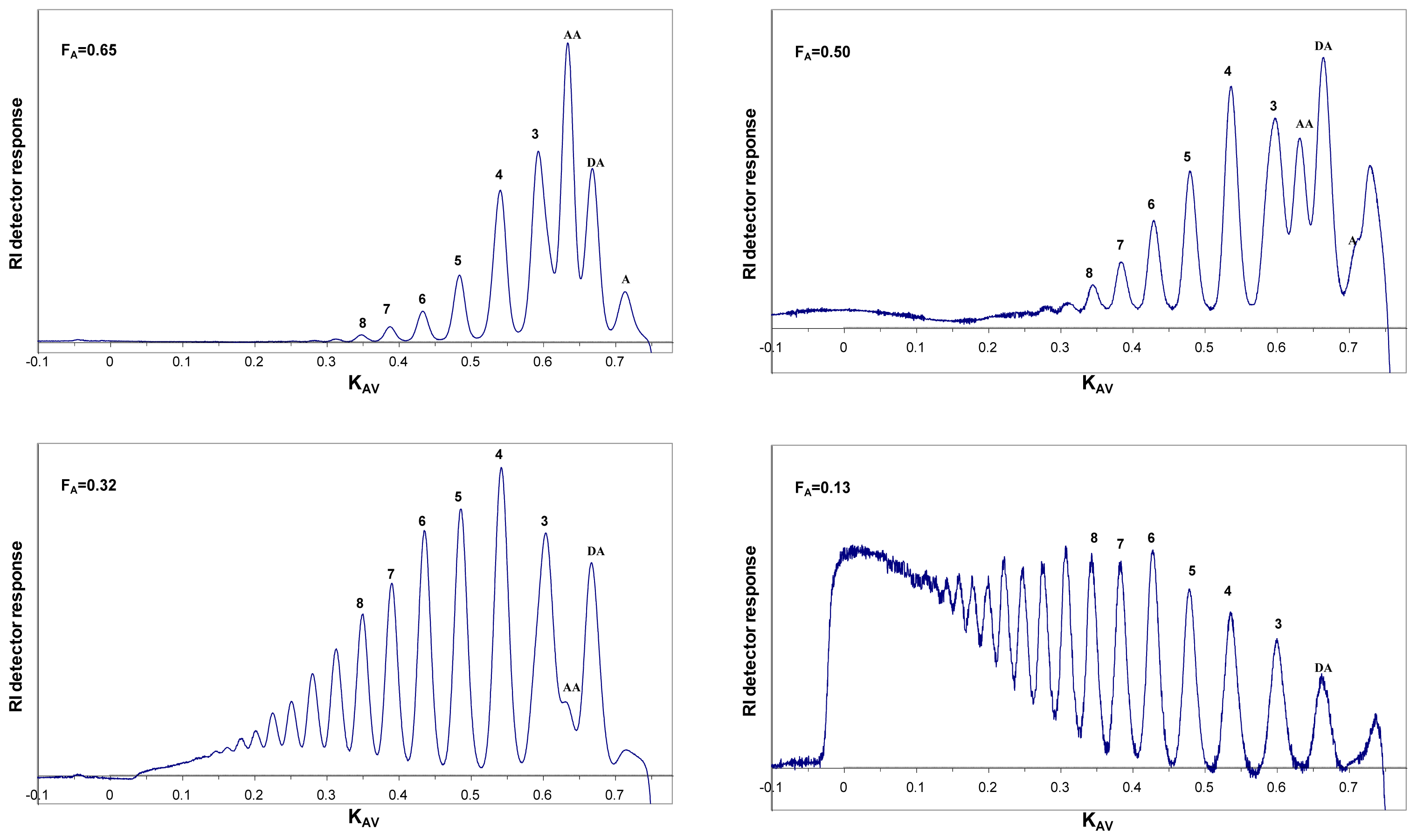
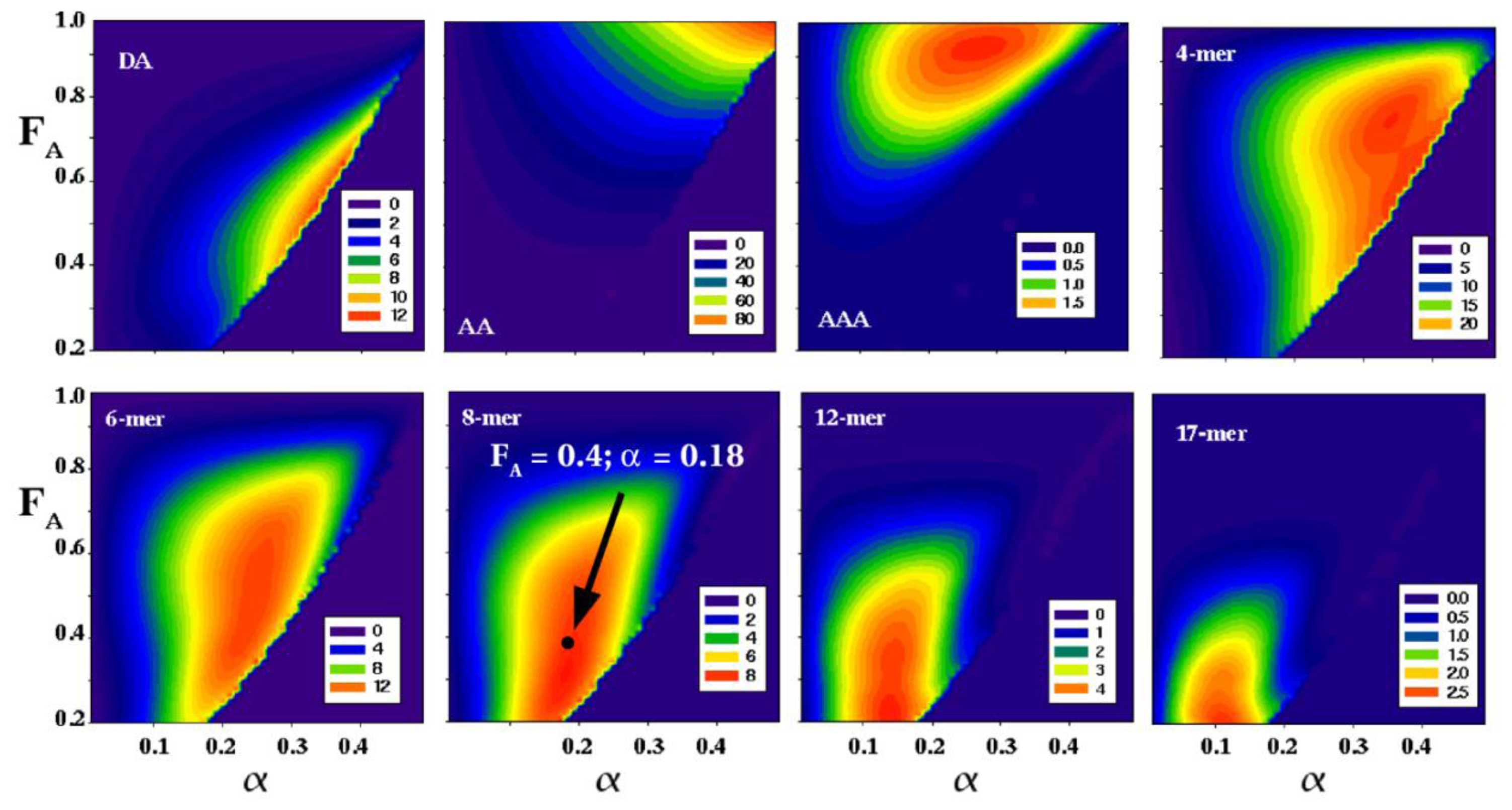
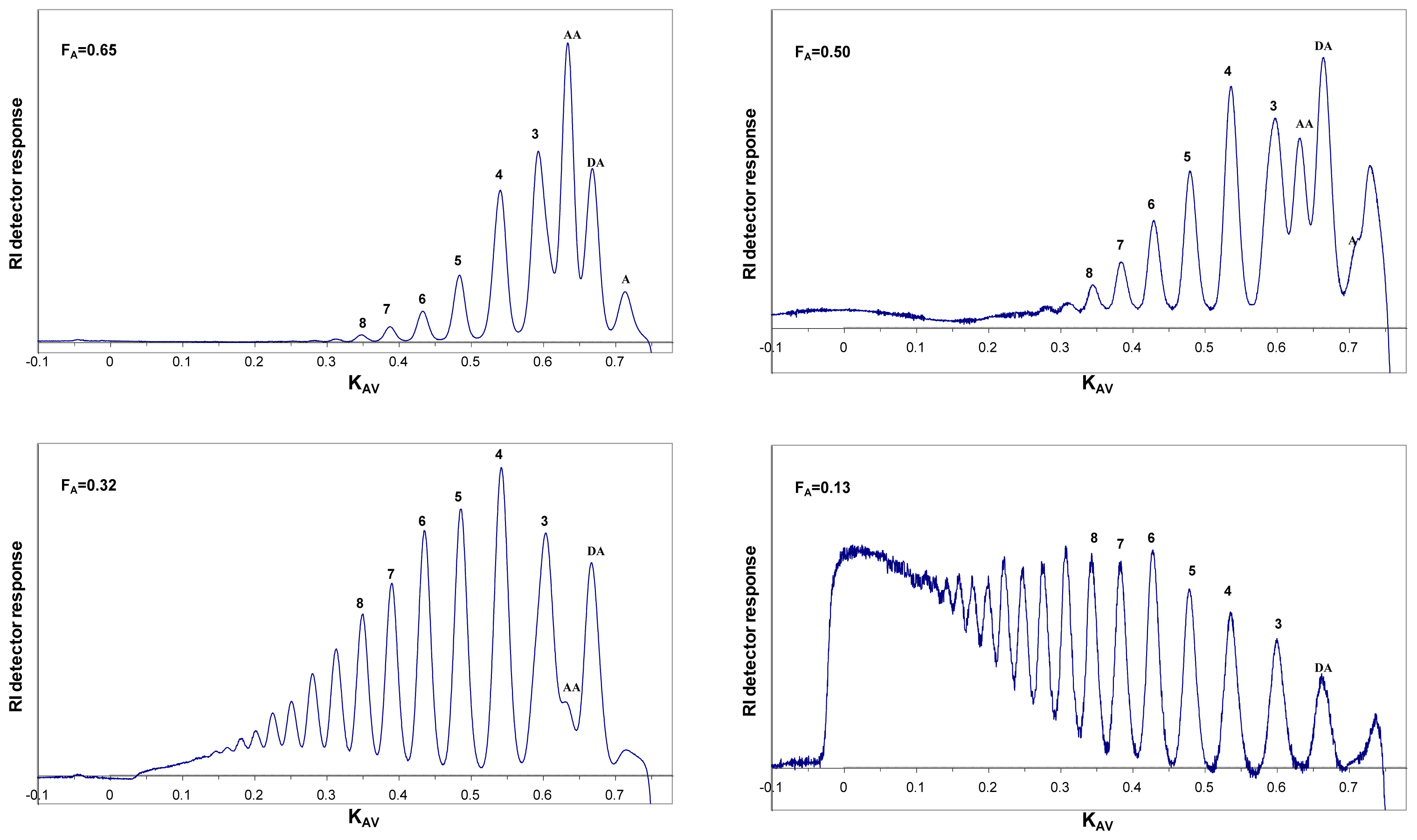
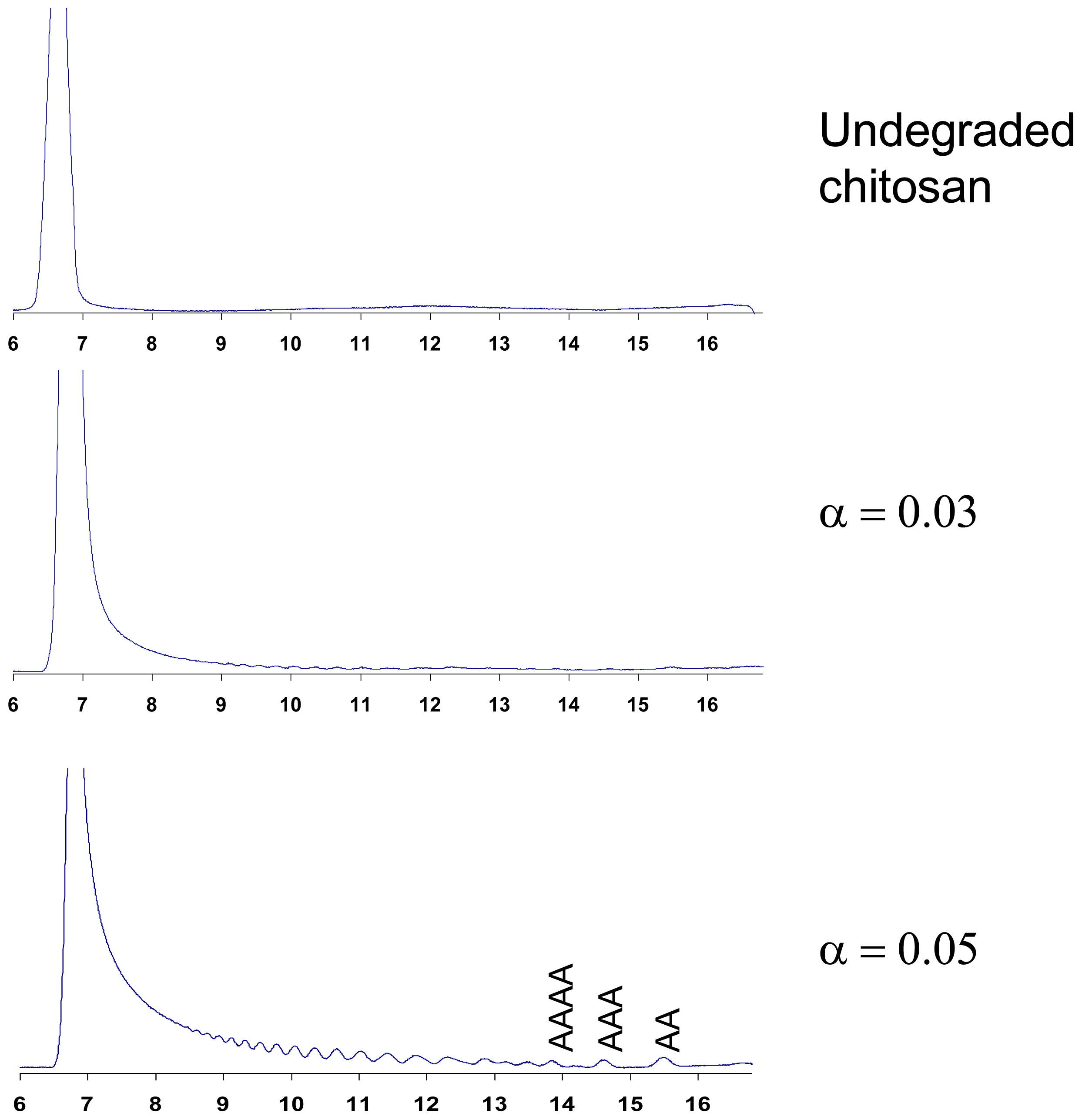
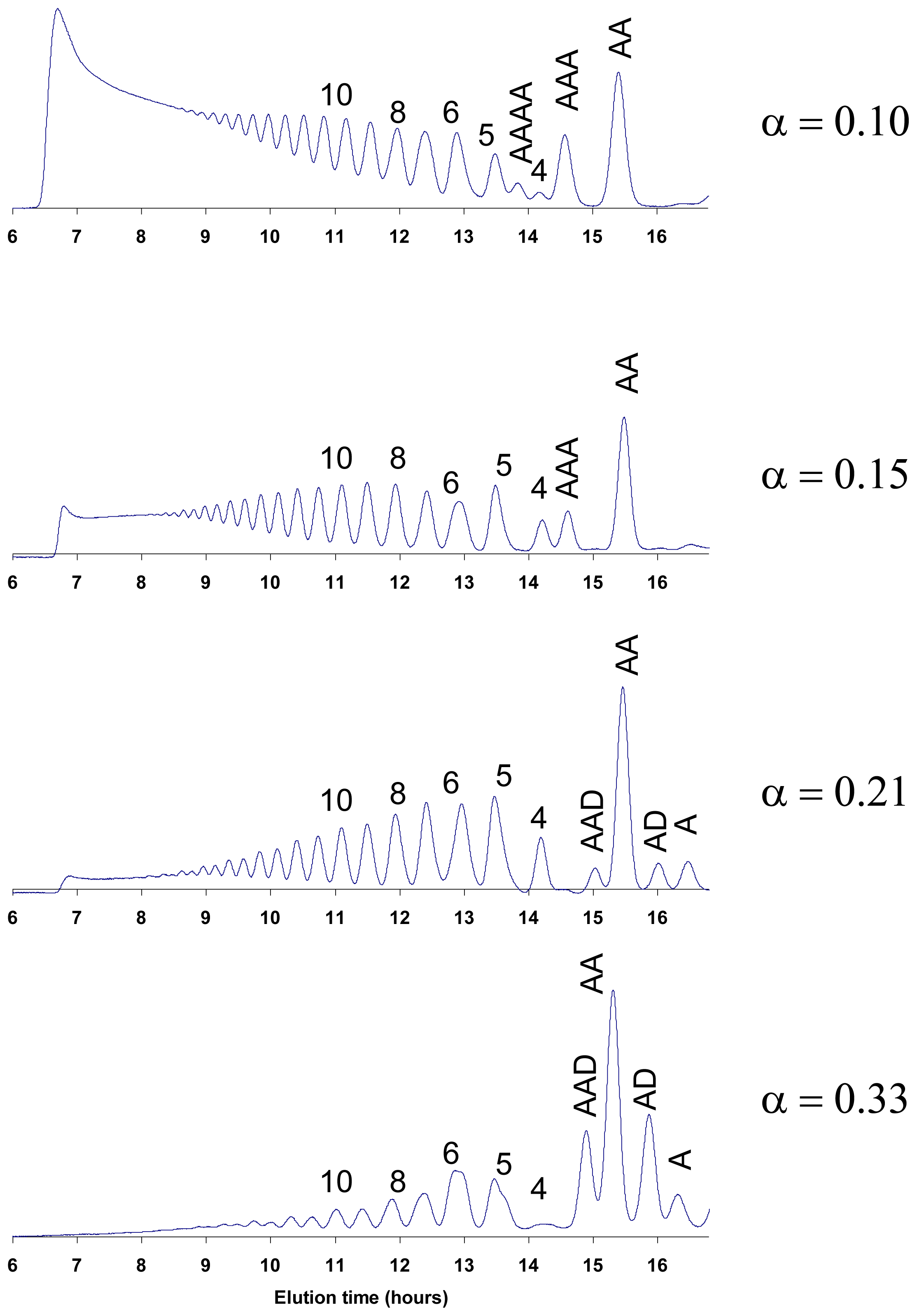
6. Purification and Characterization of CHOS
7. Applications of CHOS
7.1. Tumor growth inhibition
7.2. Asthma
7.3. Increased bone strength
7.4. CHOS in gene therapy
7.5. Prevention of bacterial adhesion to human cells
7.6. CHOS as a chitinase-inhibitor for preventing malaria
7.7. Applications of chitosan/CHOS in wound-dressings
7.8. Antifungal effects
8. Concluding Remarks and Future Perspectives
References
- Minke, R; Blackwell, J. The structure of alpha-chitin. J Mol Biol 1978, 120, 167–181. [Google Scholar]
- Gardner, KH; Blackwell, J. Refinement of structure of beta-chitin. Biopolymers 1975, 14, 1581–1595. [Google Scholar]
- Bhatnagar, A; Sillanpää, M. Applications of chitin- and chitosan-derivatives for the detoxification of water and wastewater - A short review. Adv Colloid Interface Sci 2009, 152, 26–38. [Google Scholar]
- Dodane, V; Vilivalam, VD. Pharmaceutical applications of chitosan. Pharm Sci Tech Today 1998, 1, 246–253. [Google Scholar]
- Harish Prashanth, KV; Tharanathan, RN. Chitin/chitosan: modifications and their unlimited application potential -an overview. Trends Food Sci Technol 2007, 18, 117–131. [Google Scholar]
- Kim, S-K; Rajapakse, N. Enzymatic production and biological activities of chitosan oligosaccharides (COS): A review. Carbohydr Polym 2005, 62, 357–368. [Google Scholar]
- Vårum, KM; Smidsr⊘d, O. Dumitriu, S, Ed.; Structure-property relationship in chitosans. In Polysaccharides: Structural Diversity and Functional Versatility; Marcel Dekker: New York, NY, USA, 2005; pp. 625–642. [Google Scholar]
- Yin, H; Du, YG; Zhang, JZ. Low molecular weight and oligomeric chitosans and their bioactivities. Curr Top Med Chem 2009, 9, 1546–1559. [Google Scholar]
- Donnelly, LE; Barnes, PJ. Acidic mammalian chitinase - a potential target for asthma therapy. Trends Pharmacol Sci 2004, 25, 509–511. [Google Scholar]
- Elias, JA; Homer, RJ; Hamid, Q; Lee, CG. Chitinases and chitinase-like proteins in Th2 inflammation and asthma. J Allergy Clin Immunol 2005, 116, 497–500. [Google Scholar]
- Kawada, M; Hachiya, Y; Arihiro, A; Mizoguchi, E. Role of mammalian chitinases in inflammatory conditions. Keio J Med 2007, 56, 21–27. [Google Scholar]
- Zhu, Z; Zheng, T; Homer, RJ; Kim, YK; Chen, NY; Cohn, L; Hamid, Q; Elias, JA. Acidic mammalian chitinase in asthmatic Th2 inflammation and IL-13 pathway activation. Science 2004, 304, 1678–1682. [Google Scholar]
- Rhoades, J; Gibson, G; Formentin, K; Beer, M; Rastall, R. Inhibition of the adhesion of enteropathogenic Escherichia coli strains to HT-29 cells in culture by chito-oligosaccharides. Carbohydr Polym 2006, 64, 57–59. [Google Scholar]
- Ribeiro, MP; Espiga, A; Silva, D; Baptista, P; Henriques, J; Ferreira, C; Silva, JC; Borges, JP; Pires, E; Chaves, P; Correia, IJ. Development of a new chitosan hydrogel for wound dressing. Wound Repair Regen 2009, 17, 817–824. [Google Scholar]
