1-O-Sulfatobastadins-1 and -2 from Ianthella basta (Pallas).Antagonists of the yR1-FKBP12 Ca2+ Channel

Abstract
:Introduction
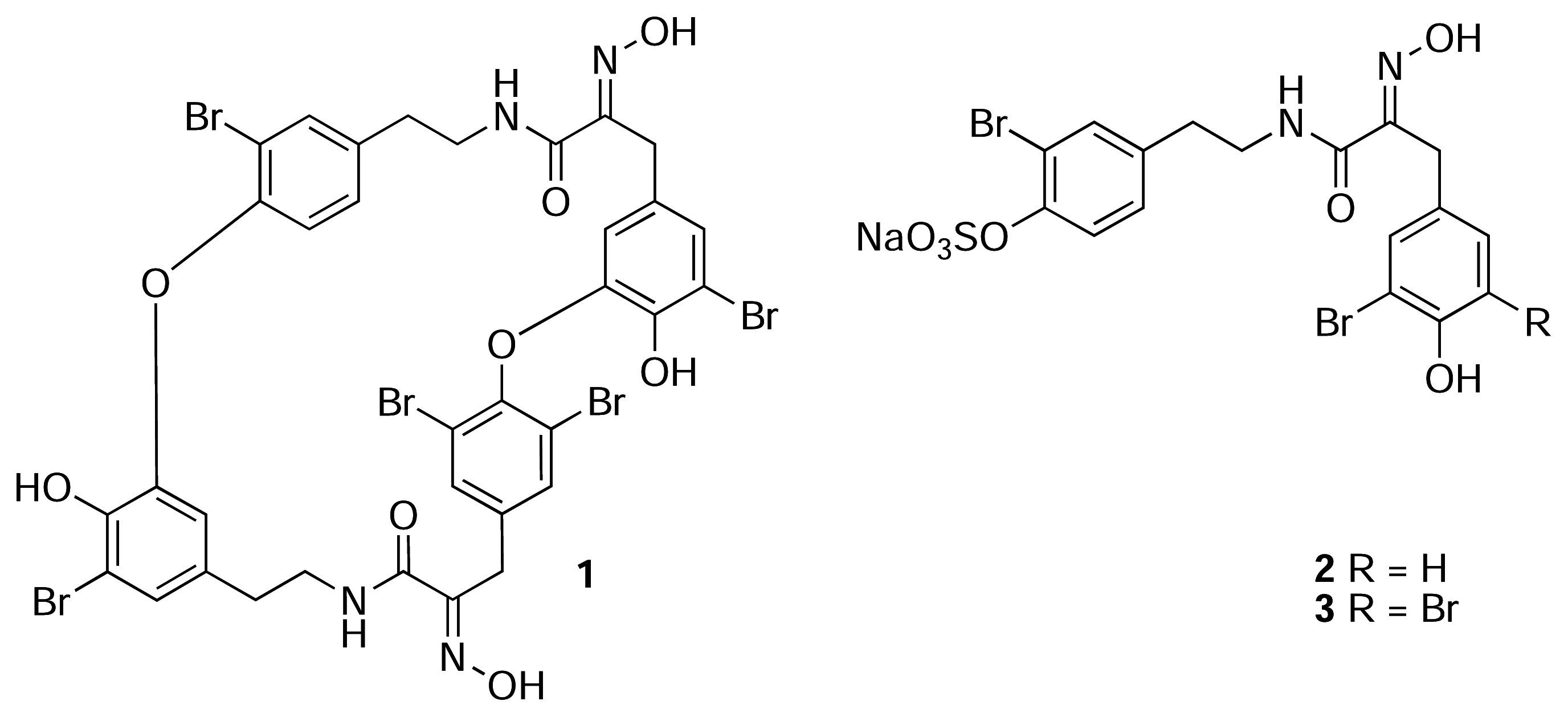
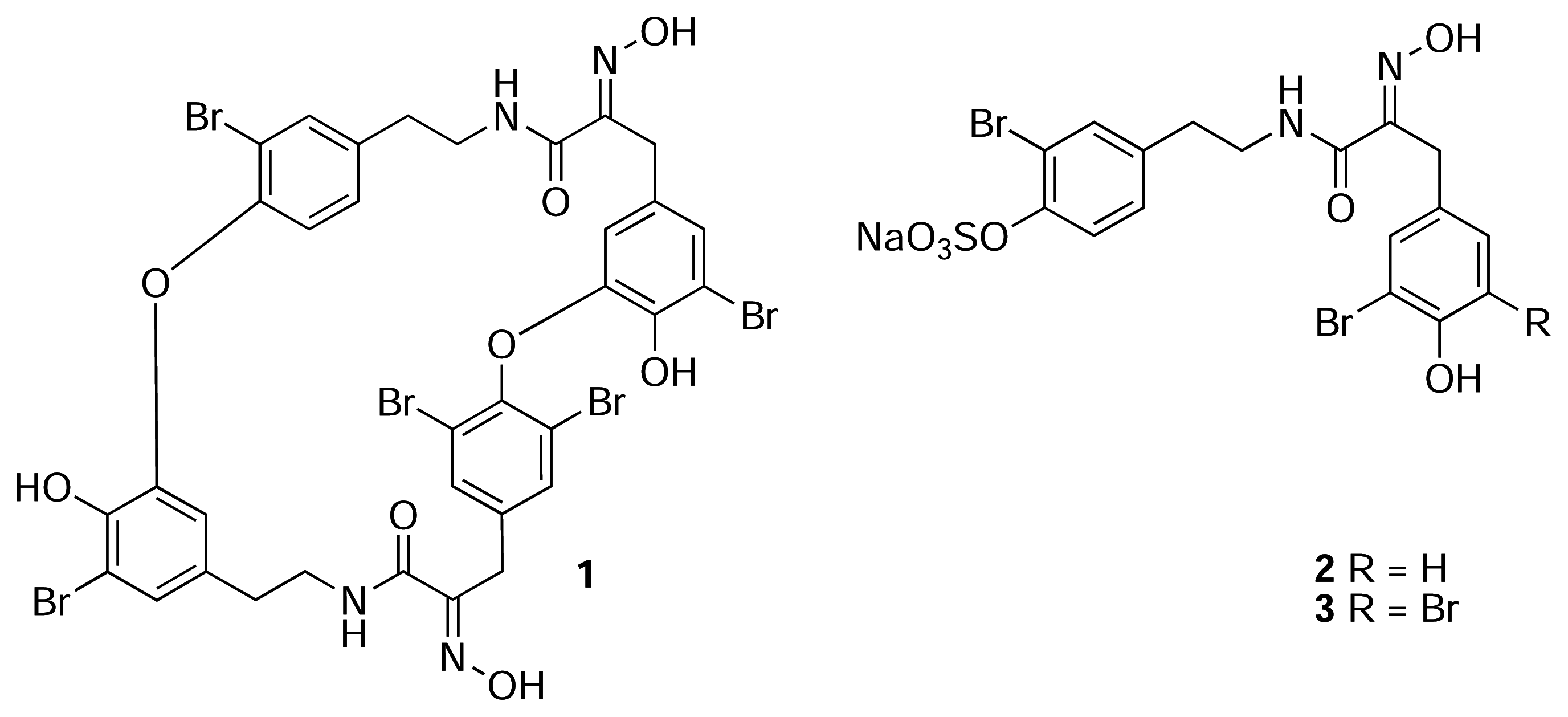
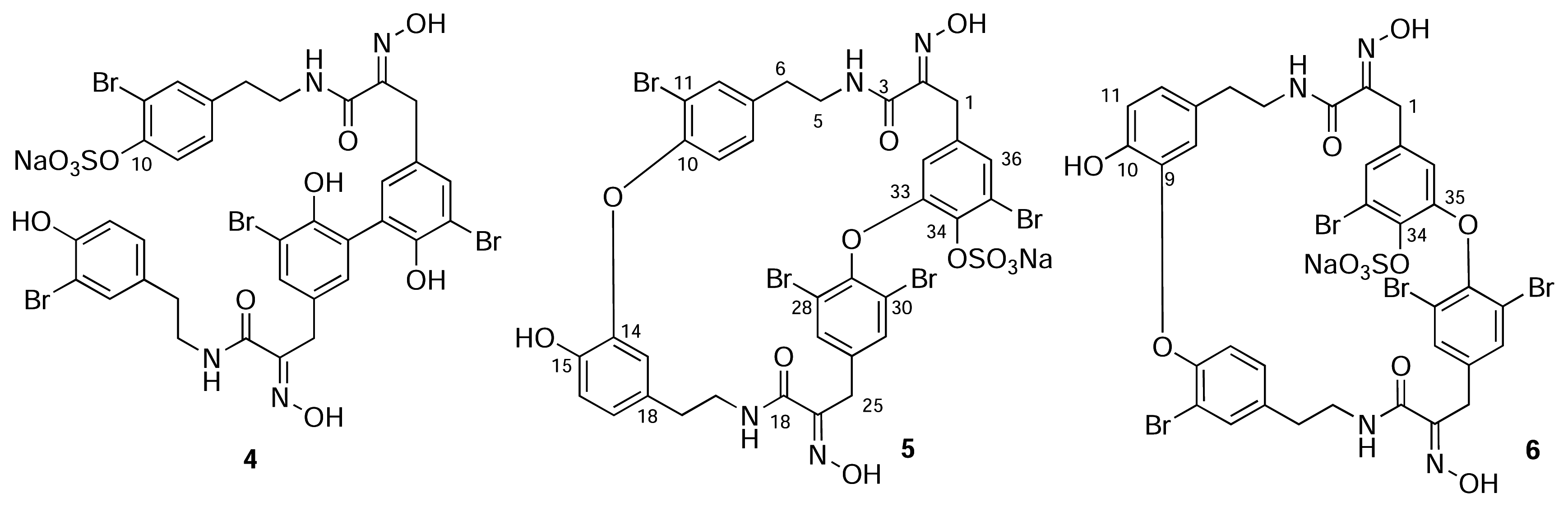
Results and Discussion
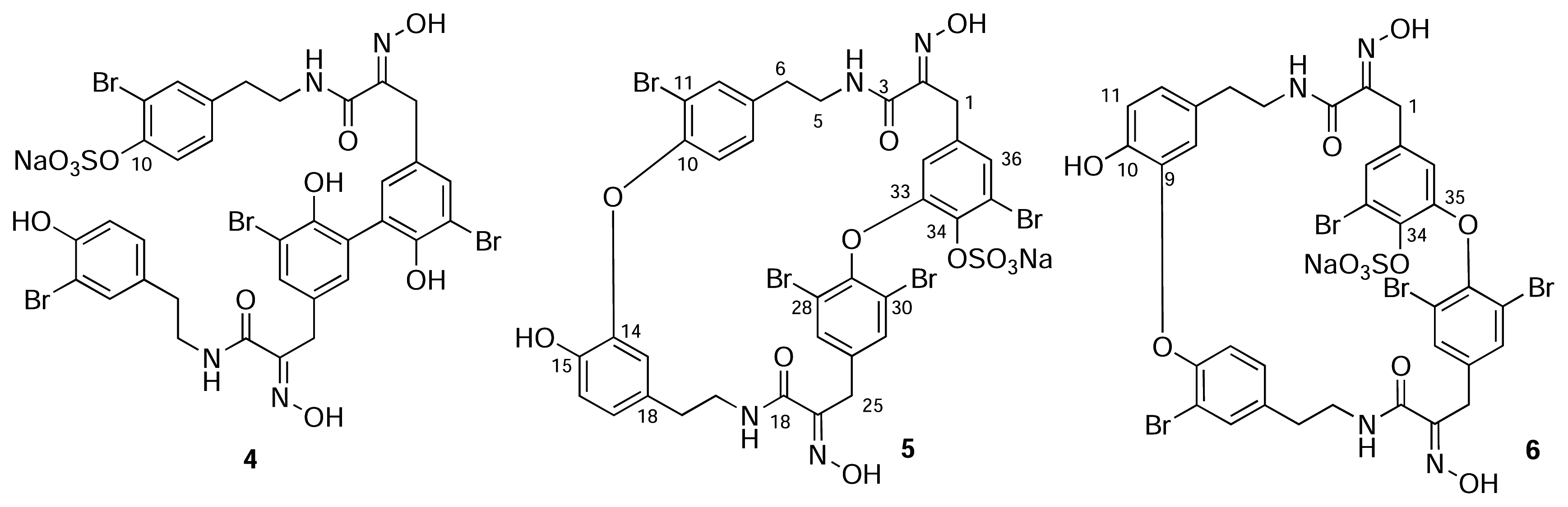
Conclusions
Experimental
General
