Recent N-Atom Containing Compounds from Indo-Pacific Invertebrates

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
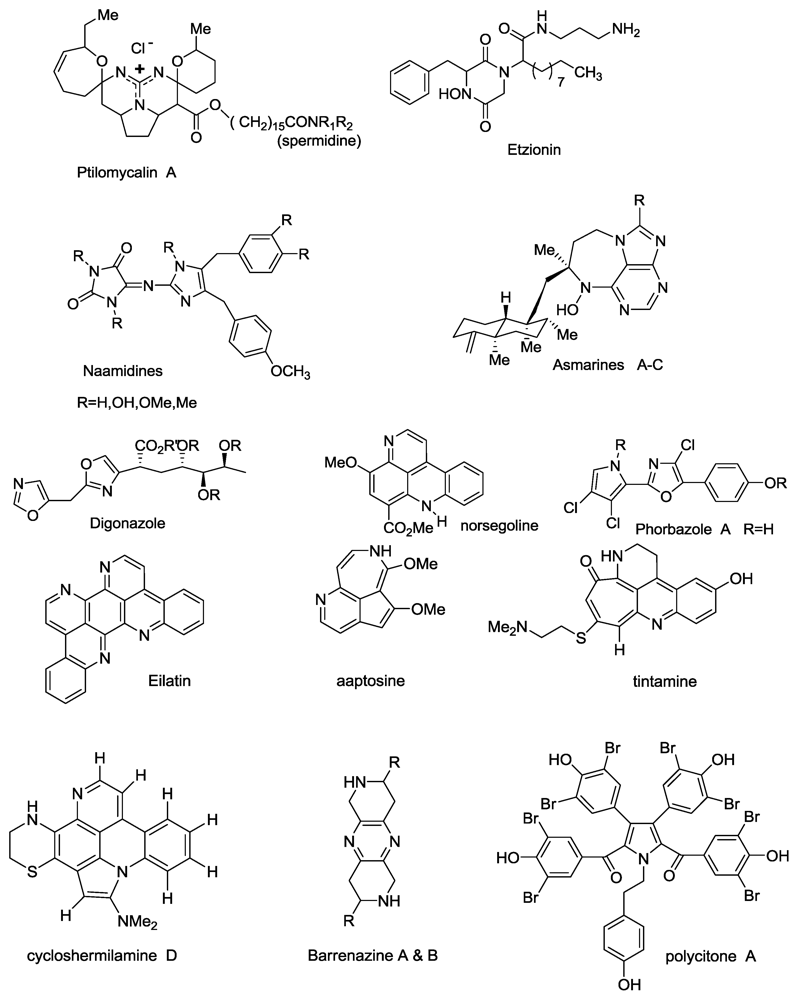
:1. Introduction
2. Results and Discussion
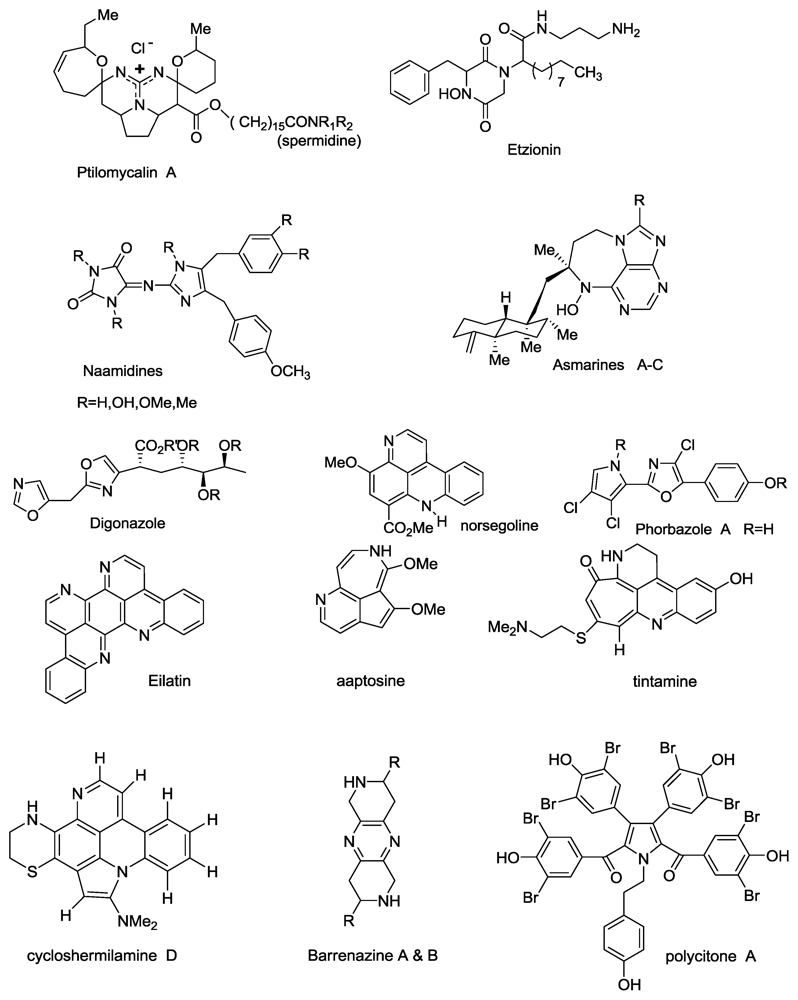
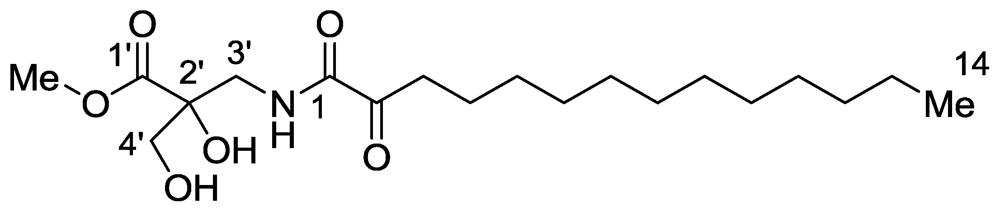
2.1. Salaramides A and B
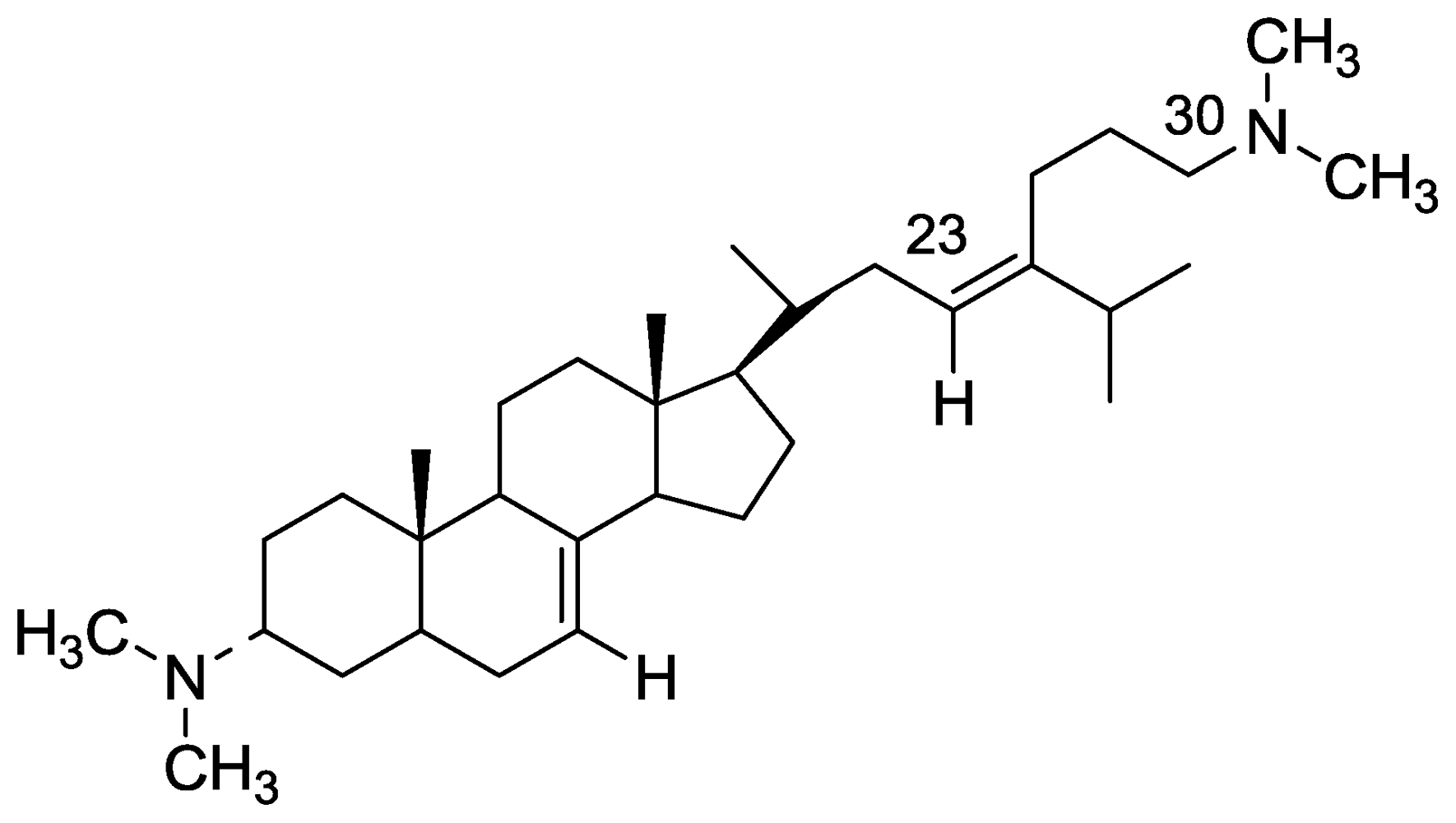