- You, Y; Park, WH; Ko, BM; Min, BM. Effects of PVA sponge containing chitooligosaccharide in the early stage of wound healing. J Mater Sci: Mater Med 2004, 15, 297–301. [Google Scholar]
- Köping-Höggård, M; Mel'nikova, YS; Vårum, KM; Lindman, B; Artursson, P. Relationship between the physical shape and the efficiency of oligomeric chitosan as a gene delivery system in vitro and in vivo. J Gene Med 2003, 5, 130–141. [Google Scholar]
- Köping-Höggård, M; Vårum, KM; Issa, M; Danielsen, S; Christensen, BE; Stokke, BT; Artursson, P. Improved chitosan-mediated gene delivery based on easily dissociated chitosan polyplexes of highly defined chitosan oligomers. Gene Ther 2004, 11, 1441–1452. [Google Scholar]
- Muzarelli, RAA. Chitin; Oxford Pergamon Press: London, UK, 1977; pp. 262–270. [Google Scholar]
- Nam, KS; Kim, MK; Shon, YH. Inhibition of proinflammatory cytokine-induced invasiveness of HT-29 cells by chitosan oligosaccharide. J Microbiol Biotechnol 2007, 17, 2042–2045. [Google Scholar]
- Shen, K-T; Chen, M-H; Chan, H-Y; Jeng, J-H; Wang, Y-J. Inhibitory effects of chitooligosaccharides on tumor growth and metastasis. Food Chem Toxicol 2009, 47, 1864–1871. [Google Scholar]
- Klokkevold, PR; Vandemark, L; Kenney, EB; Bernard, GW. Osteogenesis enhanced by chitosan (poly-N-acetyl glucosaminoglycan)in vitro. J Periodontol 1996, 67, 1170–1175. [Google Scholar]
- Ratanavaraporn, J; Kanokpanont, S; Tabata, Y; Damrongsakkul, S. Growth and osteogenic differentiation of adipose-derived and bone marrow-derived stem cells on chitosan and chitooligosaccharide films. Carbohydr Polym 2009, 78, 873–878. [Google Scholar]
- Shahabuddin, M; Toyoshima, T; Aikawa, M; Kaslow, DC. Transmission-blocking activity of a chitinase inhibitor and activation of malarial parasite chitinase by mosquito protease. Proc Natl Acad Sci USA 1993, 90, 4266–4270. [Google Scholar]
- Kim, HM; Hong, SH; Yoo, SJ; Baek, KS; Jeon, YJ; Choung, SY. Differential effects of chitooligosaccharides on serum cytokine levels in aged subjects. J Med Food 2006, 9, 427–430. [Google Scholar]
- Oliveira, EN, Jr; El Gueddari, NE; Moerschbacher, BM; Peter, MG; Franco, TT. Growth of phytopathogenic fungi in the presence of partially acetylated chitooligosaccharides. Mycopathologia 2008, 166, 163–174. [Google Scholar]
- Seyfarth, F; Schliemann, S; Elsner, P; Hipler, UC. Antifungal effect of high- and low-molecular- weight chitosan hydrochloride, carboxymethyl chitosan, chitosan oligosaccharide and N-acetyl-D-glucosamine against Candida albicans, Candida krusei and Candida glabrata. Int J Pharm 2008, 353, 139–148. [Google Scholar]
- Lee, HW; Park, YS; Choi, JW; Yi, SY; Shin, WS. Antidiabetic effects of chitosan oligosaccharides in neonatal streptozotocin-induced noninsulin-dependent diabetes mellitus in rats. Biol Pharm Bull 2003, 26, 1100–1103. [Google Scholar]
- Sato, K; Saimoto, H; Morimoto, M; Shigemasa, Y. Depolymerization of chitin and chitosan under hydrothermal conditions. Sen-I Gakkaishi 2003, 59, 104–109. [Google Scholar]
- Xing, RE; Liu, S; Yu, HH; Guo, ZY; Wang, PB; Li, CP; Li, Z; Li, PC. Salt-assisted acid hydrolysis of chitosan to oligomers under microwave irradiation. Carbohydr Res 2005, 340, 2150–2153. [Google Scholar]
