Marine Toxins Targeting Ion Channels

Department of Pharmaceutical Sciences, College of Pharmacy, Western University of Health Sciences, Pomona, California 91766, USA
Mar. Drugs 2006, 4(3), 37-69; https://doi.org/10.3390/md403037
Submission received: 10 January 2006
/
Accepted: 27 March 2006
/
Published: 6 April 2006
(This article belongs to the Special Issue Marine Drugs and Ion Channels)
Abstract
:This introductory minireview points out the importance of ion channels for cell communication. The basic concepts on the structure and function of ion channels triggered by membrane voltage changes, the so-called voltage-gated ion channels (VGICs), as well as those activated by neurotransmitters, the so-called ligand-gated ion channel (LGICs), are introduced. Among the most important VGIC superfamiles, we can name the voltage-gated Na+ (NaV), Ca2+ (CaV), and K+ (KV) channels. Among the most important LGIC super families, we can include the Cys-loop or nicotinicoid, the glutamate-activated (GluR), and the ATP-activated (P2XnR) receptor superfamilies. Ion channels are transmembrane proteins that allow the passage of different ions in a specific or unspecific manner. For instance, the activation of NaV, CaV, or KV channels opens a pore that is specific for Na+, Ca2+, or K+, respectively. On the other hand, the activation of certain LGICs such as nicotinic acetylcholine receptors, GluRs, and P2XnRs allows the passage of cations (e.g., Na+, K+, and/or Ca2+), whereas the activation of other LGICs such as type A γ-butyric acid and glycine receptors allows the passage of anions (e.g., Cl− and/or HCO3−). In this regard, the activation of NaV and CaV as well as ligand-gated cation channels produce membrane depolarization, which finally leads to stimulatory effects in the cell, whereas the activation of KV as well as ligand-gated anion channels induce membrane hyperpolarization that finally leads to inhibitory effects in the cell. The importance of these ion channel superfamilies is emphasized by considering their physiological functions throughout the body as well as their pathophysiological implicance in several neuronal diseases. In this regard, natural molecules, and especially marine toxins, can be potentially used as modulators (e.g., inhibitors or prolongers) of ion channel functions to treat or to alleviate a specific ion channel-linked disease (e.g., channelopaties).
1. Introduction
The communication between individual neurons is performed at a morphologically and functionally highly specialized region named the synapse (reviewed in [111]). The name synapse was coined from the Greek syn, meaning “together”, and haptein, meaning “to clasp”. The synapse is structurally shaped by the presynaptic and the postsynaptic membrane both separated by the synaptic cleft. Figure 1 shows a simple scheme of the synapse. A very interesting review on the mechanisms of vertebrate synaptogenesis has been recently published [104].
An important concept on the synaptic mechanism is that the transference of information between cells is achieved neither by a direct contact nor electrically but chemically. The chemo-electrical communication mechanism starts with the elicited action potential on the nerve ending which activates voltage-gated Na+ (NaV) channels (reviewed in [81]). The opening of voltage-gated Na+ channels depolarizes the presynaptic membrane. The depolarization process activates both voltage-gated Ca2+ (CaV) and K+ (KV) channels. On one hand, the opening of CaV channels enhances Ca2+ permeation in the presynaptic membrane (reviewed in [84]). The raised intracellular Ca2+ concentration triggers a series of lipid membrane-vesicle fusion processes (reviewed in [91,101]) that finally produce neurotransmitter release. More specifically, the increase in intracellular Ca2+ activates calmodulin, and subsequently protein kinase C, which in turn phosphorylates other proteins that are involved in the process of vesicle mobilization, docking, and priming (reviewed in [104, 111]). On the other hand, the activation of KV channels provokes membrane hyperpolarization, stopping finally, the presynaptic action potential and thus, neurotransmitter release. Some ligand-gated ion channels (LGICs) with high permeability to Ca2+ (e.g., N-methyl-D-aspartate-subtype glutamate receptor and the α7 nicotinic acetylcholine receptor subtype) can also induce these intracellular processes (reviewed in [77]). A specialized region of the presynaptic membrane called “active zone” fuses with the neurotransmitter-containing vesicles by means of different fusion proteins. Among these proteins we can name the synaptophysin-synaptobrevin complex, a hallmark of synaptic vesicle maturation (e.g., see [15]). The vesicles contain several additional fusion proteins located within its lipid membrane bilayer. After several decades of study, the fusion process is being understood (reviewed in [91,101,111]). Most steps after vesicle-membrane fusion are actually rationalized (reviewed in [29,39]). For example, it is known that after the vesicle content is released into the synaptic cleft the time course of neurotransmitter clearance is between 0.1 and 2.0 ms. The neurotransmitter molecules diffuse through the synaptic space in less than 0.2 ms reaching concentrations of 1–5 mM. Nevertheless, these parameters vary between synapses on a single neuron as well as among synapses from different neurons.
Neurotransmitter release can be modulated by different molecular entities: (1) extracellular and intracellular messengers (reviewed in [42]), and/or (2) presynaptic receptors (reviewed in [49,59,74]). For instance, M2-type muscarinic receptors inhibit the release of the neurotransmitter acetylcholine (ACh) in the presynaptic membrane from the neuromuscular junction. The modulation of neurotransmitter release may lead to short- or long-term adaptations of the synapse to altered environmental signals (reviewed in [59,76]). Finally, the chemical information is converted into electrical currents on the postsynaptic membrane.
The idea of chemical transmission started in the first decade of 1900 (e.g., see [61]), however, it took about 50 more years to be finally established. The molecules bearing chemical information were called neurotransmitters. Up to date, hundreds of neurotransmitters and neuropeptides have been identified as responsible for chemical signaling. In addition, it has been found that a single neuron can synthesize and liberate several of these substances at the same time. Different neurotransmitters operate at different parts of the nervous system and have different effects. Some promote the transmission of impulses while others inhibit it. At least three mechanisms modulating the neurotransmitter concentration in the synaptic cleft are well known: (a) enzymatic inactivation. For instance, the neurotransmitter ACh is catabolized to choline and acetate by neuronal acetylcholinesterases; (b) presynaptic membrane re-uptake by specific neurotransmitter transporters. Figure 1 shows neurotransmitter transporters. This mechanism is used for the re-uptake of monoamine neurotransmitters such as norepinephrine, epinephrine, dopamine, and serotonin (5-hydroxytryptamine; 5-HT); and (c) soluble neurotransmitter-binding proteins. Several proteins homologous to the soluble ACh-binding protein (AChBPs) secreted by Lymnaea stagnalis glial cells [90] have been characterized. For instance, AChBPs from mollusks such as Aplysia californica[47] and Bulinus truncatus[24]. The secretion of these proteins is modulated by the neurotransmitter itself [90] (reviewed in [89]). Functionally, they act as neurotransmitter carriers and they may buffer the neurotransmitter concentration in the synapse. Another potential function is that they may transport neurotransmitter molecules to extrasynaptic sites, allowing the interaction with, for instance, glial cells or other adjacent neurons. Similar binding proteins for other neurotransmitters might also exist in the central nervous system (CNS) from vertebrates.
The postsynaptic membrane is highly specialized in the recognition and binding of neurotransmitters by means of lipid-embedded protein receptors. On the basis of their elicited physiological response two main receptor classes have been distinguished: ionotropic receptors and metabotropic receptors. The binding of one kind of neurotransmitter to its specific metabotropic receptor produces a slow biological response. The mechanism by which the activated receptor produces its final effect on the cell is mediated not directly but indirectly by the enzymatic interaction with one of the members of the G-protein family. An example of metabotropic receptor is the adrenergic receptor. On the other hand, the specific binding of a ligand to its ionotropic receptor induces a fast opening of the ion channel intrinsically coupled to the receptor. Figure 1 shows the location of postsynaptic ionotropic receptors. Interestingly, perisynaptic or extracellular ionotropic receptors have also been found (reviewed in [35,49,59]). The function of these receptors is not totally clear, but some perisynaptic receptor types may produce separate and sometimes opposite effects as its postsynaptic counterpart (reviewed [39,59]).
2. Ion channels
Ion flux across excitable cells mediated by activation of ion channels is a signaling mechanism that it is extensively used by different organisms from the three major superkindoms of life. Ion channels are responsible for generating and orchestrasting the electrical signals throughout the body. These signals ultimately modulate the thinking process in the brain, the heartbeat in the heart, and the muscle contraction in the muscle, to name only a few important physiological functions. Using methods such as molecular biology and electrophysiology, researchers have cloned, expressed, and characterized genes encoding many of these ion channels.
Ion channels may be gated by different signals, including membrane voltage changes, mechanical forces, and chemicals. Ion channels that are triggered by specific endogenous ligands (e.g., neurotransmitters, neuropeptides, etc.) are called LGICs. LGICs are genetically, structurally, and functionally different from voltage-gated ion channels (VGICs). One of the main distinctions is that VGICs are gated not by endogenous ligands but by membrane voltage changes (reviewed in [17,81,95]).
Several ion channels from different organs and tissues have been implicated in the pathophysiology of distinct diseases (reviewed in [2]). For instance, mutations in the epithelial chloride channel, CFTR (the cystic fibrosis transmembrane regulator), are responsible for cystic fibrosis, mutations in the renal chloride channel are responsible for hypercalciuric nephrolithiasis (Dent’s disease), truncation in the ATP-sensitive pancreatic potassium channel is involved in familial persistent hyperinsulinemic hypoglycemia from the infancy, mutations in the epithelial sodium channel are related with Liddle’s syndrome (produces hereditary hypertension and pseudoaldosteronism), mutations in the skeletal- muscle chloride channel are involved with different myotonias (e.g., Becker’s or Thomsen’s myotonia), mutations in the ryanodine calcium channel are related with the central core storage disease and with malignant hyperthermia. The list of the most important diseases related with VGICs or LGICs are provided in the following sections.
In addition, ion channels are the targets of several marine toxins. One of the most important toxins from the point of view of their diversity and specificity are the snail toxins obtained from different Conus species. In this regard, Layer and McIntosh review, in this special issue, the most important structural details of these conotoxins as well as their therapeutical potential for the treatment of different diseases.
3. Voltage-Gated Ion Channels
Voltage-gated ion channels are complex proteins that are embedded in the lipid membrane of the cell. These channels conduct ions at very high rates (~1 million ions per second) and are regulated by the voltage across the membrane. The best known VGICs are NaV, KV, and CaV channels, as well as voltage-gated Cl− channels. This classification corresponds to the type of ion that each channel allows to pass. Subunits homologous to subunit α from the different VGICs form the structure of the ion pore. Subunit α also bears the voltage sensor that allows the channel to detect and gate in response to changes in the transmembrane voltage (reviewed in [17,95]). The opening of only one of these ion channels allows the passage of about 10 million ions per second (reviewed in [2]). In this regard, every time that a channel is open, a current of few picoamperes (pA) is generated (1 Ampere = 1 coulomb/sec = 6.24×1018 electrons moving through a surface in one second). Since these channels are very efficient, there are only few thousand per cell of a given type. Consistent with the normal electrochemical gradients across the cell membrane for these ions, the opening of NaV or CaV channels induces membrane depolarization by allowing positive Na+ or Ca2+ ions flow into the cell. In contrast, the opening of KV or voltage-gated Cl− channels induces membrane hyperpolarization (K+ exits from, whereas Cl− enters, the cell, increasing the number of negative charges at the cytoplasmic surface of the membrane). Additional subunits (e.g., α2, β1, β2, and γ) from these ion channels have accessory functions. For instance, they modulate ion channel function, and interact with cytoskeleton proteins for anchoring as well as with protein kinases for phosphorylation processes.
Given their physiological importance, VGICs are the targets for numerous small molecules and toxins of natural origin. Malfunctioning of these VGICs is implicated in many important diseases, and these ion channels are under intense scrutiny as potential targets for drugs for the treatment of different diseases. In this regard, Messerli and Greenberg, in this volume, review the effects of Cnidarian toxins (marine toxins) in VGICs.
3.1. The Voltage-Gated Na+ Channel Superfamily
Voltage-gated Na+ channels were purified from Electrophorus electricus electric organs in 1978 [3]. Since then, a good deal of information on the structure and function of different NaV channels has been obtained. Mammalian NaV channels from brain are structurally formed by three different subunits, α, β1, and β2, whereas channels formed by subunits α and β3 are present in dorsal root ganglia, and those comprised by subunits α and β1 are found in skeletal muscle (reviewed in [36,110]). There are at least nine α subunits forming channels NaV1.1–NaV1.9. The Nomenclature Committee from the International Union of Pharmacology has recently reviewed and accepted a new nomenclature for NaV channel types [22,110].
In the NaV channel structure, the α subunit, which makes up the core structure, is formed by four domains (I-IV), each containing six transmembrane segments (S1–S6) (reviewed in [17,110]). In contrast, each β1 and β2 subunit has just one transmembrane segment. Figure 2 depicts the schematic structure of the NaV channel. Interestingly, the ion-conducting pore of the channel is supported by the S5 and S6 transmembrane segments from the α subunit. The outer part of the pore is lined by a reentrant loop (the P loop) that links the transmembrane segments S5 and S6, while the C-terminal ends of the S6 segments line the cytoplasmic section of the pore. Certain amino acid residues in the C-terminal segment of the P loop are critical for determining the conductance and ion selectivity of the Na+ channel. The activation gate is probably formed by the S6 segments at a point where they come into close apposition in the cytoplasmic half of the pore. The conformational change from closed to open is ultimately dependent on the motion of the positively charged, S4 segments, which act as voltage sensors by responding to changes in the electric field within the membrane. The transmembrane segment S4 is positively-charged and functions as a sensor for voltage changes across the lipid membrane. The activation of this sensor induces complex conformational changes in the protein that finally open the pore allowing the flux of Na+ into the cytoplasm of the cell. In vivo, Na+ influx produces a regenerative membrane depolarization in the millisecond time frame. The rapid depolarization activates both KV channels, which contribute to the rapid repolarizing phase of the action potential, and CaV channels. The activation of presynaptic CaV channels, in turn, induces neurotransmitter release. Figure 3B shows a voltage-clamp recording of NaV channel activity in response to a step depolarization of the membrane.
NaV channels undergo cyclic conformational changes among at least three functionally different states. Figure 3A shows the three main conformational states. In the resting state, the ion channel is nonconducting. Membrane voltage changes trigger ion channel opening in the millisecond time regime. This conducting state is called the activated state. A few milliseconds after the initial activation of NaV channels, an intrinsic process of inactivation takes place in which the short intracellular loop connecting domains III and IV serves as the fast inactivation gate of the ion channel. This loop folds into the intracellular mouth of the ion pore and occludes it, ending the process of membrane depolarization as well as of neurotransmitter release. The hydrophobic residues isoleucinephenylalanine- methionine (IFM) are considered to serve as the inactivating domain. The inactivated state is a closed state which is distinct from the resting state. Although the channel inactivation process is mechanistically different from the desensitization found in LGICs, both processess produce similar modulatory effects on these ion channels. Finally, the gating process can be modulated by different protein kinases and phosphatases. Phosphorylation of the inactivation gate by protein kinase C slows inactivation, whereas phosphorylation of the intracellular loop between domains I and II by either protein kinase C or A reduces channel activation. Such modulations modulate patterns of electrical activity in neurons and other excitable cells.
Different mutations in NaV channels are implicated in several diseases, the so-called “channelopathies”, from the muscle, heart, or nervous system (reviewed in [2,41,110]). For example, mutations in cardiac NaV1.5 channels are responsible for long-QT type 3 and Brugada syndromes which produce cardiac arrhythmias, whereas mutations in the skeletal-muscle NaV1.4 channel are implicated in “hyperkalemic periodic paralysis” (a muscle weakness disorder aggravated by high K+), in Masseter-muscle rigidity (succinylcholine-induced), as well as in different kinds of myotonias (e.g., paramyotonia congenital, a syndrome of cold- and exercise-induced muscle stiffness). In addition, mutations in the neuronal NaV1.1 channel produce “generalized epilepsy with febrile seizures plus, type II”, whereas mutations in the β1 subunit produce “generalized epilepsy with febrile seizures plus, type I”. Finally, knockout mice lacking the NaV1.8 channel are viable but their response to pain is not normal.
NaV channels are targets for the action of natural as well as synthetic drugs (reviewed in [2,36,37]). Among clinically important synthetic drugs we can name anticonvulsants (e.g., carbamazepine, phenytoin, and valproic acid), local anesthetics (e.g., bupivacaine, cocaine, lidocaine, mepivacaine, and tetracaine), as well as Class IA (e.g., disopyramide, procainamide, and quinidine), Class IB (e.g., lidocaine, mexiletine, phenytoin, and tocainide), and Class IC antiarrhythmics (e.g., encainide, flecainide, and propafenone). Recent research efforts have focused on the development of analgesic agents which selectively block dorsal root ganglion sodium channels that are involved in transmission of pain sensation.