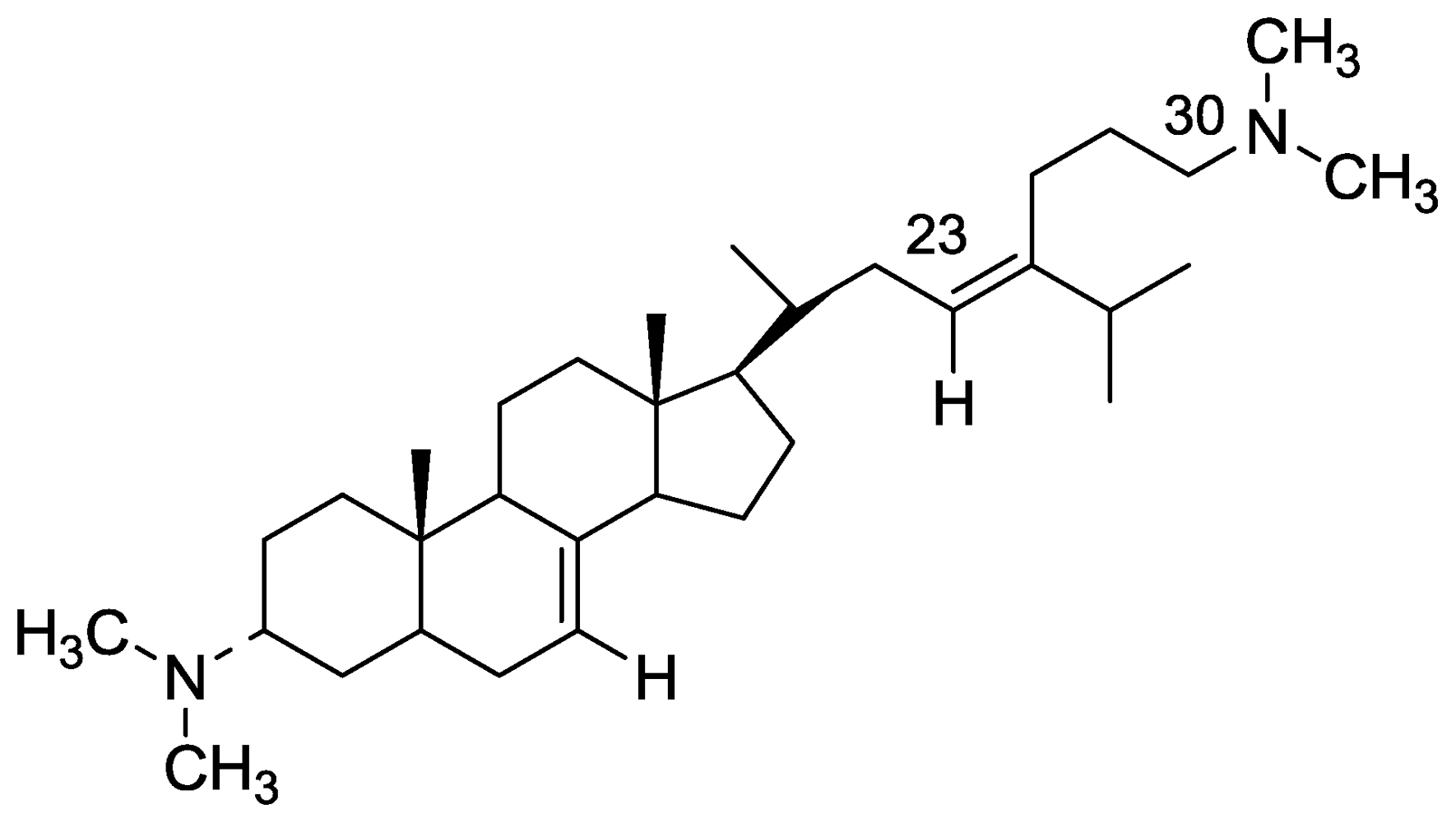
2.2. Plakinamine L
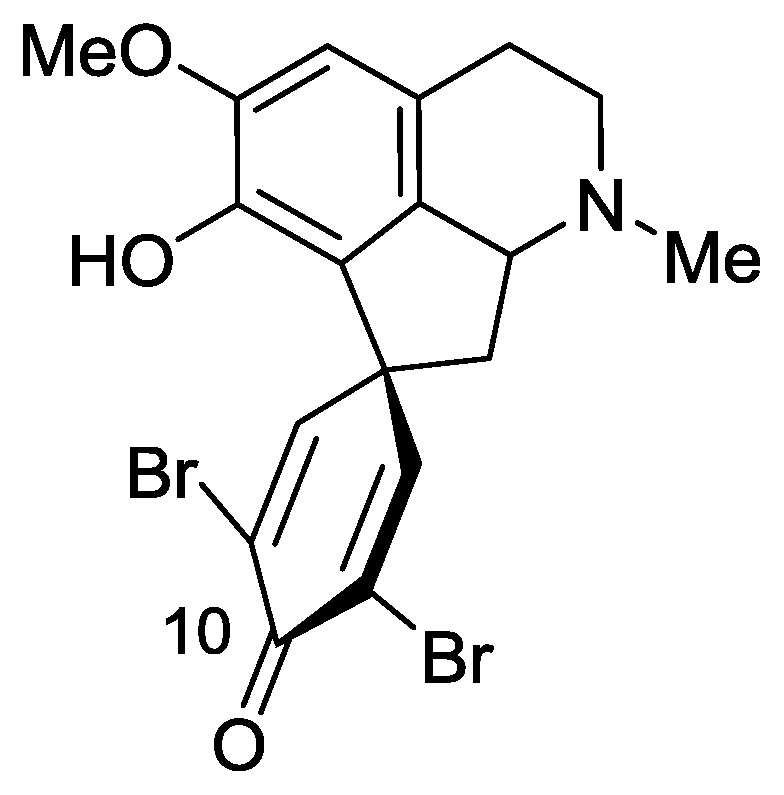
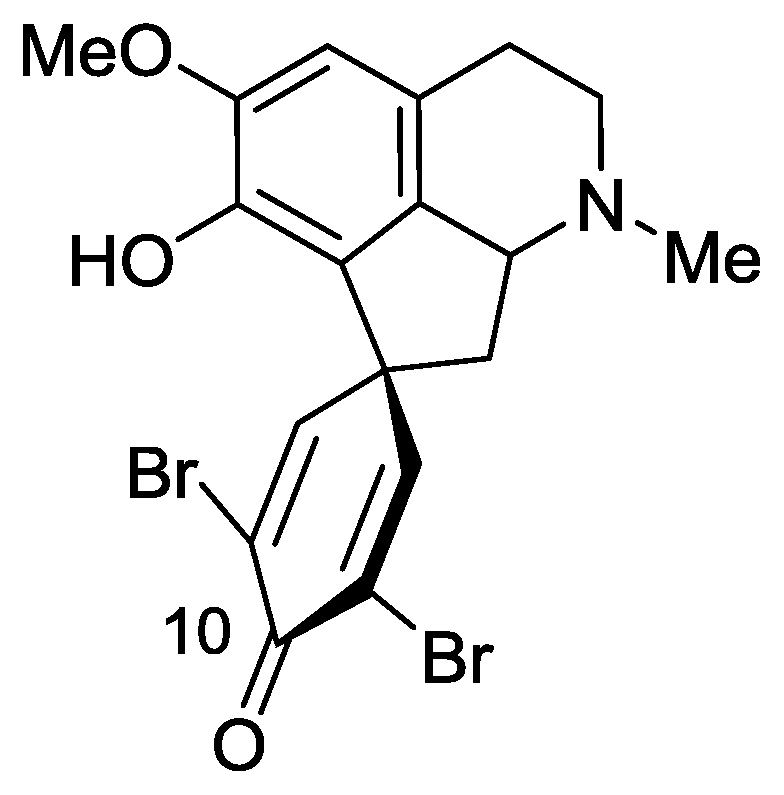
2.3. Saldedines A and B
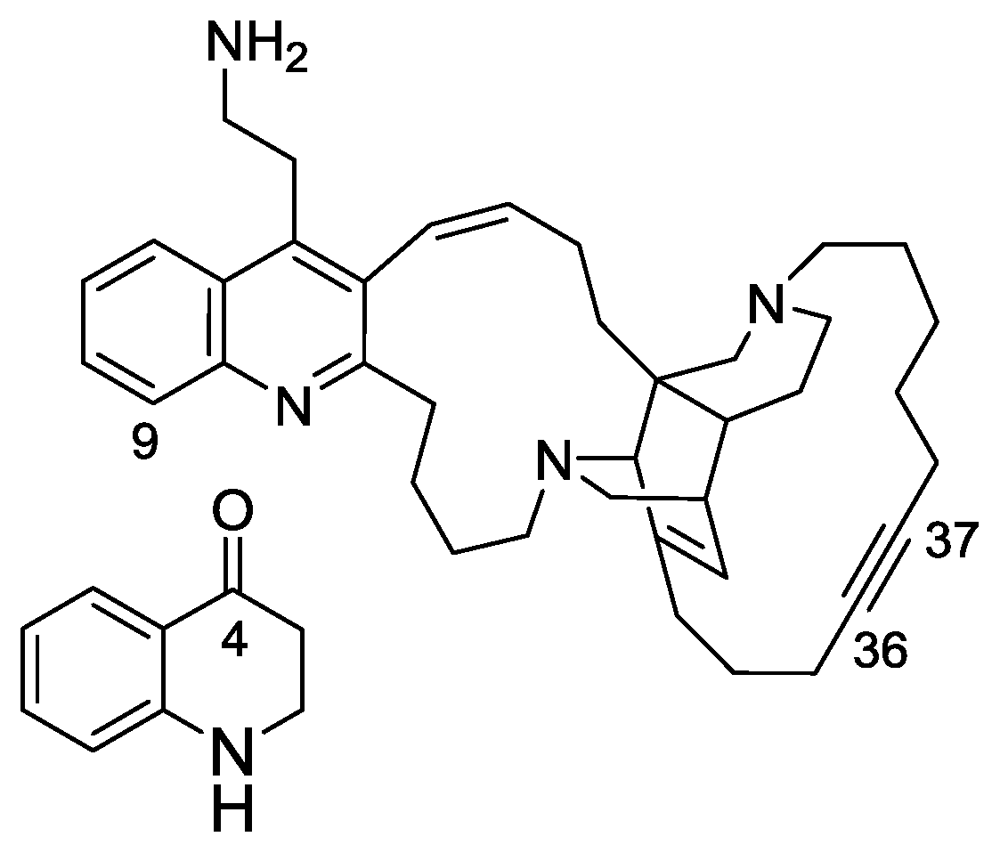
2.4. Njaoamines G and H
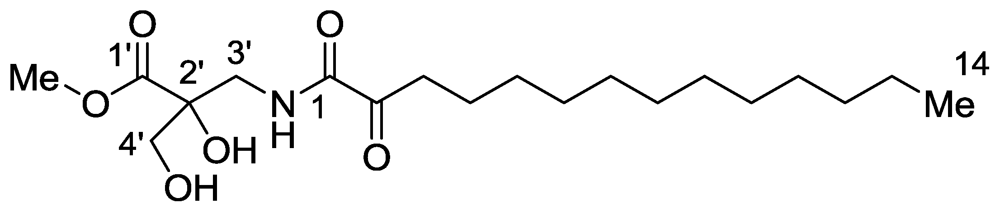
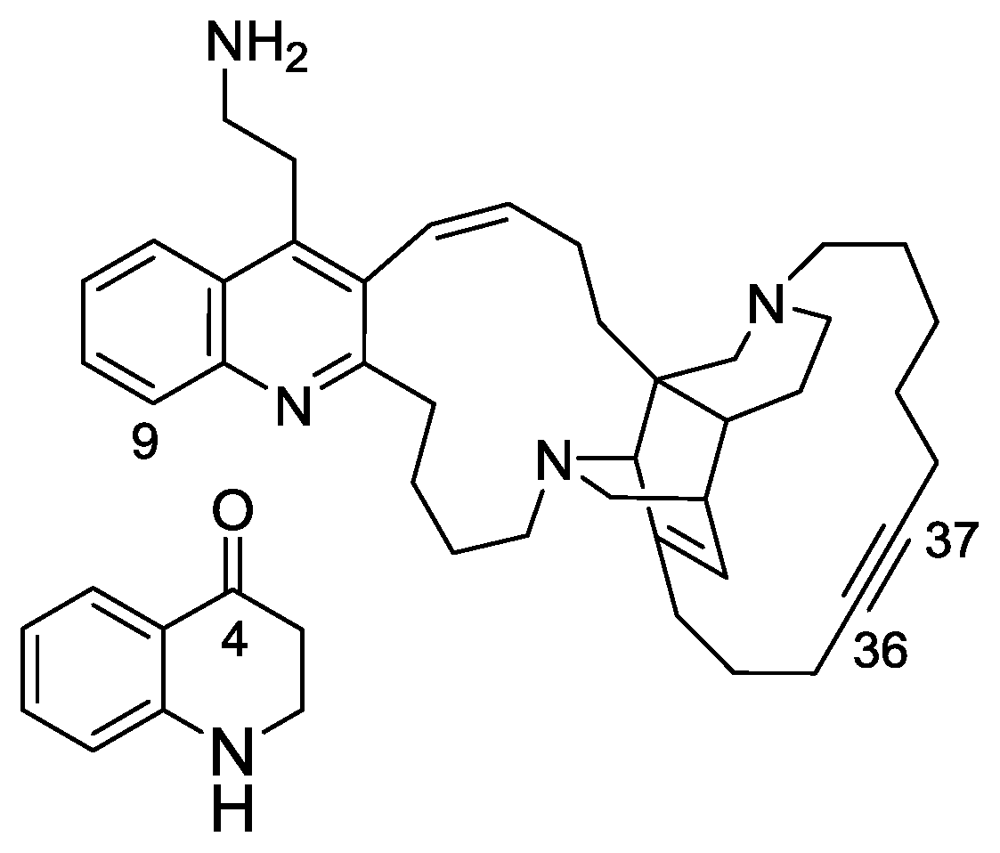
