Philinopgenin A, B, and C, Three New Triterpenoid Aglycones from the Sea Cucumber Pentacta quadrangulasis

Abstract
:Introduction
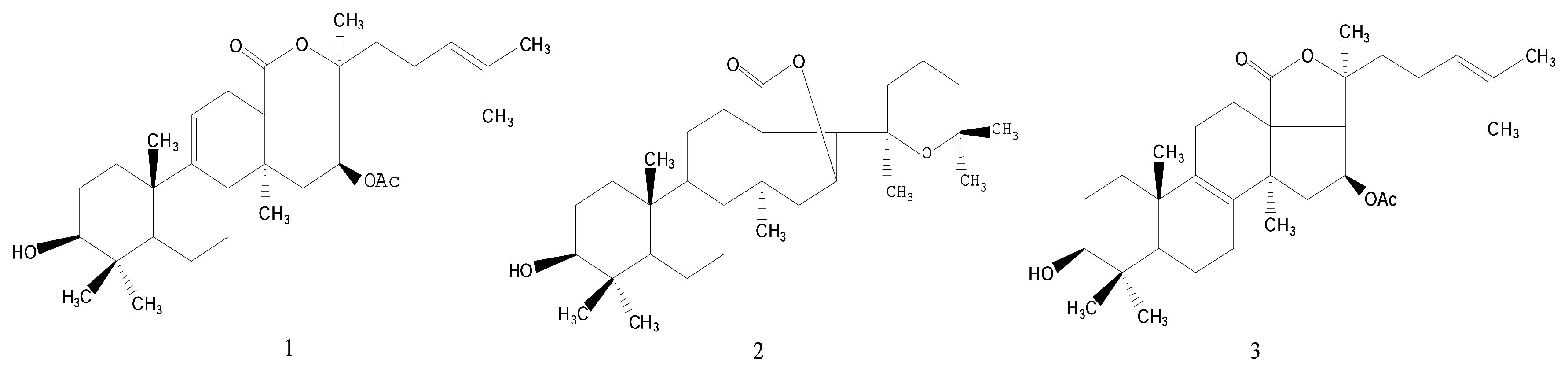
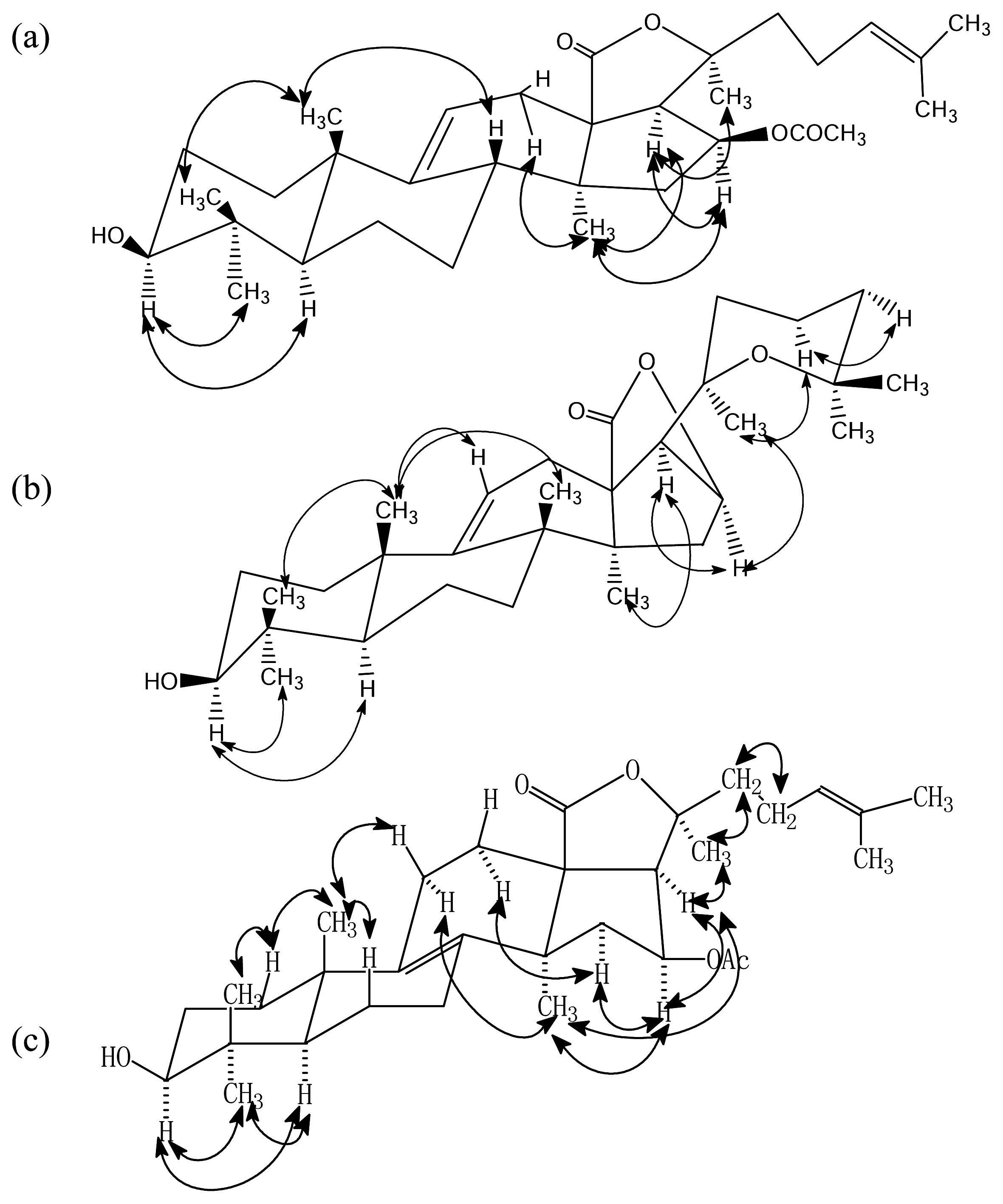