- Wu, T; Zivanovic, S; Hayes, DG; Weiss, J. Efficient reduction of chitosan molecular weight by high-intensity ultrasound: Underlying mechanism and effect of process parameters. J Agric Food Chem 2008, 56, 5112–5119. [Google Scholar]
- Yoksan, R; Akashi, M; Miyata, M; Chirachanchai, S. Optimal gamma-ray dose and irradiation conditions for producing low-molecular-weight chitosan that retains its chemical structure. Radiat Res 2004, 161, 471–480. [Google Scholar]
- Domard, A; Cartier, N. Glucosamine oligomers: 4. solid state-crystallization and sustained dissolution. Int J Biol Macromol 1992, 14, 100–106. [Google Scholar]
- Einbu, A; Vårum, KM. Depolymerization and de-N-acetylation of chitin oligomers in hydrochloric acid. Biomacromolecules 2007, 8, 309–314. [Google Scholar]
- Lin, F; Jia, XG; Lei, WX; Li, ZJ; Zhang, TY. Spectra Analyses of Chitosans Degraded by Hydrogen Peroxide under Optimal Conditions. Spectrosc Spectr Anal 2009, 29, 43–47. [Google Scholar]
- Morris, VB; Neethu, S; Abraham, TE; Pillai, CKS; Sharma, CP. Studies on the Condensation of Depolymerized Chitosans With DNA for Preparing Chitosan-DNA Nanoparticles for Gene Delivery Applications. J Biomed Mater Res Part B 2009, 89B, 282–292. [Google Scholar]
- Roberts, GAF. Chitin Chemistry; The Macmillan Press Ldt: Hong Kong, 1992. [Google Scholar]
- Sannan, T; Kurita, K; Iwakura, Y. Studies on Chitin, 2. Effect of deacetylation on solubility. Macromol Chem 1976, 177, 3589–3600. [Google Scholar]
- Kurita, K; Sannan, T; Iwakura, Y. Studies on Chitin, 4. Evidence for formation of block and random copolymers of N-Acetyl-D-Glucosamine and D-Glucosamine by heterogeneous and homogeneous hydrolyses. Macromol Chem 1977, 178, 3197–3202. [Google Scholar]
- Vårum, KM; Anthonsen, MW; Grasdalen, H; Smidsr⊘d, O. 13C-n.m.r. studies of the acetylation sequences in partially N-deacetylated chitins (chitosans). Carbohydr Res 1991, 217, 19–27. [Google Scholar]
- Vårum, KM; Anthonsen, MW; Grasdalen, H; Smidsr⊘d, O. Determination of the degree of N-acetylation and the distribution of N-acetyl groups in partially N-deacetylated chitins (chitosans) by high-field n.m.r. spectroscopy. Carbohydr Res 1991, 211, 17–23. [Google Scholar]
- Weinhold, MX; Sauvageau, JCM; Kumirska, J; Thöming, J. Studies on acetylation patterns of different chitosan preparations. Carbohydr Polym 2009, 78, 678–684. [Google Scholar]
- Gooday, GW. The ecology of chitin degradation. Adv Microb Ecol 1990, 11, 387–430. [Google Scholar]
- Sandford, PA. Vårum, KM, Domard, A, Smidsr⊘d, O, Eds.; Commercial sources of chitin & chitosan and their utilization. In Advances in Chitin Science; Tapir: Trondheim, Norway, 2002; Volume VI. [Google Scholar]
- Chitin; Chitosan. A Global Strategic Business Report MCP-2039; Global Industry Analysts Inc. Available online: http://www.marketresearch.com (accessed on 27 April 2010).
- Cantarel, BL; Coutinho, PM; Rancurel, C; Bernard, T; Lombard, V; Henrissat, B. The Carbohydrate-Active enZYmes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res 2009, 37, D233–D238. [Google Scholar]
- CAZy The Carbohydrate-Active enZYmes database. Available online: http://www.cazy.org (accessed on 27 April 2010).