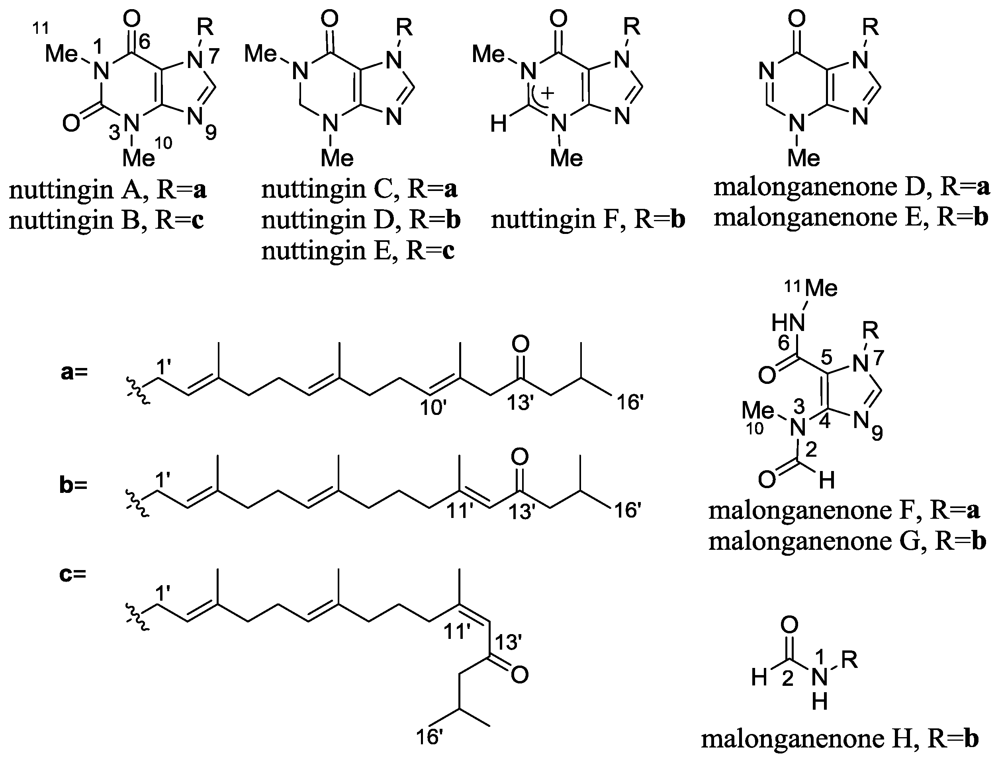
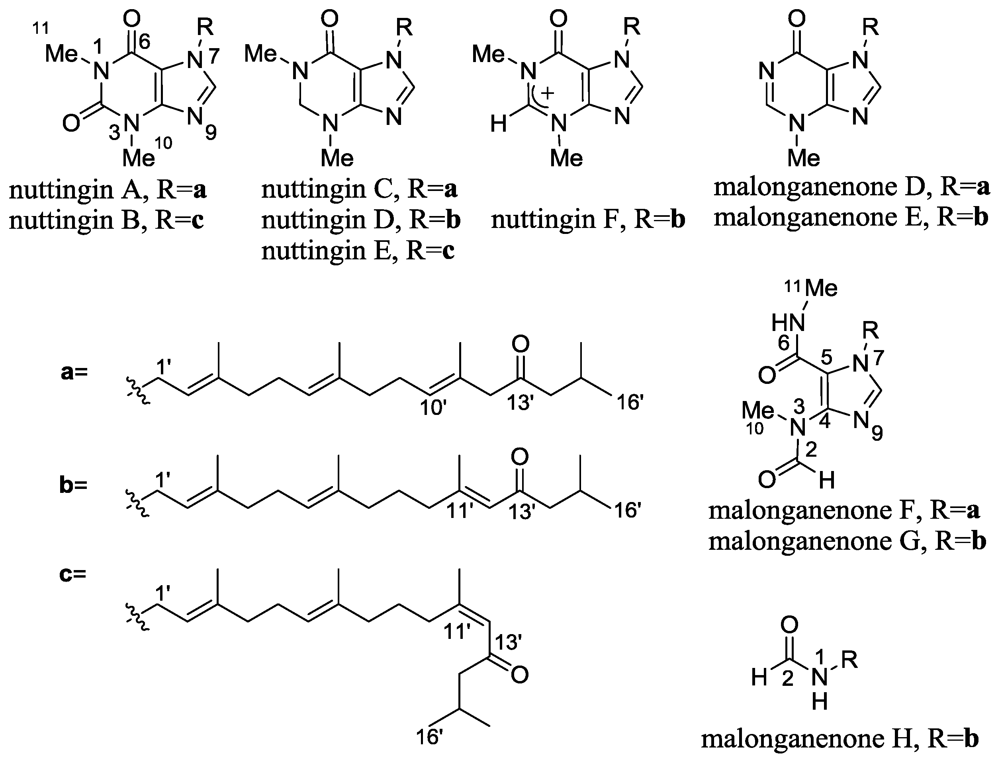
2.5. Nuttingins A–F and Malonganenones D–H
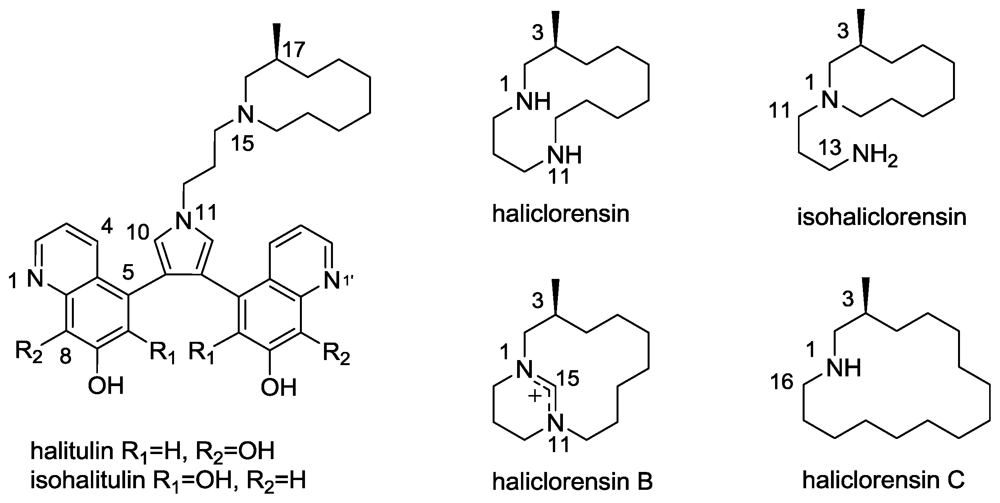
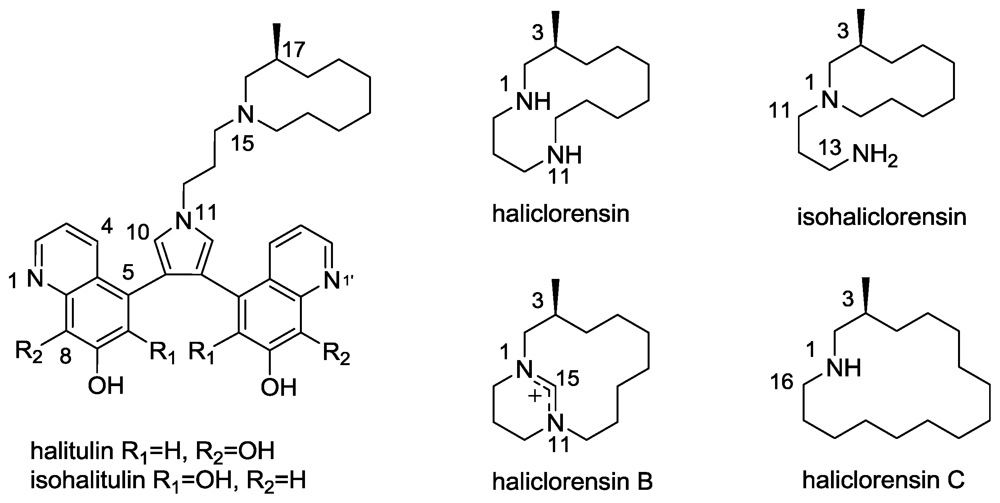
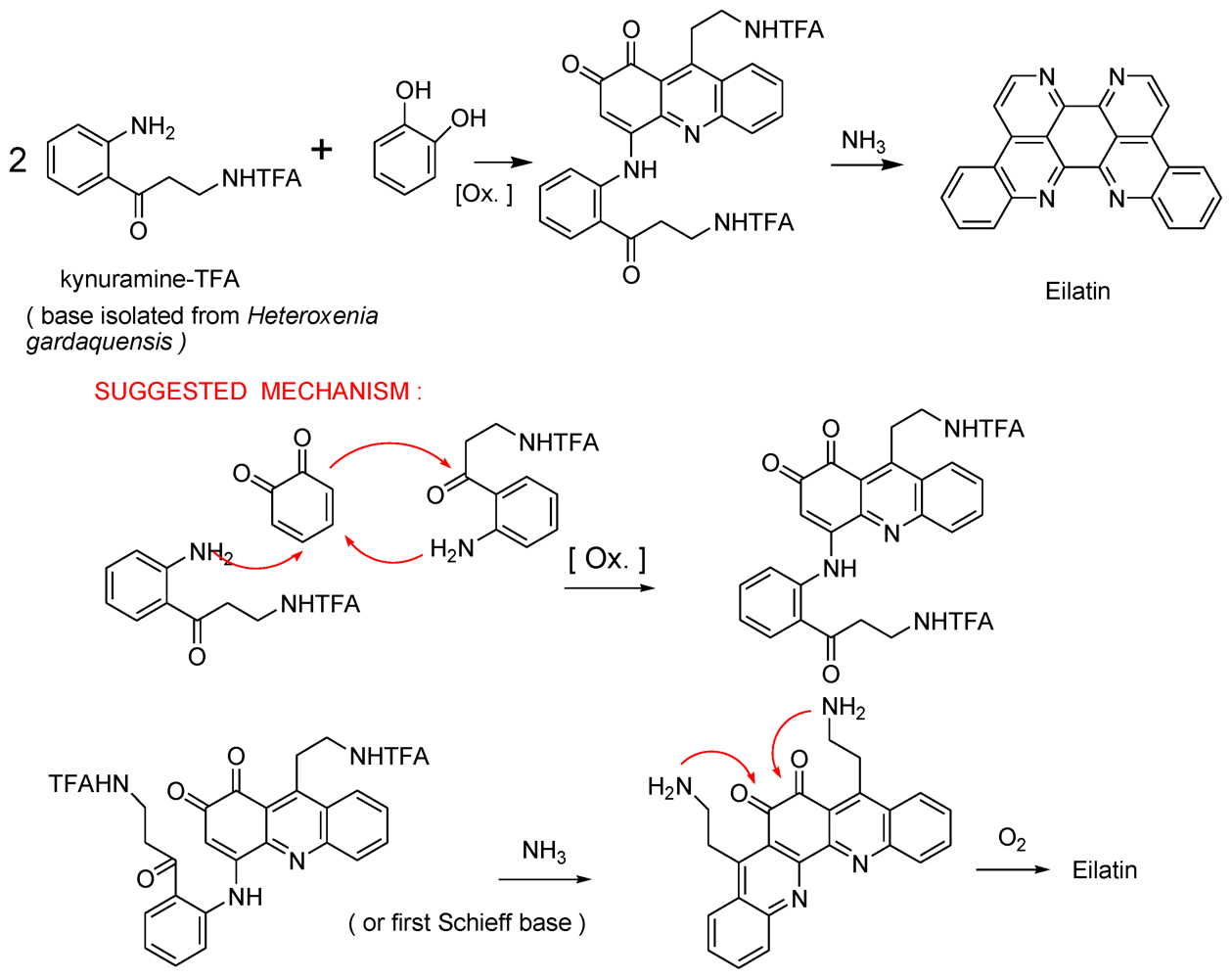
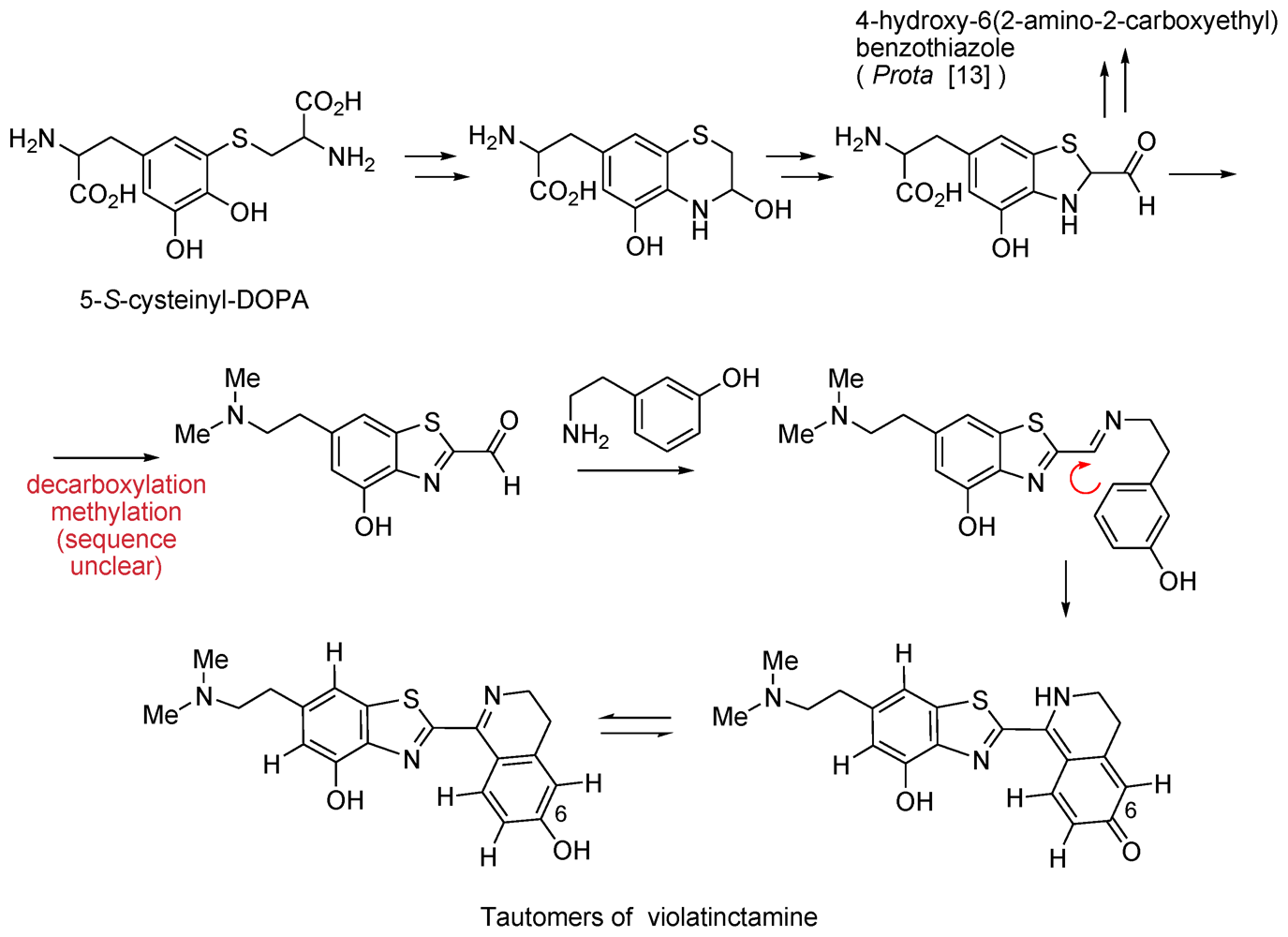
2.6. Isohalitulin and Haliclorensins B and C