NaV channels present at least six binding sites for synthetic and natural molecules (Sites 1–6, as well as the binding site for local anesthetics) that are located in different domains throughout the protein (reviewed in [36]). Table 1 shows the most important natural molecules including marine toxins that affect NaV channels. Natural drugs targeting NaV channels are chemically very diverse and include polypeptides, alkaloids, cyclic polyethers, esters, and heterocycles. For example, the reversible blockers tetrodotoxin (from puffer fish and related species from the Tetraodontidae family), maculotoxin (actually tetrodotoxin present in the blue-ringed octopus Hapalochlaena maculosa), saxitoxin (from different algal species of the order Dinoflagellata such as Alexandrium tamarense, Gymnodinium catenatum, and Pyrodinium bahamense, as well as from certain freshwater cyanobacteria), and μ–conotoxins GIIIA, GIIIB, and GIIIC (polypeptides from the marine snail Conus geographus) and PIIIA (from C. purpurascens) bind to Site 1. Certain amino acids from the pore lining in the extracellular mouth of the ion channel are critical for the binding of these toxins. The main pharmacological action of these toxins is to block current flow through the pore thus, decreasing membrane depolarization and, as a consequence, also inhibiting neurotransmitter release. The toxic effects induced by these toxins include diarrhea, vomiting, paralysis, paraesthesia, dyspnea, hypotention, convulsions, mental impairments, cardiac arrhythmia, respiratory failure, and finally death.
Local anesthetics also block the ion channel but its binding site is located in a different domain from that for Site 1. Interestingly, local anesthetics pharmacologically behave as noncompetitive antagonists (NCAs) of nicotinic acetylcholine receptors (AChRs) (reviewed in [9,12]). Batrachotoxin (alkaloid from the skin of Colombian arrow poison frogs of the genus Phyllobates), veratridine (alkaloid from plants of the Liliaceae family), aconitine (alkaloid from the plant Aconitum napellus), and grayanotoxin (diterpenes from several Rhododendron species) bind to Site 2. The main action elicited by these drugs is to produce persistent channel activation. In the heart, batrachotoxin, veratridine, and grayanotoxin, but not aconitine, produce positive ionotropic effects (i.e., increased contraction force). Class 1 scorpion α-toxins (from the genera Buthus, Androctonus, and Leiurus), δ-atracotoxins (from Australian funnel web spiders of the Atracinae family such as Atrax robustus which produces robustoxin and Hadronyche versutus that produces versutoxin), μ-agatoxin-I and -IV (from the web spider Agelenopsis aperta), sea anemone toxins (from the genera Anthopleura and Anemonia), several toxins from the spider Phoneutria nigriventer, and δ–conotoxins such as PVIA (from C. purpurascens) as well as TxVIA (also called King-Kong peptide TxIA), TxIB, and TxIIA (from C. textile neovicarius) bind to Site 3. This site is located in the loop formed between the transmembrane segments S3 and S4 from domain IV. The main pharmacological effects of these toxins are to slow inactivation, and/or enhance or prolong activation. Persistent channel activation sustains membrane depolarization, enhancing neurotransmitter release, leading to rigid paralysis and, finally, death. Toxins that bind to Site 3 enhance the activity of molecules acting on Site 2. Scorpion β-toxins from the genus Centuroides bind to Site 4. Class 2 Scorpion β-toxins shift the voltage-dependence of activation to more negative membrane potentials without modifying the channel inactivation process, whereas Class 3 Scorpion β-toxins alter the voltage-dependence of both activation and inactivation. The lipid-soluble polyether marine toxins brevetoxins (produced by the dinoflagellate Karenia brevis) and ciguatoxins (from several dinoflagellate species including Gambierdiscus toxicus) bind to Site 5. The effects elicited by these NaV channel activators include repetitive neuronal firing, shifting the voltage-dependence, and blocking the channel inactivation process. Finally, insecticides such as pyrethroids (from the flowers of certain Chrysanthemum species) and dichlorodiphenyltrichloroethane (DDT) bind to Site 6, a poorly defined binding domain. These compounds produce hyperexcitability and paralysis preferentially in invertebrates rather than in mammals. Sites of action of fast inactivation inhibitors such as pronase, N-bromoacetamide and chloramine-T have not been positively identified as well as for prolongers including DPI 201-106 and BDF9148 that are in experimental stage as therapeutics for the treatment of congestive heart failure. Finally, the location of binding sites for other natural toxins are still under debate. These include homobatrachotoxin (steroidal alkaloids from birds of the genus Pitohui) which activates NaV channels, and phoneutriatoxins (from the Banana spider Phoneutria nigriventer) as well as α- and β-pompilidotoxins (from the solitary wasps Anoplius samariensis and Batozonellus maculifrons) which slow the NaV channel inactivation process.
In this special issue, French and collaborators review the general aspects of the structural and functional properties of marine toxins that interact with NaV channels. In addition, Geffeny and Ruben cover the information regarding the specific interaction of tetrodotoxin with NaV channels, and finally Nicholson and Lewis present updated information regarding the pharmacological effects of ciguatoxins on NaV channels.
3.2. The Voltage-Gated Ca2+ Channel Superfamily
CaV channels were first identified in crustacean muscle fibers by Fatt and Katz [40]. The use of molecular biology has helped to identify subclasses of these channels in all types of excitable cells in vertebrates and invertebrates, and even in plants. CaV channels can be classified depending on their activity or site of activity. For instance, in neurons, CaV2.1 and CaV2.2 channels are located presynaptically and are involved in neurotransmitter release. Almost all other types have been found in cell bodies and dendrites. The currently accepted nomenclature for CaV channels is found in Catterall et al. [23], and summarized here in Table 2.
CaV channels are structurally formed by several subunits including α1, α2, δ, β, and γ (reviewed in [95,109]). The α1 subunit is formed by six transmembrane segments, and contains the ion channel, the voltage sensor, and the gate. This structural arrangement is shown in Figure 4, and it is similar to NaV channels (see Fig. 2). The β subunit is located intracellularly and it is involved in membrane trafficking of α1 subunits. The α2 subunit is a highly glycosylated extracellular protein that is attached to the membrane-spanning δ subunit by means of disulfide bonds. The α2 subunit provides structural support, whereas the δ subunit modulates the voltage dependent activation and steady-state inactivation processes. The γ subunit is a glycoprotein having four transmembrane segments.
The biophysical properties of these channels were studied by using voltage- and patch-clamp methods. CaV channels are normally closed at resting membrane potentials, and open upon membrane depolarization. The resultant current through the cell membrane can be characterized by a number of properties including the membrane potential range over which the channel reaches its maximum open probability as well as the current kinetics. The use of selective drugs and toxins has helped to identify specific current components observed in an entire cell that correspond to a particular channel type.
There is accumulating evidence to support the view that CaV channels are implicated in several diseases and channelopathies (reviewed in [2]). For instance, autoantibodies against P/Q-type CaV channels are produced in the Lambert-Eaton syndrome, reducing the number of these channels in the presynaptic cell and provoking myasthenic syndrome (i.e., muscle weakness). Mutations in dihydropiridine-sensitive CaV channels, more specifically in the S4 region, are related with the muscle disorder “hypokalemic period paralysis”. Other mutations have been implicated in diseases such as spino-cerebellar ataxia (type 6), familial hemiplegic migraine, episodic ataxia (type 2), and X-linked congenital night blindness among others.
Different CaV channel families are targets for distinct therapeutically relevant drugs (reviewed in [2,37,109]). Table 2 shows the pharmacological effect of several drugs including marine toxins on different CaV channels. For instance, the CaV1 channel family (L-type currents) is the target for the pharmacological action of different drugs used in the treatment of cardiovascular diseases including phenylalkylamines, dihydropyridines, and benzothiazepines (class IV antiarrhythmics). Interestingly, these classes of drugs bind to different sites. Phenylalkylamines (e.g., verapamil, devapamil, gallopamil, and thiapamil) enter the pore from the cytoplasmic side and bind to a site located at segment S5 from domain IV, similar to that for local anesthetics in the NaV channel, and occlude the pore. Dihydropyridines (e.g., nifedipine, nicardipine, nitrendipine, nimodipine, nisoldipine, and felodipine) inhibit the ion channel by an allosteric mechanism. This site is formed by segment S6 from domains III and IV and segment S5 from domain III. Other dihydripyridine derivatives (e.g., Bay K8644) activate CaV channels producing vasoconstriction and positive ionotropic effects. Benzothiazepines (e.g., diltiazem) access the channel from the extracellular side and bind to a site located close to the extracellular mouth. Snake toxins such as calcicludine and calciseptine (from Dendroaspis angusticeps) as well as TaiCatoxin (from Oxyuranus scutellatus scutellatus) are also potent and selective blockers of L-type channels. The CaV2 channel family is specifically blocked by spider and marine snail toxins. For example, ω-agatoxins IVA and IVB (from the funnel web spider Agelenopsis asperta) as well as ω-conotoxins MVIIC and MVIIA (from C. magus) inhibit the gating process in CaV2.1 channels (P/Q-type currents), whereas ω-conotoxins GVIA (from C. geographus) blocks CaV2.2 channels (N-type currents). Since SNX-482 (from the tarantula Hysterocrates gigas) specifically inhibits CaV2.3 channels, this toxin was crucial for the characterization of R-type currents. A polyamine with analgesic properties obtained from the fishing spider Dolomedes okefinokensii reversibly blocks L-and R-type CaV channels.
On the other hand, the activation gating on CaV3 channels (T-type currents) is inhibited by kurtoxin (obtained from South African scorpions). Mibefradil and pimozide block CaV3 channels slightly more strongly than either CaV1 or CaV2 channels. In addition, ω-grammotoxin SIA (obtained from the South American rose tarantula Grammostola spatulata), HWTX-I (obtained from the Chinese bird spider), taicatoxin (obtained from the Australian Taipan snake), and several spider toxins from Phoneutria ngriventer, block different CaV channels as well.
Regarding CaV channel marine toxins, Schroeder and Lewis cover the effect of ω-conotoxins and their therapeutical applications, whereas Zhang is reviewing different aspects of the interaction of SO- 3 conotoxins with N-type CaV channels.
3.3. The Voltage-Gated K+ Channel Superfamily
The first KV channel sequence was determined from the Shaker mutant of Drosophila melanogaster in 1987 [94]. Since then, a great effort has been done to elucidate the structural arrangement of this channel family that recently culminated in the crystallographic structure of the mammalian Shaker KV channel [71]. KV channels are more diverse and widespread than other ion channel families (reviewed in [76]). In this regard, KV channels have been divided in nine families (e.g., KV1–KV9) (reviewed in [95]). KV1–KV4 channels are also called Shaker, Shab, Shaw, and Shal, respectively. The current nomenclature for these channels is found in Gutman et al. [46]. Regarding their assembly properties, families KV1–KV4 can form homomeric or heteromeric channels, whereas families KV5–KV9 cannot form homomeric channels but interact with members from the KV2 family to form heteromeric channels. This is why subunits KV5–KV9 are called modulatory subunits.
Figure 5 depicts the most important structural features of a prototypic KV channel. KV channels are composed of four separate α subunits, each containing six transmembrane segments (S1–S6) (reviewed in [95]). The α subunits are assembled as a tetramer to form a central pore that is specific for K+ ions. Four accessory β subunits (e.g., β1 and β2) may be complexed with the α subunits in a stoichiometry α4β4 to form the functional KV channel. The α subunit determines the basic gating properties. More specifically, the peptide chain (H5 or loop P) between segments S5 and S6 lines the water-filled channel pore. This region is highly conserved in all VGICs. Mutations in this region alter the permeation properties of the ion channel. As for NaV channels, KV channels have an S4 segment, bearing a cluster of positively-charged residues (e.g., lysines and arginines), that serves as the voltage sensor. Voltage-dependent fast inactivation (N-type) of the channel is mediated by a long chain of amino acids with a ball in the amino terminal that swings into the mouth of the ion channel and blocks the permeation pathway from the cytoplasmic side (i.e., the “ball and chain” model). Several residues in S6 are involved in the slow inactivation process (C-type). Figure 5 illustrates the basic “ball and chain” model.
KV channel mutations are implicated in several important diseases (reviewed in [76]). Among them we can name cardiac arrythmias (e.g., long-QT syndromes), congenital deafness, epilepsy, diabetes, misregulation of blood pressure, multiple sclerosis, Alzheimer’s disease and other dementias, migraine, pain, anxiety and bipolar disorder.
KV channels are also the target of several synthetic and natural drugs (reviewed in [2,37,54,86]). Among synthetic drugs we can name antidiabetic drugs (e.g., glipizide, glyburide, and tolazamide), antihypertensive drugs (e.g., diazoxide and minoxidil), and Class III antiarrhythmics (e.g., amiodarone, clofillium, dofetilide, and sotalol). Among nonselective blockers we can include inorganic cations (e.g., Li+, Cs+, Ba+, Hg+, and Zn2+) and quaternary ammonium derivatives (e.g., tetramethy-, tetrahethyl-, tetrabutyl-ammonium derivatives). Aminopyridines (e.g., 4-aminopyridine, and 3,4-diaminopyridine) are more selective drugs in clinical trial for the treatment of long-standing spinal cord injury. These drugs assist recovery of sensory and motor functions, enhance pulmonary functions, and diminish muscle spasticity.
Regarding natural toxins, Scorpionidae toxins including charybdotoxin (obtained from Leiurus quinquestriatus hebraeus), agitoxin-2 (obtained from Leiurus quinquestriatus hebraeus), margatoxin (obtained from Centruroides margaritatus), noxiustoxin (obtained from Centruroides noxius), anuroctoxin (obtained from Anuroctonus phaiodactylus), tityustoxin-Kα and -Kβ (obtained from the Brazilian scorpion Tityus serrulatus), hongotoxin-1 (isolated from Centruroides limbatus), and parabutoxin-3 (obtained from Parabuthus transvaalicus) selectively block KV channels. For instance, parabutoxin 3 and hongotoxin-1 block several KV1 channels, whereas margatoxin, noxiustoxin, and agitoxin-2 prefer the KV1.3 subtype. Agitoxin-2 interacts with the outer vestibule of the ion channel. Several snake toxins such as α- and β-dendrodotoxin [obtained from green (Dendroaspis viridis) and black mamba (Dendroaspis polylepis)] are KV channel blockers as well. Unlike scorpion toxins which act by pore occlusion, spider toxins appear to bind to four equivalent sites at the surface of the KV channel (one per subunit) close to the voltage sensor and thus, modify the gating process. Among them, we can include phrixotoxin-1 (obtained from the Chilean fire tarantula Phrixotrichus auratus), stromatoxin (obtained from Stromatopelma calceata), hanatoxin-1 and -2 (obtained from the Chilean rose tarantula Grammostola spatulata), heteropodatoxin-2 (obtained from the huntsman spider Heteropoda venatoria), as well as heteroscodratoxin-1 and -2 (obtained from Heteroscodra maculata). These toxins inhibit KV2 and KV4 channel families. Other natural toxins include MCD peptide (obtained from the honeybee Apis mellifera) which has high affinity for KV1.1, KV1.2, and KV1.6 channel subtypes, stichodactyla toxin (obtained from the sea anemone Stichodactyla helianthus), phalloidine (alkaloid obtained from the death-cap mushroom Amanita phalloides), and correolide derivatives (triterpenes purified from the root and bark of the Costa Rican tree Spachea correae) that are selective blockers of KV1 channels and may be used as immunosuppressants by inhibiting the lymphocyte KV1.3 channel. Among marine toxins we can name κ-conotoxin PVIIA (obtained from C. purpurascens) which is the first identified marine toxin that blocks Drosophila KV channels. However, no mammalian channel has been shown to be sensitive to the action of this conotoxin. Interestingly, a new member from the I-conotoxin superfamily, conotoxin ViTx (obtained from C. virgo), inhibits the vertebrate channels KV1.1 and KV1.3 but not KV1.2 [53], whereas conkunitzin-S1 (obtained from C. striatus) and 6-bromo-2-mercaptotryptamine (obtained from the marine snail Calliostoma canaliculatum) interact with the Shaker KV channel. Ciguatoxin-1 (obtained from the dinoflagellate Gambierdiscus toxicus) is also a blocker from these ion channels. In general, these blockers prolong the duration of presynaptic action potentials in neurons, increasing Ca2+ influx by indirect CaV activation, enhancing neurotransmitter release and subsequently, provoking strong convulsions.
4. Ligand-Gated Ion Channels
Among LGIC receptors, four different superfamilies have been described in detail, namely the Cysloop or nicotinicoid receptor superfamily, the ionotropic glutamate receptor (GluR) superfamily, the ATP-activated receptor (P2XnR) superfamily (reviewed in [7,8,69]), and the transient receptor potential-canonical channels (reviewed in [102]). These receptor superfamilies are considered different from each other based upon genetical, structural, and functional properties. An updated data base for the LGIC receptor superfamilies can be found in the following web page: www.pasteur.fr/recherche/banques/LGIC/LGIC.html (see [64,65]).
4.1. Structural and Functional Similitude Among LGIC Superfamilies
Although the Cys-loop receptor, the GluR, and the P2XnR superfamily are genetically and structurally different among them, all receptor members present at least some common features. Figure 3 shows the most important structural characteristics that are preserved throughout the different LGIC receptor superfamilies. From the structural point of view, we can generalize that these LGIC receptor superfamilies are formed by three main modules: a large extracellular portion, the transmembrane portion, and a smaller cytoplasmic portion. The extracellular domain contains the neurotransmitter binding sites and several consensus sequences involved in glycosylation. The transmembrane domain has two faces, one internal face that is mainly related with the formation of the intrinsic ion channel and another external face which interacts with the lipid membrane. The aqueous pore allows the passage of different cations or anions across the cell membrane finally producing excitatory (for cation flux) or inhibitory (for anion flux) cell responses. The cytoplasmic portion serves as a structural link to cytoskeleton proteins for receptor anchorage, clustering, stabilization, and assembly (reviewed in [50]). This domain is also related with receptor modulation (e.g., desensitization) by phosphorylationdephosphorylation processes (e.g., reviewed in [19,49,56,58,93]).