Isolation of 1–5
Spectroscopic Data
[3H]-Ryanodine Binding Assay
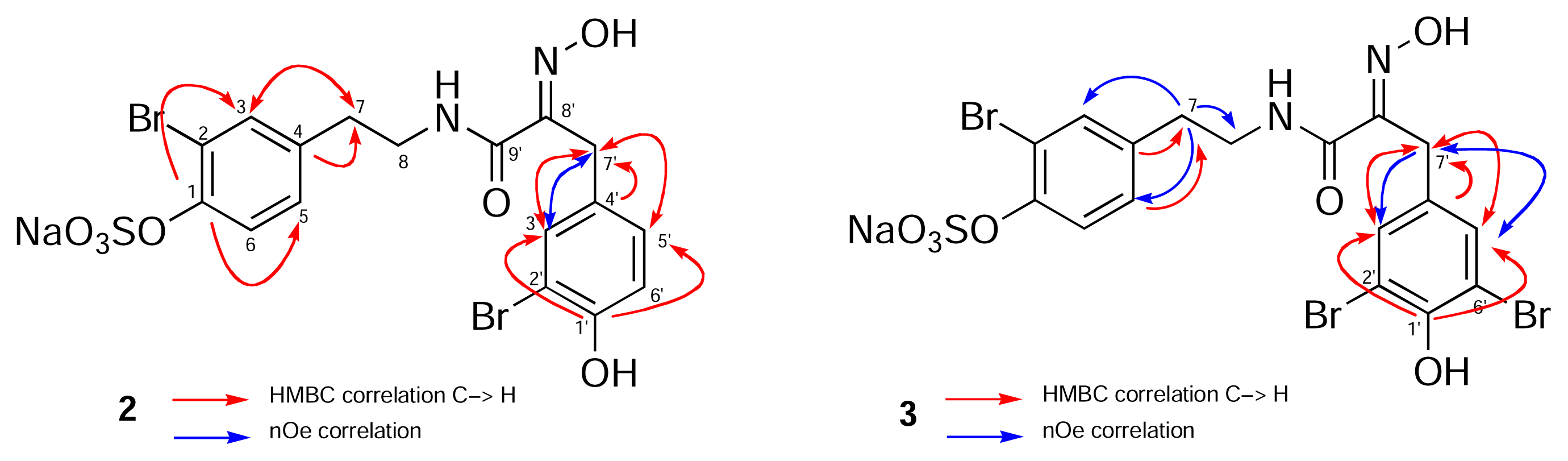

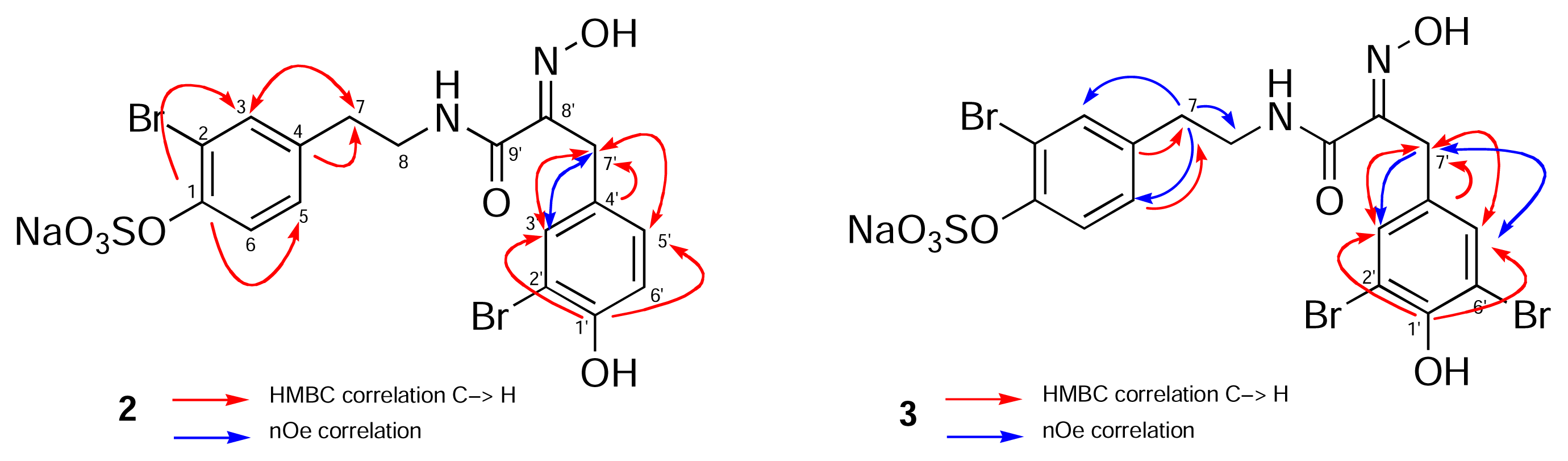{kind=link}
{kind=link}
{kind=link}
| # | 13C-NMR (2), δ | 1H-NMR (2) δ (mult., J Hz, Int.) | HMBC | 13C-NMR (3) δ | 1H-NMR (3) δ (mult., J Hz, Int.) | HMBC |
|---|---|---|---|---|---|---|
| 1b | 149.7 | H3, H5, H6 | 149.8 | |||