3. Introduction to the Fascaplysinopsis sp. Metabolites
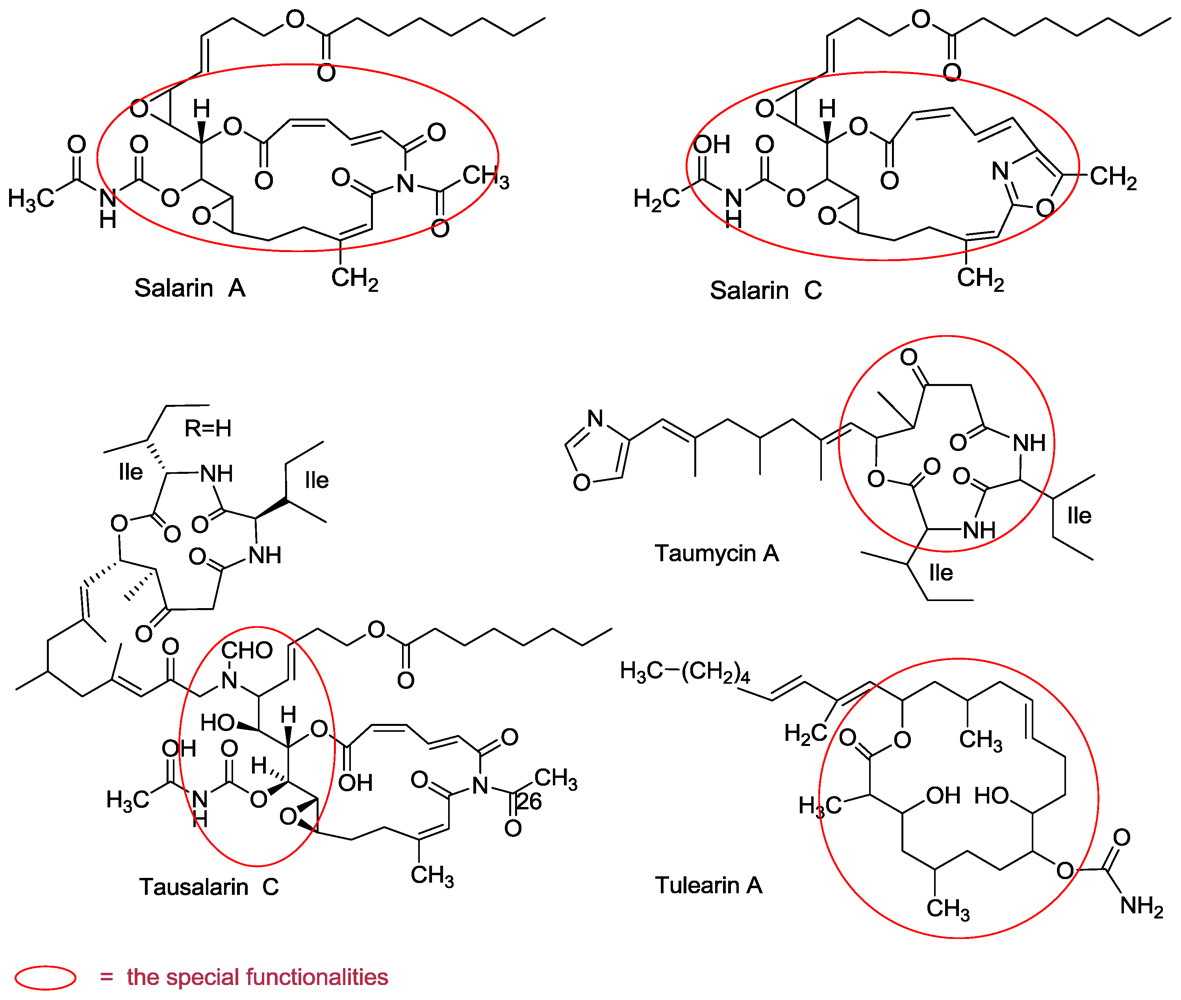
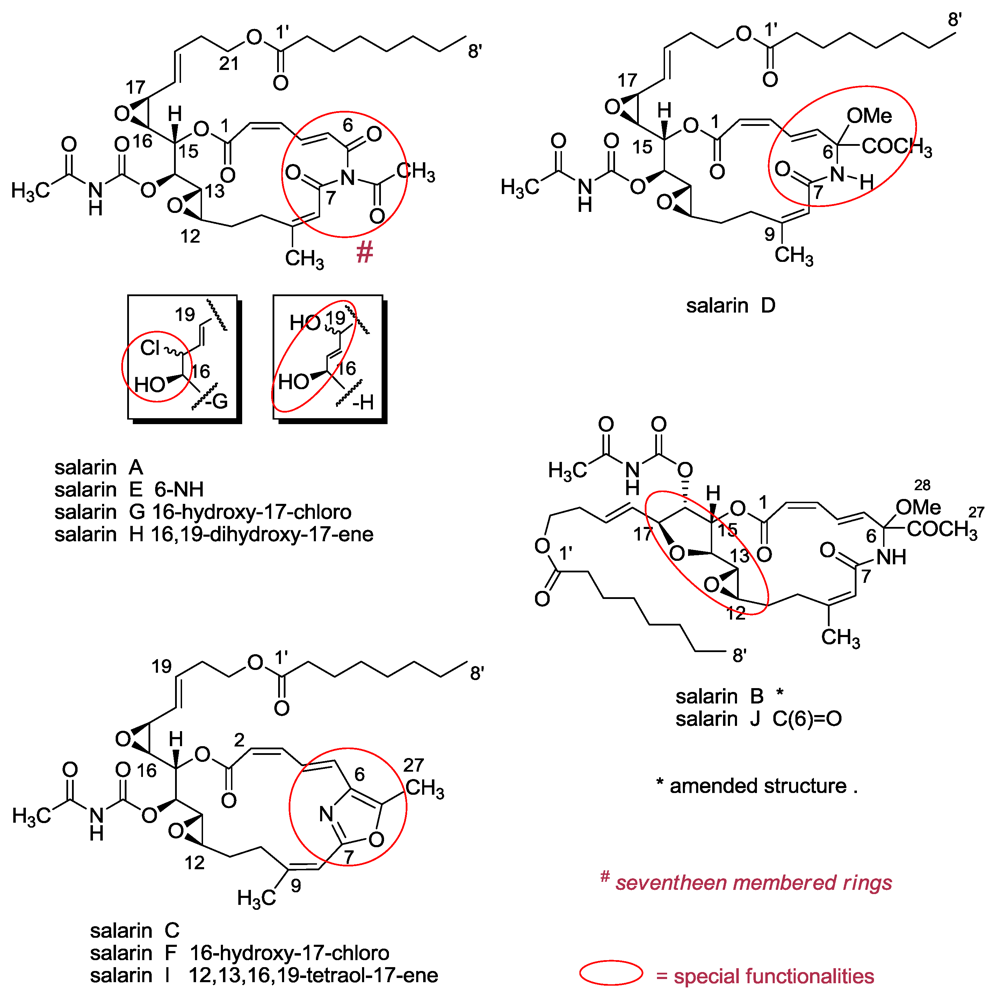
3.1. Salarins A–J
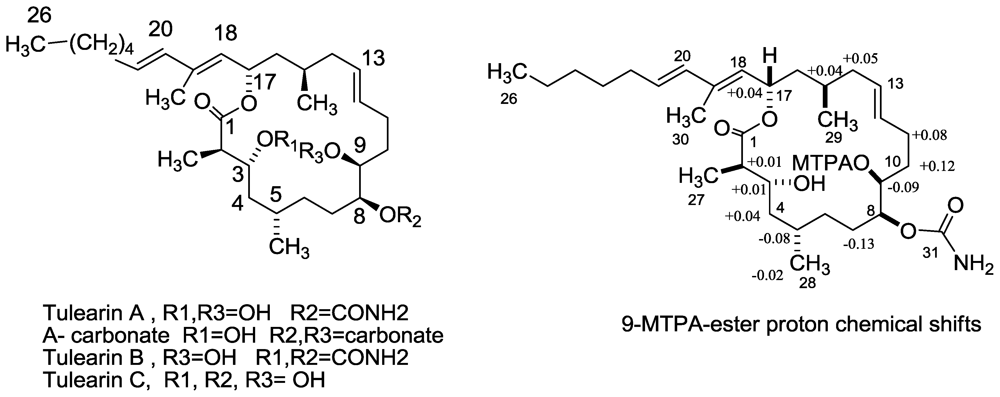
3.2. Tulearins A–C
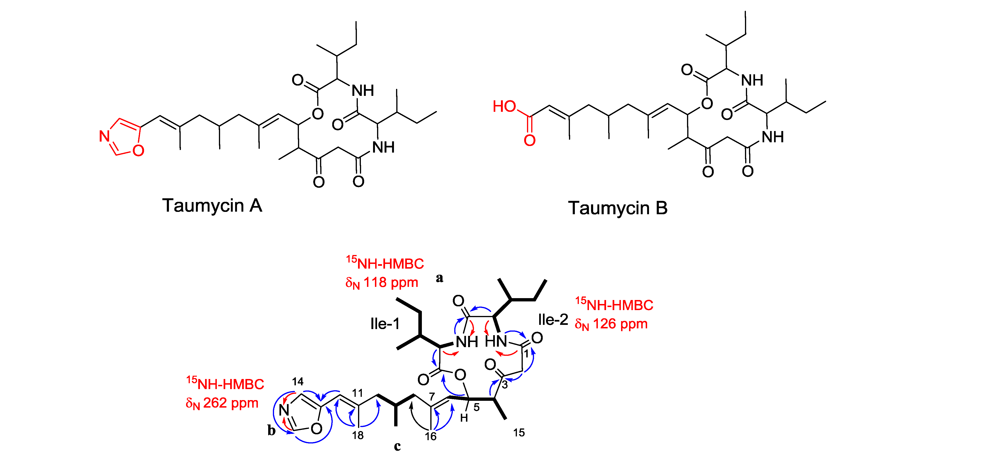
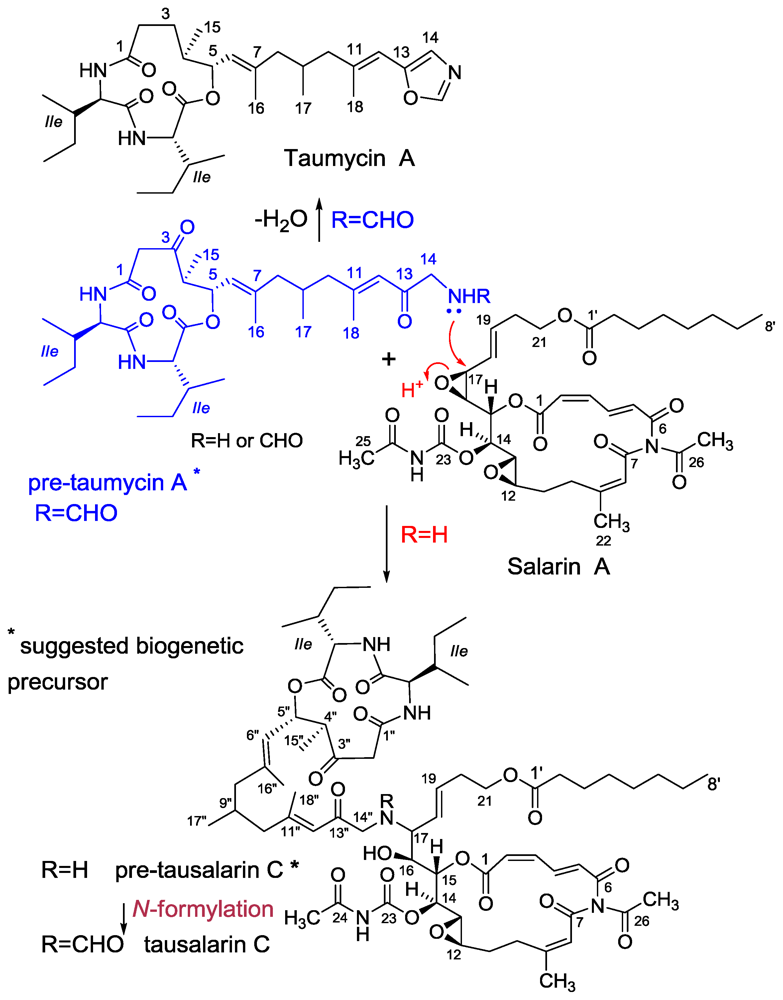
3.3. Taumycin A and B
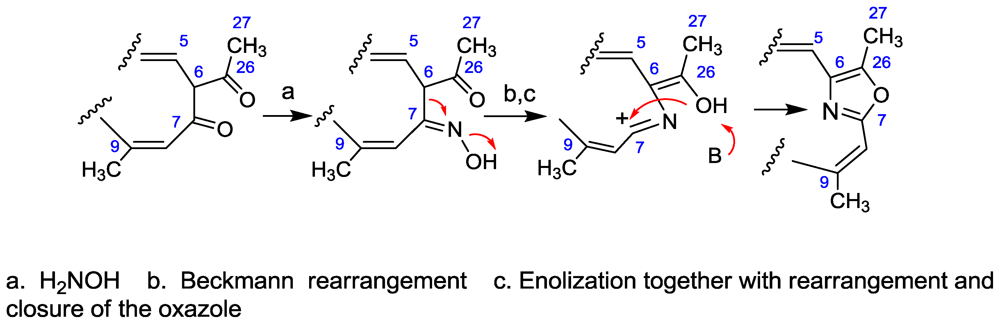
3.4. Tausalarin C
4. Conclusions
Acknowledgements
References and Notes
- Kashman, Y; Rudi, A; Pappo, D. Recent heterocyclic compounds from marine invertebrates: Structure and synthesis. Pure Appl Chem 2007, 79, 491–505. [Google Scholar]
- Kashman, Y; Fishelson, L; Neeman, I. N-acyl-2-methylene-β-alanine methy esters from the Sponge Fasciospongia cavernosa. Tetrahedron 1973, 29, 3655–3657. [Google Scholar]
- Pelletier, SW (Ed.) Alkaloids; John Wiley and Sons: New York, NY, USA, 1983; pp. 25–27.
- The most “accurate” definition is may be still the following one: “an alkaloid is like my wife, I can recognize her when I see her, but I can not define her”.