- Davies, G; Henrissat, B. Structures and mechanisms of glycosyl hydrolases. Structure 1995, 3, 853–859. [Google Scholar]
- Henrissat, B; Bairoch, A. Updating the sequence-based classification of glycosyl hydrolases. Biochem J 1996, 316, 695–696. [Google Scholar]
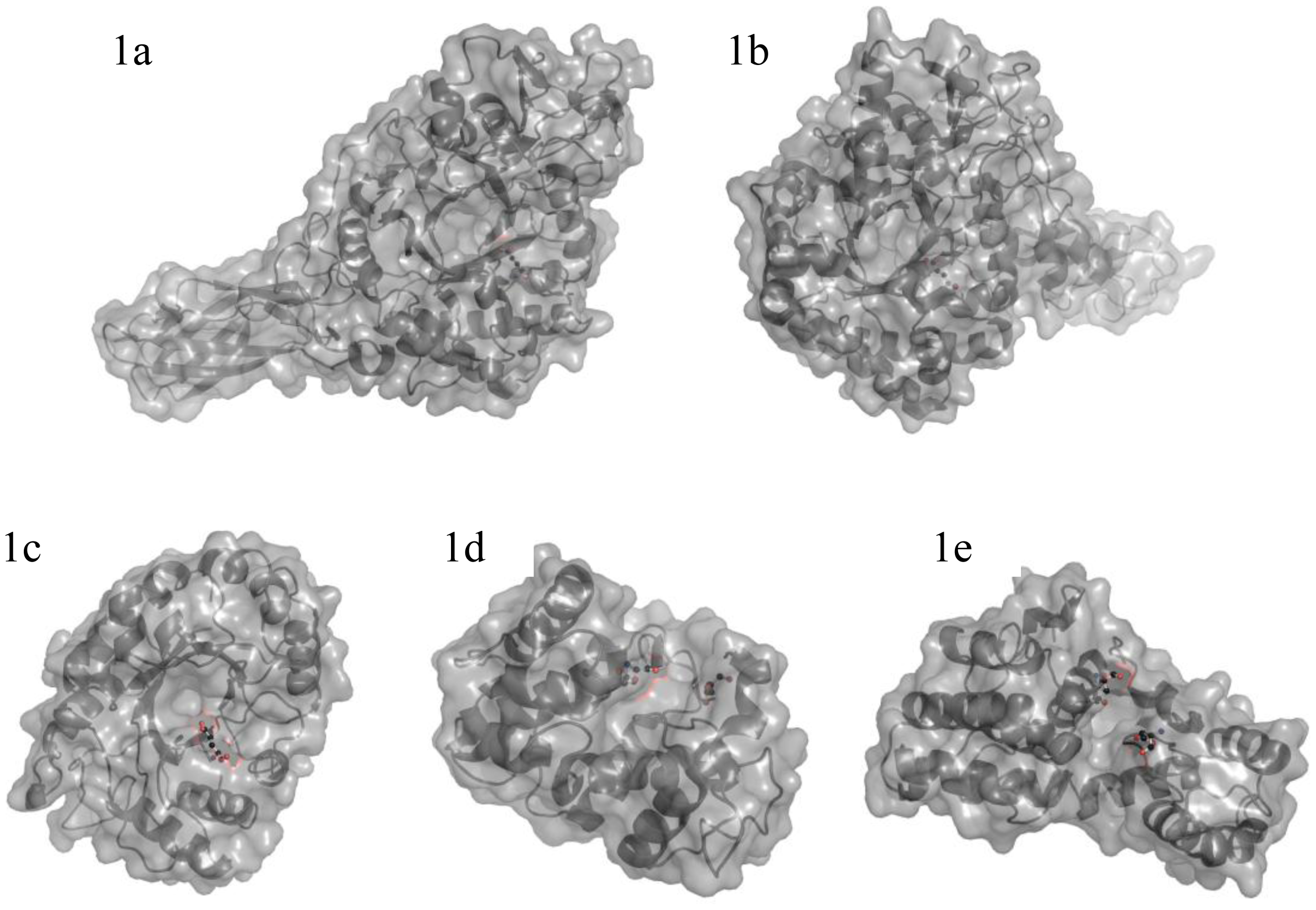
- Adachi, W; Sakihama, Y; Shimizu, S; Sunami, T; Fukazawa, T; Suzuki, M; Yatsunami, R; Nakamura, S; Takenaka, A. Crystal structure of family GH-8 chitosanase with subclass II specificity from Bacillus sp. K17. J Mol Biol 2004, 343, 785–795. [Google Scholar]
- Lacombe-Harvey, ME; Fukamizo, T; Gagnon, J; Ghinet, MG; Dennhart, N; Letzel, T; Brzezinski, R. Accessory active site residues of Streptomyces sp N174 chitosanase. FEBS J 2009, 276, 857–869. [Google Scholar]
- Marcotte, EM; Monzingo, AF; Ernst, SR; Brzezinski, R; Robertus, JD. X-ray structure of an anti-fungal chitosanase from Streptomyces N174. Nat Struct Biol 1996, 3, 155–162. [Google Scholar]
- Saito, J; Kita, A; Higuchi, Y; Nagata, Y; Ando, A; Miki, K. Crystal structure of chitosanase from Bacillus circulans MH-K1 at 1.6-angstrom resolution and its substrate recognition mechanism. J Biol Chem 1999, 274, 30818–30825. [Google Scholar]
- Fukamizo, T; Ohkawa, T; Ikeda, Y; Goto, S. Specificity of chitosanase from Bacillus pumilus. Biochim Biophys Acta, Prot Struct Mol Enzym 1994, 1205, 183–188. [Google Scholar]
- Cheng, CY; Chang, CH; Wu, YJ; Li, YK. Exploration of glycosyl hydrolase family 75, a chitosanase from Aspergillus fumigatus. J Biol Chem 2006, 281, 3137–3144. [Google Scholar]
- Fukamizo, T; Brzezinski, R. Chitosanase from Streptomyces sp. strain N174: a comparative review of its structure and function. Biochem Cell Biol 1997, 75, 687–696. [Google Scholar]
- Horn, SJ; S⊘rbotten, A; Synstad, B; Sikorski, P; S⊘rlie, M; Vårum, KM; Eijsink, VG. Endo/exo mechanism and processivity of family 18 chitinases produced by Serratia marcescens. FEBS J 2006, 273, 491–503. [Google Scholar]
- Koshland, DE, Jr; Stein, SS. Correlation of bond breaking with enzyme specificity; cleavage point of invertase. J Biol Chem 1954, 208, 139–148. [Google Scholar]
- Sinnott, ML. Catalytic mechanisms of enzymatic glycosyl transfer. Chem Rev 1990, 90, 1171–1202. [Google Scholar]
- Brameld, KA; Goddard, WA. The role of enzyme distortion in the single displacement mechanism of family 19 chitinases. Proc Natl Acad Sci USA 1998, 95, 4276–4281. [Google Scholar]
- Fukamizo, T; Honda, Y; Goto, S; Boucher, I; Brzezinski, R. Reaction mechanism of chitosanase from Streptomyces sp. N174. Biochem J 1995, 311 Pt 2, 377–383. [Google Scholar]
- Iseli, B; Armand, S; Boller, T; Neuhaus, JM; Henrissat, B. Plant chitinases use two different hydrolytic mechanisms. FEBS Lett 1996, 382, 186–188. [Google Scholar]
- Vocadlo, DJ; Davies, GJ; Laine, R; Withers, SG. Catalysis by hen egg-white lysozyme proceeds via a covalent intermediate. Nature 2001, 412, 835–838. [Google Scholar]
- Brameld, KA; Goddard, WA. Substrate distortion to a boat conformation at subsite -1 is critical in the mechanism of family 18 chitinases. J Am Chem Soc 1998, 120, 3571–3580. [Google Scholar]
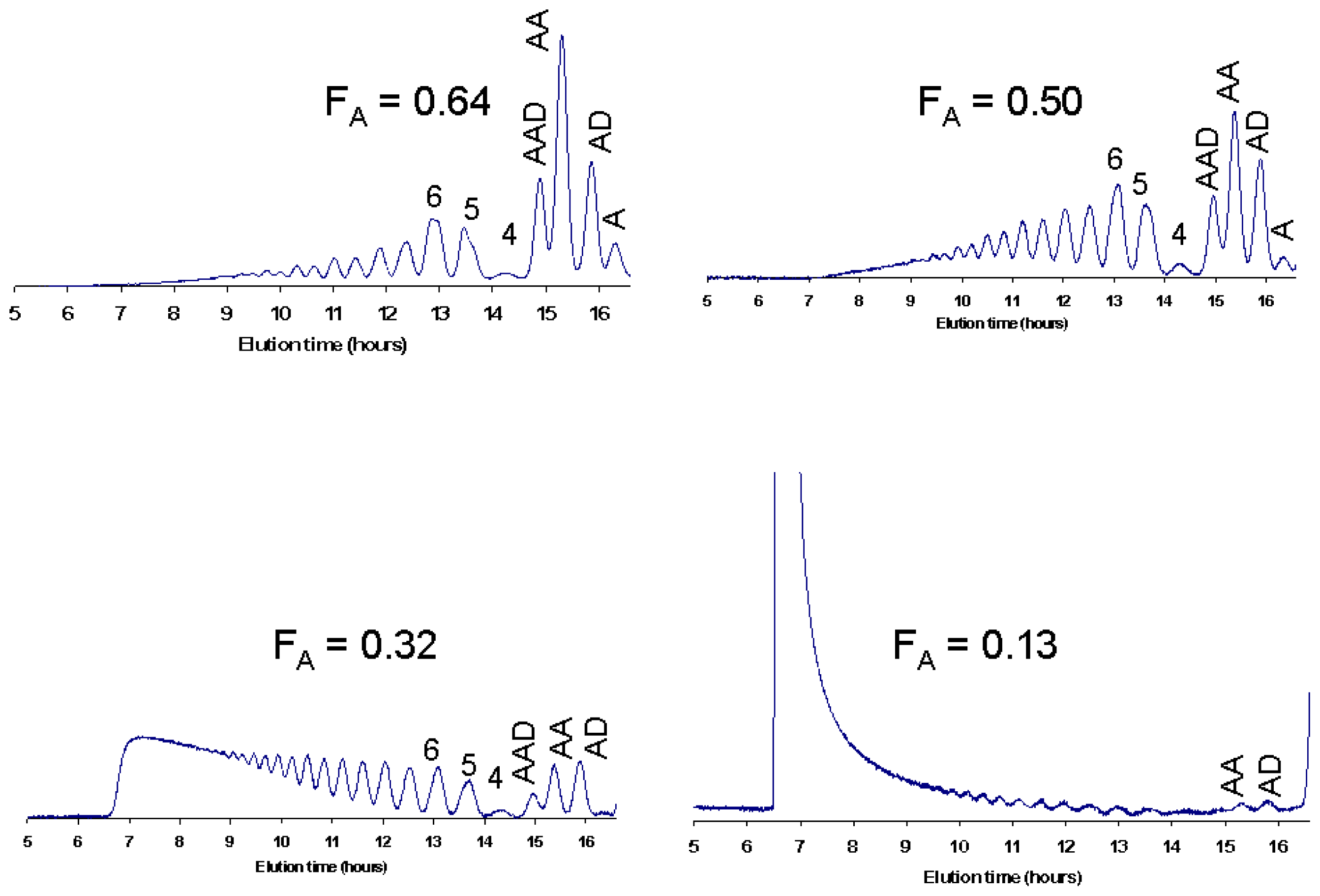
- S⊘rbotten, A; Horn, SJ; Eijsink, VG; Vårum, KM. Degradation of chitosans with chitinase B from Serratia marcescens. Production of chito-oligosaccharides and insight into enzyme processivity. FEBS J 2005, 272, 538–549. [Google Scholar]
- Terwisscha van Scheltinga, AC; Armand, S; Kalk, KH; Isogai, A; Henrissat, B; Dijkstra, BW. Stereochemistry of chitin hydrolysis by a plant chitinase/lysozyme and X-ray structure of a complex with allosamidin: evidence for substrate assisted catalysis. Biochemistry 1995, 34, 15619–15623. [Google Scholar]
- Tews, I; Terwisscha van Scheltinga, AC; Perrakis, A; Wilson, KS; Dijkstra, BW. Substrate-assisted catalysis unifies two families of chitinolytic enzymes. J Am Chem Soc 1997, 119, 7954–7959. [Google Scholar]
- van Aalten, DM; Komander, D; Synstad, B; Gåseidnes, S; Peter, MG; Eijsink, VG. Structural insights into the catalytic mechanism of a family 18 exo-chitinase. Proc Natl Acad Sci USA 2001, 98, 8979–8984. [Google Scholar]
- Eijsink, VG; Vaaje-Kolstad, G; Vårum, KM; Horn, SJ. Towards new enzymes for biofuels: lessons from chitinase research. Trends Biotechnol 2008, 26, 228–235. [Google Scholar]
- Horn, SJ; Sikorski, P; Cederkvist, JB; Vaaje-Kolstad, G; S⊘rlie, M; Synstad, B; Vriend, G; Vårum, KM; Eijsink, VGH. Costs and benefits of processivity in enzymatic degradation of recalcitrant polysaccharides. Proc Natl Acad Sci USA 2006, 103, 18089–18094. [Google Scholar]
- Zakariassen, H; Aam, BB; Horn, SJ; Vårum, KM; S⊘rlie, M; Eijsink, VG. Aromatic residues in the catalytic center of chitinase A from Serratia marcescens affect processivity, enzyme activity, and biomass converting efficiency. J Biol Chem 2009, 284, 10610–10617. [Google Scholar]
- Bussink, AP; Speijer, D; Aerts, JMFG; Boot, RG. Evolution of mammalian chitinase(-like) members of family 18 glycosyl hydrolases. Genetics 2007, 177, 959–970. [Google Scholar]
- Hollak, CE; van, WS; van Oers, MH; Aerts, JM. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J Clin Invest 1994, 93, 1288–1292. [Google Scholar]
- Bussink, AP; van Eijk, M; Renkema, GH; Aerts, JM; Boot, RG; Kwang, WJ. The biology of the gaucher cell: the cradle of human chitinases. In International Review of Cytology A Survey of Cell Biology; Academic Press: San Diego, CA, USA, 2006; Volume 252, pp. 71–128. [Google Scholar]