From the functional point of view, LGIC receptor superfamilies also have some basic aspects that are common among them. LGIC superfamilies present a very simple repertoire of functional properties. Each receptor recognizes its specific neurotransmitter, and upon binding, the intrinsically-coupled ion channel is opened, augmenting in turn the possibility of ions to cross the lipid membrane. Thus, after channel opening, a new ionic concentration is found at the aqueous solutions bathing the opposed faces of the lipid bilayer of the cell. In particular, the cytoplasmic compartment will have a higher content of Na+ or Ca2+ (for cation channels) or of Cl− or HCO3− (for anion channels). The increased cation concentration in the cytoplasm produces membrane depolarization, whereas the increased anion concentration in the cytoplasm induces membrane hyperpolarization.
Considering the high level of complexity of the neuronal network, the activation of certain cation ion channels can result however in either neuronal inhibition or disinhibition (reviewed in [4]). The opposite is also true, the activation of certain anionic channels may finally produce membrane depolarization in immature neurons (e.g., see review by [39]).
In general, these receptor superfamilies are functionally involved in higher order brain mechanisms such as cognition, learning, memory, and behavior, as well as in peripheral functions including muscle contraction, bone formation, cardiac and gastrointestinal modulation, embryonic development, sensory perception, nociception, and pain processing (reviewed in [38,43,49,70,103]). The malfunctioning of these receptors has been considered as the origin of several neurological disorders as well as drug addiction (e.g., see [38,43,49,70,103]).
All these biologically relevant receptor properties are triggered by the binding of the specific neurotransmitter. Upon binding, the receptor protein undergoes several conformational changes. Several lines of experimental evidence suggest the existence of a high number of intermediate states (reviewed in [8,26,30,82]). To make it simpler, however, we will assume that receptors exist in a minimum of three interconvertible states. The diagram indicating the existence of at least three receptor states is shown in Fig. 6. In the absence of the specific neurotransmitter, most receptors are in the resting state (R). The diagram shows a generalized receptor with just one neurotransmitter binding site. However, up to five binding sites may exist for pentameric receptors formed by just one subunit subtype (e.g., homomeric receptors). The resting state is defined by the existence of a closed ion channel. Nevertheless, the receptor is still activatable. In this regard, the receptor is activated (A) in the presence of the neurotransmitter, and the ion channel is opened in a very fast time frame (R → A). The time regime is different among receptor subtypes. Although ion channels open with higher probability in the presence of agonists, thermodynamics fluctuations also allow ion channels to be open but with much lower probability (e.g., see review by [39]). In the prolonged presence of the neurotransmitter, the activated receptor is concomitantly commuted to a desensitized state (D) in a timescale that in general is slower than the activation process (A → D). Neither a specific energy source nor an ionic gradient is needed to induce the R → A → D conformational shift. The desensitized state is refractory to the pharmacological action of the neurotransmitter or other agonists, and in this case the ion channel remains closed. In addition, the desensitized state has a relatively higher affinity for the specific neurotransmitter as well as for other agonists and some antagonists than that in the resting state. Different receptor subtypes have distinct desensitized rates and each receptor may present various desensitized states. For instance, the AChR α7 subtype presents a fast desensitization rate, whereas the α2β2 subtype practically does not desensitize. In general, the desensitization process is a reversible process: after agonist washout, the receptor can be activated again (D → R). The receptor can recover from the desensitized state by means of at least two pathways: a ligand-bound receptor can reopen, or the neurotransmitter dissociates first and then the receptor can return to the resting state. Desensitization is a classical example of allosteric protein behavior that considers the previous existence of discrete conformational states. Physiologically, desensitization might be a major determinant of synaptic efficacy by controlling the number of available receptors (reviewed in [82]).
5. The Cys-Loop or Nicotinicoid Receptor Superfamily
This superfamily is called “Cys-loop receptor superfamily” because all receptor members conserve a 15-residue-spaced disulfide loop between amino acids 128 and 142 (corresponding to the AChR α1 subunit) located in the extracellular domain of the receptor. Figure 7 shows the most important structural characteristics of the Cys-loop receptor superfamily, and its distinction with the ionotropic GluR and P2XnR superfamilies. More recently, the term “nicotinicoid receptor superfamily” has been imprinted. Examples of this kind of receptors are the members of four families of ion channel-coupled receptors which are gated by specific neurotransmitters, including type A and C GABA receptors (GABAAR and GABACR), glycine receptors (GlyRs), type 3 5-HT receptors (5-HT3Rs), and both neuronal- and muscle-type nicotinic receptors (AChRs) (reviewed in [7,8,13,14,30,32,55,63,67,69,75,78]). The hirerarchy for the different LGIC receptor superfamilies is summarized in Table 3.
Apart from the fact that all receptors from this superfamily have a 15-residue Cys-loop, they also share some structural and functional features that make them a true superfamily of receptors. Among the most important structural and functional features, we can name:
- (1)
- High homology in amino acid [63,66] and pair base [78] sequence. Homology of ~30% is observed among different subunits, whereas ~70% homology is obtained considering just one subunit from different species. The genetic evidence also suggests that all members have evolved from the same ancestor protein [63,66,67,78].
- (2)
- A pentameric quaternary structure. This structural characteristic has been confirmed by electron microscopy of tubular crystals of native membranes containing high density of AChRs prepared from Torpedo electric organs. The enduring effort from Unwin’s laboratory has recently rendered a receptor three-dimensional image with a resolution of 4 Å [99].
- (3)
- A large N-terminal domain formed by ~220 residues located extracellularly, bearing the neurotransmitter binding sites. This funnel-shaped domain has an outer diameter of 70–80 Å and protrudes ~60 Å above the lipid membrane surface. This domain had been previously modeled mainly as antiparallel β-sheet secondary structures arranged perpendicular to the membrane surface [66]. This model was recently confirmed by determining the crystallographic structure of the AChBP from L. stagnalis[20], which resembles the extracellular portion of the AChR, in particular the α7 homomeric receptor, by 24%.
- (4)
- An interfacial location for the neurotransmitter binding sites. The comparison with the different AChBP structures, which also bind AChR agonists (e.g., ACh, CCh, nicotine, etc.) and competitive antagonists [e.g., α-bungarotoxin (α-BTx), d-tubocurarine, etc.], have confirmed that ligand sites are located between two subunits [18,20,24,25,27,48,87,106] (reviewed in [89]). Additional comparisons with other members of the superfamily including GABAARs [28,33,52,96], GlyRs [62], and 5-HT3Rs [83,84] also support such interfacial location.
- (5)
- An agonist binding site comprised by several loops (A-F) (reviewed in [7]), which have been re-named as “binding segments A-F” because some segments are actually in β-sheet rather than a loop conformation (reviewed in [69]). Binding segments A-C form the principal component of the agonist binding domain (which is located in the α subunit from AChRs and homologous subunits from other receptors), whereas binding segments D-F form the secondary component (which is located in the non-α subunit from AChRs and homologous subunits from other receptors). The evidence that supports this structural feature was provided by photoaffinity labeling and sitedirected mutagenesis experiments (reviewed in [7]), and more lately by cysteine- [92] and lysinescanning mutagenesis [88], as well as by comparative studies using the recently found AChBP structures as templates [see paragraph (4)].
- (6)
- Four transmembrane segments (M1–M4) per subunit (twenty transmembrane segments in total), where the M2 transmembrane segment from each subunit (five M2 segments in total) forms the wall of the ion channel. The ion channel is the main domain for the action of noncompetitive antagonists from endogenous as well as exogenous origin (reviewed in [5,6,9,10,12–14]). The transmembrane domain (including the phospholipid headgroup region) measure about 40 Å. Earlier hydropathy analysis suggested that the AChR have four transmembrane segments and that each segment is mainly in an α-helix secondary structure. This result has been confirmed by electron microscopy aided by computational methods [99,100], photoaffinity labeling (reviewed in [13,14]), substituted cysteine accessibility method (reviewed in [21,51], and references therein), tryptophan-scanning mutagenesis, as well as by circular dichroism and nuclear magnetic resonance spectroscopy (reviewed in [8,21]). However, these ion channels contain significant amounts of non-helical structures as well. For instance, β-sheet secondary structures have been observed in the external portion of M1 and other segments of M3.
- (7)
- A “hydrophobic girdle”, the so-called “kink”, in the middle of the M2 segment that is formed by residues that are conserved through all superfamily member subunits. This hydrophobic girdle constitutes the “gate” that is occluding ion flux in the resting (closed) state. In the activated (open) state, the rotation of one or two of the M2 segments would disrupt the hydrophobic interactions with adjacent helices allowing the passage of ions through the channel. In this regard, anticlockwise rotations in the extracellular domain as well as in the M2 domain were observed in refined electron microscopy images when the AChR was activated [100].
- (8)
- A dynamic mechanism, called “ion channel gating”, where agonist binding provokes a conformational change in the protein that finally leads the opening of the intrinsically-associated ion channel that is located far apart from the proper ligand binding domain. This modular mechanism has been studied in detail in the AChR by Auerbach’s laboratory. The main conclusion from these studies is that the chemical signal triggered by ACh binding is locally transmitted to the extracellular loop formed between M2 and M3 segments, the M2–M3 linker (reviewed in [21,30,31]), as a “conformational wave” that finally reaches the ion channel [45] (reviewed in [44]).Based on the evidence provided by low resolution electron microscopy, a concerted anticlockwise rotation of all extracellular domains has been hypothetized as a plausible mechanism for gating [98]. Additional evidence has been shown in other receptor members as well. For instance, electrostatic interactions were found between a Lys located in the M2–M3 linker and acidic residues located in the Cys-loop and in the loop 2 from the GABAAR [52]. Homologous residues in the GlyR [1] and the 5-HT3R (reviewed in [72]) were also found to be critical in the gating process.
- (9)
- A desensitization process, where the prolonged exposure of the receptor to agonists as well as some competitive and noncompetitive antagonists produces a conformational change in the protein that finally leads to a refractory state where the agonist does not open the ion channel (reviewed in [82]). This conformational state is depicted in Fig. 6. Qualitatively, the desensitization process is similar among the members of the Cys-loop receptor superfamily. Desensitization might be very important in the prevention of citotoxicity, in the process of nicotine addiction, or in other pathological states (reviewed in [82]). For instance, a Ser248 to Phe mutation in the pore domain of the α4 subunit results in faster desensitization of α4β2 AChRs causing a type of human cortical epilepsy [107].
- (10)
- A small cytoplasmic domain formed by the loop between M3 and M4 of variable length (from 80 to 265 amino acids). This region contains the greatest sequence divergence among the members of this receptor superfamily. This region is very important for receptor modulation by phosphorylation-deposphorylation processes (reviewed in [21,49,93]). For instance, tyrosine kinases have been found to reduce nicotine-induced currents, whereas phosphotyrosine phosphatases have been found to enhance nicotine-induced currents in chromaffin cells [105]. Receptor phosphorylation by serine/threonine and/or tyrosine kinases also increases the desensitization rate in several members of this LGIC superfamily (reviewed in [49,93]).The anchorage of certain receptors to the cytoskeleton is also mediated by the interaction of non-receptor proteins with the cytoplasmic domain. Among them we can name rapsyn for AChRs (reviewed in [108]) and the tubulin-binding element gephyrin for both GABAARs and GlyRs (reviewed in [21,57,73]). Protein-protein contacts may affect receptor assembly, trafficking, clustering, targeting, modulation, and turnover, among other processes.Finally, the cytoplasmic domain is considered an additional determinant for ion channel conductance (reviewed in [80]). Using different 5-HT3R chimaeras, a novel determinant of single-channel conductance was located to a putative amphipathic helix (the ‘HA stretch’), apart from the M2 domain. Key residues may be components of narrow openings within the inner vestibule of the channel, which contribute to the permeation pathway.
- (11)
- And finally, an extracellular location for the very short C-terminal. This small area is involved, at least in certain neuronal-type AChRs (e.g., α4β2 and α4β4), in the binding of neurosteroids [34,79] (reviewed in [5,6,9,10,13,14]). Neurosteroids are molecules synthesized in the CNS that modulate some of the functions of this receptor superfamily. For instance, 17β-estradiol may enhance the activity of α4β2 and α4β4 neuronal AChRs [34,79] (reviewed in [5,6,9,10,13,14,49,60]). GABAAR function is also enhanced by different neurosteroids (reviewed in [14,16]), which presents therapeutical opportunities for the treatment of several mood disorders. Opposite to that, neurosteroids other than 17β-estradiol (e.g., progesterone, hydrocortisone, etc.) inhibit noncompetitively several AChRs by interacting with the lipid-protein interface (reviewed in [5,6,9,10,13,14,49]).
Since one of the topics covered in this special issue includes the action of different marine drugs on distinct AChRs, we will give more detailed information on the structure and function of the AChR family.
5.1. The Nicotinic Acetylcholine Receptor Family
The AChR family can be subdivided in two main receptor types (see Table 4): (a) the muscle-type AChR that is formed by a combination of four different subunits in a stoichiometry that depends on the developmental stage of the muscle. The muscle in the embryonic stage has receptors (as well as the Torpedo or Electrophorus receptor) with the stoichiometry α12β1γδ, whereas muscle cells in the adult stage has receptors with the stoichiometry α12β1ɛδ. Both receptors are arranged in an anticlockwise manner as αγ/ɛαδβ; and (b) neuronal-type AChRs which can be formed by the combination of several α (α2–α10) and β (β2–β4) subunits. Functional, active homomeric AChRs are formed only by subunits α7, α8, or α9, whereas heteromeric AChRs can be shaped by a combination of two, three, or even four different subunits (reviewed in [7,75]).
From the functional point of view, the activation of AChRs provokes an excitatory response on the cell because these receptors are connected to a cation-selective channel that allows the passage of cations such as Na+, K+, or Ca2+. The activation of these ion channel receptors increases cation permeability which subsequently produces membrane depolarization. Depolarization of the membrane provokes specific physiological responses on each involved cell. For instance, if the muscle membrane depolarization is large enough, an action potential is elicited. This action potential propagates from the neuromuscular junction all over the muscle fiber. During the propagating action potential the release of Ca2+ ions from intracellular stores in the muscle cell (e.g., ER) is stimulated. The final response in the muscle is the contraction of its myofibrils. On the other hand, in neurons, the excitatory signal provided by the activation of the cation channel is summed with other signals (including inhibitory ones provided by the activation of anionic channels) and subsequently re-directed to another neuron or to an endocrine gland cell as a nerve action potential.
The AChR family is implicated in several important physiological functions (reviewed in [70]). For instance, AChRs may directly or indirectly act on cognition, learning, memory and attention, neurodegeneration, pain perception, reward, anxiety and depression control, body temperature, auditory function development, control of voluntary motion, respiration, sleep and wakefulness, protective responses to stress (e.g., hypoxia), cardiovascular and gastrointestinal tract, as well as on muscle contraction and tone. From the pathophysiological point of view, AChRs are involved in different neurological disorders including Alzheimer’s and Parkinson’s disease, schizophrenia, drug addiction, Tourette’s syndrome, myasthenia gravis and myasthenic syndromes, depression, anxiety, ulcerative colitis, attention-deficit hyperactivity disorder, and neuroinflammation [97], as well as on different types of epilepsies, including nocturnal lobe, juvenile myoclonic, and catamenial epilepsy (reviewed in [49,68,70]).
Several members from the AChR family are targets for the pharmacological action of different natural toxins (reviewed in [7,11]). The list of alkaloids that act as agonists includes (-)nicotine (obtained from the leaves of the tobacco plant Nicotiana tabacum), (+)anatoxin-a (obtained from the blue-green algae Anabena flos-aquae), (-)cytisine (obtained from the plant Thermopsis alterniflora), (±)epibatidine (obtained from the skin of the Ecuadorian frog Epipedobates tricolor), lobeline (obtained from the plant Lobelia inflata), and anabaseine (obtained from Nemertine marine worm species and from venom glands of certain ants). Among competitive antagonists we can name alkaloids such as d-tubocurarine (obtained from the bush Chondrodendron tomentosum), methyllycaconitine (obtained from the seeds of the plant Delphinium brownii), and dihydro-β-erythroidine (obtained from different Erythrina plant species), snake polypeptides such as α- and κ-bungarotoxins [obtained from different Elapidae (e.g., the many-banded krait Bungarus multicinctus) and Hydrophiidae (sea snakes) species] and cobrotoxins (e.g., from Naja naja siamensis), as well as marine toxins such as lophotoxins (obtained from gorgonian corals such as Lophogorgia and Pseudopterogorgia), neosurugatoxin (obtained from the Japanese ivory shell Babylonia japonica), as well as α- and αA-conotoxins (obtained from several Conus species). Finally, among noncompetitive antagonists we can quote the alkaloid ibogaine (obtained from the shrub Tabernanthe iboga), and the psi-conotoxin PIIIE (obtained from C. purpurascens). For more details on conotoxins see the minireview by Layer and McIntosh in this special issue.
Although some toxins bind to different AChRs in a nonspecific manner, several toxins present very high affinity and high specificity for certain receptor types, pointing out the importance for potential therapeutic uses. In this regard, Kem and colleagues review the activity of benzylidene-anabaseine derivatives as partial specific agonists of the α7 AChR as well as the noncompetitive mechanism for these compounds in muscle-type AChRs. In addition, Wonnacott and Gallagher cover the structurefunction relationship for anatoxin-a.