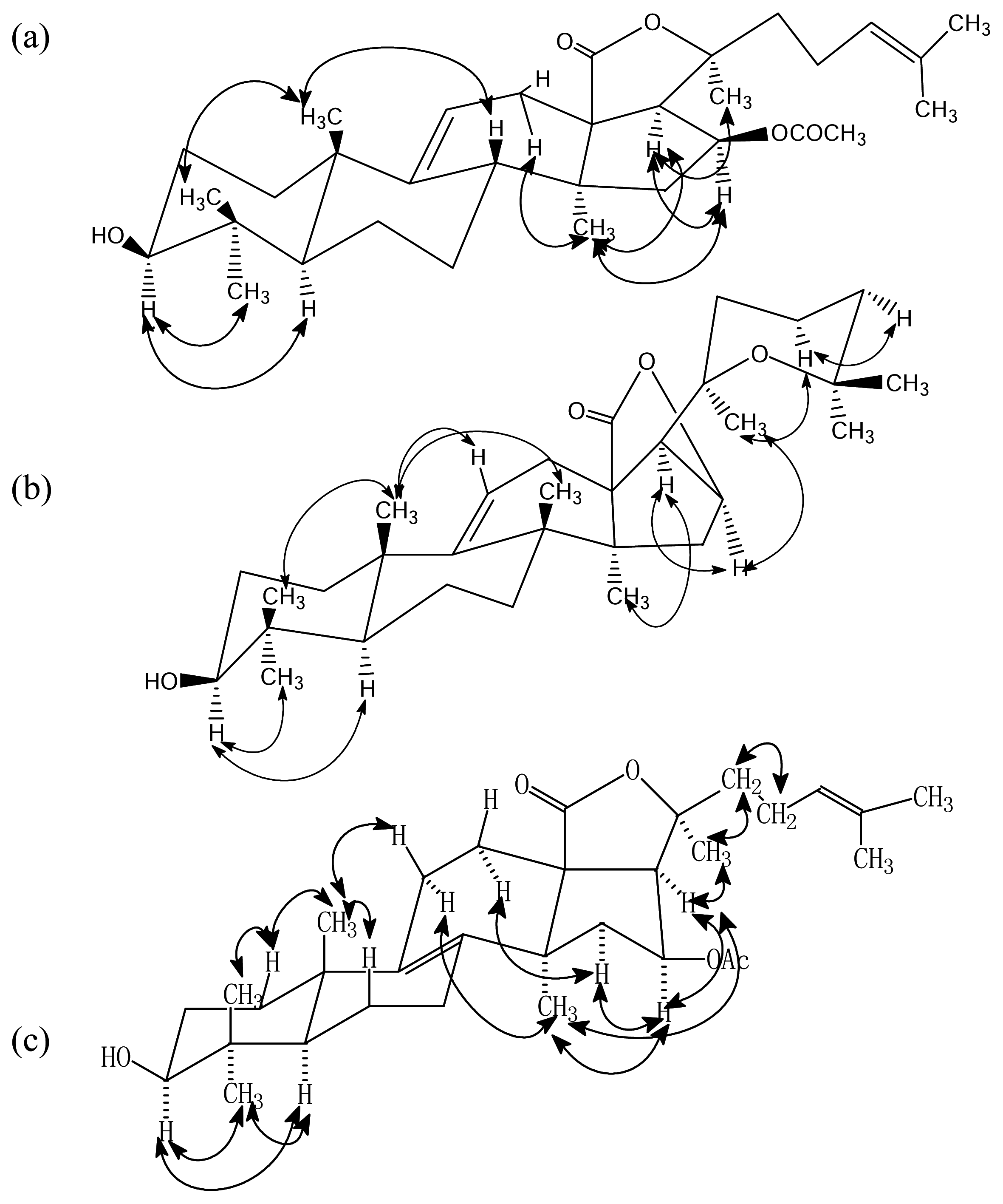
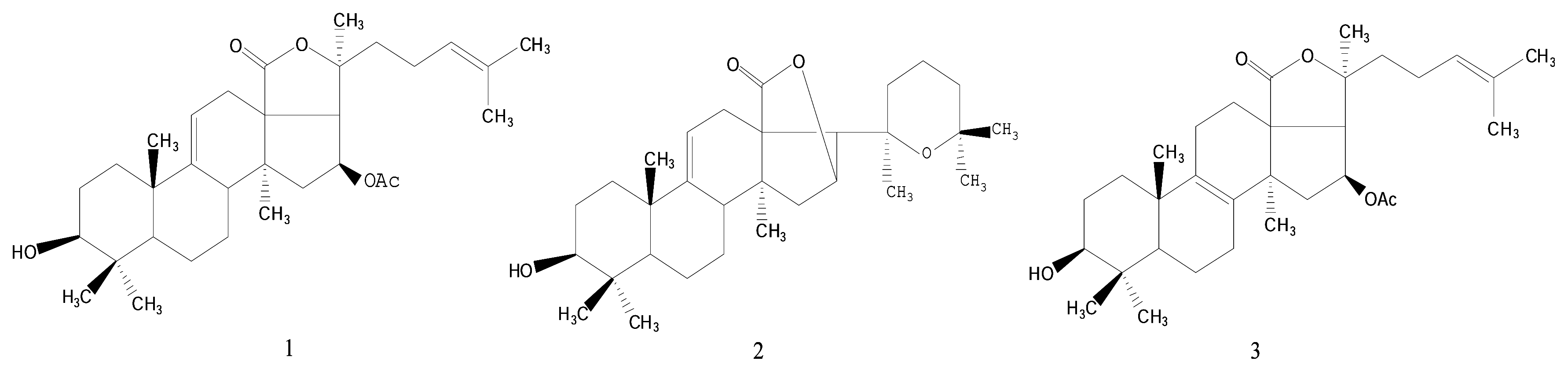
Results and Discussion
Experimental
General
Animal material
Extraction and Purification
Spectral Data
- Philinopgenin A (1): a white powder, mp 208.5–208.5 °C; IR (KBr): ν = 3443, 2970, 2937, 2869, 1770, 1748, 1378, 1240 cm−1; EIMS: m/z = 512 [M]+, 493[M-H2O, A], 479 [A-CH3, B], 435, 69 [C5H9, base peak]; 13C-NMR and 1H-NMR: see Table 1.