- Barone, R; Simpore, J; Malaguarnera, L; Pignatelli, S; Musumeci, S. Plasma chitotriosidase activity in acute Plasmodium falciparum malaria. Clin Chim Acta 2003, 331, 79–85. [Google Scholar]
- Labadaridis, I; Dimitriou, E; Theodorakis, M; Kafalidis, G; Velegraki, A; Michelakakis, H. Chitotriosidase in neonates with fungal and bacterial infections. Arch Dis Child-Fetal Neonatal Ed 2005, 90, F531–F532. [Google Scholar]
- Bargagli, E; Margollicci, M; Perrone, A; Luddi, A; Perari, MG; Bianchi, N; Refini, RM; Grosso, S; Volterrani, L; Rottoli, P. Chitotriosidase analysis in bronchoalveolar lavage of patients with sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2007, 24, 59–64. [Google Scholar]
- Brunner, J; Scholl-B, S; Prelog, M; Zimmerhackl, LB. Chitotriosidase as a marker of disease activity in sarcoidosis. Rheumatol Int 2007, 27, 1185–1186. [Google Scholar]
- Artieda, M; Cenarro, A; Ganan, A; Lukic, A; Moreno, E; Puzo, J; Pocovi, M; Civeira, F. Serum chitotriosidase activity, a marker of activated macrophages, predicts new cardiovascular events independently of C-reactive protein. Cardiology 2007, 108, 297–306. [Google Scholar]
- Karadag, B; Kucur, M; Isman, FK; Hacibekiroglu, M; Vural, VA. Serum chitotriosidase activity in patients with coronary artery disease. Circ J 2008, 72, 71–75. [Google Scholar]
- Kucur, M; Isman, FK; Balci, C; Onal, B; Hacibekiroglu, M; Ozkan, F; Ozkan, A. Serum YKL-40 levels and chitotriosidase activity as potential biomarkers in primary prostate cancer and benign prostatic hyperplasia. Urol Oncol 2008, 26, 47–52. [Google Scholar]
- Malaguarnera, L; Rosa, MD; Zambito, AM; dell'Ombra, N; Marco, RD; Malaguarnera, M. Potential role of chitotriosidase gene in nonalcoholic fatty liver disease evolution. Am J Gastroent 2006, 101, 2060–2069. [Google Scholar]
- Sotgiu, S; Barone, R; Arru, G; Fois, ML; Pugliatti, M; Sanna, A; Rosati, G; Musumeci, S. Intrathecal chitotriosidase and the outcome of multiple sclerosis. Mult Scler 2006, 12, 551–557. [Google Scholar]
- Wajner, A; Michelin, K; Burin, MG; Pires, RF; Pereira, ML; Giugliani, R; Coelho, JC. Biochemical characterization of chitotriosidase enzyme: comparison between normal individuals and patients with Gaucher and with Niemann-Pick diseases. Clin Biochem 2004, 37, 893–897. [Google Scholar]
- Boot, RG; Blommaart, EFC; Swart, E; Ghauharali-van der Vlugt, K; Bijl, N; Moe, C; Place, A; Aerts, JMFG. Identification of a novel acidic mammalian chitinase distinct from chitotriosidase. J Biol Chem 2001, 276, 6770–6778. [Google Scholar]
- Musumeci, M; Bellin, M; Maltese, A; Aragona, P; Bucolo, C; Musumeci, S. Chitinase levels in the tears of subjects with ocular allergies. Cornea 2008, 27, 168–173. [Google Scholar]
- Ramanathan, M, Jr; Lee, WK; Lane, AP. Increased expression of acidic mammalian chitinase in chronic rhinosinusitis with nasal polyps. Am J Rhinol 2006, 20, 330–335. [Google Scholar]
- Herrera-Estrella, A; Chet, I. Chitinases in biological control. EXS 1999, 87, 171–184. [Google Scholar]
- Palli, SR; Retnakaran, A. Molecular and biochemical aspects of chitin synthesis inhibition. EXS 1999, 87, 85–98. [Google Scholar]
- Shibata, Y; Foster, LA; Bradfield, JF; Myrvik, QN. Oral administration of chitin down-regulates serum IgE levels and lung eosinophilia in the allergic mouse. J Immunol 2000, 164, 1314–1321. [Google Scholar]
- van Eijk, M; van Roomen, CPAA; Renkema, GH; Bussink, AP; Andrews, L; Blommaart, EFC; Sugar, A; Verhoeven, AJ; Boot, RG; Aerts, JMFG. Characterization of human phagocyte-derived chitotriosidase, a component of innate immunity. Int Immun 2005, 17, 1505–1512. [Google Scholar]
- Chupp, GL; Lee, CG; Jarjour, N; Shim, YM; Holm, CT; He, S; Dziura, JD; Reed, J; Coyle, AJ; Kiener, P; Cullen, M; Grandsaigne, M; Dombret, M; Aubier, M; Pretolani, M; Elias, JA. A chitinase-like protein in the lung and circulation of patients with severe asthma. N Engl J Med 2007, 357, 2016–2027. [Google Scholar]
- Rathcke, CN; Vestergaard, H. YKL-40-an emerging biomarker in cardiovascular disease and diabetes. Cardiovasc Diabet 2009, 8. article number 61. [Google Scholar]
- Johansen, JS; Schultz, NA; Jensen, BV. Plasma YKL-40: a potential new cancer biomarker? Future Oncology 2009, 5, 1065–1082. [Google Scholar]
- Kim, PJ; Hong, DG; Park, JY; Cho, YL; Park, IS; Lee, YS. Immunohistochemical expression of YKL-40 in peritoneal endometriosis. Gynecol Endocrinol 2010, 26, 58–62. [Google Scholar]
- Hempen, M; Kopp, HP; Elhenicky, M; Hobaus, C; Brix, JM; Koppensteiner, R; Schernthaner, G; Schernthaner, GH. YKL-40 is elevated in morbidly obese patients and declines after weight loss. Obesity Surg 2009, 19, 1557–1563. [Google Scholar]
- Huang, K; Wu, LD. YKL-40: a potential biomarker for osteoarthritis. J Int Med Res 2009, 37, 18–24. [Google Scholar]
- Schiavon, LL; Narciso-Schiavon, JL; Filho, RJC; Sampaio, JP; Medina-Pestana, JO; Lanzoni, VP; Silva, AEB; Ferraz, MLG. Serum levels of YKL-40 and hyaluronic acid as noninvasive markers of liver fibrosis in haemodialysis patients with chronic hepatitis C virus infection. J Viral Hepatitis 2008, 15, 666–674. [Google Scholar]