Since this special issue includes marine drugs interacting neither with 5-HT3Rs, GABAARs, GABACRs, and GlyRs (members from the Cys-loop receptor superfamily), nor with members from the GluR or P2XnR superfamily, we will not cover these receptors any further.
6. Conclusions
Since the decade of 1900, where the concept of neurotransmission was first defined, until now, a great deal of information on the synapse structure and function has been accumulated with the help of different approaches from different research areas such as molecular and cell biology, biochemistry, electrophysiology, biophysics, and genetically-altered animals. One of the most important entities for synapse functioning, ion channels, is the focus of this special issue. Since the structural characterization of AChRs, the archetype of the LGIC receptor superfamilies, until today, additional LGIC as well as VGIC superfamilies have been extensively studied. In this regard, our actual knowledge on the structure and function of different marine drugs acting on LGIC as well as on VGIC superfamilies will pave the way to new therapies for the treatment of distinct neurological diseases.
Acknowledgements
This review was supported by Western University Health Sciences Intramural Grants (to HRA). I thank to Dr. Robert French for critical reading of the manuscript and valuable suggestions.
- Samples Availability: Available from the authors.
References
- Absalom, N. L.; Lewis, T. M.; Kaplan, W.; Pierce, K. D.; Schofield, P. R. Role of charged residues in coupling ligand binding and channel activation in the extracellular domain of the glycine receptor. J. Biol. Chem 2003, 278, 50151–50157. [Google Scholar]
- Ackerman, M. J.; Clapham, D. E. Ion channels − Basic science and clinical disease. N. Engl. J. Med 1997, 336, 1575–1586. [Google Scholar]
- Agnew, W. S.; Levinson, S. R.; Brabson, J. S.; Raftery, M. A. Purification of the tetrodotoxin-binding component associated with the voltage-sensitive sodium channel from Electrophorus electricus electroplax membranes. Proc. Natl. Acad. Sci. USA 1978, 75, 2602–2610. [Google Scholar]
- Alkondon, M.; Albuquerque, E. X. The nicotinic acetylcholine receptor subtypes and their function in the hippocampus and cerebral cortex. Prog. Brain Res 2004, 145, 109–120. [Google Scholar]
- Arias, H. R. Noncompetitive inhibition of nicotinic acetylcholine receptors by endogenous molecules. J. Neurosci. Res 1998, 52, 369–379. [Google Scholar]
- Arias, H. R. Binding sites for exogenous and endogenous non-competitive inhibitors of the nicotinic acetylcholine receptor. Biochim. Biophys. Acta Rev. Biomembr 1998, 1376, 173–220. [Google Scholar]
- Arias, H. R. Localization of agonist and competitive antagonist binding sites on nicotinic acetylcholine receptors. Neurochem. Int 2000, 36, 595–645. [Google Scholar]
- Arias, H. R. Biological and Biophysical Aspects of Ligand-Gated Ion Channel Receptor Superfamilies; Arias, H. R., Ed.; Research Signpost: India, 2006; Volume Chapter 1, pp. 1–25. [Google Scholar]
- Arias, H. R.; Bhumireddy, P. Anesthetics as chemical tools to study the structure and function of nicotinic acetylcholine receptors. Curr. Protein Pept. Sci 2005, 6, 451–472. [Google Scholar]
- Arias, H. R.; Bhumireddy, P.; Bouzat, C. Molecular mechanisms and binding site locations for noncompetitive antagonists of nicotinic acetylcholine receptors. Int. J. Biochem. Cell Biol. 2006, in press.. [Google Scholar]
- Arias, H. R.; Blanton, M. P. α-Conotoxins. Int. J. Biochem. Cell Biol 2000, 32, 1017–1028. [Google Scholar]
- Arias, H. R.; Blanton, M. P. Molecular and physicochemical aspects of local anesthetics acting on nicotinic acetylcholine receptor-containing membranes. Mini Rev. Med. Chem 2002, 2, 385–410. [Google Scholar]
- Arias, H. R.; Kem, W. R.; Trudell, J. R.; Blanton, M. P. Unique general anesthetic binding sites within distinct conformational states of the nicotinic acetylcholine receptor. Int. Rev. Neurobiol 2002, 54, 1–50. [Google Scholar]
- Arias, H. R.; Machu, T. K.; Trudell, J. R.; Blanton, M. P. Recent Research Developments in Biophysical Chemistry; Condat, C. A., Baruzzi, A., Eds.; Research Signpost: India, 2002; pp. 123–154. [Google Scholar]
- Becher, A.; Drenckhahn, A.; Pahner, I.; Margittai, M.; Jahn, R.; Ahnert-Hilger, G. The synaptophysin-synaptobrevin complex is developmentally upregulated in cultivated neurons but is absent in neuroendocrine cells. J. Neurosci 1999, 19, 1922–1931. [Google Scholar]
- Belelli, D.; Lambert, J. J. Neurosteroids: endogenous regulators of the GABAA receptor. Nat. Rev. Neurosci 2005, 6, 565–575. [Google Scholar]
- Bezanilla, F. Voltage-gated ion channels. IEEE Trans. Nanobiosci 2005, 4, 34–48. [Google Scholar]
- Bourne, Y.; Talley, T. T.; Hansen, S. B.; Taylor, P.; Marchot, P. Crystal structure of a Cbtx- AChBP complex reveals essential interactions between snake α-neurotoxins and nicotinic receptors. EMBO J 2005, 24, 1512–1522. [Google Scholar]
- Brandon, N.; Jovanovic, J.; Moss, S. Multiple roles of protein kinases in the modulation of γ-aminobutyric acidA receptor function and cell surface expression. Pharmacol. Ther 2002, 94, 113–122. [Google Scholar]
- Brejc, K.; van Dijk, W. J.; Klassen, R.V.; Schuumans, M.; van Der Oost, J.; Smit, A. B.; Sixma, T. K. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 2001, 403, 269–276. [Google Scholar]
- Cascio, M. Structure and function of the glycine receptor and related nicotinicoid receptors. J. Biol. Chem 2004, 279, 19383–19386. [Google Scholar]
- Catterall, W. A.; Goldin, A. L.; Waxman, S. G. International Union of Pharmacology. International Union of Pharmacology. XXXIX. Compendium of voltage-gated ion channels: sodium channels. Pharmacol. Rev 2003, 55, 575–578. [Google Scholar]
- Catterall, W. A.; Striessnig, J.; Snutch, T. P.; Perez-Reyes, E. International Union of Pharmacology. International Union of Pharmacology. XL. Compendium of voltage-gated ion channels: calcium channels. Pharmacol. Rev 2003, 55, 579–581. [Google Scholar]
- Celie, P. H. N.; Klaassen, R.V.; van Rossum-Fikkert, S. E.; van Elk, R.; van Nierop, P.; Smit, A. B.; Sixma, T. K. Crystal structure of acetylcholine-binding protein from Bulinus truncatus reveals the conserved structural scaffold and sites of variation in nicotinic acetylcholine receptors. Biol. Chem 2005, 280, 26457–26466. [Google Scholar]
- Celie, P. H. N.; van Rossum-Fikkert, S. E.; van Dijk, W. J.; Brejc, K.; Smit, A. B.; Sixma, T. K. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 2004, 41, 907. [Google Scholar]
- Changeux, J-P.; Edelstein, S. J. Allosteric receptors after 30 years. Neuron 1998, 21, 959–980. [Google Scholar]
- Chou, K-C. Insights from modelling the 3D structure of the extracellular domain of α7 nicotinic acetylcholine receptor. Biochem. Biophys. Res. Commun 2004, 319, 433–438. [Google Scholar]
- Chou, K-C. Modelling extracellular domains of GABA-A receptors: subtypes 1, 2, 3, and 5. Biochem. Biophys. Res. Commun 2004, 316, 636–642. [Google Scholar]
- Clement, J. G. Toxicology and pharmacology of bispyridium oximes-insight into the mechanism of action vs Soman poisoning in vivo. Fundam. Appl. Toxicol 1981, 1, 193–202. [Google Scholar]
- Colquhoun, D.; Sivilotti, L. G. Function and structure in glycine receptors and some of their relatives. Trends Neurosci 2003, 27, 337–344. [Google Scholar]
- Condat, C. A.; Lamberti, P. W.; Arias, H. R. Biological and Biophysical Aspects of Ligand- Gated Ion Channel Receptor Superfamilies; Arias, H. R., Ed.; Research Signpost: India, 2006; Volume Chapter 4. [Google Scholar]
- Connolly, C. N.; Wafford, K. A. The Cys-loop superfamily of ligand-gated ion channels: the impact of receptor structure on function. Biochem. Soc. Trans 2004, 32, 529–534. [Google Scholar]
- Cromer, B. A.; Morton, C. J.; Parker, M. W. Anxiety over GABAA receptor structure relieved by AChBP. Trends Biochem. Sci 2002, 27, 280–287. [Google Scholar]
- Curtis, L.; Buisson, B.; Bertrand, S.; Bertrand, D. Potentiation of human α4β2 neuronal nicotinic acetylcholine receptor by estradiol. Mol. Pharmacol 2002, 61, 127–135. [Google Scholar]
- Dahan, M.; Levi, S.; Luccardini, C.; Rostaing, P.; Riveau, B.; Triller, A. Diffusion dynamics of glycine receptors revealed by single-quantum dot tracking. Science 2003, 302, 442–445. [Google Scholar]
- Denac, H.; Mevissen, M.; Scholtysik, G. Structure, function and pharmacology of voltage-gated sodium channels. Naunyn-Schmiedeberg’s Arch. Pharmacol 2000, 362, 453–479. [Google Scholar]
- Escoubas, P.; Diochot, S.; Corzo, G. Structure and pharmacology of spider venos neurotoxins. Biochimie 2000, 82, 893–907. [Google Scholar]
- Farber, L.; Haus, U.; Spath, M.; Drechsler, S. Physiology and pathophysiology of the 5-HT3 receptor. Scand. J. Rheumatol. Suppl 2004, 119, 2–8. [Google Scholar]
- Farrant, M.; Nusser, Z. Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat. Rev. Neurosci 2005, 6, 215–229. [Google Scholar]
- Fatt, P.; Katz, B. The electrical properties of crustacean muscle fibers. J. Physiol 1953, 120, 171–204. [Google Scholar]
- George, A. L., Jr. Inherited disorders of voltage-gated sodium channels. J. Clin. Invest 2005, 115, 1990–1999. [Google Scholar]
- Ghijsen, W. E.; Leenders, A. G. M. Differential signaling in presynaptic neurotransmitter release. Cell Mol. Life Sci 2005, 62, 937–954. [Google Scholar]
- Gotti, C.; Clementi, F. Neuronal nicotinic receptors: from structure to pathology. Progr. Neurobiol 2004, 74, 363–396. [Google Scholar]
- Grosman, C. Linear free-energy relationships and the dynamics of gating in the acetylcholine receptor channel. A Φ-value analysis of an allosteric transition at the single-molecule level. J. Biol. Phys 2002, 28, 267–277. [Google Scholar]
- Grosman, C.; Zhou, M.; Auerbach, A. Mapping the conformational wave of acetylcholine receptor channel gating. Nature 2000, 403, 773–776. [Google Scholar]
- Gutman, G. A.; Chandy, K. G.; Adelman, J. P.; Aiyar, J.; Bayliss, D. A.; Clapham, D. E.; Covarriubias, M.; Desir, G. V.; Furuichi, K.; Ganetzky, B.; Garcia, M. L.; Grissmer, S.; Jan, L. Y.; Karschin, A.; Kim, D.; Kuperschmidt, S.; Kurachi, Y.; Lazdunski, M.; Lesage, F.; Lester, H. A.; McKinnon, D.; Nichols, C. G.; O’Kelly, I.; Robbins, J.; Robertson, G. A.; Rudy, B.; Sanguinetti, M.; Seino, S.; Stuehmer, W.; Tamkun, M. M.; Vandenberg, C. A.; Wei, A.; Wulff, H.; Wymore, R. S. International Union of Pharmacology. International Union of Pharmacology. XLI. Compendium of voltage-gated ion channels: potassium channels. Pharmacol. Rev 2003, 55, 583–586. [Google Scholar]
- Hansen, S. B.; Talley, T. T.; Radić, Z.; Taylor, P. Structural and ligand recognition characteristics of an acetylcholine-binding protein from Aplysia californica. J. Biol. Chem 2004, 279, 24197–24202. [Google Scholar]
- Harel, M.; Kasher, R.; Nicolas, A.; Guss, J. M.; Balass, M.; Fridkin, M.; Smit, A.B.; Brejc, K.; Sixma, T. K.; Katchalski-Katzir, E.; Sussman, J. L.; Fuchs, S. The binding site of acetylcholine receptor as visualized in the X-Ray structure of a complex between α-bungarotoxin and a mimotope peptide. Neuron 2001, 32, 265. [Google Scholar]
- Hogg, R. C.; Raggenbass, M.; Bertrand, D. Nicotinic acetylcholine receptors: from structure to brain function. Rev. Physiol. Biochem. Pharmacol 2003, 147, 1–46. [Google Scholar]
- Huh, K. H.; Fuhrer, C. Clustering of nicotinic acetylcholine receptors: from the neuromuscular junction to interneuronal synapses. Mol. Neurobiol 2002, 25, 79–112. [Google Scholar]
- Karlin, A. Emerging structure of the nicotinic acetylcholine receptors. Nat. Rev. Neurosci 2002, 3, 102–114. [Google Scholar]
- Kash, T. L.; Trudell, J. R.; Harrison, N. L. Structural elements involved in activation of the gamma-aminobutyric acid type A (GABAA) receptor. Biochem. Soc. Trans 2004, 32, 540–546. [Google Scholar]
- Kauferstein, S.; Huys, I.; Lamthanh, H.; Stöcklin, R.; Sotto, F.; Menez, A.; Tytgat, J.; Mebs, D. A novel conotoxin inhibiting vertebrate voltage-sensitive potassium channels. Toxicon 2003, 42, 43–52. [Google Scholar]
- Kažić, T.; Gojković-Bukarica, L. Ion channels and drug development. Focus on potassium channels and their modulators. Facta Universitatis 1999, 6, 23–30. [Google Scholar]
- Keramidas, A.; Moorhouse, A. J.; Schofield, P. R.; Barry, P. H. Ligand-gated ion channels: mechanisms underlying ion selectivity. Progr. Biophys. Mol. Biol 2004, 86, 161–204. [Google Scholar]
- Kittler, J. T.; Moss, S. J. Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: implications for the efficacy of synaptic inhibition. Curr. Opin. Neurobiol 2003, 13, 341–347. [Google Scholar]
- Kneussel, M.; Betz, H. Receptors, gephyrin and gephyrin-associated proteins: novel insights into the assembly of inhibitory postsynaptic membrane specializations. J. Physiol 2000, 525, 1–9. [Google Scholar]
- Koles, L.; Wirkner, K.; Illes, P. Modulation of ionotropic glutamate receptor channels. Neurochem. Res 2001, 26, 925–932. [Google Scholar]