- Faulkner, DJ. Marine natural products. Nat Prod Rep 2002, 19, 1–48, and previous reports in this series. [Google Scholar]
- Carrole, AR; Feng, Y; Coll, JC; Bowden, BF. Studies of Australian ascidians. 5. virenamides A-C, new cytotoxic linear peptides from the colonial didemnid ascidian Diplosoma virens. J Nat Prod 1996, 61, 4059–4061. [Google Scholar]
- Rudi, A; Aknin, M; Gaydou, EM; Kashman, Y. Four new cytotoxic cyclic hexa- and heptapeptides from the marine ascidian Didemnum molle. Tetrahedron 1998, 54, 13203–13210. [Google Scholar]
- Sorek, H. Isolation, structure elucidation and biological activity of natural products from marine organisms. PhD thesis, Tel Aviv University, Israel, 2010. [Google Scholar]
- Rudi, A; BenayahuYGoldberg, I; Kashman, Y. Eilatin, a novel alkaloid from the marine tunicate Eudistoma sp. Tetrahedron Lett 1988, 29, 6655–6656. [Google Scholar]
- Gellerman, G; Babad, M; Kashman, Y. A two step biomimetic total synthesis of eilatin. Tetrahedron Lett 1993, 34, 1827–1830. [Google Scholar]
- Chill, L; Rudi, A; Benayahu, Y; Kashman, Y. Violatinctamine, a new heterocyclic compound from the marine tunicate Cystodytes cf violatinctus. Tetrahedron Lett 2004, 45, 7925–7928. [Google Scholar]
- Berrer, N; Rudi, A; Goldberg, I; Benayahu, Y; Kashman, Y. Callynormine A, a new marine cyclic peptide of a novel class. Org Lett 2004, 6, 2543–2545. [Google Scholar]
- Di Donato, P; Napolitano, A; Prota, G. Metal ions as potential regulatory factors in the biosynthesis of red hair pigments: a new benzothiazole intermediate in the iron or copper assisted oxidation of 5-S-cysteinyldopa. Biochim Biophys Acta 2002, 1571, 157–166. [Google Scholar]
- Stierle, AC; Cardellina, JH, II; Singelton, FL. Benzothiazoles from a putative bacterial symbiont of the marine sponge Tedania ignis. Tetrahedron Lett 1991, 32, 4847–4848. [Google Scholar]
- Susuki, H; Shindo, K; Ueno, A; Miura, T; Takei, M; Sakakibara, M; Fukamachi, H; Tanaka, J; Higa, T. S1319: a novel beta2-andrenoceptor agonist from a marine sponge Dysidea sp. Bioorg Med Chem Lett 1999, 9, 1361–1364. [Google Scholar]
- Dierks, T; Miech, C; Hummerjohann, J; Schmidt, B; Kertesz, MA; von Figura, K. Posttranslational formation of formylglycine in prokaryotic sulfatases by modification of either cysteine or serine. J Biol Chem 1998, 273, 25560–25564. [Google Scholar]
- Chruszcz, M; Laidler, P; Monkiewicz, M; Ortlund, E; Lebioda, L; Lewinski, K. Crystal structure of a covalent intermediate of endogenous human arylsulfatase A. J Inorg Biochem 2003, 96, 386–392. [Google Scholar]
- Pappo, D; Vartanian, M; Lang, S; Kashman, Y. Synthesis of cyclic endiamino peptides. J Am Chem Soc 2005, 127, 7682–7683. [Google Scholar]
- Nakazawa, T; Suzuki, T; Ishii, M. Synthesis and characterization of β-O-tosyldehydroserine as a precursor of dehydroamino acids. Tetrahedron Lett 1997, 38, 8951–8954. [Google Scholar]
- Goren, L; Pappo, D; Goldberg, I; Kashman, Y. Acyclic and cyclic thioenamino cyclic peptides; Solution and solid phase synthesis. Tetrahedron Lett 2009, 50, 1048–1050. [Google Scholar]
- Bensemhoun, J; Rudi, A; Kashman, Y; Gaydou, EM; Vacelet, J; Aknin, M. Salaramides A and B; Two α-oxoamides isolated from the marine sponge Hippospongia sp.(Porifera, Dictyoceratida). Nat Prod Commun 2010, 5, 259–260. [Google Scholar]
- Borbone, N; De Marino, S; Zollo, F; Iorizzi, M; Debitus, C; Esposito, G; Luvonne, T. Minor steroidal alkaloids from the marine sponge Corticium sp. J Nat Prod 2002, 65, 1206–1209, references there in. [Google Scholar]
- Ridley, CP; Faulkner, DJ. New cytotoxic steroidal alkaloids from the Philippine sponge Corticium niger. J Nat Prod 2003, 66, 1536–1539. [Google Scholar]
- Jurek, J; Scheuer, PJ; Kelly-Borges, M. Two steroidal alkaloids from a sponge, Corticium sp. J Nat Prod 1994, 57, 1004–1007. [Google Scholar]
- Aknin, M; Rudi, A; Kashman, Y; Vacelet, J; Gaydou, EM. Plakinamine L: A new steroidal alkaloid from the marine sponge Corticium sp. Nat Prod Commun 2010, 5, 33–34. [Google Scholar]
- Sorek, H; Rudi, A; Goldberg, I; Aknin, M; Kashman, Y. Saldedines A and B, dibromo proaporphine alkaloids from a Madagascan tunicate. J Nat Prod 2009, 72, 784–786. [Google Scholar]
- Phillipson, JD; Gray, AI; Askari, AR; Khalil, AA. Alkaloids from Iraqi species of Papaveraceae. J Nat Prod 1981, 44, 296–307. [Google Scholar]
- Honda, T; Shigehisa, H. Novel and efficient synthetic path to proaporphine alkaloids: total synthesis of (+/−)-stepharine and (+/−)-pronuciferine. Org Lett 2006, 8, 657–659, references therein. [Google Scholar]
- Pathirana, C; Andersen, RJ. Imbricatine, an unusual benzyltetrahydroisoquinoline alkaloid isolated from the starfish Dermasterias imbricata. J Am Chem Soc 1986, 108, 8288–8289. [Google Scholar]
- Sorek, H; Rudi, A; Benayahu, Y; Kashman, Y. Njaoamines G and H, two new cytotoxic polycyclic alkaloids from the marine sponge Neopetrosia sp. Tetrahedron Lett 2007, 48, 7691–7694. [Google Scholar]
- Reyes, F; Fernandez, R; Urda, C; Francesch, A; Bueno, S; de Eguilior, C; Cuevas, C. Njaoamines A–F, new cytotoxic polycyclic alkaloids from the haplosclerid sponge Reniera sp. Tetrahedron 2007. [Google Scholar]
- Kong, F; Andersen, RJ; Allen, TM. Ingenamine, a novel pentacyclic alkaloid from the marine sponge Xestospongia ingens. Tetrahedron Lett 1994, 35, 1643–1646. [Google Scholar]
- Kong, F; Andersen, RJ. Ingenamine alkaloids isolated from sponge Xestospongia ingens: structures and absolute configurations. Tetrahedron 1995, 51, 2895–2906. [Google Scholar]
- de Oliveira, JHHL; Grube, A; KoÅNck, M; Berlinck, RGS; Macedo, ML; Ferreira, AG; Hadju, E. Ingenamine G and cyclostellettamines G-I, K, and L from the new Brazilian species of marine sponge Pachychalina sp. J Nat Prod 2004, 67, 1685–1689. [Google Scholar]
- Blunt, JW; Copp, BR; Munro, MHG; Northcote, PT; Prinsep, MR. Marine natural products. Nat Prod Rep 2006, 23, 26–78, earlier reports in the series. [Google Scholar]
- Munro, MHG; Blunt, JW. Marine literature database; Department of Chemistry, University of Canterbury: New Zealand, 2009. [Google Scholar]
- Keyzers, RA; Gray, CA; Schleyer, MH; Whibley, CE; Hendricks, DT; Davies-Coleman, M. Malonganenones A–C, novel tetraprenylated alkaloids from the Mozambique gorgonian Leptogorgia gilchristi. Tetrahedron 2006, 62, 2200–2206. [Google Scholar]
- Martin, EG; Hadden, EC. Long range 1H-15N Heteronuclear shift correlation at natural abundance. J Nat Prod 2000, 63, 543–585. [Google Scholar]
- Laxer, A; Major, DT; Gottlieb, HE; Fischer, B. (15N5)-Labeled adenine derivatives: synthesis and studies of tautomerism by 15N NMR spectroscopy and theoretical calculations. J Org Chem 2001, 66, 5463–5481. [Google Scholar]
- Pappo, D; Kashman, Y. Synthesis of 9-substituted tetrahydrodiazepinones-asmarine A analogues. Tetrahedron 2003, 59, 6493–6501. [Google Scholar]
- Pappo, D; Shimony, S; Kashman, Y. Synthesis of 9-Substituted tetrahydrodiazepinopurines: Studies toward the total synthesis of asmarines. J Org Chem 2005, 70, 199–206. [Google Scholar]
- Sorek, H; Rudi, A; Benayahu, Y; Ben-Califa, N; Neumann, D; Kashman, Y. Nuttingins A–F and Malonganenones D–H, tetraprenylated alkaloids from the Tanzanian gorgonian Euplexaura nuttingi. J Nat Prod 2008, 70, 1104–1109. [Google Scholar]
- Lozzio, CB; Lozzio, BB. Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood 1975, 45, 321–334. [Google Scholar]
- Komatsu, N; Nakauchi, H; Miwa, A; Ishihara, T; Eguchi, M; Moroi, M; Okada, M; Sato, Y; Wada, H; Yawata, Y; Suda, T; Niura, Y. Establishment and characterization of a human leukemic cell line with megakaryocytic features: dependency on granulocyte-macrophage colony-stimulating factor, interleukin 3, or erythropoietin for growth and survival. Cancer Res 1991, 51, 341–348. [Google Scholar]