- Philinopgenin B (2): a white powder, mp 212.5–213.5°C; IR (KBr): ν = 3446, 2968, 2934, 2869, 1770, 1649 cm−1; EIMS: m/z = 470 [M+], 452 [m+-H2O], 391, 109, 69 [C5H9, base peak]; 13C-NMR and 1H- NMR: see Table 1.
- Philinopgenin C (3): a white powder, mp 216.5–217.5 °C; IR (KBr): ν = 3466, 2968, 2935, 1869, 1770, 1748, 1649, 1030 cm−1; EIMS: m/z = 513 [M+1], 494 [M+1-H2O, A], 479 [A-CH3, B], 435, 69 [C5H9, base peak]. 13C-NMR and 1H-NMR: see Table 1.

{kind=link}
{kind=link}
| Compound | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| Position | δC multa | δH multb (J in Hz) | δC multa | δH multb (J in Hz) | δC multa | δH multb (J in Hz) |
| 1 | 36.6t | Hα 1.44 m | 35.9t | Hα 1.43 m | 36.0t | Hα 1.45 m |
| Hβ 1.80 | Hβ 1.87 m | Hβ 1.84 m | ||||
| 2 | 28.0t | Hα | 27.8t | Hα1.68 m | 27.8t | Hα |
| Hβ | Hβ 1.72 m | Hβ | ||||
| 3 | 79.1d | 3.34, dd (5.2, 11.6) | 78.3d | 3.20 dd, (4.6, 11.6) | 79.0d | 3.21 dd, (5.2, 11.6) |
| 4 | 40.0s | 39.2s | 38.5s | |||
| 5 | 52.6d | 0.90, d, 11.0 | 52.6d | 0.86 tr | 52.5d | 0.90 m |
| 6 | 21.4t | Hα 1.58 m | 21.1t | Hα 1.46 m | 21.0t | Hα 2.07 m |
| Hβ 1.75 m | Hβ 1.70 m | Hβ 2.07 m | ||||
| 7 | 28.0t | Hα | 28.3t | Hα 1.78 m | 27.8t | Hα 1.36 m |
| Hβ | Hβ 1.78 m | Hβ 1.62 m | ||||
| 8 | 39.6d | 3.22, dd (2.4, 13.2) | 43.4d | 130.2s | ||
| 9 | 151.2s | 151.3s | 135.6s | |||
| 10 | 40.5s | 39.4s | 39.3s | |||
| 11 | 110.8d | 5.16s | 113.4d | 5.40 d, (6.0) | 27.8t | |
| 12 | 34.0t | Hα 2.36 m | 24.9t | Hα 2.33 m | 32.3t | Hα 2.40 m |
| Hβ 2.36 m | Hβ 2.40 m | Hβ 2.45 m | ||||
| 13 | 58.6s | 54.8s | 59.6s | |||
| 14 | 43.2s | 40.4s | 44.7s | |||
| 15 | 44.1t | Hα 2.19 m | 42.0t | Hα1.56 m | 41.1t | Hα 2.22 m |
| Hβ 1.43m | Hβ 1.74 m | Hβ 1.42m | ||||
| 16 | 75.0d | 5.76 m | 79.2d | 4.77 tr | 75.0d | 5.63 m |
| 17 | 52.9d | 2.57 d (9.6) | 63.5d | 2.25 m | 52.3d | 2.51 d, (9.6) |
| 18 | 176.7s | 178.1s | 176.8s | |||
| 19 | 22.3q | 1.30 s | 21.5q | 1.25 s | 21.8q | 1.19 s |
| 20 | 84.8s | 73.0s | 84.4s | |||
| 21 | 28.8q | 1.36 s | 26.0s | 1.25 s | 28.2q | 1.50 s |
| 22 | 38.5t | Hα 1.96 m | 33.8t | Hα1.48 m | 38.4t | Hα 1.76 m |
| Hβ 2.30m | Hβ 1.48 m | Hβ 2.42 m | ||||
| 23 | 23.8t | Hα 1.98 m | 16.3t | Hα 1.58 m | 23.5t | Hα 1.92 m |
| Hβ 2.02 m | Hβ 1.76 m | Hβ 2.11 m | ||||