- Kullmann, D. M.; Ruiz, A.; Rusakov, D. M.; Scott, R.; Semyanov, A.; Walker, M. C. Presynaptic, extrasynaptic and axonal GABAA receptors in the CNS: where and why? Prog. Biophys. Mol. Biol 2005, 87, 33–46. [Google Scholar]
- Lambert, J. J.; Belelli, D.; Harney, S. C.; Peters, J. A.; Frenguelli, B. G. Modulation of native and recombinant GABAA receptors by endogenous and synthetic neuroactive steroids. Brain Res. Brain Res. Rev 2001, 37, 68–80. [Google Scholar]
- Langley, J. N. On the reaction of cells and of nerve endings to certain poisons, chiefly as regards the reaction of striated muscle to nicotine and to curare. J. Physiol (London) 1905, 33, 374–413. [Google Scholar]
- Laube, B.; Maksay, G.; Schemm, R.; Betz, H. Modulation of glycine receptor function: a novel approach for therapeutic intervention at inhibitory synapses? Trends Pharmacol. Sci 2002, 23, 519–527. [Google Scholar]
- Le Novére, N.; Changeux, J-P. Molecular evolution of the nicotinic acetylcholine receptor: an example of multigene family in excitable cells. J. Mol. Evol 1995, 40, 155–172. [Google Scholar]
- Le Novére, N.; Changeux, J-P. LGICdb: the ligand-gated ion channel database. Nucleic Acids Res 2001, 29, 294–295. [Google Scholar]
- Le Novére, N.; Changeux, J-P. The ligand gated ion channel database: an example of a sequence database in neuroscience. Philos. Trans. R. Soc. Lond. B Biol. Sci 2001, 356, 1121–1130. [Google Scholar]
- Le Novére, N.; Corringer, P. J.; Changeux, J-P. Improved secondary structure predictions for a nicotinic receptor subunit: incorporation of solvent accessibility and experimental data into a twodimensional representation. Biophys. J 1999, 76, 2329–2345. [Google Scholar]
- Le Novére, N.; Corringer, P. J.; Changeux, J-P. The diversity of subunit composition in nAChRs: evolutionary origins, physiologic and pharmacologic consequences. J. Neurobiol 2002, 53, 447–456. [Google Scholar]
- Leonard, S.; Bertrand, D. Neuronal nicotinic receptors: from structure to function. Nicotine Tobacco Res 2001, 3, 203–223. [Google Scholar]
- Lester, H. A.; Dibas, M. I.; Dahan, D. S.; Leite, J. F.; Dougherty, D. A. Cys-loop receptors: new twists and turns. Trends Neurosci 2004, 27, 329–336. [Google Scholar]
- Lloyd, G. K.; Williams, M. Neuronal nicotinic acetylcholine receptors as novel drug targets. J. Pharmacol. Exp. Ther 2000, 292, 461–467. [Google Scholar]
- Long, S. B.; Campbell, E. B.; Mackinnon, R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 2005, 309, 897–903. [Google Scholar]
- Lummis, S. C. R. The transmembrane domain of the 5-HT3 receptor: its role in selectivity and gating. Biochem. Soc. Trans 2004, 32, 535–539. [Google Scholar]
- Lynch, J. W. Molecular structure and function of the glycine receptor chloride channel. Physiol. Rev 2004, 84, 1051–1095. [Google Scholar]
- MacDermott, A. B.; Role, L. W.; Siegelbaum, S. A. Presynaptic ionotropic receptors and the control of transmitter release. Annu. Rev. Neurosci 1999, 22, 443–485. [Google Scholar]
- Millar, N. S. Assembly and subunit diversity of nicotinic acetylcholine receptors. Biochem. Soc. Trans 2003, 31, 869–874. [Google Scholar]
- Miller, C. An overview of the potassium channel family. Genome Biol. 2000, 4, 0004:1–0004:5. [Google Scholar]
- Miller, R. J. Presynaptic receptors. Annu. Rev. Pharmacol. Toxicol 1998, 38, 201–227. [Google Scholar]
- Ortells, M. O.; Lunt, G. G. Evolutionary history of the ligand-gated ion-channel superfamily of receptors. Trends Neurosci 1995, 18, 121–127. [Google Scholar]
- Paradiso, K.; Zhang, J.; Steinbach, J. H. The C terminus of the human nicotinic α4β2 receptor forms a binding site required for potentiation by an estrogenic steroid. J. Neurosci 2001, 21, 6561–6568. [Google Scholar]
- Peters, J. A.; Kelley, S. P.; Dunlop, J. I.; Kirkness, E. F.; Hales, T. G.; Lambert, J. J. The 5- hydroxytryptamine type 3 (5-HT3) receptor reveals a novel determinant of single-channel conductance. Biochem. Soc. Trans 2004, 32, 547–552. [Google Scholar]
- Pichon, Y.; Prime, L.; Benquet, P.; Tiaho, F. Some aspects of the physiological role of ion channels in the nervous system. Eur. Biophys. J 2004, 33, 211–226. [Google Scholar]
- Quick, M. W.; Lester, R. A. J. Desensitization of neuronal nicotinic receptors. J. Neurobiol 2002, 53, 457–478. [Google Scholar]
- Reeves, D. C.; Lummis, S. C. R. The molecular basis of the structure and function of the 5-HT3 receptor: a model ligand-gated ion channel. Mol. Membr. Biol 2002, 19, 11–26. [Google Scholar]
- Reeves, D. C.; Sayed, M. F. R.; Chau, P. L.; Price, K. L.; Lummis, S. C. R. Prediction of 5-HT3 receptor agonist-binding residues using homology modeling. Biophys. J 2003, 84, 2338–2344. [Google Scholar]
- Reid, C. A.; Bekkers, J. M.; Clements, J. D. Presynaptic Ca2+ channels: a functional patchwork. Trends Neurosci 2003, 26, 683–687. [Google Scholar]
- Roux, B. Ion conduction and selectivity in K+ channels. Annu. Rev. Biophys. Biomol. Struct 2005, 34, 153–171. [Google Scholar]
- Schapira, M.; Abagyan, R.; Totrov, M. Structural model of nicotinic acetylcholine receptor isotypes bound to acetylcholine and nicotine. BMC Struct. Biol 2002, 2, 1–8. [Google Scholar]
- Sine, S. M.; Wang, H. L.; Bren, N. Lysine scanning mutagenesis delineates structural model of the nicotinic receptor ligand binding domain. J Biol Chem. 2002, 277, 29210–29223. [Google Scholar]
- Sixma, T. K.; Smit, A.B. Acetylcholine binding protein (AChBP): a secreted glial protein that provides a high-resolution model for the extracellular domain of pentameric ligand-gated ion channels. Annu. Rev. Biophys. Biomol. Struct 2003, 32, 311–334. [Google Scholar]
- Smit, A. B.; Syed, N. I.; Schaap, D.; van Minnen, J.; Klumperman, J.; Kits, K. S.; Lodder, H.; van der Schors, R. C.; van Elk, R.; Sorgedrager, B.; Brejc, K.; Sixma, T. K.; Geraerts, W. P. A. gliaderived acetylcholine-binding protein that modulates synaptic transmission. Nature 2001, 411, 261–268. [Google Scholar]
- Stevens, C. F. Presynaptic function. Curr. Opin. Neurobiol 2004, 14, 341–345. [Google Scholar]
- Sullivan, D.; Chiara, D. C.; Cohen, J. B. Mapping the agonist binding site of the nicotinic acetylcholine receptor by cysteine scanning mutagenesis: antagonist footprint and secondary structure prediction. Mol. Pharmacol 2002, 61, 463–472. [Google Scholar]
- Swope, S. L.; Moss, S. J.; Raymond, L. A.; Huganir, R. L. Regulation of ligand-gated ion channels by protein phosphorylation. Adv. Second Messenger Phosphoprot. Res 1999, 33, 49–78. [Google Scholar]
- Tempel, T. M.; Papazian, D. M.; Schwarz, T. L.; Jan, Y. N.; Jan, L.Y. Sequence of a probable potassium channel component encoded at Shaker locus in Drosophila. Science 1987, 237, 770–775. [Google Scholar]
- Trimmer, J. S.; Rhodes, K. J. Localization of voltage-gated ion channels in mammalian brain. Annu. Rev. Physiol 2004, 66, 477–519. [Google Scholar]
- Trudell, J. Unique assignment of inter-subunit association in GABAA α1β3γ2 receptors determined by molecular modeling. Biochim. Biophys. Acta 2002, 1565, 91–96. [Google Scholar]
- Tuppo, E. E.; Arias, H. R. The role of inflammation in Alzheimer’s disease. Int. J. Biochem. Cell Biol 2005, 37, 289–305. [Google Scholar]
- Unwin, N. The Croonian Lecture 2000. Nicotinic acetylcholine receptor and the structural basis of fast synaptic transmission. Philos. Trans. R. Soc. London Ser B 2000, 355, 1813–1829. [Google Scholar]
- Unwin, N. Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J. Mol. Biol 2005, 346, 967–989. [Google Scholar]
- Unwin, N.; Miyazawa, A.; Li, J.; Fujiyoshi, Y. Activation of the nicotinic acetylcholine receptor involves a switch in conformation of the α subunits. J. Mol. Biol 2002, 319, 1165–1176. [Google Scholar]
- Van der Kloot, W.; Molgó, J. Quantal acetylcholine release at the vertebrate neuromuscular junction. Physiol. Rev 1994, 74, 899–991. [Google Scholar]
- Vazquez, G.; Wedel, B. J.; Aziz, O.; Trebak, M.; Putney, J.W., Jr. The mammalian TRPC cation channels. Biochim. Biohphys. Acta 2004, 1742, 21–36. [Google Scholar]
- Vial, C.; Roberts, J.A.; Evans, R.J. Molecular properties of ATP-gated P2X receptor ion channels. Trends Pharmacol. Sci 2004, 25, 487–493. [Google Scholar]
- Waites, C. L.; Craig, A. M.; Garner, C. C. Mechanisms of vertebrate synaptogenesis. Annu. Rev. Neurosci 2005, 28, 251–274. [Google Scholar]
- Wang, K.; Hackett, J. T.; Cox, M. E.; van Hoek, M.; Lindstrom, J. M.; Parsons, S. J. Regulation of the neuronal nicotinic acetylcholine receptor by SRC family tyrosine kinases. J. Biol. Chem 2004, 279, 8779–8786. [Google Scholar]
- Wei, D.; Sirois, S.; Du, Q-S.; Arias, H.R.; Chou, K-C. Theoretical studies of Alzheimer’s disease drug candidate 3-[(2,4-dimethoxy)benzylidene]-anabaseine (GTS-21) and its derivatives. Biochem. Biophys. Res. Commun. 2005, 338, 1059–1064. [Google Scholar]
- Weiland, S.; Witzemann, V.; Villarroel, A.; Propping, P.; Steinlein, O. An amino acid exchange in the second transmembrane segment of a neuronal nicotinic receptor causes partial epilepsy by altering its desensitization kinetics. FEBS Lett 1996, 398, 91–96. [Google Scholar]
- Willmann, R.; Fuhrer, C. Neuromuscular synaptogenesis: clustering of acetylcholine receptors revisited. Cell Mol. Life Sci 2002, 59, 1296–1316. [Google Scholar]
- Yamakage, M.; Namiki, A. Calcium channels - basic aspects of their structure, function and gene encoding; anesthetic action on the channels - a review. Can. J. Anesth 2002, 49, 151–164. [Google Scholar]
- Yu, F. H.; Catterall, W. A. Overview of the voltage-gated sodium channel family. Genome Biol 2003, 4, 207. [Google Scholar]
- Ziv, N. E.; Garner, C. C. Cellular and molecular mechanisms of presynaptic assembly. Nat. Rev. Neurosci 2004, 5, 385–399. [Google Scholar]
Figure 1.
Diagram showing the most important components of the synapse, including the pre- (axon terminal) and the post-synaptic cells (dendritic spine) and the synaptic cleft (taken from www.en.wikipedia.org/wiki/Synapse). CaV channels involved in neurotransmitter release, vesicles for neurotransmitter storage, and neurotransmitter re-uptake transporters are depicted in the presynaptic cell. Neurotransmitters are released from the presynaptic membrane to the synaptic cleft. Neurotransmitters reach the postsynaptic membrane by diffusion, and then activate neurotransmitter receptors. Neurotransmitter transporters are also shown in the presynaptic membrane.
Figure 1.
Diagram showing the most important components of the synapse, including the pre- (axon terminal) and the post-synaptic cells (dendritic spine) and the synaptic cleft (taken from www.en.wikipedia.org/wiki/Synapse). CaV channels involved in neurotransmitter release, vesicles for neurotransmitter storage, and neurotransmitter re-uptake transporters are depicted in the presynaptic cell. Neurotransmitters are released from the presynaptic membrane to the synaptic cleft. Neurotransmitters reach the postsynaptic membrane by diffusion, and then activate neurotransmitter receptors. Neurotransmitter transporters are also shown in the presynaptic membrane.
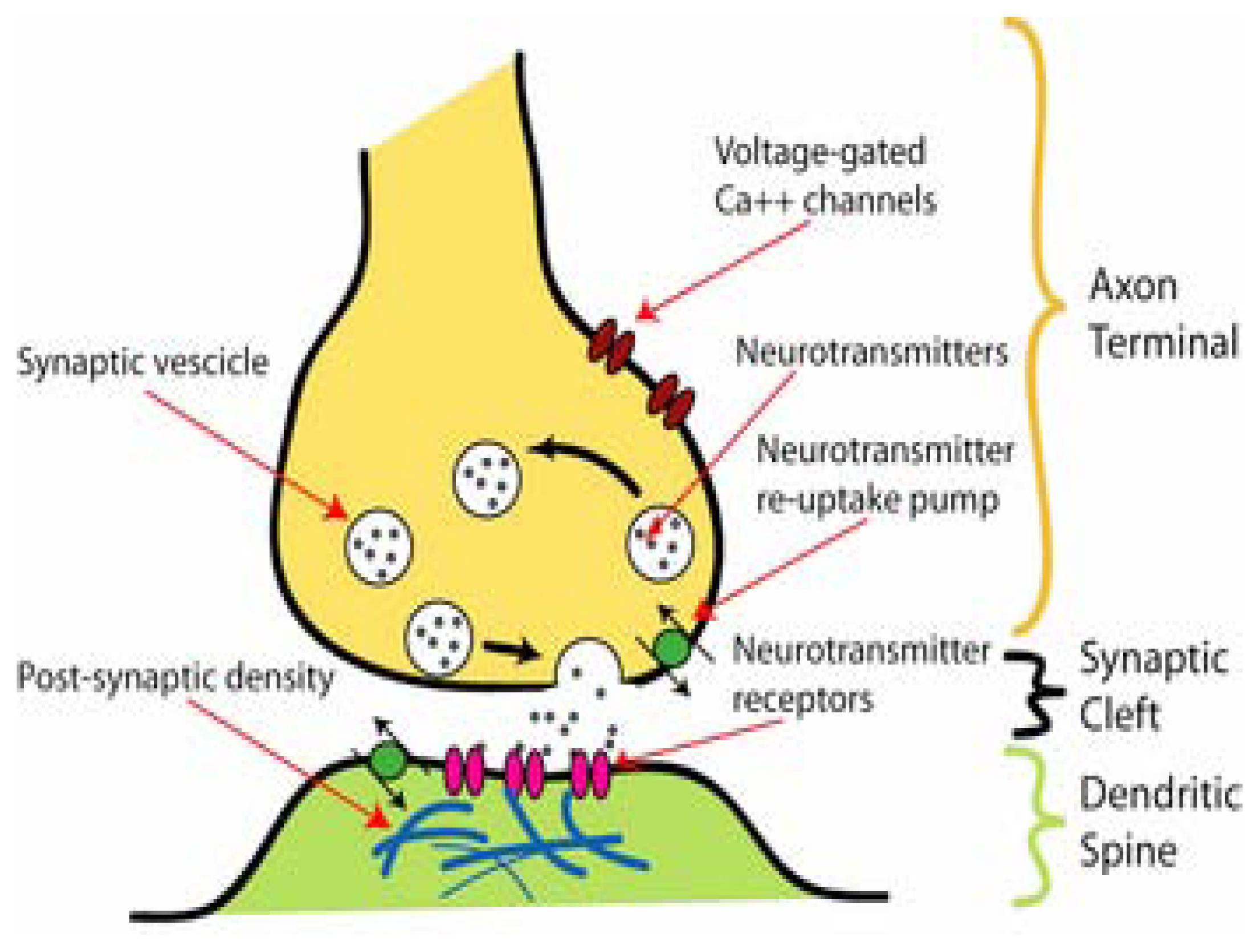
Figure 2.
Schematic representation of the structure of the NaV channel (modified from [36]). (A) The α subunit consists of four homologous domains (I-IV), each containing six transmembrane segments (S1–S6). Positively-charged S4 segments [S4(+)] form the voltage sensor. Single segments and domains are connected by intracellular and extracellular loops. The intracellular loop between domain I and II presents several phosphorylation sites (P). Extracellular loops contain several glycosylation sites (Y). The isoleucine-phenylalanine-methionine (IFM) motif has a crucial role in the process of channel inactivation. The inactivation gate is depicted in red and includes the IFM motif. (B) Transmembrane arrangement of domains I-IV (DI-DIV) around the ion pore, seen from the cytoplasmic end. The inactivation gate is depicted in red, close to the IFM motif [see (A)]. (C) Binding sites (1–5) for different ligands [e.g., tetrodotoxin (TTX)] as well as for local anesthetics (LAs). The selectivity filter is depicted in blue.
Figure 2.
Schematic representation of the structure of the NaV channel (modified from [36]). (A) The α subunit consists of four homologous domains (I-IV), each containing six transmembrane segments (S1–S6). Positively-charged S4 segments [S4(+)] form the voltage sensor. Single segments and domains are connected by intracellular and extracellular loops. The intracellular loop between domain I and II presents several phosphorylation sites (P). Extracellular loops contain several glycosylation sites (Y). The isoleucine-phenylalanine-methionine (IFM) motif has a crucial role in the process of channel inactivation. The inactivation gate is depicted in red and includes the IFM motif. (B) Transmembrane arrangement of domains I-IV (DI-DIV) around the ion pore, seen from the cytoplasmic end. The inactivation gate is depicted in red, close to the IFM motif [see (A)]. (C) Binding sites (1–5) for different ligands [e.g., tetrodotoxin (TTX)] as well as for local anesthetics (LAs). The selectivity filter is depicted in blue.
