Thiazole and Oxazole Alkaloids: Isolation and Synthesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. New Thiazoles Isolated from Marine Sources
3. Thiazolines Isolated from Marine Sources
4. Synthesis of Thiazoles of Marine Origin and Analogues
5. Synthesis of Thiazolines of Marine Origin and Analogues
6. New Oxazoles Isolated from Marine Sources
7. Synthesis of Oxazoles of Marine Origin and Analogues
8. Synthesis of Oxazolines of Marine Origin and Analogues
Acknowledgements
References
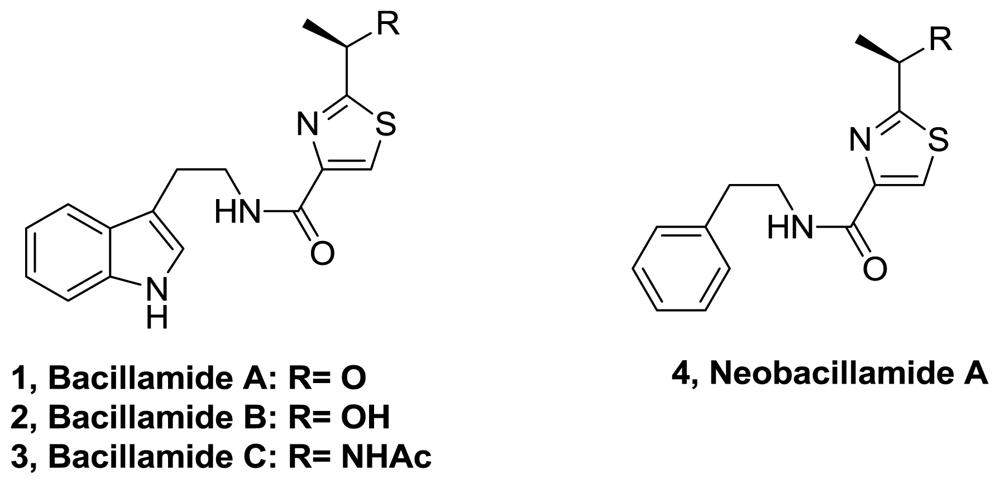
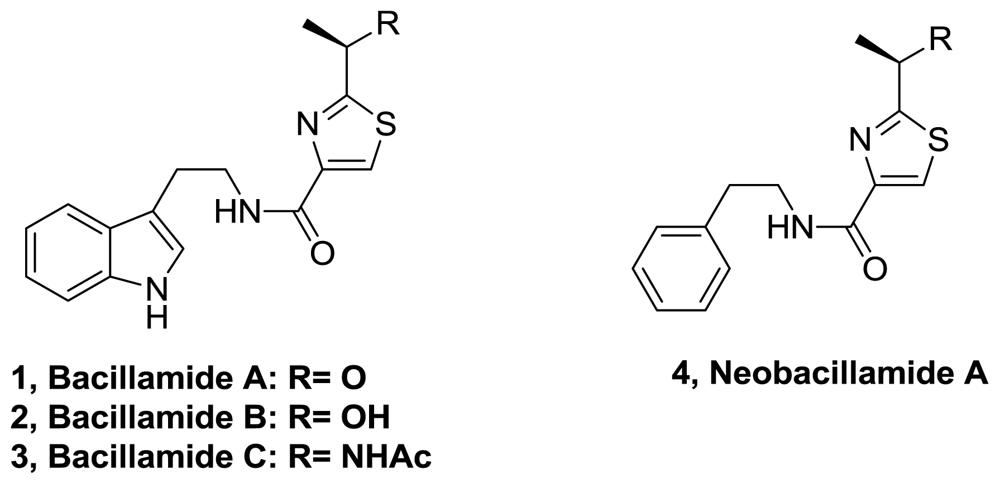
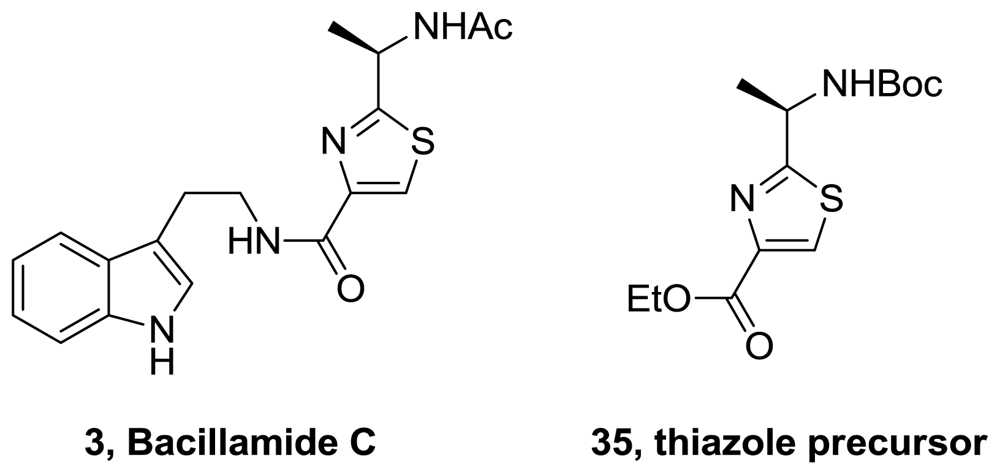
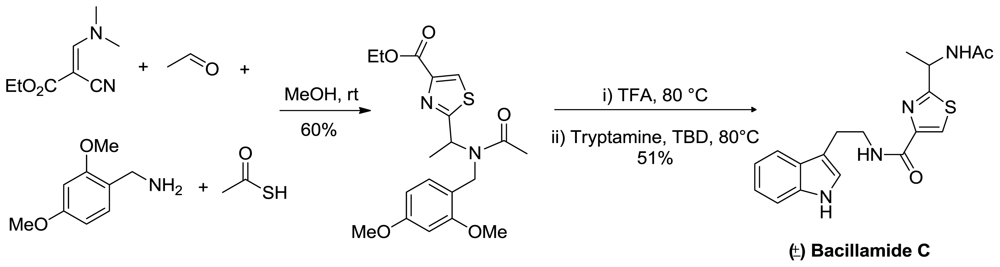
- Aaron, MS; Richard, AL; David, CR. Bacillamides from a hypersaline microbial mat bacterium. J Nat Prod 2007, 70, 1793–1795. [Google Scholar]
- Yu, L; Li, Z; Peng, C; Li, Z; Guo, Y. Neoobacillamide A, a novel thiazole-containing alkaloid from the marine bacterium bacillus vallismortis C89, associated with south china sea sponge Dysidea avara. Helv Chim 2009, 92, 607–612. [Google Scholar]
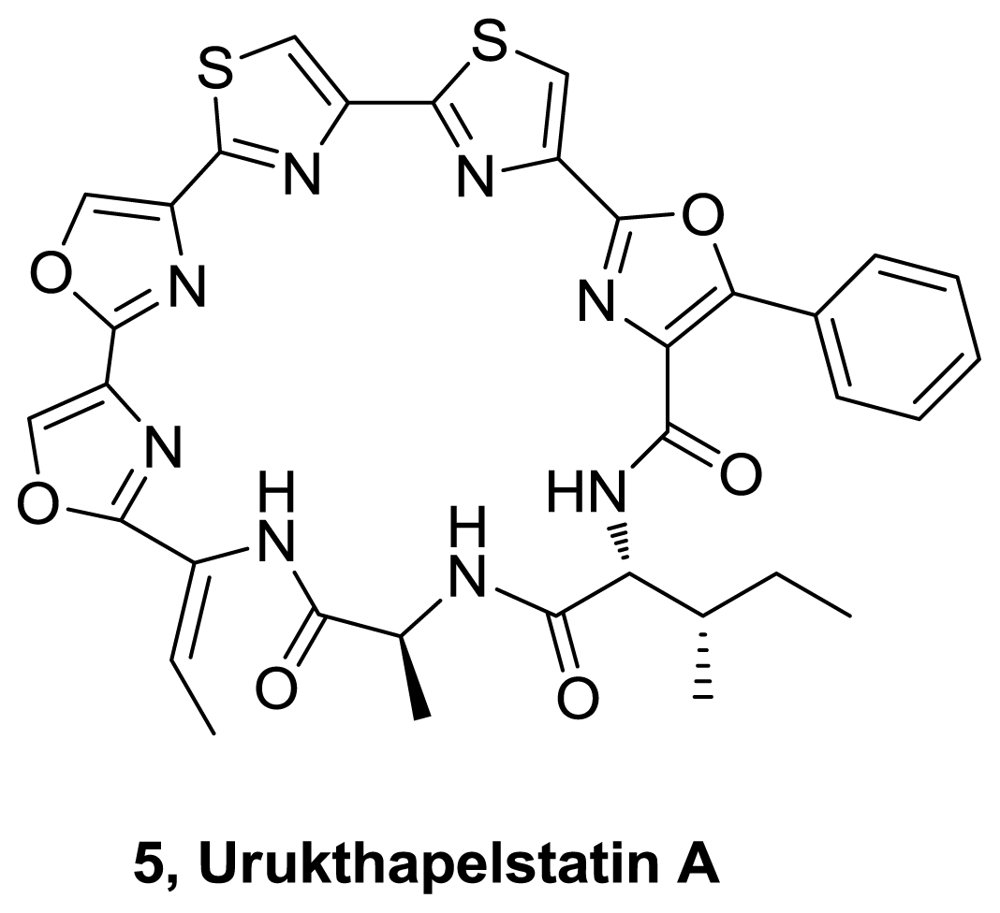
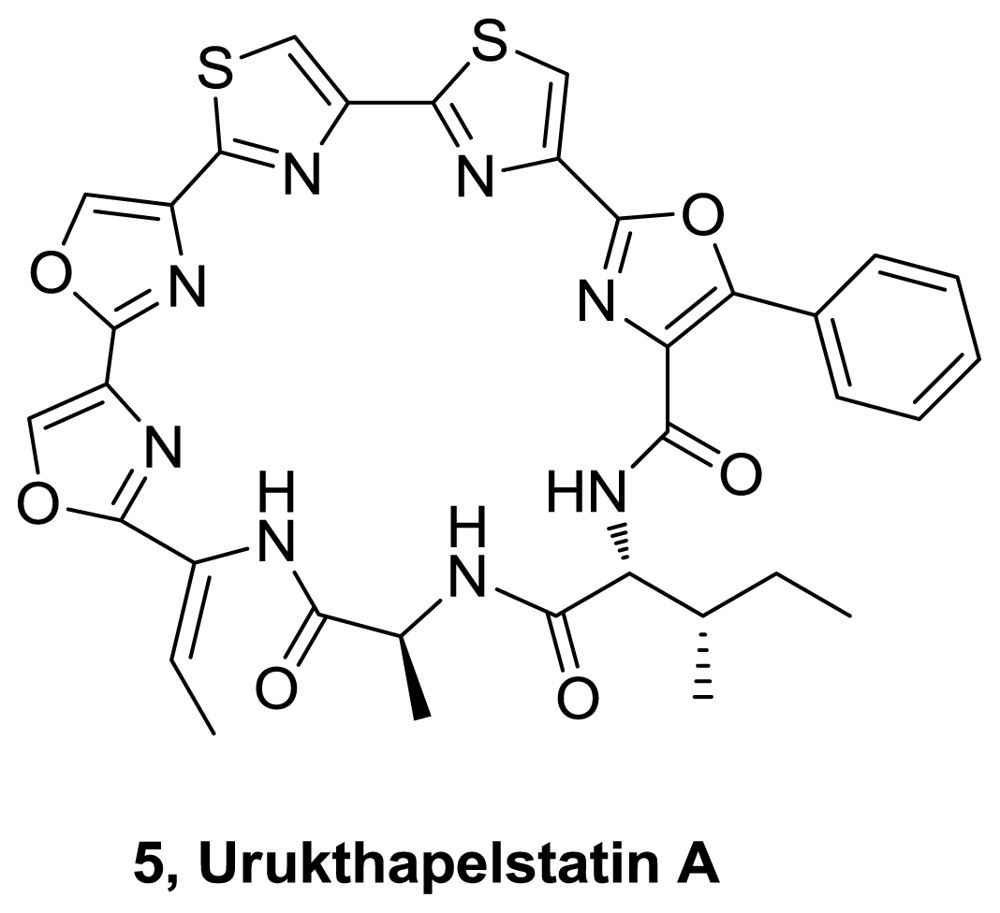
- Matsuo, Y; Kanoh, K; Imagawa, H; Adachi, K; Nishizawa, M; Shizuri, Y. Urukthapelstatin A, a novel cytotoxic substance from marine-derived Mechercharimyces asporophorigenens YM11-542. J Antib 2007, 60, 256–260. [Google Scholar]
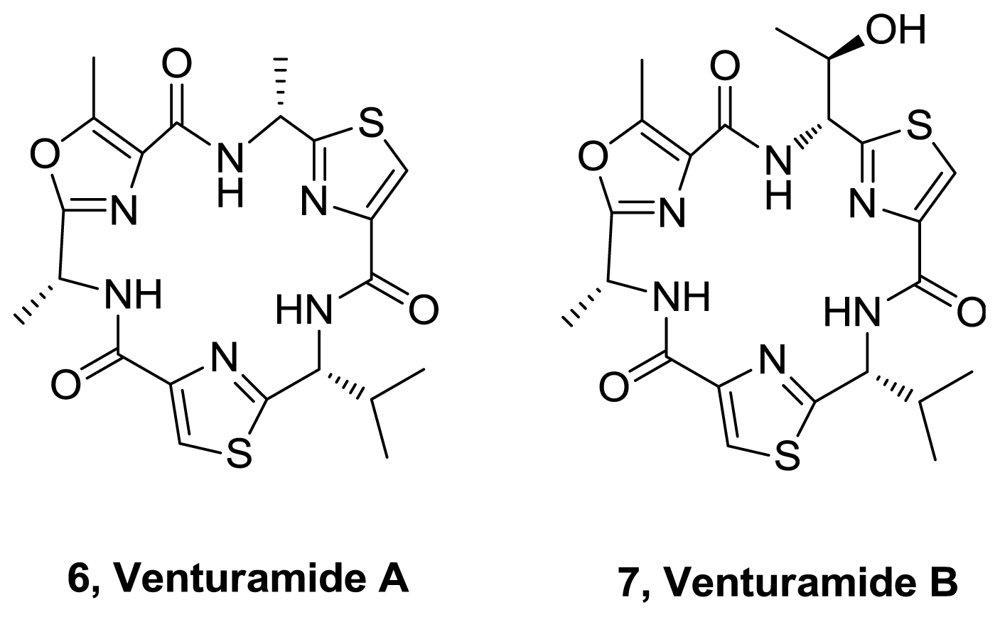
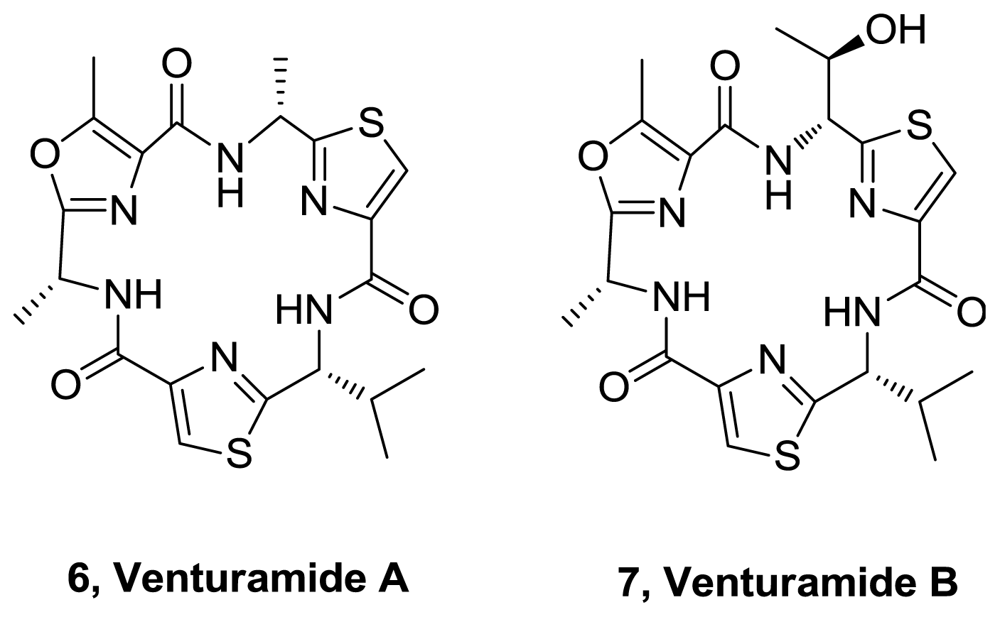
- Linington, RG; González, J; Urena, L; Romero, LI; Ortega-Barría, E; Gerwick, WH. Venturamides A and B: antimalarial constituents of the panamanian marine cyanobacterium Oscillatoria sp. J Nat Prod 2007, 70, 397–401. [Google Scholar]
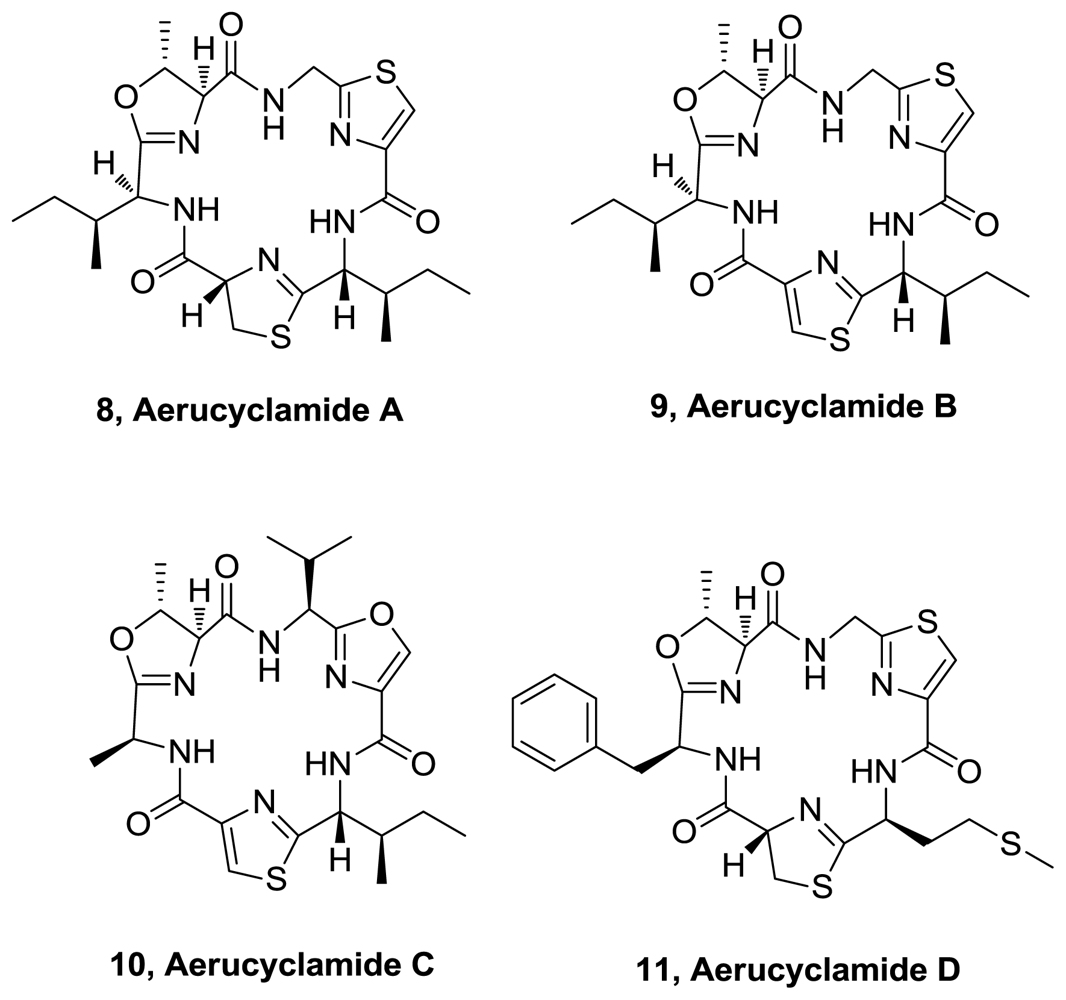
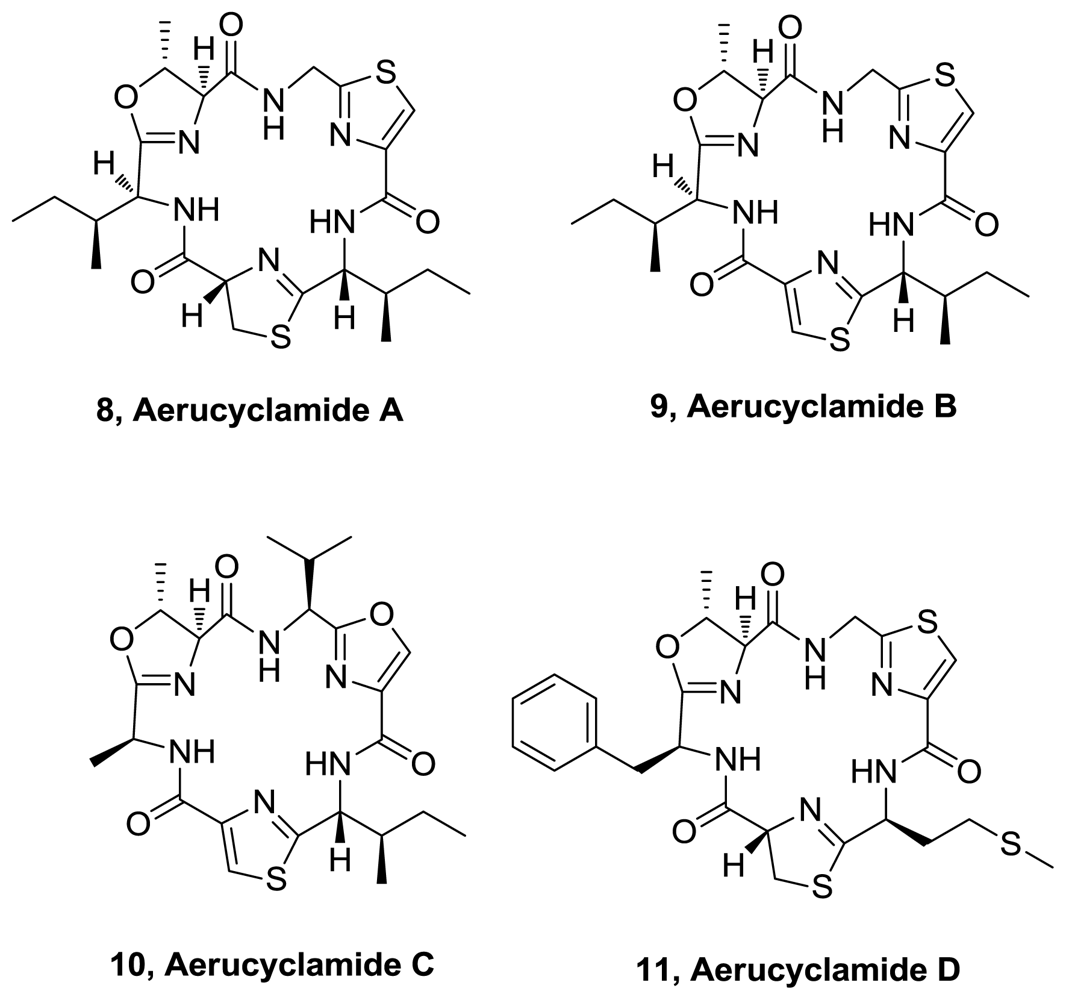
- Portmann, C; Blom, JF; Gademann, K; Jüttner, F. Aerucyclamides A and B: isolation and synthesis of toxic ribosomal heterocyclic peptides from the cyanobacterium Microcystis aeruginosa PCC 7806. J. Nat. Prod 2008, 71, 1193–1196. [Google Scholar]
- Portmann, C; Blom, JF; Kaiser, M; Brun, R; Jüttner, F; Gademann, K. Isolation of Aerucyclamides C and D and structure revision of Microcyclamide 7806A: heterocyclic ribosomal peptides from Microcystis aeruginosa PCC 7806 and their antiparasite evaluation. J Nat Prod 2008, 71, 1891–1896. [Google Scholar]
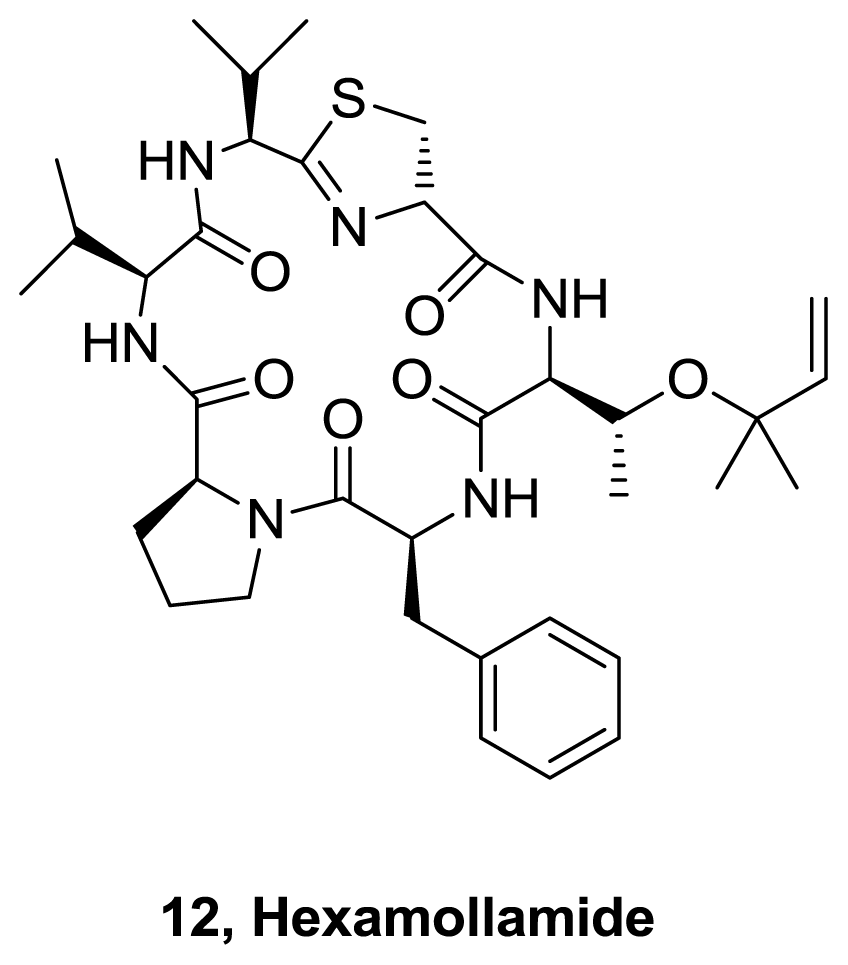
- Teruya, T; Sasaki, H; Suenaga, K. Hexamollamide, a hexapeptide from an Okinawan ascidian Didemnum molle. Tetraedron Lett 2008, 49, 5297–5299. [Google Scholar]