| 2 | 116.7 | H3, H6 | 116.7 | |||
| 3 | 134.5 | 7.42 (d, J=2.0, 1H) | H5, H7 | 134.5 | 7.42 (d, J=2.0, 1H) | - |
| 4 | 138.5 | H6, H7, H8 | 138.5 | H7, H8 | ||
| 5 | 129.7 | 7.09 (dd, J=8.4,2.0, 1H) | H3, H7 | 129.7 | 7.11 (dd, J=8.4,2.0, 1H) | H7 |
| 6 | 123.4 | 7.49 (d, J=8.4, 1H) | 123.5 | 7.50 (d, J=8.4, 1H) | ||
| 7 | 35.4 | 2.75 (t, J=7.2, 2H) | H3, H5, H8 | 35.4 | 2.76 (t, 2H, J=7.2) | H8 |
| 8 | 41.7 | 3.43 (t, J=7.2, 2H) | H7 | 41.8 | 3.43 (t, 2H, J=7.2) | H7 |
| 1′b | 153.8 | H3′, H5′ | 150.7 | H3′, H5′ | ||
| 2′ | 110.5 | H3′, H6′ | 112.0 | H3′ | ||
| 3′ | 134.5 | 7.36 (d, J=2.0, 1H) | H5′, H7′ | 133.9 | 7.38 (s, 2H) | H7′ |
| 4′ | 130.6 | H6′, H7′ | 132.3 | H7′ | ||
| 5′ | 130.2 | 7.02 (dd, J=8.0, 2.0, 1H) | H3′, H7′ | 133.9 | 7.38 (s, 2H) | H7′ |
| 6′ | 117.1 | 6.76 (d, J=8.0, 1H) | 112.0 | H5′ | ||
| 7′ | 28.6 | 3.78 (s, 2H) | H3′, H5′ | 28.4 | 3.78 (s, 1H) | H3′, H5′ |
| 8′ | 153.2 | H7′ | 152.5 | H7′ | ||
| 9′ | 165.8 | H7′, H8 | 165.6 | H8, H7′ |
Acknowledgements
- Sample Availability: Contact authors.