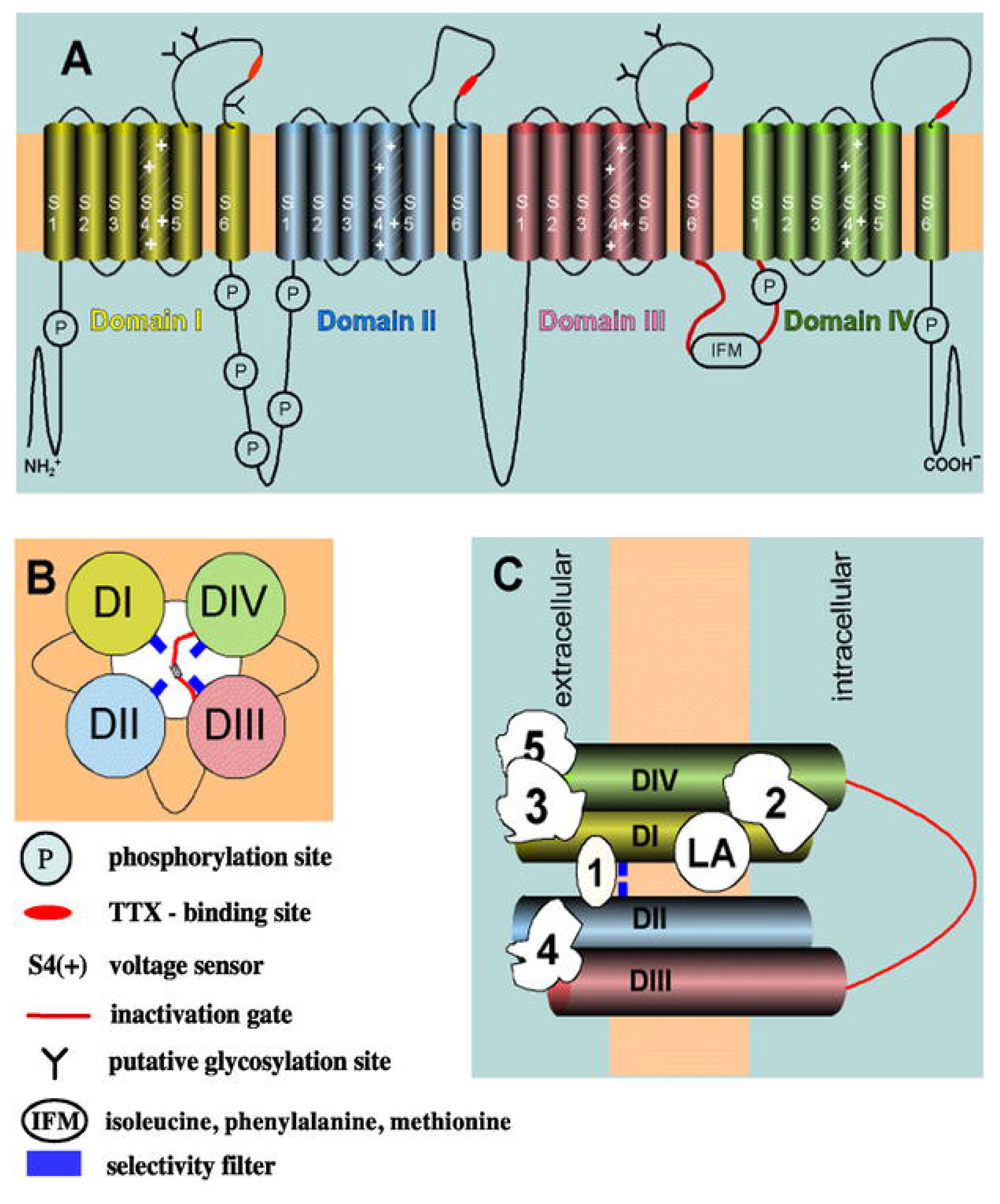
Figure 3.
Functional properties of NaV channels (modified from [41]). (A) Schematic representation of a NaV channel undergoing the major gating transitions (resting ↔ open ↔ inactivated). (B) Voltage-clamp recording of NaV channel activity in response to membrane depolarization. Downward deflection of the current trace (red) corresponds to inward movement of Na+.
Figure 3.
Functional properties of NaV channels (modified from [41]). (A) Schematic representation of a NaV channel undergoing the major gating transitions (resting ↔ open ↔ inactivated). (B) Voltage-clamp recording of NaV channel activity in response to membrane depolarization. Downward deflection of the current trace (red) corresponds to inward movement of Na+.
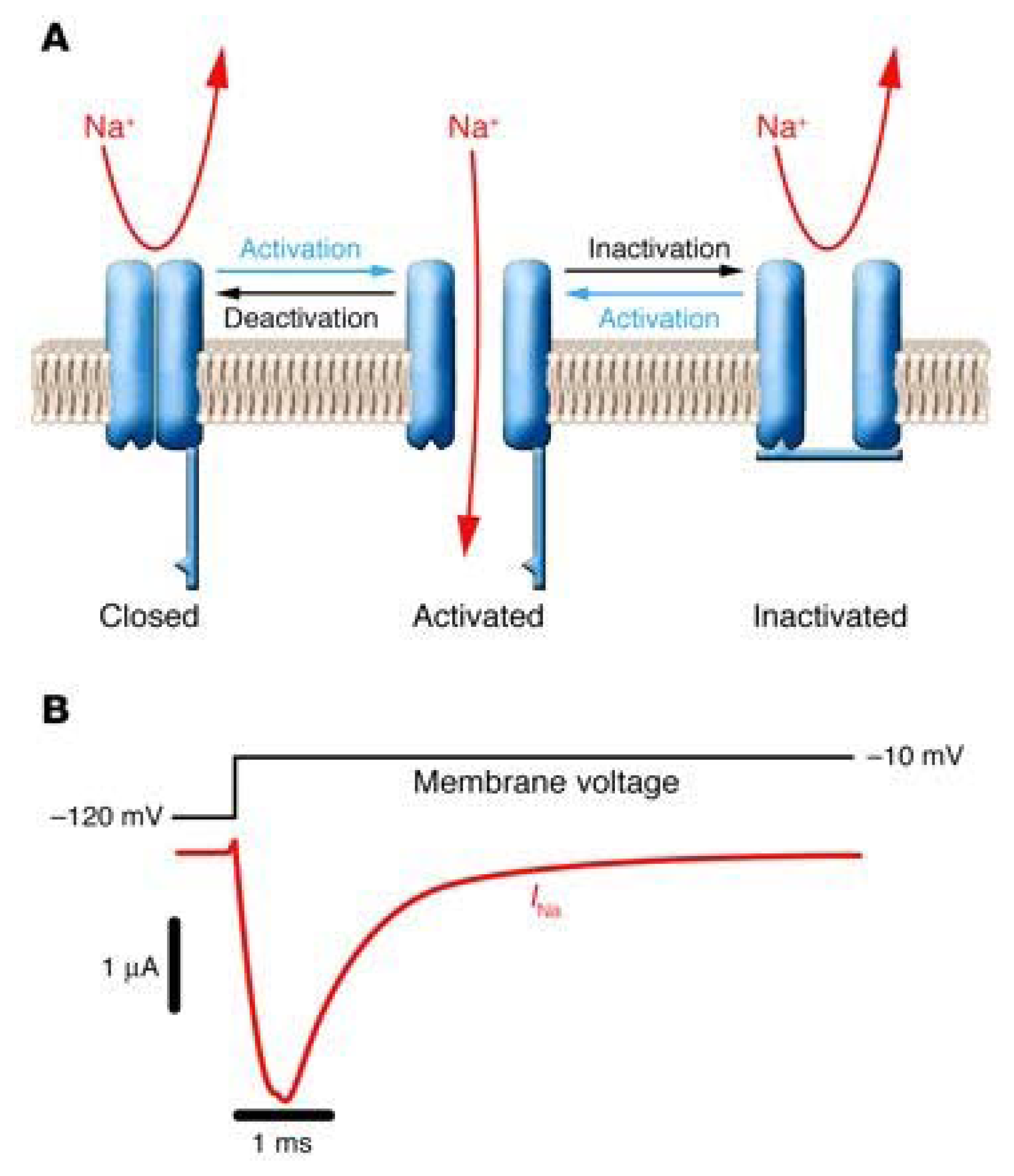
Figure 4.
Schematic representation of the structure of the CaV channel (taken from www.omedon.co.uk/ionchan/channels/calcium). The α1 subunit consists of four homologous domains (I-IV; depicted in different colors), each containing six transmembrane segments (S1–S6). This subunit contains the ion conducting pore, the voltage sensor, and the gate. The β subunit is located intracellularly and it is involved in membrane trafficking of α1 subunits. The α2 subunit is attached to the membrane-spanning δ subunit by means of disulfide bonds. The α2 subunit provides structural support, whereas the δ subunit modulates the voltage-dependent activation and steady-state inactivation processes. The γ subunit is a glycoprotein having four transmembrane segments.
Figure 4.
Schematic representation of the structure of the CaV channel (taken from www.omedon.co.uk/ionchan/channels/calcium). The α1 subunit consists of four homologous domains (I-IV; depicted in different colors), each containing six transmembrane segments (S1–S6). This subunit contains the ion conducting pore, the voltage sensor, and the gate. The β subunit is located intracellularly and it is involved in membrane trafficking of α1 subunits. The α2 subunit is attached to the membrane-spanning δ subunit by means of disulfide bonds. The α2 subunit provides structural support, whereas the δ subunit modulates the voltage-dependent activation and steady-state inactivation processes. The γ subunit is a glycoprotein having four transmembrane segments.
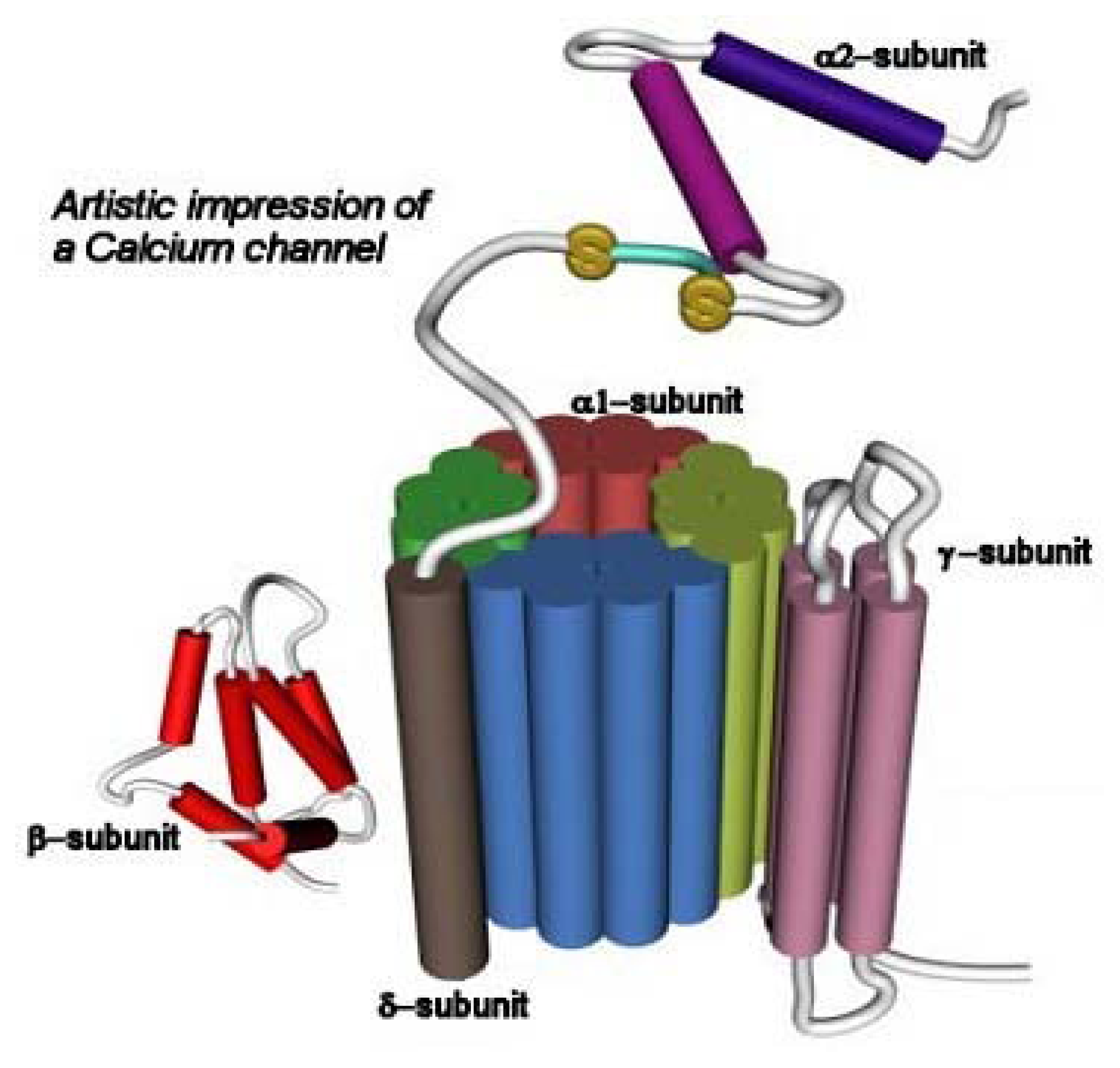
Figure 5.
Schematic representation of the structure of the KV channel (modified from [2]). (A) Each α subunit consists of six transmembrane segments (S1–S6). Positively-charged amino acids (e.g., lysine and arginine) in the S4 segment form the voltage sensor. Key residues lining the channel pore (H5 or P loop) are found between S5 and S6. The “ball and chain” structure at the N-terminal participates in N-type “fast inactivation”, occluding the permeation pathway, whereas certain residues at S6 are involved in the C-type “slow inactivation” process. Several phosphorylation sites (P) are present in the C-terminal domain. (B) The proper KV channel is assembled by the interaction of four α subunits. The pore region appears to have wide intracellular and extracellular vestibules (~28–34 Å wide and 4–8 Å deep) that lead to a constricted pore of 9–14 Å in diameter at its entrance, tapering to a diameter of 4–5 Å at a depth of ~6 Å from the vestibule. K+ ions (small green balls) are passing through the ion filter from the cytoplasmic to the extracellular side of the ion channel (efflux).
Figure 5.
Schematic representation of the structure of the KV channel (modified from [2]). (A) Each α subunit consists of six transmembrane segments (S1–S6). Positively-charged amino acids (e.g., lysine and arginine) in the S4 segment form the voltage sensor. Key residues lining the channel pore (H5 or P loop) are found between S5 and S6. The “ball and chain” structure at the N-terminal participates in N-type “fast inactivation”, occluding the permeation pathway, whereas certain residues at S6 are involved in the C-type “slow inactivation” process. Several phosphorylation sites (P) are present in the C-terminal domain. (B) The proper KV channel is assembled by the interaction of four α subunits. The pore region appears to have wide intracellular and extracellular vestibules (~28–34 Å wide and 4–8 Å deep) that lead to a constricted pore of 9–14 Å in diameter at its entrance, tapering to a diameter of 4–5 Å at a depth of ~6 Å from the vestibule. K+ ions (small green balls) are passing through the ion filter from the cytoplasmic to the extracellular side of the ion channel (efflux).
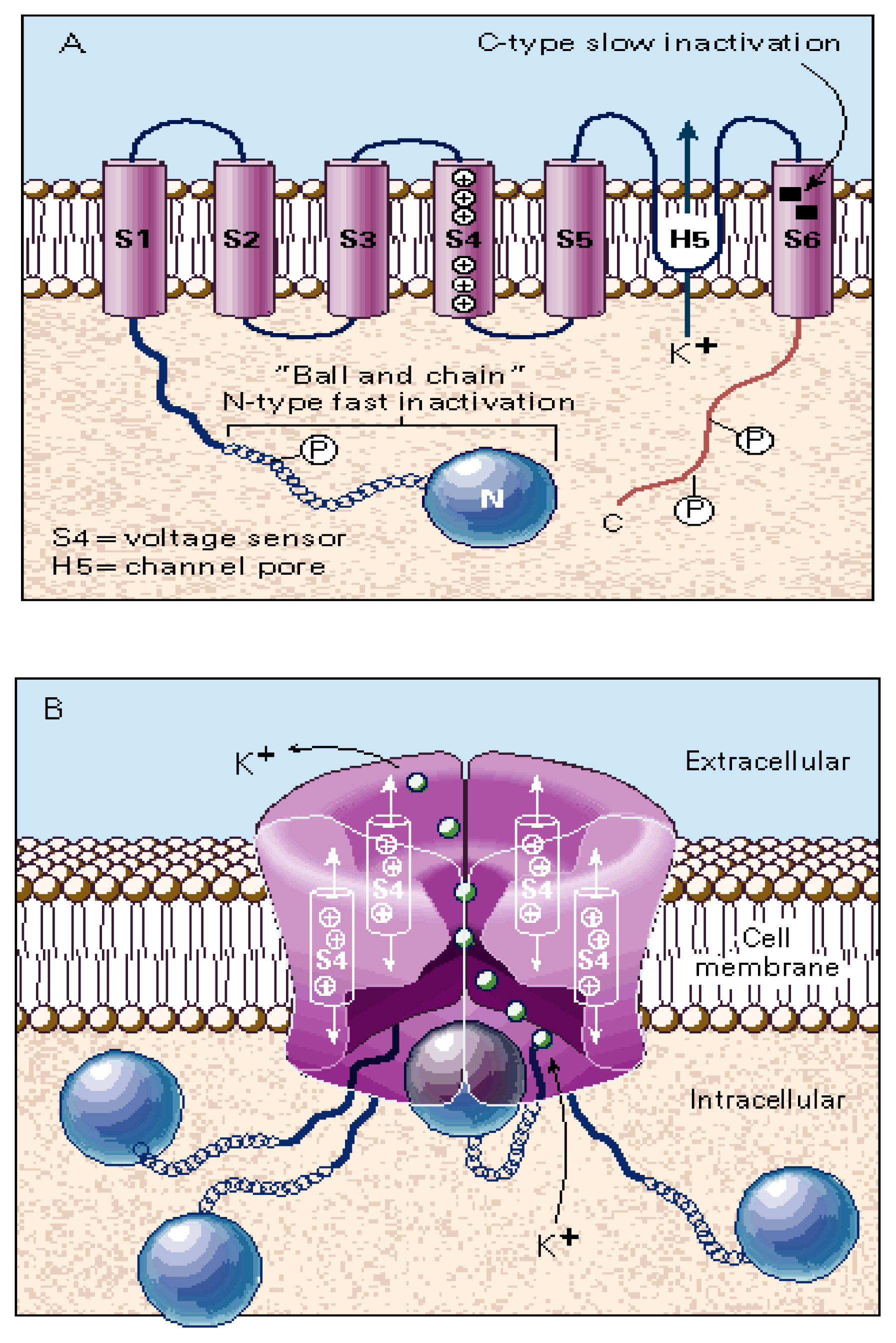
Figure 6.