- Zabriskie, TM; Foster, MP; Stout, TJ; Clardy, J; Ireland, CM. Studies on the solution- and solid-state structure of patellin 2. J Am Chem Soc 1990, 112, 8080–8084. [Google Scholar]
- Carroll, AR; Coll, JC; Bourne, DJ; MacLeod, JK; Ireland, CM; Bowden, BF. Patellins 1–6 and trunkamide A: novel cyclic hexa-, hepta- and octa-peptides from colonial ascidians, Lissoclinum sp. Aust J Chem 1996, 49, 659–667. [Google Scholar]
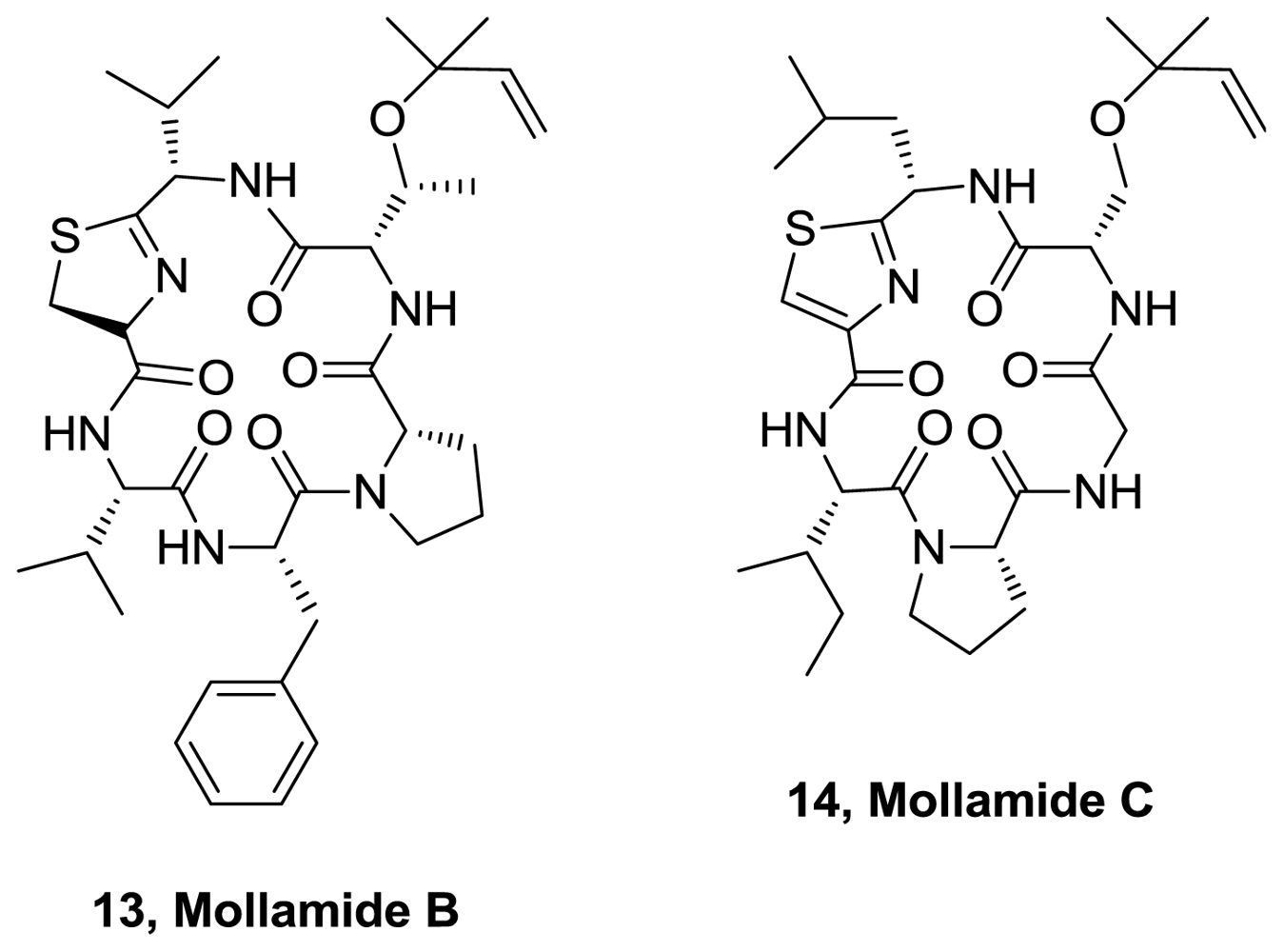
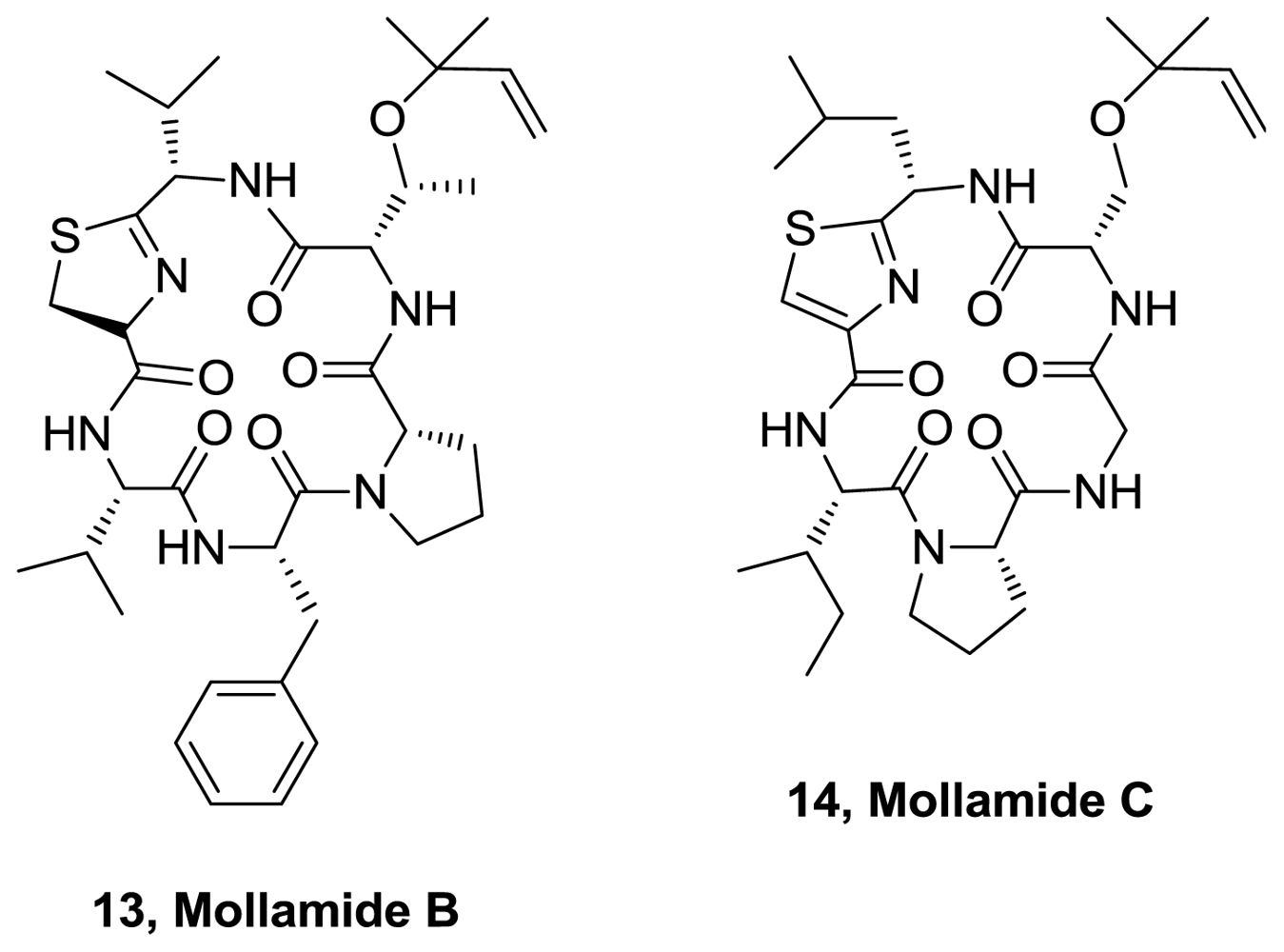
- Donia, MS; Wang, B; Dunbar, DC; Desai, PV; Patny, A; Avery, M; Hamann, MT. Mollamides B and C, Cyclic Hexapeptides from the Indonesian Tunicate Didemnum molle. J Nat Prod 2008, 71, 941–945. [Google Scholar]
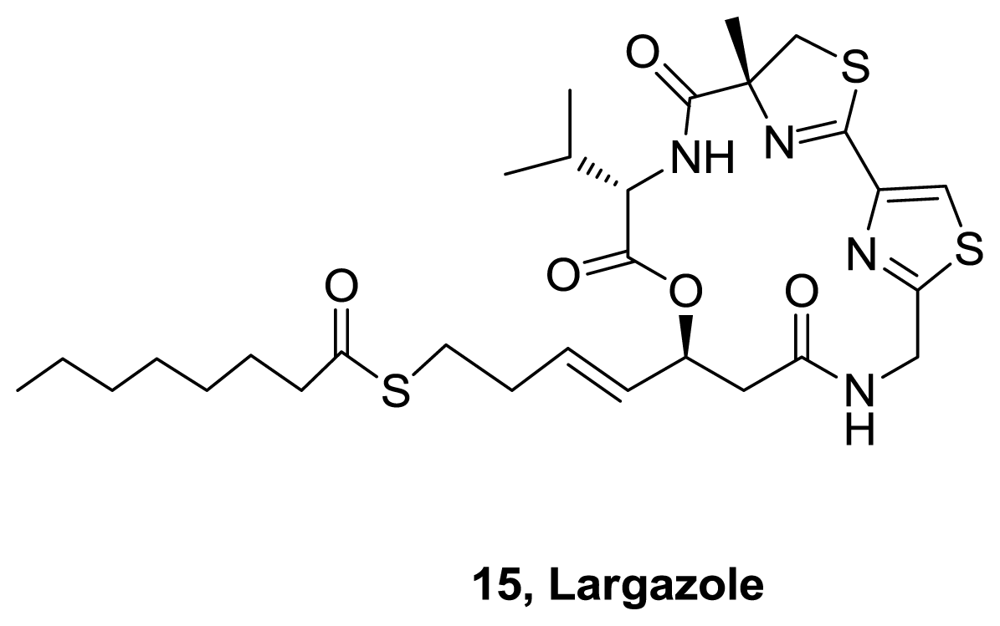
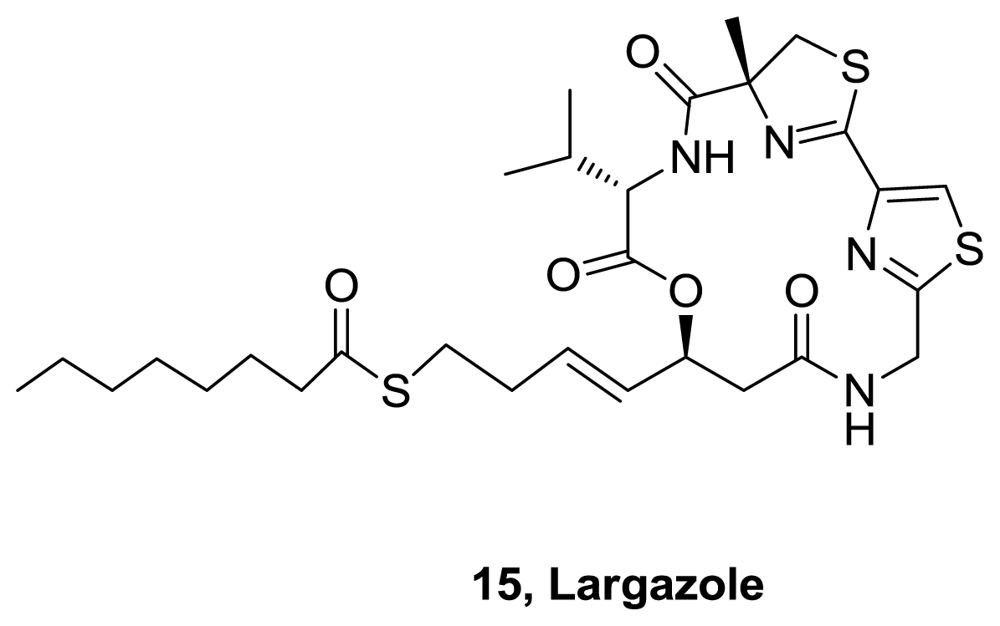
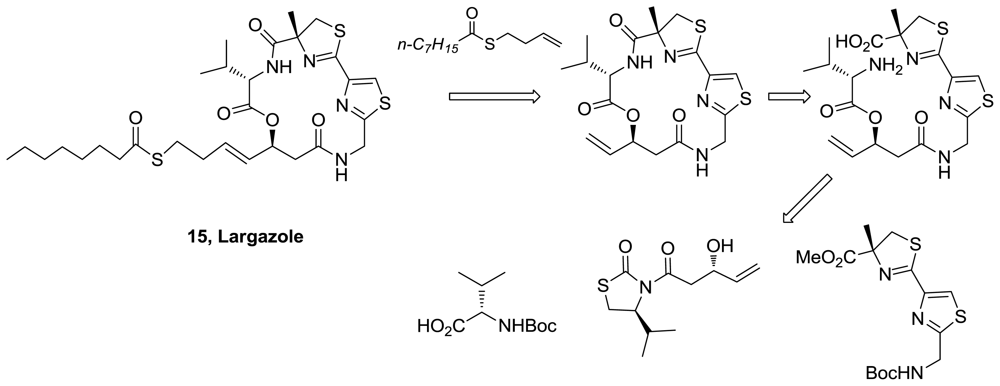
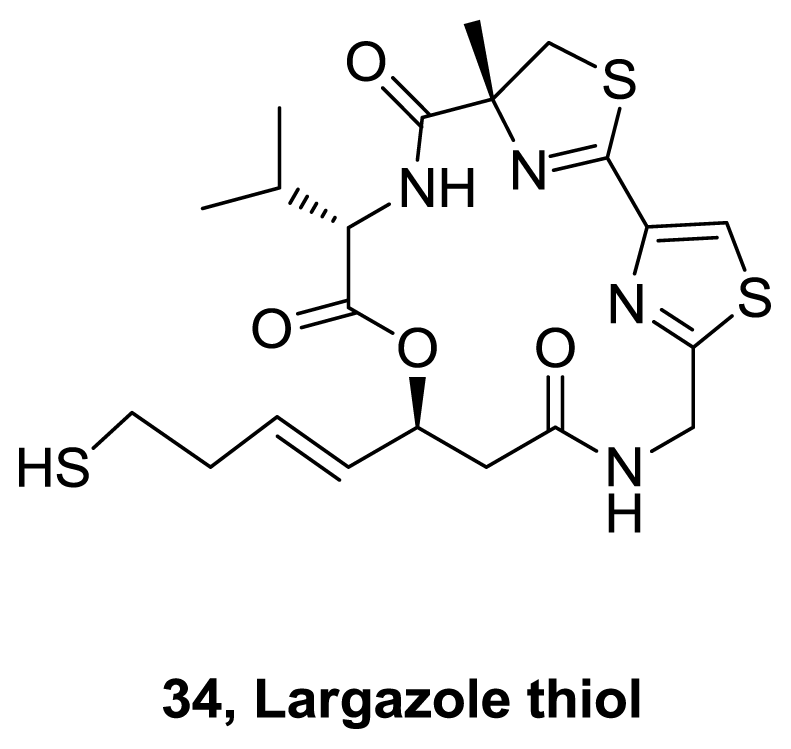
- Taori, K; Paul, VJ; Luesch, H. Structure and activity of largazole, a potent antiproliferative agent from the floridian marine cyanobacterium Symploca sp. J Am Chem Soc 2008, 130, 1806–1807. [Google Scholar]
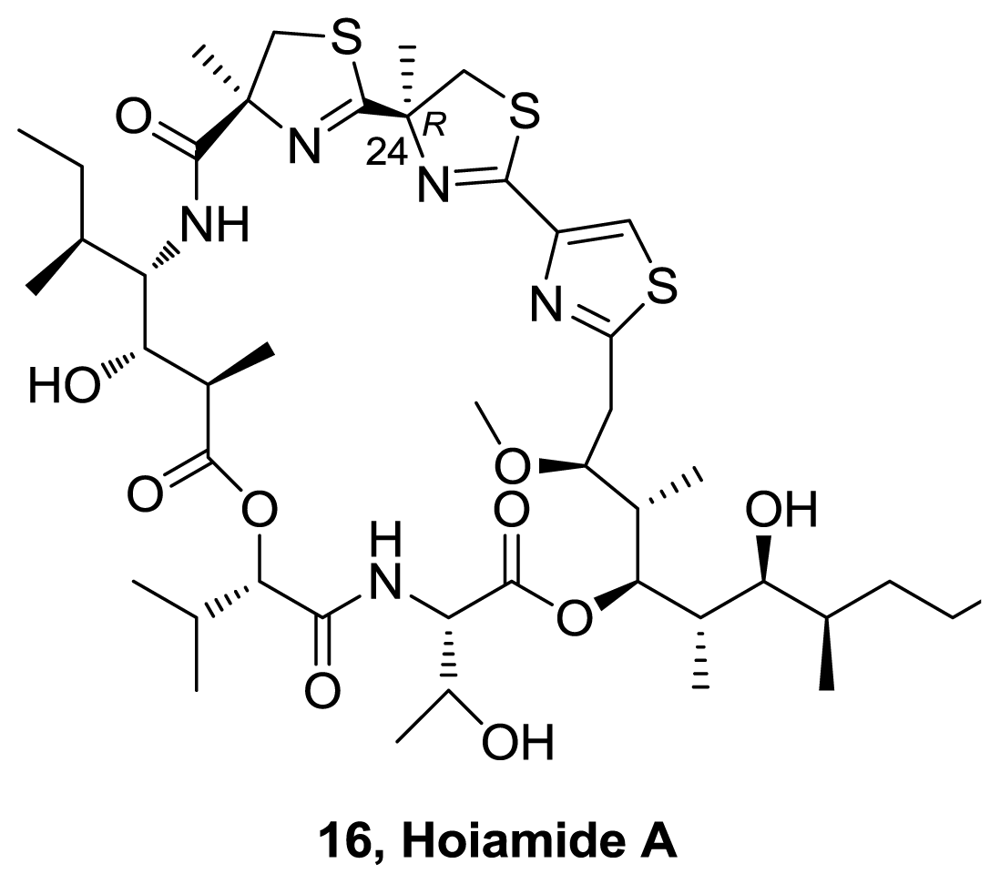
- Pereira, A; Cao, Z; Murray, TF; Gerwick, WH. Hoiamide A, a sodium channel activator of unusual architecture from a consortium of two papua new guinea cyanobacteria. Chem Biol 2009, 16, 893–906. [Google Scholar]
- Pereira, A; Cao, Z; Murray, TF; Gerwick, WH. Hoiamide A, a sodium channel activator of unusual architecture from a consortium of two papua new guinea cyanobacteria. Chem Biol 2009, 16, 1208. [Google Scholar]
- Raveh, A; Moshe, S; Evron, Z; Flescher, E; Carmeli, S. Novel thiazole and oxazole containing cyclic hexapeptides from a waterbloom of the cyanobacterium Microcystis sp. Tetrahedron 2010, 66, 2705–2712. [Google Scholar]
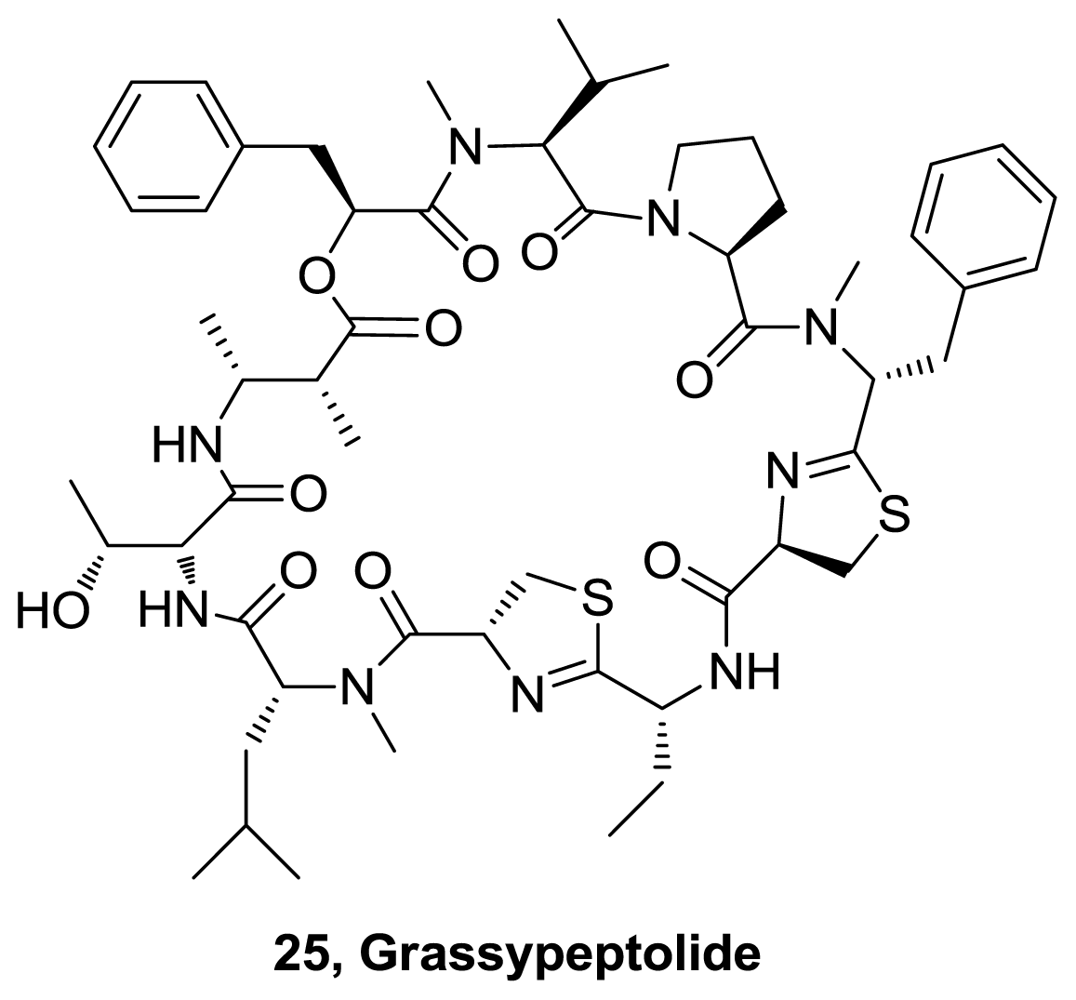
- Kwan, J; Rocca, JR; Abboud, KA; Paul, VJ; Luesch, H. Total structure determination of grassypeptolide, a new marine cyanobacterial cytotoxin. Org Lett 2008, 10, 789–792. [Google Scholar]
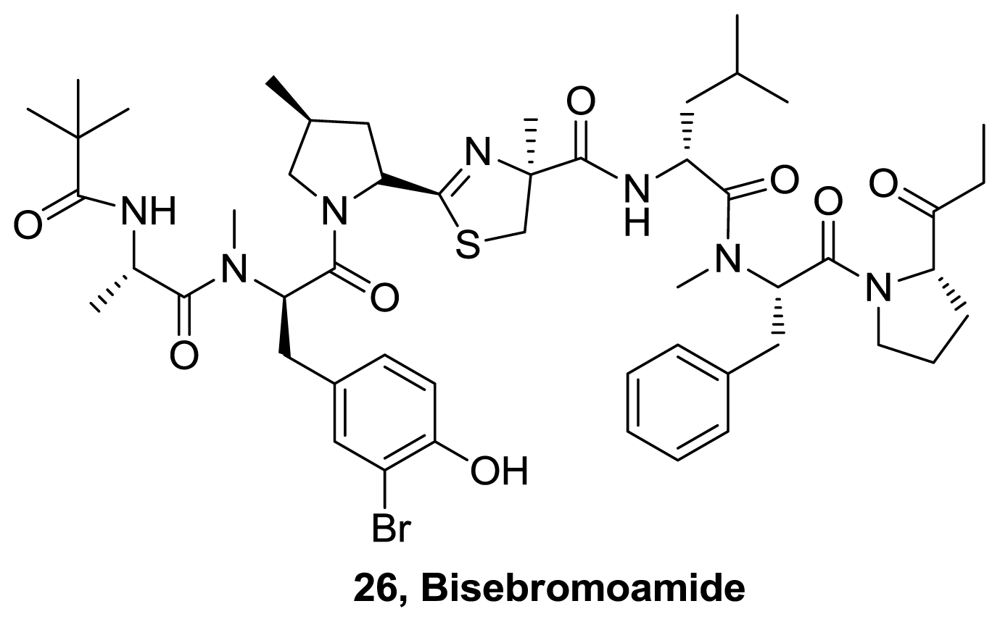
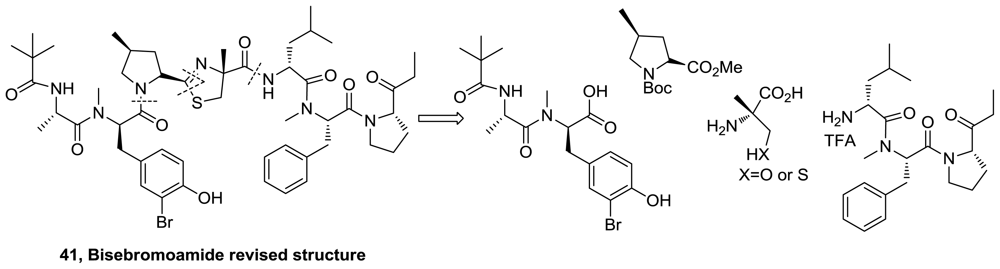
- Teruya, T; Sasaki, H; Fukazawa, H; Suenaga, K. Bisebromoamide, a potent cytotoxic peptide from the marine cyanobacterium Lyngbya sp. Isolation, stereostructure, and biological activity. Org Lett 2009, 11, 5062–5065. [Google Scholar]
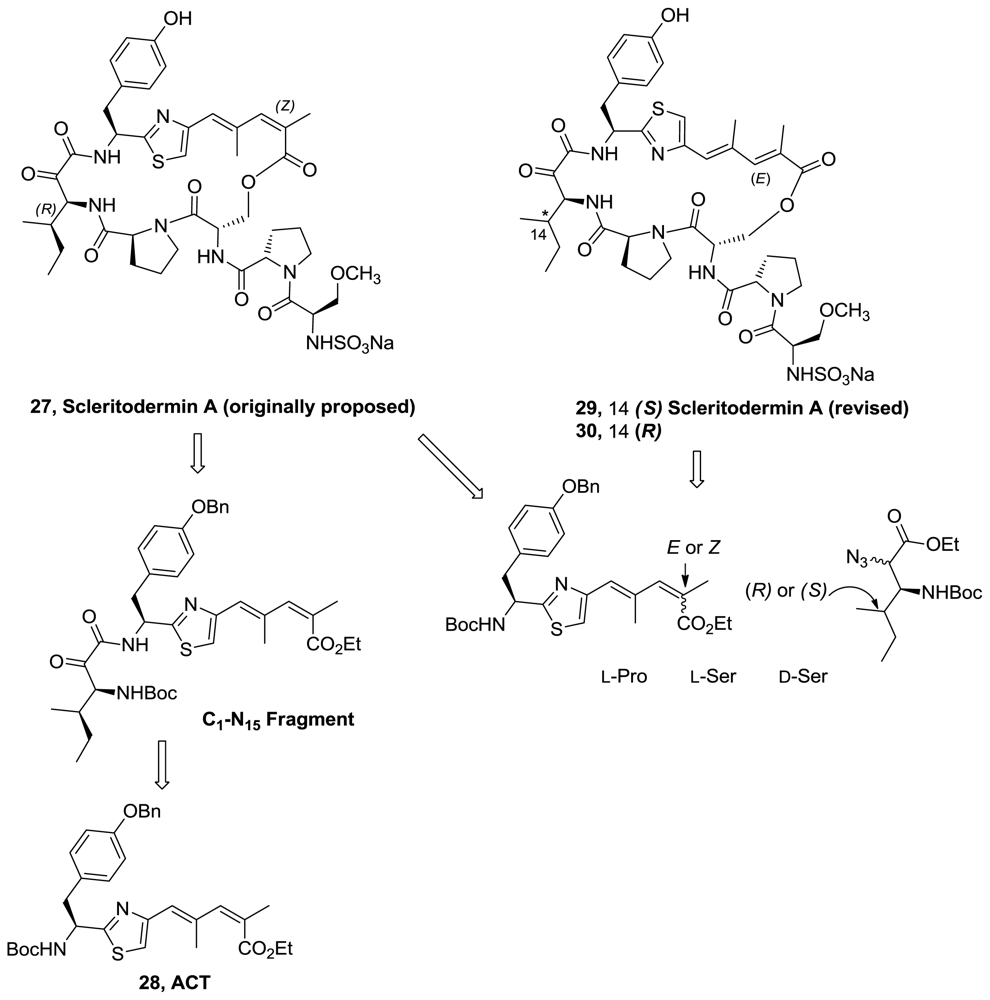