| 24 | 124.4d | 5.04 d (11.2) | 36.5t | Hα 1.36 m) | 123.7d | 5.05 d, (11.2) |
| 25 | 131.8s | 71.8s | 132.1s | |||
| 26 | 25.5q | 1.57 s | 32.7q | 1.13 s | 25.5q | 1.69 s |
| 27 | 17.7q | 1.50 s | 28.4q | 1.20 s | 18.6q | 1.60 s |
| 30 | 16.4q | 0.98 s | 15.2q | 0.80 s | 15.4q | 0.85 s |
| 31 | 28.6q | 1.10 s | 28.3q | 0.98 s | 28.1q | 1.02 s |
| 32 | 21.4q | 0.80 s | 21.7q | 1.07 s | 27.0q | 1.10 s |
| CH3COO | 170.0s | 170.3s | ||||
| CH3COO | 21.4q | 1.93 s | 21.0q | 2.02 s | ||
Acknowledgments
- Sample Availability: Samples are available from the authors.
References
- Katagawa, T.; Sugawara, I.; Yosioka, K. Saponin and Sapogenol. XIV. Antifungal Glycosides from the Sea Cucumber Stichopus japonicus Selenka. Chem. Pharm. Bull 1976, 24, 266–274. [Google Scholar]
- Girard, M.; Bélanger, J.; ApSimon, J.W.; Garneau, F-X.; Harvey, C.; Brisson, J-R. Frondoside A. A novel triterpene glycoside from the holothurian Cucumaria frondosa. Can. J. Chem 1990, 68, 11–18. [Google Scholar]
- Avilov, S.A.; Antonov, A.S.; Drozdova, O.A.; Kalinin, V.I.; Kalinovsky, A.I.; Stonik, V.A.; Riguera, R.; Lenis, L.A.; Jiménez, C. Triterpene Glycosides from the Far-East Sea Cucumber Pentamera calcigera. 1. Monosulfated Glycosides and Cytotoxicity of Their Unsulfated. J. Nat. Prod 2000, 65–71. [Google Scholar]
- Burnell, DJ; Apsimon, JW. Marine Natural Products — Chemical and Biochemical Perspectives; Scheuer, P.J., Ed.; Academic Press: New York, 1983; Volume 5, pp. 287–389. [Google Scholar]
© 2004 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
Zhang, S.-L.; Li, L.; Yi, Y.-H.; Zou, Z.-R.; Sun, P. Philinopgenin A, B, and C, Three New Triterpenoid Aglycones from the Sea Cucumber Pentacta quadrangulasis. Mar. Drugs 2004, 2, 185-191. https://doi.org/10.3390/md204185
Zhang S-L, Li L, Yi Y-H, Zou Z-R, Sun P. Philinopgenin A, B, and C, Three New Triterpenoid Aglycones from the Sea Cucumber Pentacta quadrangulasis. Marine Drugs. 2004; 2(4):185-191. https://doi.org/10.3390/md204185
Chicago/Turabian StyleZhang, Shi-Long, Ling Li, Yang-Hua Yi, Zheng-Rong Zou, and Peng Sun. 2004. "Philinopgenin A, B, and C, Three New Triterpenoid Aglycones from the Sea Cucumber Pentacta quadrangulasis" Marine Drugs 2, no. 4: 185-191. https://doi.org/10.3390/md204185