- Knorr, T; Obermayr, F; Bartnik, E; Zien, A; Aigner, T. YKL-39 (chitinase 3-like protein 2), but not YKL-40 (chitinase 3-like protein 1), is up regulated in osteoarthritic chondrocytes. Ann Rheum Dis 2003, 62, 995–998. [Google Scholar]
- Steck, E; Breit, S; Breusch, SJ; Axt, M; Richter, W. Enhanced expression of the human chitinase 3-like 2 gene (YKL-39) but not chitinase 3-like 1 gene (YKL-40) in osteoarthritic cartilage. Biochem Biophys Res Commun 2002, 299, 109–115. [Google Scholar]
- Houston, DR; Recklies, AD; Krupa, JC; van Aalten, DMF. Structure and ligand-induced conformational change of the 39-kDa glycoprotein from human articular chondrocytes. J Biol Chem 2003, 278, 30206–30212. [Google Scholar]
- Fukamizo, T; Sasaki, C; Schelp, E; Bortone, K; Robertus, JD. Kinetic properties of chitinase- 1 from the fungal pathogen Coccidioides immitis. Biochemistry 2001, 40, 2448–2454. [Google Scholar]
- Biarnes, X; Ardevol, A; Planas, A; Rovira, C; Laio, A; Parrinello, M. The conformational free energy landscape of beta-D-glucopyranose. Implications for substrate preactivation in beta-glucoside hydrolases. J Am Chem Soc 2007, 129, 10686–10693. [Google Scholar]
- Letzel, M; Synstad, B; Eijsink, VGH; Peter-Katalinic, J; Peter, MG. Libraries of chito-oligosaccharides of mixed acetylation patterns and their interactions with chitinases. Adv Chitin Sci 2000, 4, 545–552. [Google Scholar]
- Cederkvist, F; Zamfir, AD; Bahrke, S; Eijsink, VGH; S⊘rlie, M; Peter-Katalinic, J; Peter, MG. Identification of a high-affinity-binding oligosaccharide by (+) nanoelectrospray quadrupole time-of-flight tandem mass spectrometry of a noncovalent enzyme-ligand complex. Angewandte Chemie-Int Ed 2006, 45, 2429–2434. [Google Scholar]
- Cederkvist, FH; Parmer, MP; Vårum, KM; Eijsink, VGH; S⊘rlie, M. Inhibition of a family 18 chitinase by chitooligosaccharides. Carbohydr Polym 2008, 74, 41–49. [Google Scholar]
- Schindler, M; Assaf, Y; Sharon, N; Chipman, DM. Mechanism of lysozyme catalysis - role of ground-state strain in subsite D in hen eggwhite and human lysozymes. Biochemistry 1977, 16, 423–431. [Google Scholar]
- Amano, K; Ito, E. The action of lysozyme on partially deacetylated chitin. Eur J Biochem 1978, 85, 97–104. [Google Scholar]
- Vårum, KM; Holme, HK; Izume, M; Stokke, BT; Smidsr⊘d, O. Determination of enzymatic hydrolysis specificity of partially N-acetylated chitosans. Biochim Biophys Acta Gen Subj 1996, 1291, 5–15. [Google Scholar]
- Blair, DE; Hekmat, O; Schuttelkopf, AW; Shrestha, B; Tokuyasu, K; Withers, SG; van Aalten, DM. Structure and mechanism of chitin deacetylase from the fungal pathogen Colletotrichum lindemuthianum. Biochemistry 2006, 45, 9416–9426. [Google Scholar]
- Hekmat, O; Tokuyasu, K; Withers, SG. Subsite structure of the endo-type chitin deacetylase from a deuteromycete, Colletotrichum lindemuthianum: an investigation using steady-state kinetic analysis and MS. Biochem J 2003, 374, 369–380. [Google Scholar]
- Tokuyasu, K; Mitsutomi, M; Yamaguchi, I; Hayashi, K; Mori, Y. Recognition of chitooligosaccharides and their N-acetyl groups by putative subsites of chitin deacetylase from a deuteromycete, Colletotrichum lindemuthianum. Biochemistry 2000, 39, 8837–8843. [Google Scholar]
- Tokuyasu, K; Ono, H; Mitsutomi, M; Hayashi, K; Mori, Y. Synthesis of a chitosan tetramer derivative, beta-D-GlcNAc-(1->4)-beta-D-GlcNAc-(1->4)-beta-D-GlcNAc-(1->4)-D-GlcN through a partial N-acetylation reaction by chitin deacetylase. Carbohydr Res 2000, 325, 211–215. [Google Scholar]
- Tsigos, I; Martinou, A; Kafetzopoulos, D; Bouriotis, V. Chitin deacetylases: new, versatile tools in biotechnology. Trends Biotechnol 2000, 18, 305–312. [Google Scholar]
- Sikorski, P; Stokke, BT; S⊘rbotten, A; Vårum, KM; Horn, SJ; Eijsink, VG. Development and application of a model for chitosan hydrolysis by a family 18 chitinase. Biopolymers 2005, 77, 273–285. [Google Scholar]
- Heggset, EB; Hoell, IA; Kristoffersen, M; Eijsink, VG; Vårum, KM. Degradation of chitosans with chitinase G from Streptomyces coelicolor A3(2): production of chito-oligosaccharides and insight into subsite specificities. Biomacromolecules 2009, 10, 892–899. [Google Scholar]
- Mitsutomi, M; Hata, T; Kuwahara, T. Purification and Characterization of Novel Chitinases from Streptomyces griseus Hut 6037. J Ferment Bioeng 1995, 80, 153–158. [Google Scholar]
- Sasaki, C; Vårum, KM; Itoh, Y; Tamoi, M; Fukamizo, T. Rice chitinases: sugar recognition specificities of the individual subsites. Glycobiology 2006, 16, 1242–1250. [Google Scholar]
- Sikorski, P; S⊘rbotten, A; Horn, SJ; Eijsink, VG; Vårum, KM. Serratia marcescens chitinases with tunnel-shaped substrate-binding grooves show endo activity and different degrees of processivity during enzymatic hydrolysis of chitosan. Biochemistry 2006, 45, 9566–9574. [Google Scholar]
- Hult, EL; Katouno, F; Uchiyama, T; Watanabe, T; Sugiyama, J. Molecular directionality in crystalline beta-chitin: hydrolysis by chitinases A and B from Serratia marcescens 2170. Biochemical Journal 2005, 388, 851–856. [Google Scholar]
- Mitsutomi, M; Ueda, M; Arai, M; Ando, A; Watanabe, T. Muzzarelli, RAA, Ed.; Chitin Enzymology; Atec Edizioni: Grottammare, Italy, 1996; pp. 273–284. [Google Scholar]
- Heggset, EB; Dybvik, AI; Hoell, IA; Norberg, AL; Eijsink, VGH; S⊘rlie, M; Vårum, KM. Degradation of Chitosans with a novel family 46 Chitosanase from/Streptomyces coelicolor/A3(2). Manuscript in Preparation.