- Sellanes, D; Manta, E; Serra, G. Toward the total synthesis of scleritodermin A: preparation of the C1-N15 fragment. Tetrahedron Lett 2007, 48, 1827–1830. [Google Scholar]
- Liu, S; Cui, Y-M; Nan, F-J. Total synthesis of the originally proposed and revised structures of scleritodermin A. Org Lett 2008, 10, 3765–3768. [Google Scholar]
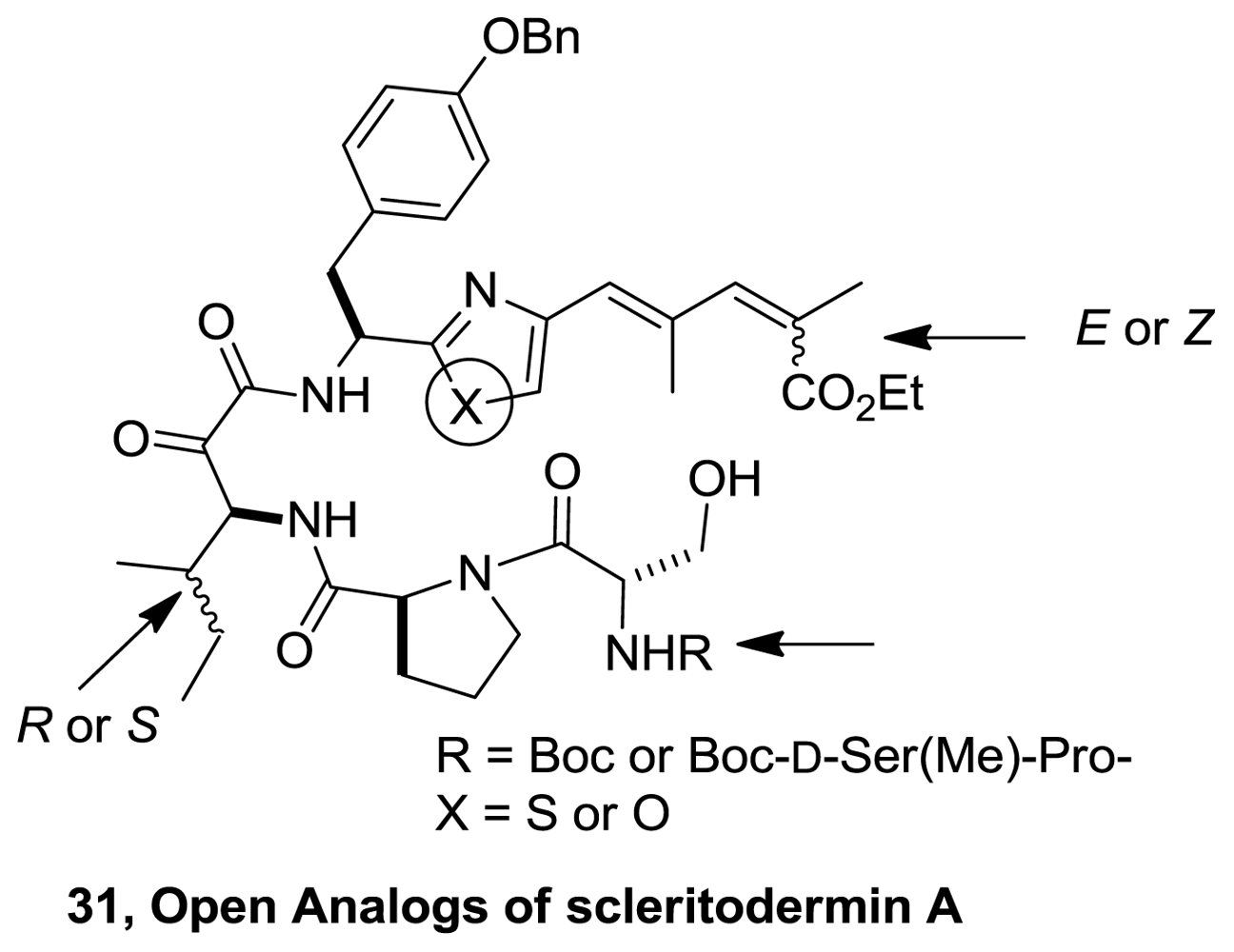
- Sellanes, D; Campot, F; Nunez, I; Lin, G; Esposito, P; Dematteis, S; Saldana, J; Dominguez, L; Manta, E; Serra, G. Preparation and biological evaluation of key fragments and open analogs of scleritodermin A. Tetrahedron 2010, 66, 5384–5395. [Google Scholar]
- Garcia-Reynaga, P; VanNieuwenhze, MS. A new total synthesis of patellamide A. Org Lett 2008, 10, 4621–4623. [Google Scholar]
- Ying, Y; Taori, K; Kim, H; Hong, J; Luesch, H. Total Synthesis and Molecular Target of Largazole, a Histone Deacetylase Inhibitor. J Am Chem Soc 2008, 130, 8455–8459. [Google Scholar]
- Nasveschuk, CG; Ungermannova, D; Liu, X; Phillips, AJ. A Concise Total Synthesis of Largazole, Solution Structure, and Some Preliminary Structure Activity Relationships. Org Lett 2008, 10, 3595–3598. [Google Scholar]
- Seiser, T; Kamena, F; Cramer, N. Synthesis and biological activity of largazole and derivatives. Angew Chem Int Ed 2008, 47, 6483–6485. [Google Scholar]
- Bowers, A; West, N; Taunton, J; Schreiber, SL; Bradner, JE; Williams, RM. Total synthesis and biological mode of action of largazole: A potent class I histone deacetylase inhibitor. J Am Chem Soc 2008, 130, 11219–11222. [Google Scholar]
- Ghosh, AK; Kulkarni, S. Enantioselective total synthesis of (+)-largazole, a potent inhibitor of histone deacetylase. Org Lett 2008, 10, 3907–3909. [Google Scholar]
- Ying, Y; Liu, Y; Byeon, SR; Kim, H; Luesch, H; Hong, J. Synthesis and activity of largazole analogues with linker and macrocycle modification. Org Lett 2008, 10, 4021–4024. [Google Scholar]
- Ren, Q; Dai, L; Zhang, H; Tan, W; Xu, Z; Ye, T. Total synthesis of largazole. Synlett 2008, 2379–2383. [Google Scholar]
- Numajiri, Y; Takahashi, T; Takagi, M; Shin-ya, K; Doi, T. Total synthesis of largazole and its biological evaluation. Synlett 2008, 2483–2486. [Google Scholar]
- Bowers, AA; Greshock, TJ; West, N; Estiu, G; Schreiber, SL; Wiest, O; Williams, RM; Bradner, JE. Synthesis and conformation-activity relationships of the peptide isosteres of FK228 and largazole. J Am Chem Soc 2009, 131, 2900–2905. [Google Scholar]