Diagram showing the general dynamics of multiple conformational states on LGIC receptor superfamilies. In the absence of neurotransmitter molecules (shown here as red triangles), the receptor is in the resting state. The receptor is shown here bearing just two neurotransmitter binding sites, but there is evidence indicating that up to five binding sites may exist. In the resting state, the ion channel is closed but the receptor is still activatable. After neurotransmitter binding, the receptor is activated and the ion channel is opened, allowing the passage of different ions across the lipid membrane. In general, the agonist affinity for the activated receptor is relatively lower (shown here as a loosed interaction between the neurotransmitter and its binding site) than that in the desensitized state (shown here as a tight interaction between the neurotransmitter and its binding site). The transition from the resting to the activated state is relatively fast, but the time regime is different among receptor subtypes. In the prolonged presence of the neurotransmitter, the receptor becomes refractive to the pharmacological action of the neurotransmitter molecule and thus, its ion channel is closed. Different receptor subtypes have distinct desensitized rates and various desensitized states. In general, the desensitization process is a reversible process: after agonist washout, the receptor can be activated again. The receptor can recover from the desensitized state by means of at least two pathways: a ligand-bound receptor can reopen, or the agonist dissociates first and then the receptor can return to the resting state.
Figure 6.
Diagram showing the general dynamics of multiple conformational states on LGIC receptor superfamilies. In the absence of neurotransmitter molecules (shown here as red triangles), the receptor is in the resting state. The receptor is shown here bearing just two neurotransmitter binding sites, but there is evidence indicating that up to five binding sites may exist. In the resting state, the ion channel is closed but the receptor is still activatable. After neurotransmitter binding, the receptor is activated and the ion channel is opened, allowing the passage of different ions across the lipid membrane. In general, the agonist affinity for the activated receptor is relatively lower (shown here as a loosed interaction between the neurotransmitter and its binding site) than that in the desensitized state (shown here as a tight interaction between the neurotransmitter and its binding site). The transition from the resting to the activated state is relatively fast, but the time regime is different among receptor subtypes. In the prolonged presence of the neurotransmitter, the receptor becomes refractive to the pharmacological action of the neurotransmitter molecule and thus, its ion channel is closed. Different receptor subtypes have distinct desensitized rates and various desensitized states. In general, the desensitization process is a reversible process: after agonist washout, the receptor can be activated again. The receptor can recover from the desensitized state by means of at least two pathways: a ligand-bound receptor can reopen, or the agonist dissociates first and then the receptor can return to the resting state.
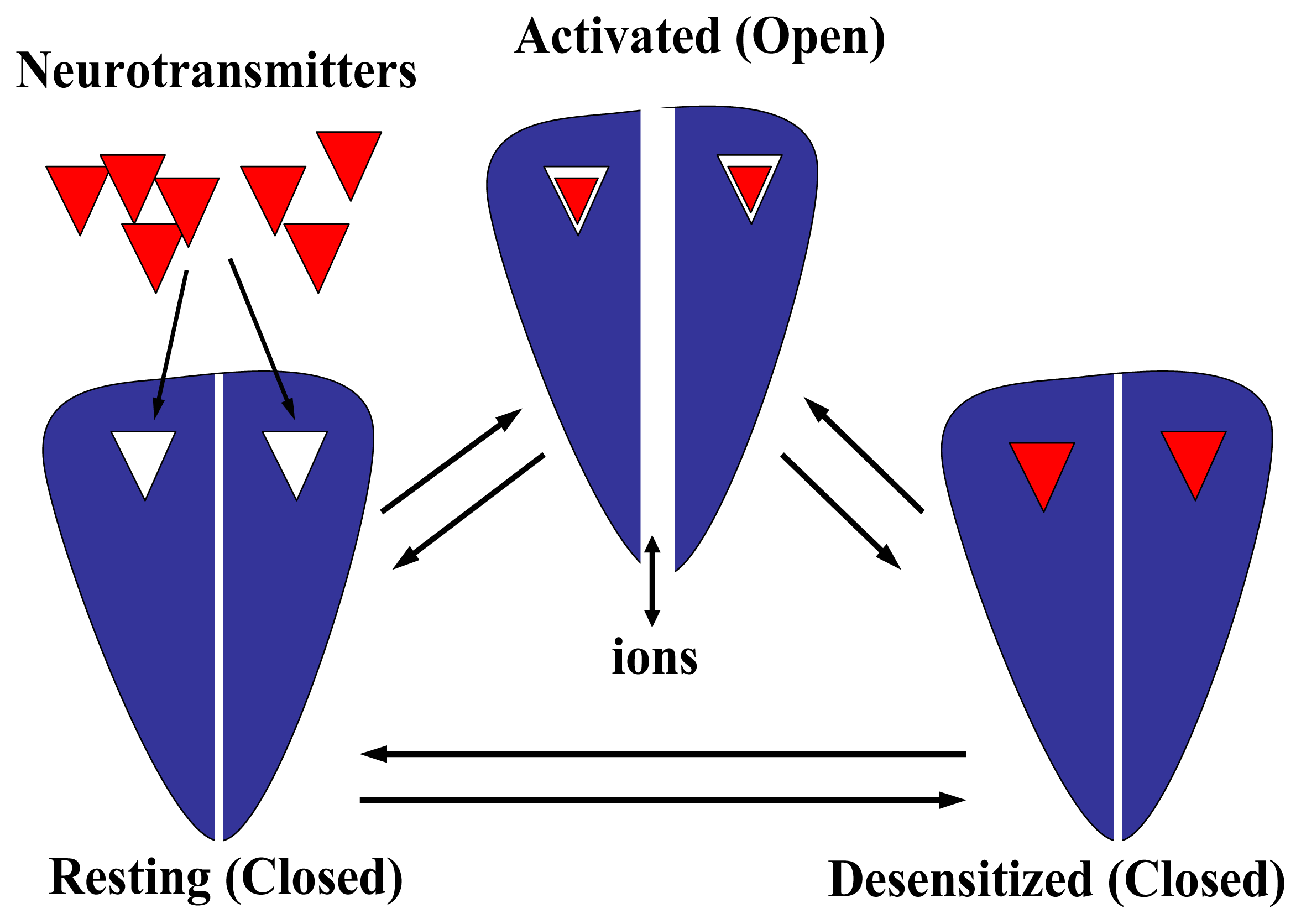
Figure 7.
Schematic representation of the structural organization of the Cys-loop receptor, GluR, and P2XnR superfamilies. (A) Schematic illustration of the primary sequence of several subunits from members of the Cys-loop receptor superfamily, including α (α1-α10) an non-α (β1-β4, γ, ɛ, and δ) subunits from the AChR, the 5-HT3AL subunit from the 5-HT3R, the α1 subunit from the GABAAR, and the α1 subunit from the GlyR. M1–M4, transmembrane domains; C—C, Cys-Cys bridge found in the Cys-loop ion-channel superfamily (homologous to Cys128 and Cys148 of the α1 AChR subunit); CC, Cys-Cys pair found in the α subunits from both muscle- and neuronal-type AChRs (corresponding to Cys192–Cys193 from the α1 AChR subunit); Y, oligosaccharide groups. (B) Diagram of the tertiary and quaternary organization of the LGIC receptor superfamilies. AChR subunits are formed by a long N-terminal hydrophilic extracellular region; four highly hydrophobic domains (M1–M4). The intrinsic ion channel is composed by five M2 segments, one from each subunit. Moreover, M1–M2 and M2–M3 are connected by minor hydrophilic stretches; and a major hydrophilic segment (M3–M4 loop) facing the cytoplasm. Additionally, the M4 domain orientates the C-terminus to the synaptic side of the membrane. On the contrary, GluR subunits present three transmembrane domains (Ml, M3, and M4) and a reentrant loop (P), as well as a cytoplasmic orientation for the COOH-terminus; while the P2XnR subunits have only two transmembrane domains (Ml and M2) and both N- and C-terminus are cytoplasm oriented. (C) Schematic representation of the oligomeric organization for LGIC receptor superfamilies. The hypothetical pentameric AChR is formed by two α subunits and three non-α chains. The two ligand binding sites (L) are located at the interfaces of one α subunit and one non-α chain. Regarding homomeric AChRs (e.g. α7, α8, and α9), up to five ligand binding sites may putatively exist. On the contrary, GluRs present four ligand binding sites, each one located in a lobe formation from each subunit, whereas P2XnRs only have three ligand binding sites, each one located on one subunit.
Figure 7.