- Sashiwa, H; Fujishima, S; Yamano, N; Kawasaki, N; Nakayama, A; Muraki, E; Sukwattanasinitt, M; Pichyangkura, R; Aiba, S. Enzymatic production of N-acetyl-D-glucosamine from chitin. Degradation study of N-acetylchitooligosaccharide and the effect of mixing of crude enzymes. Carbohydr Polym 2003, 51, 391–395. [Google Scholar]
- Terbojevich, M; Cosani, A; Muzzarelli, RAA. Molecular parameters of chitosans depolymerized with the aid of papain. Carbohydr Polym 1996, 29, 63–68. [Google Scholar]
- Xie, Y; Wei, Y; Hu, JG. Depolymerization of Chitosan with a Crude Cellulase Preparation from Aspergillus Niger. Appl Biochem Biotechnol 2010, 160, 1074–1083. [Google Scholar]
- Kuyama, H; Nakahara, Y; Nukada, T; Ito, Y; Ogawa, T. Stereocontrolled synthesis of chitosan dodecamer. Carbohydr Res 1993, 243, C1–C7. [Google Scholar]
- Aly, MRE; Ibrahim, E-SI; El Ashry, ESH; Schmidt, RR. Synthesis of chitotetraose and chitohexaose based on dimethylmaleoyl protection. Carbohydr Res 2001, 331, 129–142. [Google Scholar]
- Aly, MRE; Castro-Palomino, JC; Ibrahim, ESI; El-Ashry, ESH; Schmidt, RR. The dimethylmaleoyl group as amino protective group - Application to the synthesis of glucosamine-containing oligosaccharides. Eur J Org Chem 1998, 2305–2316. [Google Scholar]
- Trombotto, S; Ladavière, C; Delolme, F; Domard, A. Chemical preparation and structural characterization of a homogeneous series of chitin/chitosan oligomers. Biomacromolecules 2008, 9, 1731–1738. [Google Scholar]
- Ohmae, M; Fujikawa, S; Ochiai, H; Kobayashi, S. Enzyme-catalyzed synthesis of natural and unnatural polysaccharides. J Polymer Sci Part A- Polymer Chem 2006, 44, 5014–5027. [Google Scholar]
- Jahn, M; Stoll, D; Warren, RA; Szabo, L; Singh, P; Gilbert, HJ; Ducros, VM; Davies, GJ; Withers, SG. Expansion of the glycosynthase repertoire to produce defined manno-oligosaccharides. Chem Commun (Camb) 2003, 1327–1329. [Google Scholar]
- Lopatin, SA; Derbeneva, MS; Kulikov, SN; Varlamov, VP; Shpigun, OA. Fractionation of chitosan by ultrafiltration. J Anal Chem 2009, 64, 648–651. [Google Scholar]
- Haebel, S; Bahrke, S; Peter, MG. Quantitative sequencing of complex mixtures of heterochitooligosaccharides by vMALDI-linear ion trap mass spectrometry. Anal Chem 2007, 79, 5557–5566. [Google Scholar]
- Le Dévédec, F; Bazinet, L; Furtos, A; Venne, K; Brunet, S; Mateescu, MA. Separation of chitosan oligomers by immobilized metal affinity chromatography. J Chromatogr A 2008, 1194, 165–171. [Google Scholar]
- Bahrke, S; Einarsson, JM; Gislason, J; Haebel, S; Letzel, MC; Peter-Katalinic, J; Peter, MG. Sequence analysis of chitooligosaccharides by matrix-assisted laser desorption ionization postsource decay mass spectrometry. Biomacromolecules 2002, 3, 696–704. [Google Scholar]
- Okafo, G; Langridge, J; North, S; Organ, A; West, A; Morris, M; Camilleri, P. High performance liquid chromatographic analysis of complex N-linked glycans derivatized with 2-aminoacridone. Anal Chem 1997, 69, 4985–4993. [Google Scholar]
- Qin, C; Du, Y; Xiao, L; Li, Z; Gao, X. Enzymic preparation of water-soluble chitosan and their antitumor activity. Int J Biol Macromol 2002, 31, 111–117. [Google Scholar]
- Maeda, Y; Kimura, Y. Antitumor effects of various low-molecular-weight chitosans are due to increased natural killer activity of intestinal intraepithelial lymphocytes in sarcoma 180-bearing mice. J Nutr 2004, 134, 945–950. [Google Scholar]
- Harish Prashanth, KV; Tharanathan, RN. Depolymerized products of chitosan as potent inhibitors of tumor-induced angiogenesis. Biochim Biophys Acta 2005, 1722, 22–29. [Google Scholar]
- Xu, QS; Dou, HL; Wei, P; Tan, CY; Yun, XJ; Wu, YH; Bal, XF; Ma, XJ; Du, YG. Chitooligosaccharides induce apoptosis of human hepatocellular carcinoma cells via up-regulation of Bax. Carbohydrate Polymers 2008, 71, 509–514. [Google Scholar]
- Wang, Z; Zheng, L; Yang, S; Niu, R; Chu, E; Lin, X. N-acetylchitooligosaccharide is a potent angiogenic inhibitor both in vivo and in vitro. Biochem Biophys Res Commun 2007, 357, 26–31. [Google Scholar]
- Wu, H; Yao, Z; Bai, X; Du, Y; Lin, B. Anti-angiogenic activities of chitooligosaccharides. Carbohydr Polym 2008, 73, 105–110. [Google Scholar]
- Xiong, C; Wu, H; Wei, P; Pan, M; Tuo, Y; Kusakabe, I; Du, Y. Potent angiogenic inhibition effects of deacetylated chitohexaose separated from chitooligosaccharides and its mechanism of action in vitro. Carbohydr Res 2009, 344, 1975–1983. [Google Scholar]
- Muzzarelli, RAA. Chitins and Chitosans as Immunoadjuvants and Non-Allergenic Drug Carriers. Mar Drugs 2008, 8, 292–312. [Google Scholar]
- Jung, WK; Moon, SH; Kim, SK. Effect of chitooligosaccharides on calcium bioavailability and bone strength in ovariectomized rats. Life Sci 2006, 78, 970–976. [Google Scholar]
- Kim, S-K; Park, P-J; Jung, W-K; Byun, H-G; Mendis, E; Cho, Y-I. Inhibitory activity of phosphorylated chitooligosaccharides on the formation of calcium phosphate. Carbohydr Polym 2005, 60, 483–487. [Google Scholar]
- Jayakumar, R; Chennazhi, KP; Muzzarelli, RAA; Tamura, H; Nair, SV; Selvamurugan, N. Chitosan conjugated DNA nanoparticles in gene therapy. Carbohydr Polym 2010, 79, 1–8. [Google Scholar]
- Mumper, RJ; Wang, J; Claspell, JM; Rolland, AP. Novel polymeric condensing carriers for gene delivery. Proc Int Symp Control Release Bioact Mater 1995, 22, 178–179. [Google Scholar]
- Köping-Hoggard, M; Tubulekas, I; Guan, H; Edwards, K; Nilsson, M; Vårum, KM; Artursson, P. Chitosan as a nonviral gene delivery system. Structure-property relationships and characteristics compared with polyethylenimine in vitro and after lung administration in vivo. Gene Ther 2001, 8, 1108–1121. [Google Scholar]
- MacLaughlin, FC; Mumper, RJ; Wang, J; Tagliaferri, JM; Gill, I; Hinchcliffe, M; Rolland, AP. Chitosan and depolymerized chitosan oligomers as condensing carriers for in vivo plasmid delivery. J Control Release 1998, 56, 259–272. [Google Scholar]
- Roy, K; Mao, HQ; Huang, SK; Leong, KW. Oral gene delivery with chitosan-DNA nanoparticles generates immunologic protection in a murine model of peanut allergy. Nat Med 1999, 5, 387–391. [Google Scholar]
- Strand, SP; Danielsen, S; Christensen, BE; Vårum, KM. Influence of chitosan structure on the formation and stability of DNA-chitosan polyelectrolyte complexes. Biomacromolecules 2005, 6, 3357–3366. [Google Scholar]
- Strand, SP; Lelu, S; Reitan, NK; de Lange Davies, C; Artursson, P; Vårum, KM. Molecular design of chitosan gene delivery systems with an optimized balance between polyplex stability and polyplex unpacking. Biomaterials 2010, 31, 975–987. [Google Scholar]
- Sharon, N; Ofek, I. Safe as mother's milk: Carbohydrates as future anti-adhesion drugs for bacterial diseases. Glycoconjugate J 2000, 17, 659–664. [Google Scholar]
- Zopf, D; Roth, S. Oligosaccharide anti-infective agents. The Lancet 1996, 347, 1017–1021. [Google Scholar]
- WHO. World Malaria Report 2008. Available online: http://apps.who.int/malaria/wmr2008/malaria2008.pdf (accessed on 27 April 2010).
- Dessens, JT; Mendoza, J; Claudianos, C; Vinetz, JM; Khater, E; Hassard, S; Ranawaka, GR; Sinden, RE. Knockout of the rodent malaria parasite chitinase PbCHT1 reduces infectivity to mosquitoes. Infect Immun 2001, 69, 4041–4047. [Google Scholar]
- Huber, M; Cabib, E; Miller, LH. Malaria parasite chitinase and penetration of the mosquito peritrophic membrane. Proc Natl Acad Sci USA 1991, 88, 2807–2810. [Google Scholar]
- Langer, RC; Vinetz, JM. Plasmodium ookinete-secreted chitinase and parasite penetration of the mosquito peritrophic matrix. Trends Parasitol 2001, 17, 269–272. [Google Scholar]