- Bowers, AA; West, N; Newkirk, TL; Troutman-Youngman, AE; Schreiber, SL; Wiest, O; Bradner, JE; Williams, RM. Synthesis and histone deacetylase inhibitory activity of largazole analogs: alteration of the zinc-binding domain and macrocyclic scaffold. Org Lett 2009, 11, 1301–1304. [Google Scholar]
- Seiser, T; Cramer, N. Syntheses and biological activity of the HDAC class I inhibitor largazole. Chimia 2009, 63, 19–22. [Google Scholar]
- Chen, F; Gao, A-H; Li, J; Nan, F-J. Synthesis and biological evaluation of C7-demethyl largazole analogues. ChemMedChem 2009, 4, 1269–1272. [Google Scholar]
- Yan, W; O’Doherty, GA. Total synthesis of (+)-largazole, a histone deacetylase inhibitor. Chemtracts 2009, 22, 50–58. [Google Scholar]
- Wang, B; Forsyth, CJ. Total synthesis of largazole-devolution of a novel synthetic strategy. Syntesis 2009, 2873–2880. [Google Scholar]
- Zeng, X; Yin, B; Hu, Z; Liao, C; Liu, J; Li, S; Li, Z; Nicklaus, MC; Zhou, G; Jiang, S. Total synthesis and biological evaluation of largazole and derivatives with promising selectivity for cancers cells. Org Lett 2010, 12, 1368–1371. [Google Scholar]
- Souto, JA; Vaz, E; Lepore, I; Poppler, A-C; Franci, G; Alvarez, R; Altucci, L; de Lera, AR. Synthesis and biological characterization of the histone deacetylase inhibitor largazole and C7-modified analogues. J Med Chem 2010, 53, 4654–4667. [Google Scholar]
- Li, D; Yang, HS; Cui, Q; Mao, SJ; Xu, XH. Synthesis of bacillamide 3 and its analogue. Chin Chem Lett 2009, 20, 1195–1197. [Google Scholar]
- Wang, W; Joyner, S; Khoury, KAS; Doemling, A. (−)-Bacillamide C: the convergent approach. Org Biom Chem 2010, 8, 529–532. [Google Scholar]
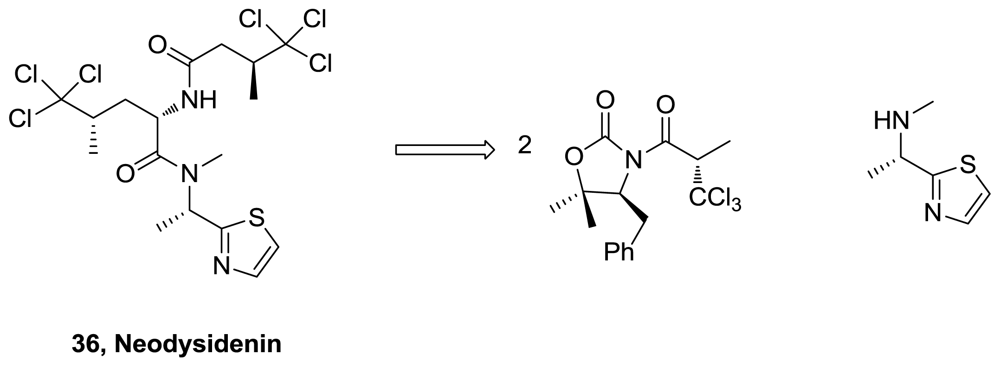
- Beaumont, S; Ilardi, EA; Monroe, LR; Zakarian, A. Valence tautomerism in titanium enolates: catalytic radical haloalkylation and application in the total synthesis of neodysidenin. J Am Chem Soc 2010, 132, 1482–1483. [Google Scholar]
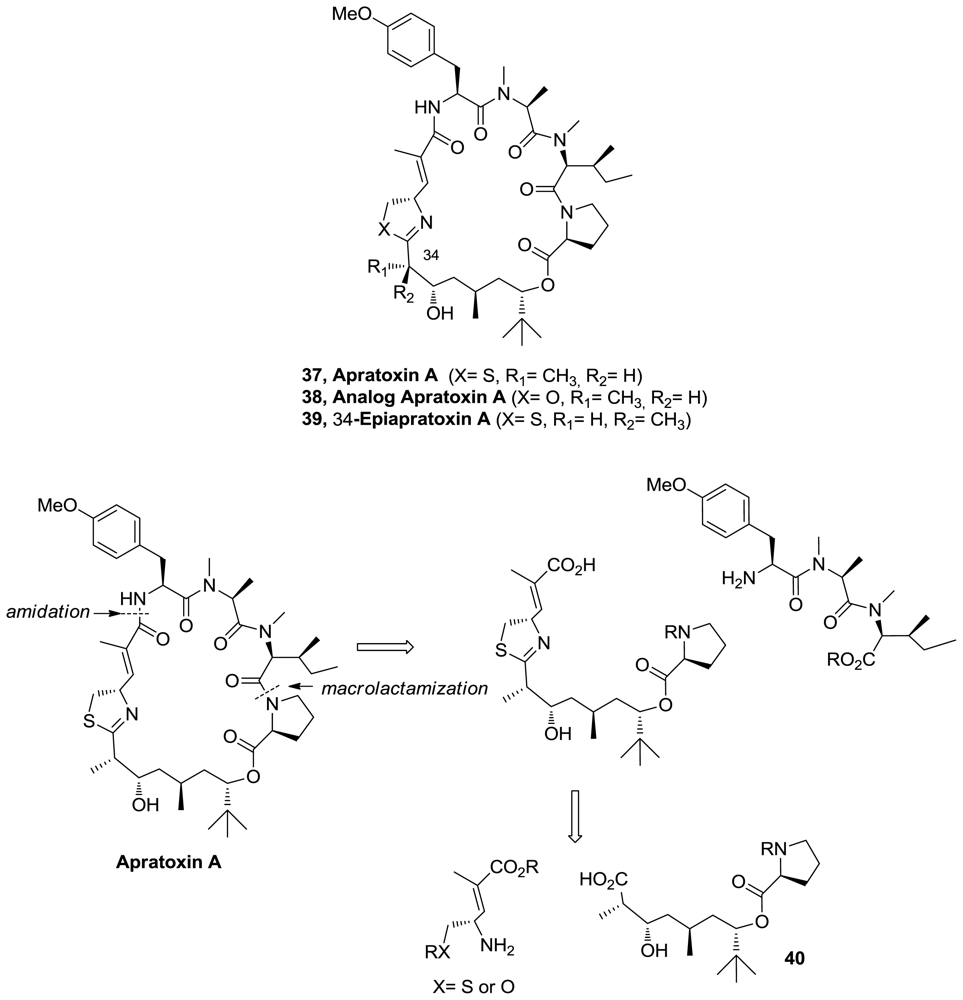
- Numajiri, Y; Takahashi, T; Doi, T. Total synthesis of (−)-apratoxin A, 34-epimer, and its oxazoline analogue. Chem Asian J 2009, 4, 111–125. [Google Scholar]
- Gao, X; Liu, Y; Kwong, S; Xu, Z; Ye, T. Total synthesis and stereochemical reassignment of bisebromoamide. Org Lett 2010, 12, 3018–3021. [Google Scholar]
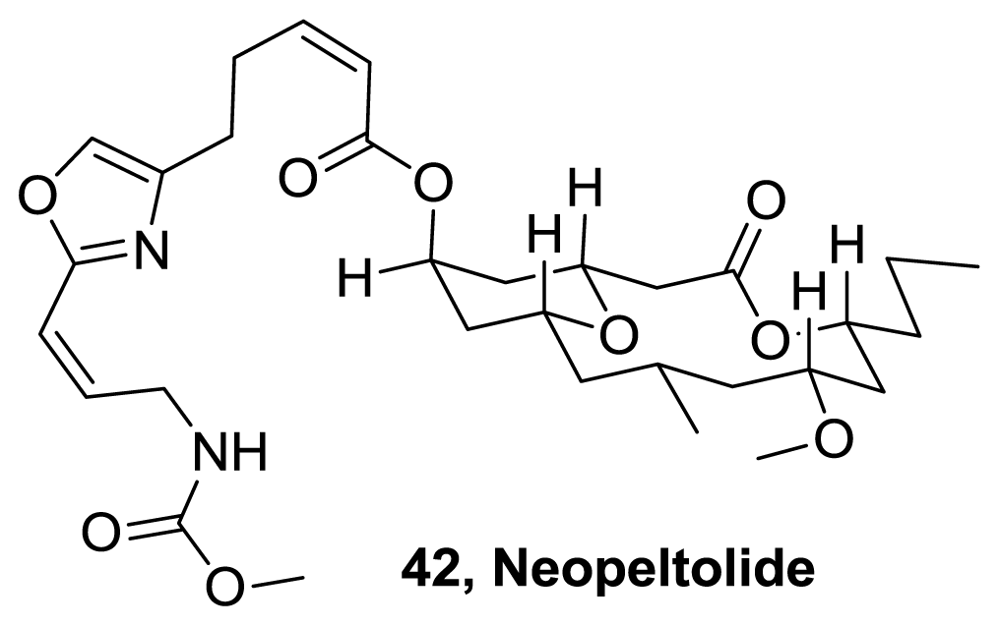
- Wright, AE; Cook Botelho, J; Guzmán, E; Harmody, D; Linley, P; McCarthy, PJ; Pitts, TP; Pomponi, SA; Reed, JK. Neopeltolide, a macrolide from a lithistid sponge of the family Neopeltidae. J Nat Prod 2007, 70, 412–416. [Google Scholar]