- Koren-Goldshlager, G; Kashman, Y; Schleyer, M. Haliclorensin, a novel diamino alkaloid from the marine sponge Haliclona tulearensis. J Nat Prod 1998, 61, 282–284. [Google Scholar]
- Kashman, Y; Koren-Goldshlager, G; Gravalos, MDG; Schleyer, M. Halitulin, a new cytotoxic alkaloid from the marine sponge Haliclona tulearensis. Tetrahedron Lett 1999, 40, 997–1000. [Google Scholar]
- Heinrich, MR; Steglich, W. Synthesis of (-)-(3S)-1-(3-aminopropyl)-3-methylazacyclodecane, the structure proposed for the marine alkaloid haliclorensin. Tetrahedron Lett 2001, 42, 3287–3289. [Google Scholar]
- Heinrich, MR; Kashman, Y; Spiteller, P; Steglich, W. Revision of the structure of haliclorensin to (S)-7-methyl-1,5-diazacyclotetradecane and confirmation of the new structure by synthesis. Tetrahedron 2001, 57, 9973–9978. [Google Scholar]
- Banwell, MG; Bray, AM; Edwards, AJ; Wong, DJ. Synthesis of the putative structure of the marine alkaloid haliclorensin. New J Chem 2001, 25, 1347–1350. [Google Scholar]
- Heinrich, MR; Steglich, W; Banwell, MG; Kashman, Y. Total synthesis of the marine alkaloid halitulin. Tetrahedron 2003, 59, 9239–9247. [Google Scholar]
- Sorek, H; Rudi, A; Aknin, M; Gaydou, EM; Kashman, Y. Isohalitulin and Haliclorensins B and C, three marine alkaloids from Haliclona tulearensis. J Nat Prod 2010, 73, 456–458. [Google Scholar]
- Tsukamoto, S; Tane, K; Ohta, T; Matsunaga, S; Fusetani, N; van Soest, RWM. Four new bioactive pyrrole-derived alkaloids from the marine sponge Axinella brevistyla. J Nat Prod 2001, 64, 1576–1578. [Google Scholar]
- Alder, RW; Blake, ME; Bufali, S; Butts, CP; Orpen, AG; Schuts, J; Williams, SJ. Preparation of tetraalkylformamidinium salts and related species as precursors to stable carbenes. J Chem Soc Perkin Trans 1 2001, 1586–1593. [Google Scholar]
- Aidouni, A; Bendahou, S; Demonceau, A; Delaude, L. Facile microwave-assisted synthesis of cyclic amidinium salts. J Comb Chem 2008, 10, 886–892. [Google Scholar]
- Tsuda, M; Kawasaki, N; Kobayashi, J. Keramaphidin C and keramamine C, plausible biogenetic precursors of manzamine C from an Okinawan marine sponge. Tetrahedron Lett 1994, 35, 4387–4388. [Google Scholar]
- Bishara, A; Rudi, A; Aknin, M; Neumann, D; Ben-Califa, N; Kashman, Y. Salarins A and B and tulearin A: new cytotoxic Sponge-derived macrolides. Org Lett 2008, 10, 153–156. [Google Scholar]
- Bishara, A; Rudi, A; Aknin, M; Neumann, D; Ben-Califa, N; Kashman, Y. Salarin C, a new cytotoxic sponge-derived nitrogenous macrolide. Tetrahedron Lett 2008, 49, 4355–4358. [Google Scholar]
- Bishara, A; Rudi, A; Aknin, M; Neumann, D; Ben-Califa, N; Kashman, Y. Taumycins A and B, Two bioactive lipodepsipeptides from the Madagascar sponge Fascaplysinopsis sp. Org Lett 2008, 10, 4307–4309. [Google Scholar]
- Bishara, A; Rudi, A; Goldberg, I; Aknin, M; Kashman, Y. Tulearins A, B, and C; structures and absolute configurations. Tetrahedron Lett 2009, 50, 3820–3822. [Google Scholar]
- Bishara, A; Rudi, A; Goldberg, I; Aknin, M; Neumann, D; Ben-Califa, N; Kashman, Y. Tausalarin C: A new bioactive marine sponge-derived nitrogenous bismacrolide. Org Lett 2009, 11, 3538–3541. [Google Scholar]
- Bishara, A; Rudi, A; Aknin, M; Neumann, D; Ben-Califa, N; Kashman, Y. Salarins D–J, seven new nitrogenous macrolides from the Madagascar sponge Fascaplysinopsis sp. Tetrahedron 2010, 66, 4339–4345. [Google Scholar]
- Ben-Califa, N; Bishara, A; Kashman, Y; Neumann, D. Salarin C, a member of the salarin superfamily of marine compounds, is a potent inducer of apoptosis. Investig New Drugs 2010, in presss. [Google Scholar]
- Klein, D; Braekman, JC; Daloze, D; Hoffmann, L; Castillo, G; Demoulin, V. Madangolide and laingolide A, two novel macrolides from Lyngbya bouillonii (Cyanobacteria). J Nat Prod 1999, 62, 934–936. [Google Scholar]
- Klein, D; Braekman, JC; Daloze, D; Hoffmann, L; Castillo, G; Demoulin, V. Laingolide, a novel 15-membered macrolide from Lyngbya bouillonii (Cyanophyceae). Tetrahedon Lett 1996, 37, 7519–7520. [Google Scholar]
- Feliu, A; Seltzer, S. Synthesis and interconversion of the four isomeric 6-oxo-2,4-heptadienoic acids. J Org Chem 1985, 50, 447–451. [Google Scholar]
- Ando, O; Salake, H; Nakajima, M; Sato, A; Nakamura, T; Kinoshita, T; Furuya, K; Hancishi, TJ. Synerazol, a new antifungal antibiotic. Antibiot 1991, 44, 382–384. [Google Scholar]
- Ichino, T; Arimoto, H; Uemura, D. Daisuke Possibility of a non-amino acid pathway in the biosynthesis of marine-derived oxazoles. Chem Commun 2006, 1742–1744. [Google Scholar]
- Martin, GE; Hadden, CE. Long-Range 1H-15N Heteronuclear shift correlation at natural abundance. J Nat Prod 2000, 63, 543–585. [Google Scholar]
- Schneider, TL; Walsh, CT; O’Conner, SE. Utilization of alternate substrates by the first three modules of the epothilone synthetase assembly line. J Am Chem Soc 2002, 124, 11272–11273. [Google Scholar]
- Iesce, R; Graziano, ML; Cimminiello, G; Cermola, F; Parrilli, M; Scarpati, R. Route of triacylamine formation in the thermal conversion of 2,3,7-trioxa-5-azabicyclo[2.2.1]hept-5-enes investigated by nuclear magnetic resonance experiments. J Chem Soc Perkin Trans 2 1991, 1085–1089. [Google Scholar]
- Hassner, A; Fischer, B. Synthetic methods. 26. The 4,5- and 2,5-additions to oxazoles. Tetrahedron 1989, 45, 6249–6262. [Google Scholar]
- Diyabalanage, T; Amsler, CD; McClintok, JB; Baker, BJ. Palmerolide A, a cytotoxic macrolide from the Antarctic tunicate Synoicum adareanum. J Am Chem Soc 2006, 128, 5630–5633. [Google Scholar]
- Neckers, L; Schulte, TM; Mimhaugh, E. Geldanamycin as a potential anticancer agent: Its molecular target and biochemical activity. Investig New Drugs 1999, 17, 361–373. [Google Scholar]
- Ohtani, I; Kusumi, T; Kashman, Y; Kakisawa, H. High-field FT NMR application of Mosher’s method. The absolute configurations of marine terpenoids. J Am Chem Soc 1991, 113, 4092–4096. [Google Scholar]
- Hiemstra, H; Houwing, HA; Possel, O; van Leusen, AM. Carbon-13 nuclear magnetic resonance spectra of oxazoles. Can J Chem 1979, 57, 3168–3170. [Google Scholar]
- Stratmann, K; Burgoyne, DL; Moore, RE; Patterson, GML; Smith, CD. Hapalosin, a cyanobacterial cyclic depsipeptide with multidrug-resistance reversing activity. J Org Chem 1995, 60, 2950–2953. [Google Scholar]
- Ratnayake, R; Fremlin, LJ; Lacey, E; Gill, JH; Capon, RJ. Acremolides A-D, lipodepsipeptides from an Australian marine-derived fungus, Acremonium sp. J Nat Prod 2008, 71, 403–408. [Google Scholar]
- Seo, C; Yim, JH; Lee, HK; Park, SM; Sohn, JH; Oh, H. Stereocalpin A, a bioactive cyclic depsipeptide from the Antarctic lichen Stereocaulon alpinum. Tetrahedron Lett 2008, 49, 29–31. [Google Scholar]
- Costantino, V; Fattorusso, E; Mangoni, A. Isolation of five-membered cyclitol glycolipids, crasserides: unique glycerides from the sponge Pseudoceratina crassa. J Org Chem 1993, 58, 186–191. [Google Scholar]
- Lu, Q; Faulkner, DJ. Two new sesterterpenoids and a new 9,11-secosterol from Spongia matamata. J Nat Prod 1997, 60, 195–198. [Google Scholar]









© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kashman, Y.; Bishara, A.; Aknin, M. Recent N-Atom Containing Compounds from Indo-Pacific Invertebrates. Mar. Drugs 2010, 8, 2810-2836. https://doi.org/10.3390/md8112810
Kashman Y, Bishara A, Aknin M. Recent N-Atom Containing Compounds from Indo-Pacific Invertebrates. Marine Drugs. 2010; 8(11):2810-2836. https://doi.org/10.3390/md8112810
Chicago/Turabian StyleKashman, Yoel, Ashgan Bishara, and Maurice Aknin. 2010. "Recent N-Atom Containing Compounds from Indo-Pacific Invertebrates" Marine Drugs 8, no. 11: 2810-2836. https://doi.org/10.3390/md8112810
APA StyleKashman, Y., Bishara, A., & Aknin, M. (2010). Recent N-Atom Containing Compounds from Indo-Pacific Invertebrates. Marine Drugs, 8(11), 2810-2836. https://doi.org/10.3390/md8112810