- Li, FW; Templeton, TJ; Popov, V; Comer, JE; Tsuboi, T; Torii, M; Vinetz, JM. Plasmodium ookinete-secreted proteins secreted through a common micronemal pathway are targets of blocking malaria transmission. J Biol Chem 2004, 279, 26635–26644. [Google Scholar]
- Tsai, YL; Hayward, RE; Langer, RC; Fidock, DA; Vinetz, JM. Disruption of Plasmodium falciparum chitinase markedly impairs parasite invasion of mosquito midgut. Infect Immun 2001, 69, 4048–4054. [Google Scholar]
- Takeo, S; Hisamori, D; Matsuda, S; Vinetz, J; Sattabongkot, J; Tsuboi, T. Enzymatic characterization of the Plasmodium vivax chitinase, a potential malaria transmission-blocking target. Parasitology Int 2009, 58, 243–248. [Google Scholar]
- Vinetz, JM; Dave, SK; Specht, CA; Brameld, KA; Xu, B; Hayward, R; Fidock, DA. The chitinase PfCHT1 from the human malaria parasite Plasmodium falciparum lacks proenzyme and chitin-binding domains and displays unique substrate preferences. Proc Natl Acad Sci USA 1999, 96, 14061–14066. [Google Scholar]
- Vinetz, JM; Valenzuela, JG; Specht, CA; Aravind, L; Langer, RC; Ribeiro, JM; Kaslow, DC. Chitinases of the avian malaria parasite Plasmodium gallinaceum, a class of enzymes necessary for parasite invasion of the mosquito midgut. J Biol Chem 2000, 275, 10331–10341. [Google Scholar]
- Langer, RC; Li, FW; Popov, V; Kurosky, A; Vinetz, JM. Monoclonal antibody against the Plasmodium falciparum chitinase, PfCHT1, recognizes a malaria transmission-blocking epitope in Plasmodium gallinaceum ookinetes unrelated to the chitinase PgCHT1. Infect Immun 2002, 70, 1581–1590. [Google Scholar]
- Chou, T-C; Fu, E; Wu, C-J; Yeh, J-H. Chitosan enhances platelet adhesion and aggregation. Biochem Biophys Res Commun 2003, 302, 480–483. [Google Scholar]
- Minagawa, T; Okamura, Y; Shigemasa, Y; Minami, S; Okamoto, Y. Effects of molecular weight and deacetylation degree of chitin/chitosan on wound healing. Carbohydr Polym 2007, 67, 640–644. [Google Scholar]
- Muzzarelli, RAA. Chitins and chitosans for the repair of wounded skin, nerve, cartilage and bone. Carbohydr Polym 2009, 76, 167–182. [Google Scholar]
- Shi, C; Zhu, Y; Ran, X; Wang, M; Su, Y; Cheng, T. Therapeutic potential of chitosan and its derivatives in regenerative medicine. J Surgical Res 2006, 133, 185–192. [Google Scholar]
- Yang, J; Tian, F; Wang, Z; Wang, Q; Zeng, YJ; Chen, SQ. Effect of chitosan molecular weight and deacetylation degree on hemostasis. J Biomed Mater Res, Part B 2008, 84B, 131–137. [Google Scholar]
- Mori, T; Okumura, M; Matsuura, M; Ueno, K; Tokura, S; Okamoto, Y; Minami, S; Fujinaga, T. Effects of chitin and its derivatives on the proliferation and cytokine production of fibroblasts in vitro. Biomaterials 1997, 18, 947–951. [Google Scholar]
- Usami, Y; Minami, S; Okamoto, Y; Matsuhashi, A; Shigemasa, Y. Influence of chain length of N-acetyl-D-glucosamine and D-glucosamine residues on direct and complement-mediated chemotactic activities for canine polymorphonuclear cells. Carbohydr Polym 1997, 32, 115–122. [Google Scholar]
- Usami, Y; Okamoto, Y; Takayama, T; Shigemasa, Y; Minami, S. Effect of N-acetyl-d-glucosamine and D-glucosamine oligomers on canine polymorphonuclear cells in vitro. Carbohydr Polym 1998, 36, 137–141. [Google Scholar]
- Okamoto, Y; Yano, R; Miyatake, K; Tomohiro, I; Shigemasa, Y; Minami, S. Effects of chitin and chitosan on blood coagulation. Carbohydr Polym 2003, 53, 337–342. [Google Scholar]
- Clark, RAF; Denver, MD. Cutaneous tissue repair: Basic biological considerations. J Am Acad Dermatol 1985, 13, 701–725. [Google Scholar]
- Allan, CR; Hadwiger, LA. The fungicidal effect of chitosan on fungi of varying cell-wall composition. Exp Mycol 1979, 3, 285–287. [Google Scholar]
- Bautista-Baños, S; Hernández-Lauzardo, AN; Velázquez-del Valle, MG; Hernández-López, M; Ait Barka, E; Bosquez-Molina, E; Wilson, CL. Chitosan as a potential natural compound to control pre and postharvest diseases of horticultural commodities. Crop Protection 2006, 25, 108–118. [Google Scholar]
- Kendra, DF; Hadwiger, LA. Characterization of the smallest chitosan oligomer that is maximally antifungal to Fusarium solani and elicit pisatin formation in Pisum sativum. Exp Mycol 1984, 8, 276–281. [Google Scholar]
- Tikhonov, VE; Stepnova, EA; Babak, VG; Yamskov, IA; Palma-Guerrero, J; Jansson, H-B; Lopez-Llorca, LV; Salinas, J; Gerasimenko, DV; Avdienko, ID; Varlamov, VP. Bactericidal and antifungal activities of a low molecular weight chitosan and its N-/2(3)-(dodec- 2-enyl)succinoyl/-derivatives. Carbohydr Polym 2006, 64, 66–72. [Google Scholar]
- Eweis, M; Elkholy, SS; Elsabee, MZ. Antifungal efficacy of chitosan and its thiourea derivatives upon the growth of some sugar-beet pathogens. Int J Biol Macromol 2006, 38, 1–8. [Google Scholar]
- Palma-Guerrero, J; Huang, IC; Jansson, HB; Salinas, J; Lopez-Llorca, LV; Read, ND. Chitosan permeabilizes the plasma membrane and kills cells of Neurospora crassa in an energy dependent manner. Fungal Genet Biol 2009, 46, 585–594. [Google Scholar]
- Park, Y; Kim, MH; Park, SC; Cheong, H; Jang, MK; Nah, JW; Hahm, KS. Investigation of the antifungal activity and mechanism of action of LMWS-chitosan. J Microbiol Biotechnol 2008, 18, 1729–1734. [Google Scholar]
- Palma-Guerrero, J; Lopez-Jimenez, JA; Pérez-Berná, AJ; Huang, I-C; Jansson, H-B; Salinas, J; Villalaín, J; Read, ND; Lopez-Llorca, LV. Membrane fluidity determines sensitivity of filamentous fungi to chitosan. Mol Microbiol 2010. [Epub ahead of print]. [Google Scholar]
- Olland, AM; Strand, J; Presman, E; Czerwinski, R; Joseph-McCarthy, D; Krykbaev, R; Schlingmann, G; Chopra, R; Lin, L; Fleming, M; Kriz, R; Stahl, M; Somers, W; Fitz, L; Mosyak, L. Triad of polar residues implicated in pH specificity of acidic mammalian chitinase. Protein Sci 2009, 18, 569–578. [Google Scholar]
- Fusetti, F; Pijning, T; Kalk, KH; Bos, E; Dijkstra, BW. Crystal structure and carbohydrate-binding properties of the human cartilage glycoprotein-39. J Biol Chem 2003, 278, 37753–37760. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
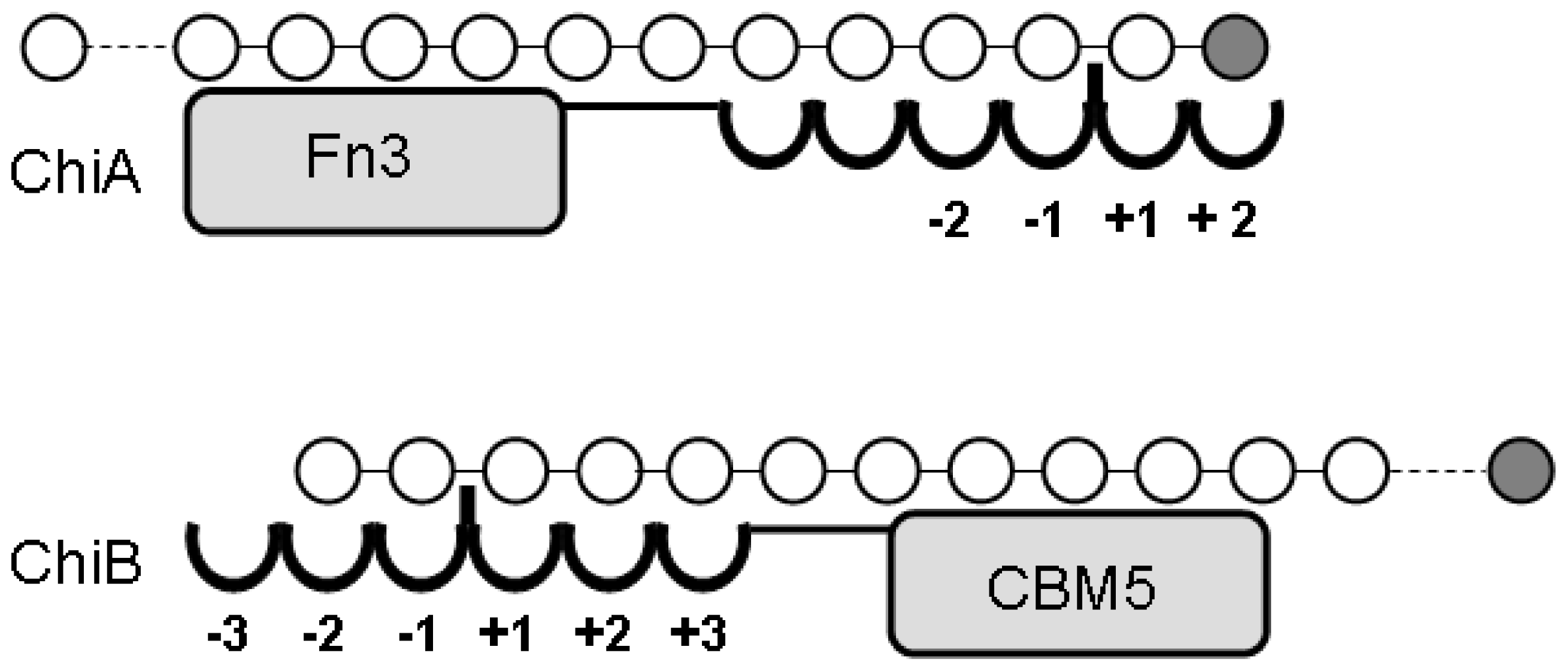
| Enzyme | GH fam | Extra CBM1 | Mechanism | Endo/Exo | Processivity | Subsite specificity | ||
|---|---|---|---|---|---|---|---|---|
| −2 | −1 | +1 | ||||||
| Chitinases: | ||||||||
| ChiA | 18 | Yes (1) | Retaining | Endo/exo2 | Yes | A/D | A | A/D |
| ChiB | 18 | Yes (1) | Retaining | Endo/exo2 | Yes | A/D | A | A/D |
| ChiC | 18 | Yes (2) | Retaining | Endo | No | A/D | A | A/D |
| ChiG | 19 | No | Inverting | Endo | No | A | A/D | A |
| Chitosanase: | ||||||||
| Csn88 | 46 | No | Inverting | Endo | No | D/A | D/A | D/A |
| Enzyme | α | Dimer | Trimer | Tetramer |
|---|---|---|---|---|
| ChiA | 0.15 | 81% AA 19% DA | 81% DAA 19% ADA | 100% -AA |
| 0.35 | 64% AA 36% DA | 51% DAA 28% ADA 21% DDA | 56% -AA 44% -DA | |
| ChiB | 0.11 | 86% AA 14% DA | 71% DDA 29% AAA | 100% -AA |
| 0.38 | 66% AA 34% DA | 95% DAA 3% DDA 2% ADA | 75% -AA 25% -DA | |
| ChiC | 0.20 | 100% AA | 66% DAA 34% AAA | 100% -AA |
| 0.38 | 81% AA 19% DA | 100% DAA | 100% -AA |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Aam, B.B.; Heggset, E.B.; Norberg, A.L.; Sørlie, M.; Vårum, K.M.; Eijsink, V.G.H. Production of Chitooligosaccharides and Their Potential Applications in Medicine. Mar. Drugs 2010, 8, 1482-1517. https://doi.org/10.3390/md8051482
Aam BB, Heggset EB, Norberg AL, Sørlie M, Vårum KM, Eijsink VGH. Production of Chitooligosaccharides and Their Potential Applications in Medicine. Marine Drugs. 2010; 8(5):1482-1517. https://doi.org/10.3390/md8051482
Chicago/Turabian StyleAam, Berit B., Ellinor B. Heggset, Anne Line Norberg, Morten Sørlie, Kjell M. Vårum, and Vincent G. H. Eijsink. 2010. "Production of Chitooligosaccharides and Their Potential Applications in Medicine" Marine Drugs 8, no. 5: 1482-1517. https://doi.org/10.3390/md8051482