- Oku, N; Adachi, K; Matsuda, S; Kasai, H; Takatsuki, A; Shizuri, Y. Ariakemicins A and B, novel polyketide-peptide Antibiotics from a marine gliding bacterium of the genus. Rapidithrix Org Lett 2008, 10, 2481–2484. [Google Scholar]
- Pettit, GR; Hogan, F; Xu, JP; Tan, R; Nogawa, T; Cichacz, Z; Pettit, RK; Du, J; Ye, QH; Cragg, GM; Herald, CL; Hoard, MS; Goswami, A; Searcy, J; Tackett, L; Doubek, DL; Williams, L; Hooper, JN; Schmidt, JM; Chapuis, JC; Tackett, DN; Craciunescu, F. Antineoplastic Agents. 536. New Sources of Naturally Occurring Cancer Cell Growth Inhibitors from Marine Organisms, Terrestrial Plants, and Microorganisms. J Nat Prod 2008, 71, 438–444. [Google Scholar]
- Bishara, A; Rudi, A; Aknin, M; Neumann, D; Ben-Califa, N; Kashman, Y. Taumycins A and B, Two Bioactive Lipodepsipeptides from the Madagascar Sponge Fascaplysinopsis sp. Org Lett 2008, 10, 4307–4309. [Google Scholar]
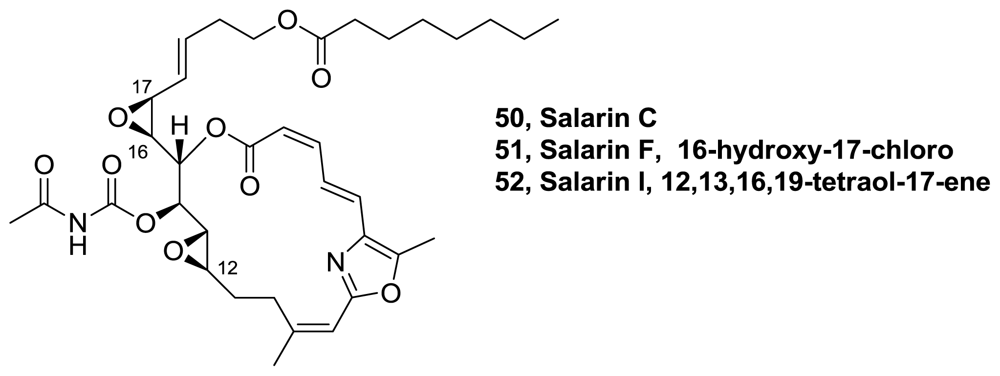
- Bishara, A; Rudi, A; Aknin, M; Neumann, D; Ben-Califa, N; Kashman, Y. Salarin C, a new cytotoxic sponge-derived nitrogenous macrolide. Tetrahedron Lett 2008, 49, 4355–4358. [Google Scholar]
- Bishara, A; Rudi, A; Aknin, M; Neumann, D; Ben-Califa, N; Kashman, Y. Salarins D–J, seven new nitrogenous macrolides from the madagascar sponge Fascaplysinopsis sp. Tetrahedron 2010, 66, 4339–4345. [Google Scholar]
- Dalisay, DS; Rogers, EW; Edison, AS; Molinski, TF. Structure elucidation at the nanomole scale. 1. Trisoxazole macrolides and thiazole-containing cyclic peptides from the nudibranch Hexabranchus sanguineus . J Nat Prod 2009, 72, 732–738. [Google Scholar]
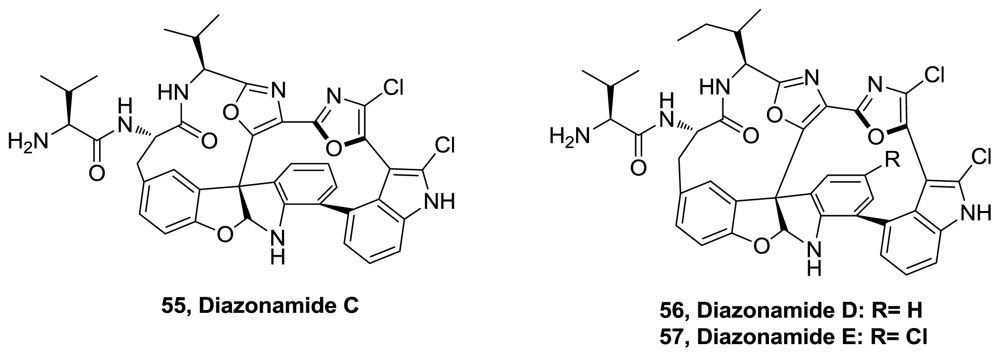
- Fernández, R; Martín, MJ; Rodríguez-Acebes, R; Reyes, F; Francesch, A; Cuevas, C. Diazonamides C–E, new cytotoxic metabolites from the ascidian Diazona sp. Tetrahedron Lett 2008, 49, 2283–2285. [Google Scholar]
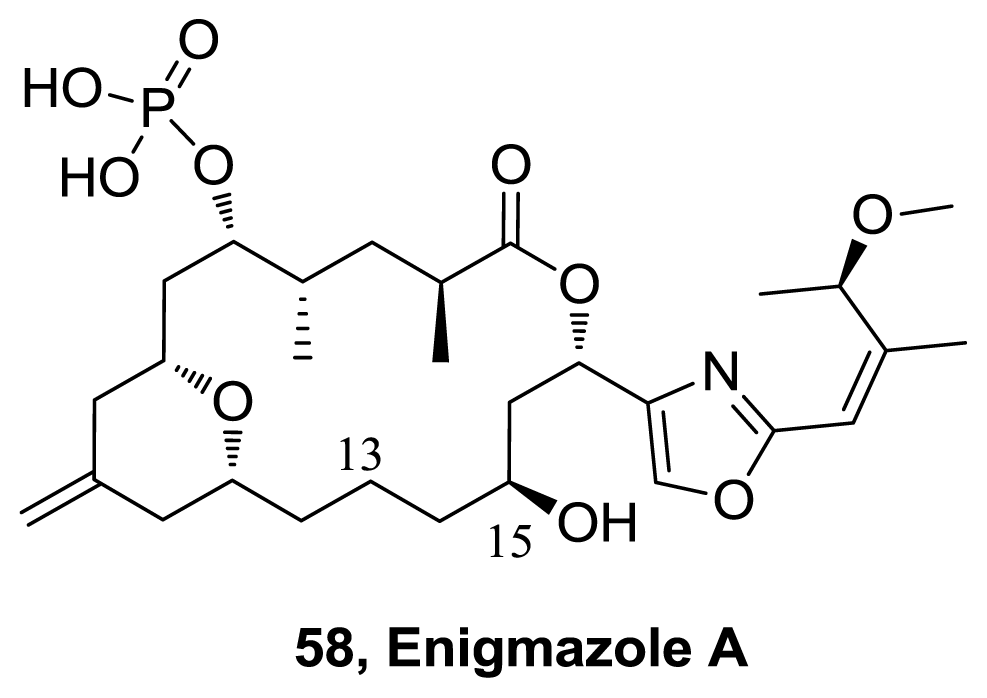
- Oku, N; Takada, K; Fuller, RW; Wilson, JA; Peach, ML; Pannell, LK; McMahon, JB; Gustafson, KR. Isolation, Structural Elucidation, and Absolute Stereochemistry of Enigmazole A, a Cytotoxic Phosphomacrolide from the Papua New Guinea Marine Sponge Cinachyrella enigmatica. J Am Chem Soc 2010, 132, 10278–10285. [Google Scholar]
- Youngsaye, W; Lowe, JT; Pohlki, F; Ralifo, P; Panek, JS. Total Synthesis and Stereochemical Reassignment of (+)-Neopeltolide. Angew Chem Int Ed 2007, 46, 9211–9214. [Google Scholar]