Schematic representation of the structural organization of the Cys-loop receptor, GluR, and P2XnR superfamilies. (A) Schematic illustration of the primary sequence of several subunits from members of the Cys-loop receptor superfamily, including α (α1-α10) an non-α (β1-β4, γ, ɛ, and δ) subunits from the AChR, the 5-HT3AL subunit from the 5-HT3R, the α1 subunit from the GABAAR, and the α1 subunit from the GlyR. M1–M4, transmembrane domains; C—C, Cys-Cys bridge found in the Cys-loop ion-channel superfamily (homologous to Cys128 and Cys148 of the α1 AChR subunit); CC, Cys-Cys pair found in the α subunits from both muscle- and neuronal-type AChRs (corresponding to Cys192–Cys193 from the α1 AChR subunit); Y, oligosaccharide groups. (B) Diagram of the tertiary and quaternary organization of the LGIC receptor superfamilies. AChR subunits are formed by a long N-terminal hydrophilic extracellular region; four highly hydrophobic domains (M1–M4). The intrinsic ion channel is composed by five M2 segments, one from each subunit. Moreover, M1–M2 and M2–M3 are connected by minor hydrophilic stretches; and a major hydrophilic segment (M3–M4 loop) facing the cytoplasm. Additionally, the M4 domain orientates the C-terminus to the synaptic side of the membrane. On the contrary, GluR subunits present three transmembrane domains (Ml, M3, and M4) and a reentrant loop (P), as well as a cytoplasmic orientation for the COOH-terminus; while the P2XnR subunits have only two transmembrane domains (Ml and M2) and both N- and C-terminus are cytoplasm oriented. (C) Schematic representation of the oligomeric organization for LGIC receptor superfamilies. The hypothetical pentameric AChR is formed by two α subunits and three non-α chains. The two ligand binding sites (L) are located at the interfaces of one α subunit and one non-α chain. Regarding homomeric AChRs (e.g. α7, α8, and α9), up to five ligand binding sites may putatively exist. On the contrary, GluRs present four ligand binding sites, each one located in a lobe formation from each subunit, whereas P2XnRs only have three ligand binding sites, each one located on one subunit.
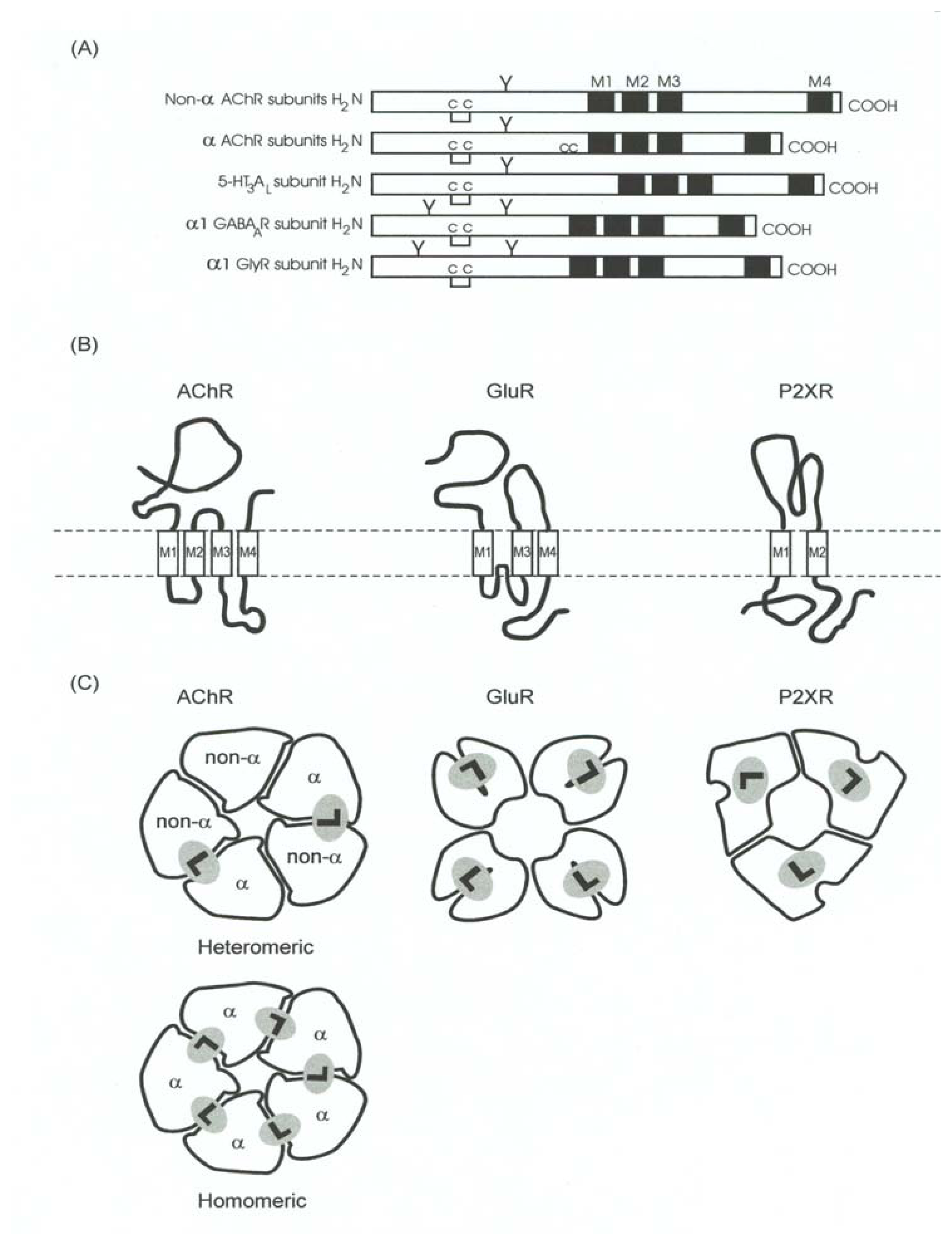

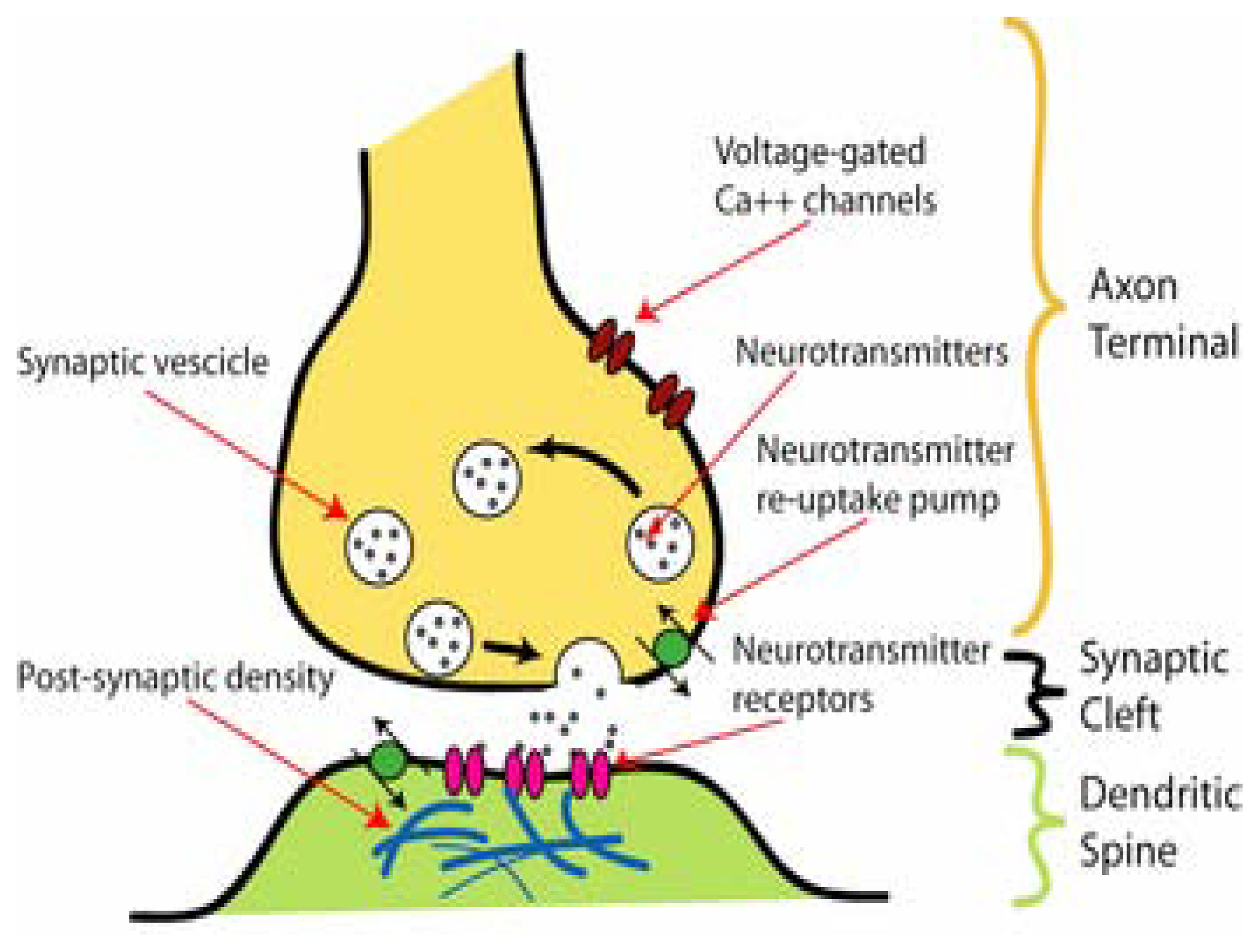{kind=link}
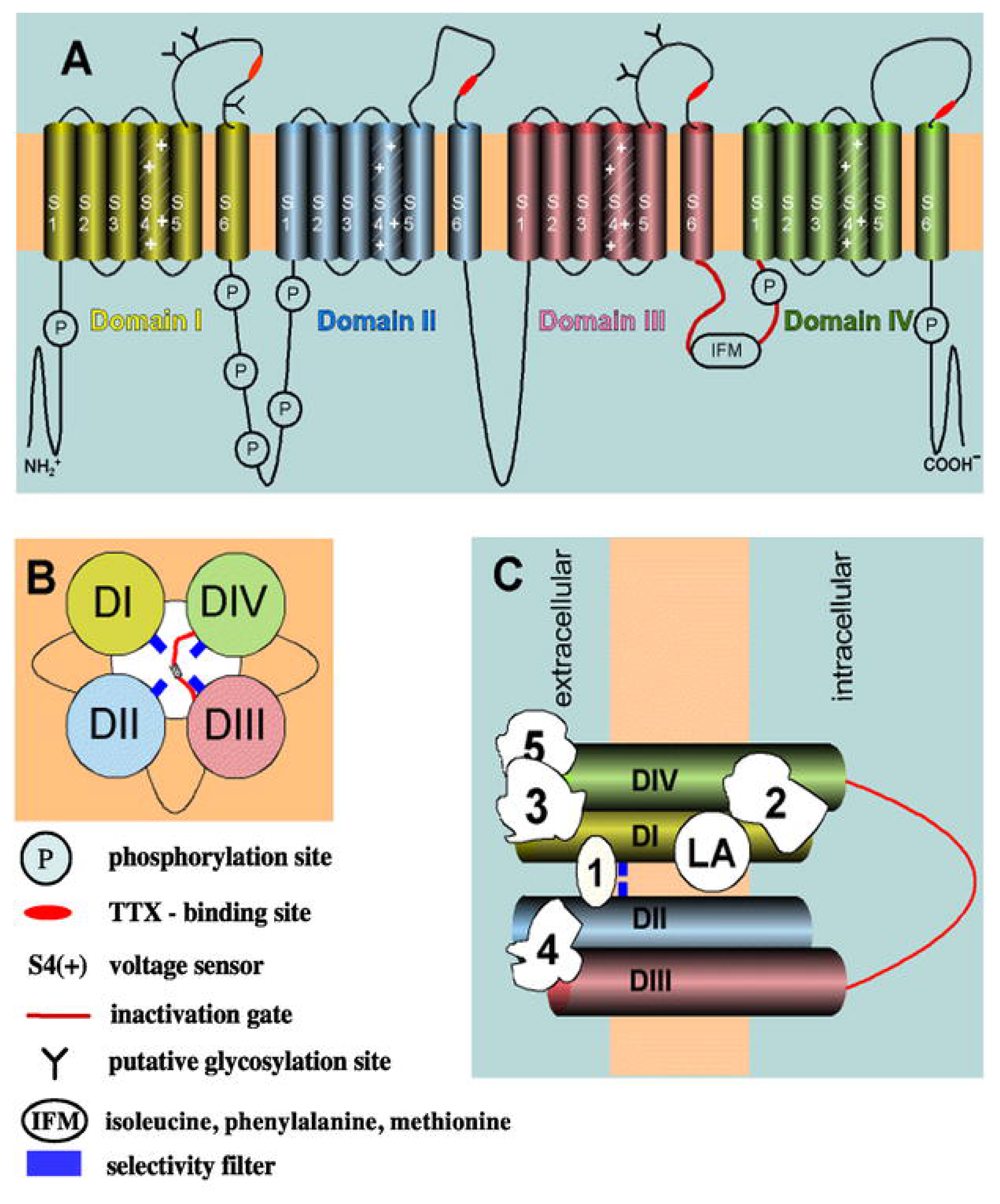{kind=link}
{kind=link}
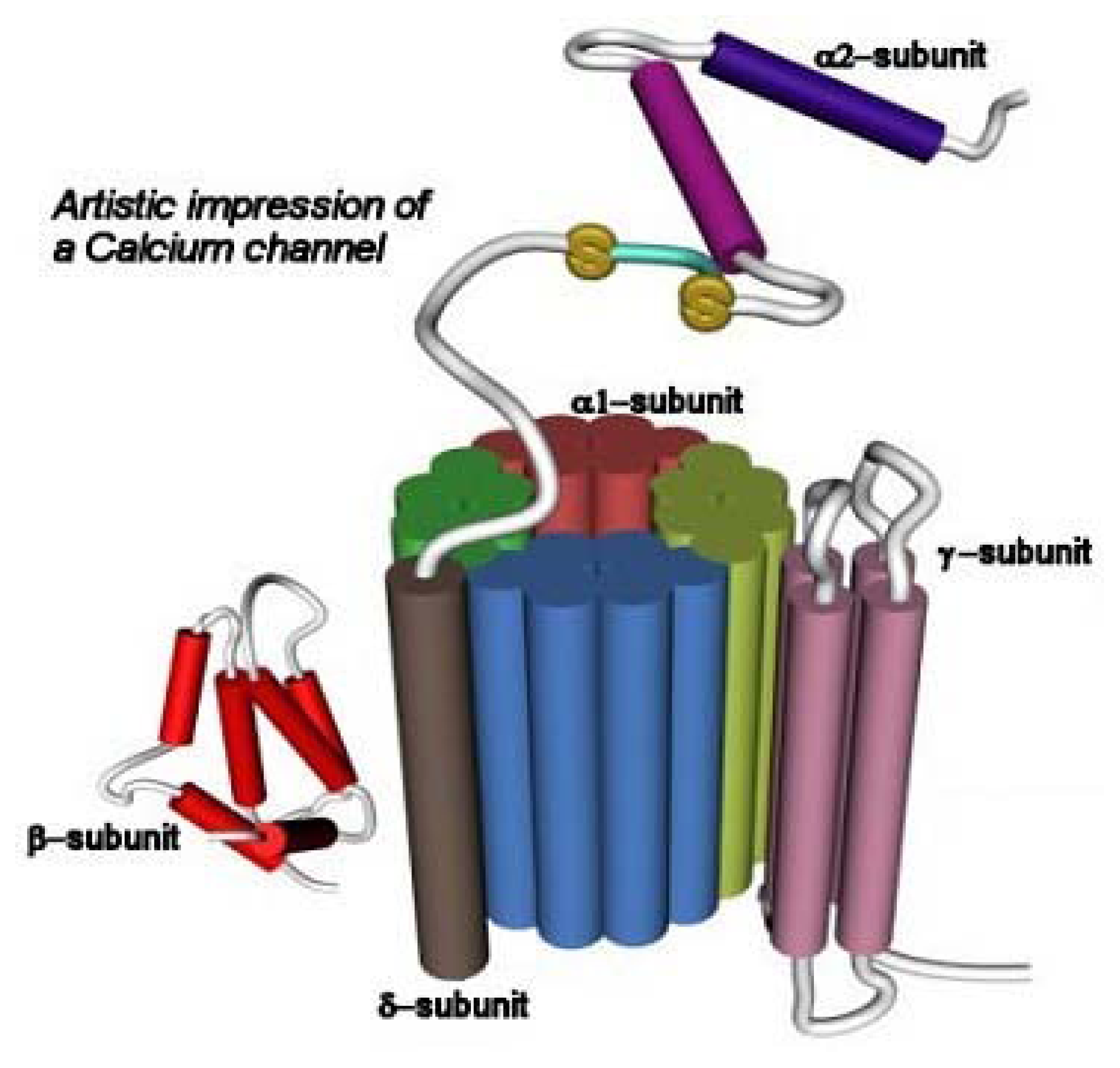{kind=link}
{kind=link}
{kind=link}
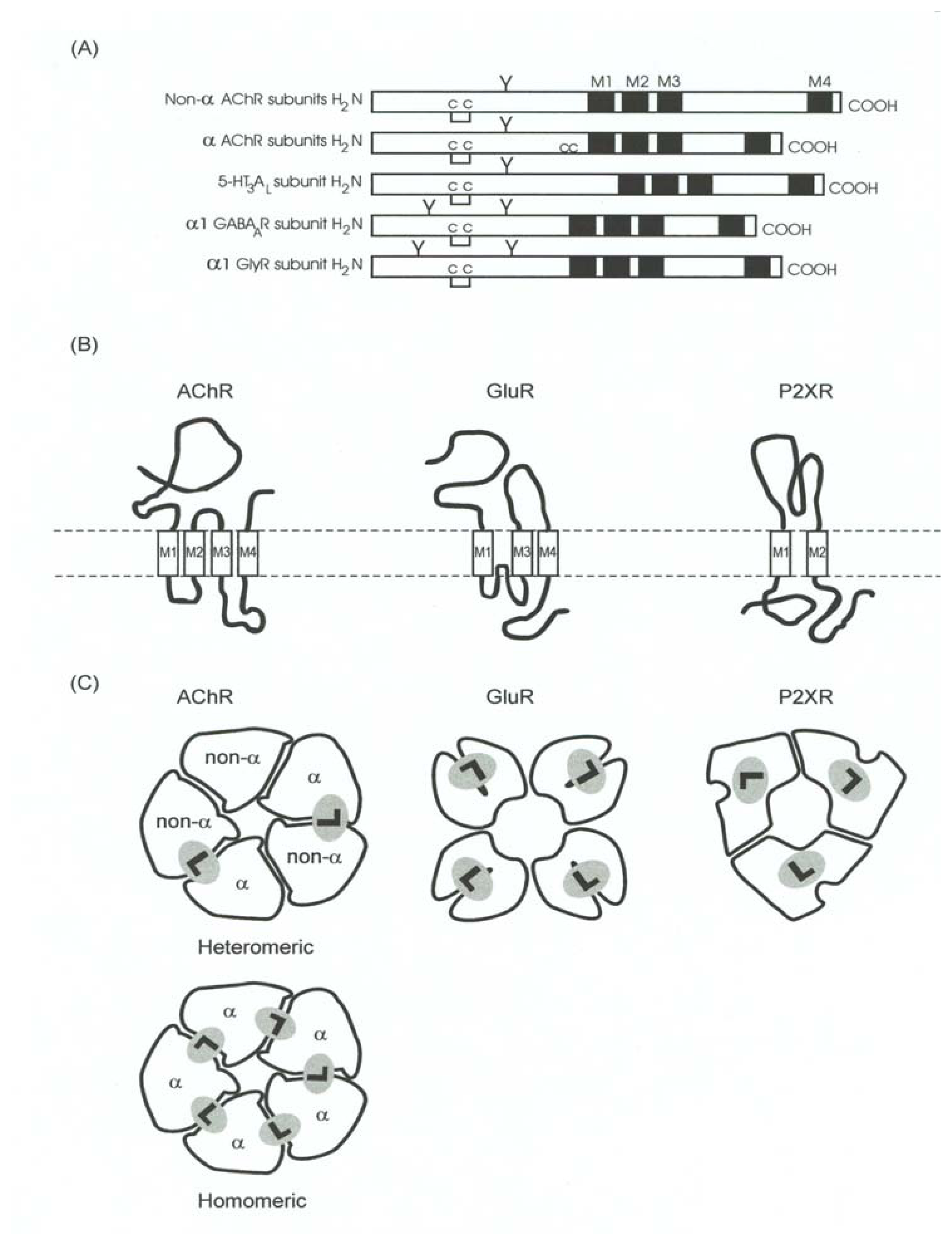{kind=link}
| Bindig Site | Toxin/Drug (origin) | Pharmacological Effect |
|---|---|---|
| 1 | Tetrodotoxin (marine) Saxitoxin (marine) μ-Conotoxins GIIIA and GIIIB (marine) | Ion channel block |
| 2 | Veratridine (plant) Batrachotoxin (animal) Aconitine (plant) Grayanotoxin (plant) | Persistent activation |
| 3 | Scorpion α-toxins (animal) Sea anemone toxins (marine) δ-Atracotoxins (animal) δ–Conotoxins PVIA, TxIA, TxIB, and TxIIA (marine) | Slow inactivation Enhancement of persistent activation |
| 4 | Scorpion β-toxins (animal) | Shift voltage-dependent activation to more negative potentials |
| 5 | Brevetoxins (marine) Ciguatoxins (marine) | Shift voltage-dependent activation to more negative potentials |
| 6 | Pyrethroids (plant) DDT (synthetic) | Slow activation, inactivation and deactivation processes |
| Local anesthetic | Local anesthetics [synthetics in general; plant product (e.g., cocaine)] | Ion channel block (use-dependent) |
| Additional binding sites | DPI 201-106 (synthetic) BDF9148 (synthetic) | Prolongation of action potential |
| Additional binding sites | Pronase (synthetic) N-bromoacetamide (synthetic) Chloramine-T (synthetic) | Disruption of fast inactivation |
| Channel Type | Current | Tissue Localization | Drugs (origin) | Pharmacological effect |
|---|---|---|---|---|
| CaV1 Channels | ||||
| CaV1.1 | L | Skeletal muscle Transverse tubules | Dihydropiridines (synthetic) Phenylalkylamines (synthetic) Benzothiazepines (synthetic) | Contraction |
| CaV1.2 | L | Cardiac myocytes Endocrine cells Neuronal cell bodies and proximal dendrites | Dihydropiridines (synthetic) Phenylalkylamines (synthetic) Benzothiazepines (synthetic) | Contraction Hormone release Synaptic integration |
| CaV1.3 | L | Endocrine cells Neuronal cell bodies and proximal dendrites | Dihydropiridines (synthetic) Phenylalkylamines (synthetic) Benzothiazepines (synthetic) | Hormone release Synaptic integration |
| CaV1.4 | L | Retina | Not established | Neurotransmitter release |
| CaV2 Channels | ||||
| CaV2.1 | P/Q | Nerve terminals and dendrites | ω-Agatoxins IVA and IVB (animal) | Inhibits neurotransmitter release |
| CaV2.2 | N | Nerve terminals and dendrites | ω-Conotoxins GVIA, MVIIC, and MVIIA (marine) | Inhibits neurotransmitter release |
| CaV2.3 | R | Neuronal cell bodies and dendrites | SNX-482 (animal) | Inhibits repetitive firing |
| CaV3 Channels | ||||
| CaV3.1 | T | Neuronal cell bodies and dendrites Cardiac myocytes | Kurtoxin (animal) Mibefradil (synthetic) Pimozide (synthetic) | Inhibits repetitive firing Inhibits pacemaker |
| CaV3.2 | T | Neuronal cell bodies and dendrites Cardiac myocytes | Kurtoxin (animal) Mibefradil (synthetic) Pimozide (synthetic) | Inhibits repetitive firing Inhibits pacemaker |
| CaV3.3 | T | Neuronal cell bodies and dendrites | Kurtoxin (animal) Mibefradil (synthetic) Pimozide (synthetic) | Inhibits repetitive firing |
L = long-lasting currents; P/Q =; R = ; T = transient currents; N = for their involvement in norepinephrine.
| Superfamily of Cys-Loop or Nicotinicoid Receptors | |||
| Family of AChRs | Family of 5-HT3Rs | Family of GABA receptors | Family of GlyRs |
| Subfamily of epithelial receptors | Subfamily of GABAARs | Subfamily of GlyRs | |
| Subfamily of neuronal α-bungarotoxin-sensitive receptors | Subfamily of GABACRs | ||
| Subfamily of muscle receptor | Subfamily of GABA-induced cation channel receptors (invertebrates) | ||
| Subfamily of heteromeric neuronal receptors | |||
| Subfamily of heteromeric protostomian receptors | |||
| Subfamily of ACh-induced anion channel receptors (invertebrates) | |||
| Superfamily of Ionotropic Glutamate Receptors | |||
| Family of excitatory GluRs | Family of inhibitory GluRs | ||
| Subfamily of NMDAR subunits | Subfamily of Glu-induced anion channel receptors (invertebrates) | ||
| Subfamily of AMPAR subunits | |||
| Subfamily of KAR subunits | |||
| Subfamily of kainate binding proteins | |||
| Subfamily of delta subunits | |||
| Superfamily of Inotropic ATP Receptors | |||
| Family of P2XnRs | |||
© 2006 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Arias, H.R. Marine Toxins Targeting Ion Channels. Mar. Drugs 2006, 4, 37-69. https://doi.org/10.3390/md403037
AMA Style
Arias HR. Marine Toxins Targeting Ion Channels. Marine Drugs. 2006; 4(3):37-69. https://doi.org/10.3390/md403037
Chicago/Turabian StyleArias, Hugo R. 2006. "Marine Toxins Targeting Ion Channels" Marine Drugs 4, no. 3: 37-69. https://doi.org/10.3390/md403037