- Custar, DW; Zabawa, TP; Scheidt, KA. Total Synthesis and Structural Revision of the Marine Macrolide Neopeltolide. J Am Chem Soc 2008, 130, 804–805. [Google Scholar]
- Custar, DW; Zabawa, TP; Hines, J; Crews, CM; Scheidt, KA. Total synthesis and structure-activity investigation of the marine natural product neopeltolide. J Am Chem Soc 2009, 131, 12406–12414. [Google Scholar]
- Woo, SK; Kwon, MS; Lee, E. Total synthesis of (+)-neopeltolide by a Prins macrocyclization. Angew Chem Int Ed 2008, 47, 3242–3244. [Google Scholar]
- Fuwa, H; Naito, S; Goto, T; Sasaki, M. Total synthesis of (+)-neopeltolide. Angew Chem Int Ed 2008, 47, 4737–4739. [Google Scholar]
- Paterson, I; Miller, NA. Total synthesis of the marine macrolide (+)-neopeltolide. Chem Comm 2008, 39, 4708–4710. [Google Scholar]
- Vintonyak, VV; Kunze, B; Sasse, F; Maier, ME. Total synthesis and biological activity of neopeltolide and analogues. Chem Eur J 2008, 14, 11132–11140. [Google Scholar]
- Kartika, R; Gruffi, TR; Taylor, RE. Concise Enantioselective Total Synthesis of Neopeltolide Macrolactone Highlighted by Ether Transfer. Org Lett 2008, 10, 5047–5050. [Google Scholar]
- Sasaki, M; Fuwa, H. Total synthesis of (+)-neopeltolide, antitumor marine macrolide. Farumashia 2009, 45, 439–443. [Google Scholar]
- Guinchard, X; Roulland, E. Total Synthesis of the Antiproliferative Macrolide (+)-Neopeltolide. Org Lett 2009, 11, 4700–4703. [Google Scholar]
- Fuwa, H; Saito, A; Naito, S; Konoki, K; Yotsu-Yamashita, M; Sasaki, M. Total Synthesis and Biological Evaluation of (+)-Neopeltolide and Its Analogues. Chem Eur J 2009, 15, 12807–12818. [Google Scholar]
- Fuwa, H; Saito, A; Sasaki, M. A Concise Total Synthesis of (+)-Neopeltolide. Angew Chem Int Ed 2010, 49, 3041–3044. [Google Scholar]
- Cui, Y; Tu, W; Floreancig, PE. Total synthesis of neopeltolide and analogs. Tetrahedron 2010, 66, 4867–4873. [Google Scholar]
- Yadav, JS; Kumar, GG. A concise stereoselective formal total synthesis of the cytotoxic macrolide (+)-Neopeltolide via Prins cyclization. Tetrahedron 2010, 66, 480–487. [Google Scholar]
- Skepper, CK; Quach, T; Molinski, TF. Total Synthesis of Enigmazole A from Cinachyrella enigmatica. Bidirectional Bond Constructions with an Ambident 2,4-Disubstituted Oxazole Synthon. J Am Chem Soc 2010, 132, 10286–10292. [Google Scholar]
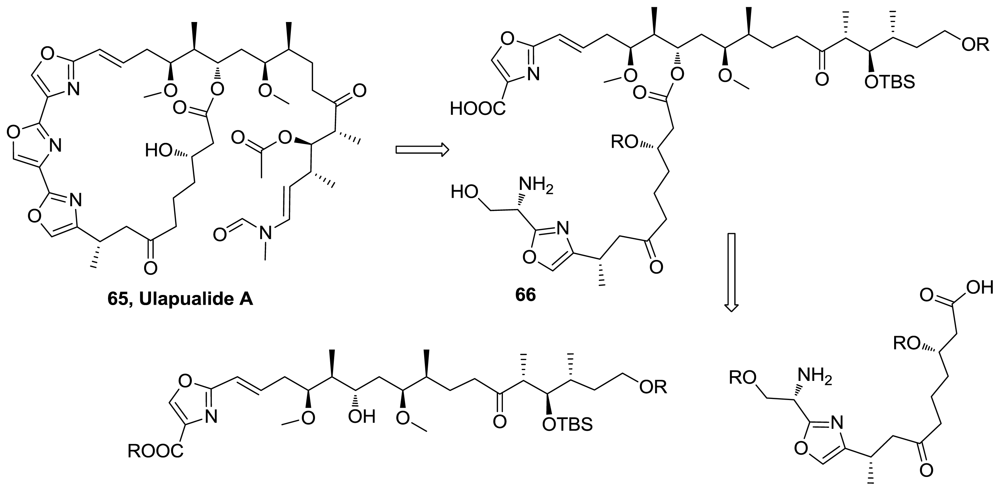
- Pattenden, G; Ashweek, NJ; Baker-Glenn, CAG; Kempson, J; Walker, GM; Yee, JGK. Total synthesis of (−)-ulapualide A, a novel tris-oxazole macrolide from marine nudibranchs, based on some biosynthesis speculation. Org Biomol Chem 2008, 6, 1478–1497. [Google Scholar]
- Pattenden, G; Ashweek, NJ; Baker-Glenn, CAG; Walker, GM; Yee, JGK. Total synthesis of (−)-ulapualide A: The danger of overdependence on NMR spectroscopy in assignment of stereochemistry. Angew Chem Int Ed 2007, 46, 4359–4363. [Google Scholar]
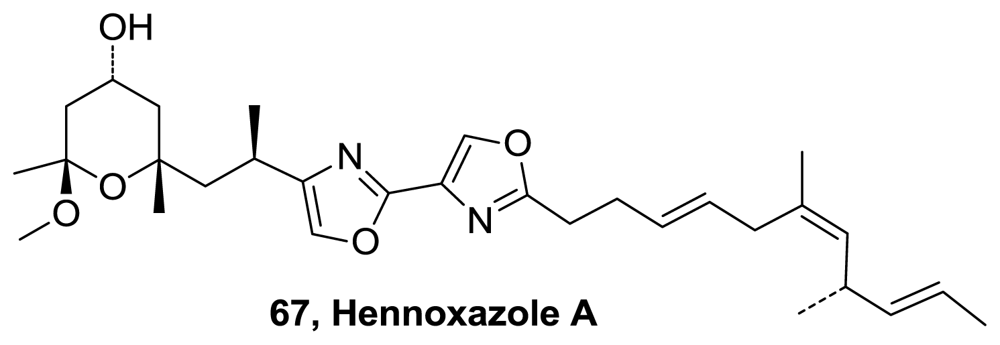
- Smith, TE; Kuo, W; Bock, VD; Roizen, JL; Balskus, EP; Theberge, AB. Total Synthesis of (−)-Hennoxazole A. Org Lett 2007, 9, 1153–1155. [Google Scholar]
- Smith, TE; Kuo, W; Balskus, EP; Bock, VD; Roizen, JL; Theberge, AB; Carroll, KA; Kurihara, T; Wessler, JD. Total Synthesis of (−)-Hennoxazole A. J Org Chem 2008, 73, 142–150. [Google Scholar]
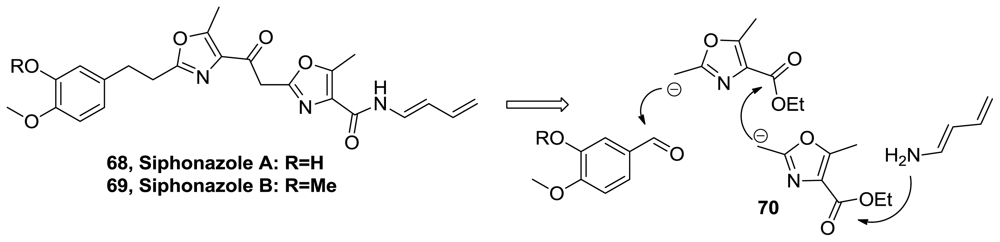
- Nett, M; Erol, O; Kehraus, S; Kock, M; Krick, A; Eguereva, E; Neu, E; König, GM. Siphonazole, an Unusual Metabolite from Herpetosiphon sp. Angew Chem Int Ed 2006, 45, 3863–3867. [Google Scholar]
- Linder, J; Moody, CJ. The total synthesis of siphonazole, a structurally unusual bis-oxazole natural product. Chem Commun 2007, 1508–1509. [Google Scholar]
- Linder, J; Blake, AJ; Moody, CJ. Total synthesis of siphonazole and its O-methyl derivative, structurally unusual bis-oxazole natural products. Org Biomol Chem 2008, 6, 3908–3916. [Google Scholar]
- Zhang, J; Polishchuk, EA; Chen, J; Ciufolini, MA. Development of an Oxazole Conjunctive Reagent and Application to the Total Synthesis of Siphonazoles. J Org Chem 2009, 74, 9140–9151. [Google Scholar]
- Chandrasekhar, S; Sudhakar, A. Total Synthesis of Bengazole A. Org Lett 2010, 12, 236–238. [Google Scholar]
- Enriquez-Garcia, A; Ley, SV. Total synthesis of the potent antifungal agents bengazole C and E Collect Czechoslovak. Chem Commun 2009, 74, 887–900. [Google Scholar]
- Bull, JA; Balskus, EP; Horan, RAJ; Langner, M; Ley, SV. Total synthesis of potent antifungal marine bisoxazole natural products benzazoles A and B. Chem Eur J 2007, 13, 5515–5538. [Google Scholar]
- Scarone, L; Fajardo, J; Saldana, J; Dominguez, L; Esposito, P; Demastteis, S; Wipf, P; Manta, E; Serra, G. Synthesis and evaluation of anthelmintic and cytotoxic properties of [2,5′]bis-1,3-azole analogs of bengazoles. Lett Drug Des Discov 2009, 6, 413–419. [Google Scholar]
- Krauss, J; Kalkbrenner, S; Schuster, A; Obainoke, A; Bracher, F. Synthesis of new bengazole analogues and their antimicrobial activity. Turk J Chem 2008, 32, 125–130. [Google Scholar]
- Mulder, RJ; Shafer, CM; Dalisay, DS; Molinski, TF. Synthesis and structure-activity relationships of bengazole A analogs. Bioorg Med Chem Lett 2009, 19, 2928–2930. [Google Scholar]
- Fresneda, PM; Castaneda, M; Blug, M; Molina, P. Iminophosphorane-based preparation of 2,5-disubstituted oxazole derivatives: synthesis of the marine alkaloid almazole C. Synlett 2007, 2, 324–326. [Google Scholar]
- N’Diaye, I; Guella, G; Mancini, I; Pietra, F. Almazole D, a New Type of Antibacterial 2,5-Disubstituted Oxazolic Dipeptide from a Red Alga of the Coast of Senegal. Tetrahedron Lett 1996, 37, 3049–3050. [Google Scholar]
- Miyake, F; Hashimoto, M; Tonsiengsom, S; Yakushijin, K; Horne, DA. Synthesis of 5-(3-indolyl) oxazole natural products. Structure revision of Almazole D. Tetrahedron 2010, 66, 4888–4893. [Google Scholar]
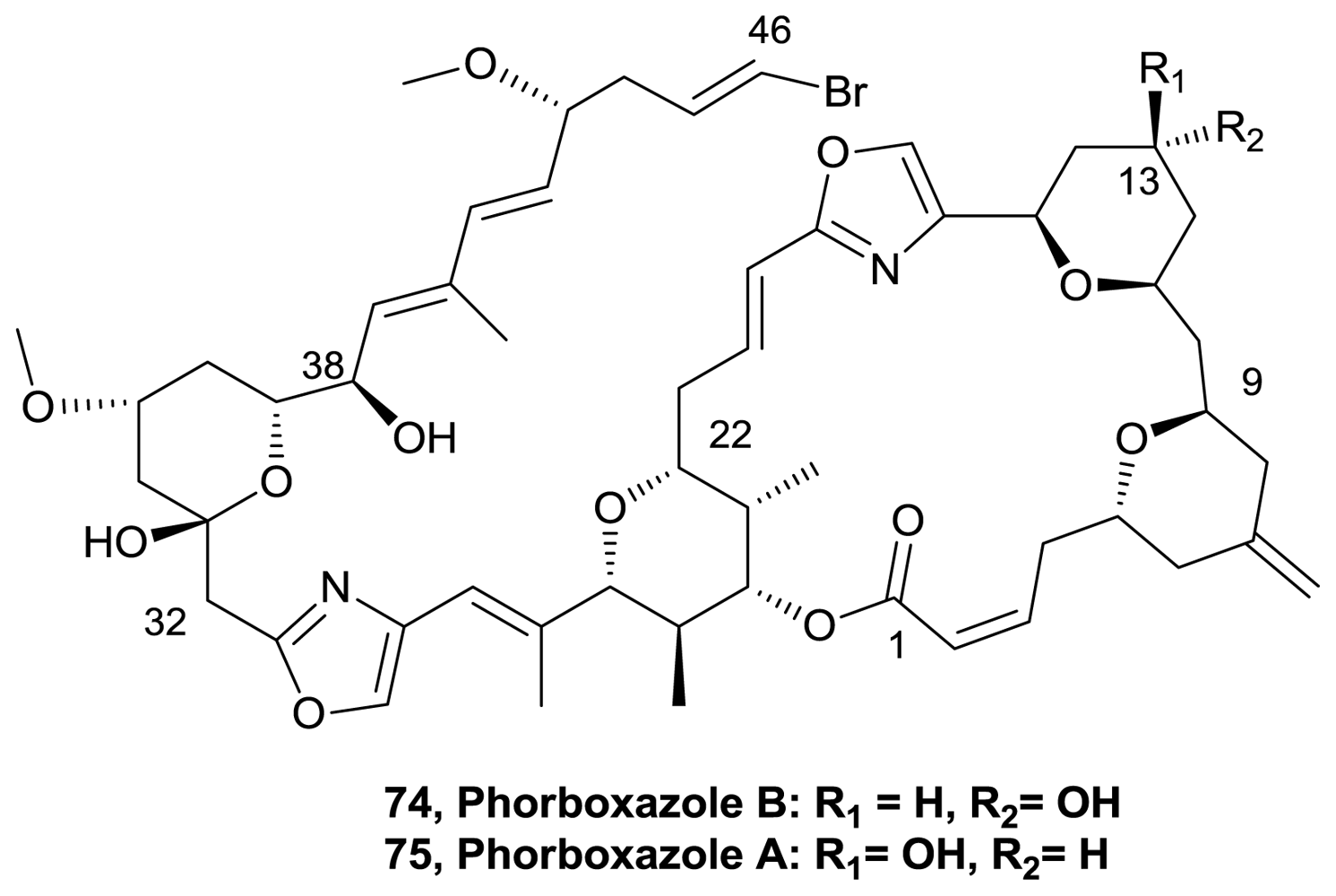
- Lucas, BS; Gopalsamuthiram, V; Burke, SD. Total synthesis of phorboxazole B. Angew Chem Int Ed 2007, 46, 769–772. [Google Scholar]
- Smith, AB, III; Razler, TM; Meis, RM; Pettit, GR. Synthesis and Biological Evaluation of Phorboxazole Congeners Leading to the Discovery and Preparative Scale Synthesis of (+)-Chlorophorboxazole A Possessing Picomolar Human Solid Tumor Cell Growth Inhibitory Activity. J Org Chem 2008, 73, 1201–1208. [Google Scholar]
- Smith, AB, III; Razler, TM; Ciavarri, JP; Hirose, T; Ishikawa, T; Meis, RM. A Second-Generation Total Synthesis of (+)-Phorboxazole A. J Org Chem 2008, 73, 1192–1200. [Google Scholar]
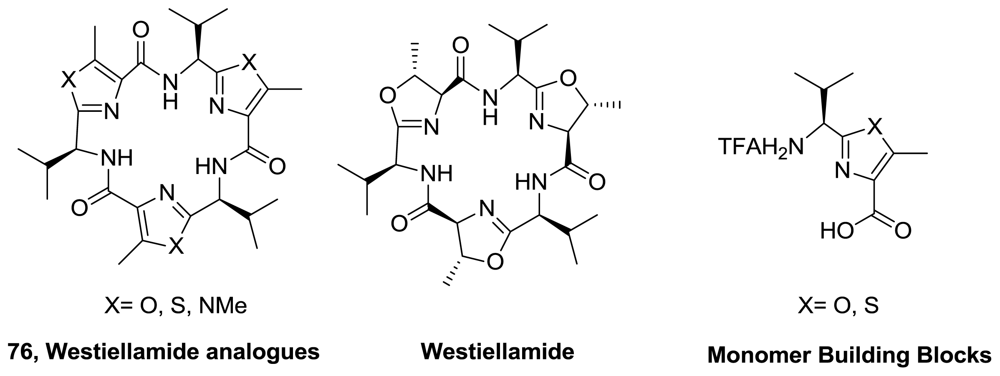
- Haberhauer, G; Drosdow, E; Oeser, T; Rominger, F. Structural investigation of westiellamide analogs. Tetrahedron 2008, 64, 1853–1859. [Google Scholar]
- Comba, P; Gahan, LR; Haberhauer, G; Hanson, GR; Noble, CJ; Seibold, B; van den Brenk, AL. Copper (II) coordination chemistry of westiellamide and its imidazole, oxazole, and thiazole analogues. Chem Eur J 2008, 14, 4393–4403. [Google Scholar]











© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Davyt, D.; Serra, G. Thiazole and Oxazole Alkaloids: Isolation and Synthesis. Mar. Drugs 2010, 8, 2755-2780. https://doi.org/10.3390/md8112755
Davyt D, Serra G. Thiazole and Oxazole Alkaloids: Isolation and Synthesis. Marine Drugs. 2010; 8(11):2755-2780. https://doi.org/10.3390/md8112755
Chicago/Turabian StyleDavyt, Danilo, and Gloria Serra. 2010. "Thiazole and Oxazole Alkaloids: Isolation and Synthesis" Marine Drugs 8, no. 11: 2755-2780. https://doi.org/10.3390/md8112755