References and Notes
- Kazlauskas, R.; Lidgard, R. O.; Murphy, P. T.; Wells, R. J.; Blount, J. F. Brominated Tyrosine-Derived Metabolites from the Sponge Ianthella basta. Aust. J. Chem 1981, 34, 765–786. [Google Scholar]
- Mack, M. M.; Molinski, T. F.; Buck, E. D.; Pessah, I. N. Novel Modulators of Skeletal Muscle FKBP12/Calcium Channel Complex from Ianthella basta: Role of FKBP in Channel Gating. J. Biol. Chem 1994, 269, 23236–23249. [Google Scholar]
- Clardy, J. The Chemistry of Signal Transduction. Proc. Nat. Acad. Sci. U.S.A 1995, 92, 56–61. [Google Scholar]
- Pordesimo, E. O.; Schmitz, F. J. New Bastadins from the Sponge Ianthella basta. J. Org. Chem 1990, 55, 4704–4709. [Google Scholar]
- Pessah, I. N.; Molinski, T. F.; Meloy, T. D.; Wong, P.; Buck, E. D.; Allen, P. D.; Mohr, F. C.; Mack, M. M. Bastadins Relate Ryanodine-Sensitive and -Insensitive Ca2+ Efflux Pathways in Skeletal SR and BC3H1 Cells. Am. J. Physiol 1997, 41, C601–C614. [Google Scholar]
- Butler, M. S.; Lim, T. K.; Capon, R. J.; Hammond, L. S. The Bastadins Revisited: New Chemistry from the Australian Marine Sponge Ianthella basta. J. Nat. Prod 1991, 44, 287–296. [Google Scholar]Pettit, G. R.; Butler, M. S.; William, M. D.; Filiatrault, M. J.; Pettit, R. K. Isolation and Structure of Hemibastadinols 1–3 from the Papua New Guinea Marine Sponge Ianthella basta. J. Nat. Prod 1996, 59, 927–934. [Google Scholar]
- For uniformity, we present the structures of all new compounds as the Na+salts of the sulfate-half esters as Na+is the most likely counter ion from extraction of marine invertebrates. The numbering scheme used here for 2 and 3 is adopted from Pettit (reference 6b) and differs from the numbering of the macrocyclic bastadins (eg. 1 reference [1]).
- Franklin, M. A.; Penn, S. G.; Lebrilla, C. B.; Lam, T. H.; Pessah, I. N.; Molinski, T. F. Bastadin 20 and Bastadin O-Sulfate Esters from Ianthella basta: Novel Modulators of the Ry(1)R FKBP12 Receptor Complex. J. Nat. Prod 1996, 59, 1121–1127. [Google Scholar]Franklin, M. A. M.Sc. Thesis, University of California, Davis, 1995.
- Unlike simple acyclic oximes which exhibit facile E-Z isomerism, the configuration of the C=N double bond in every cyclic bastadin is strictly E. A strong intramolecular H-bond between the amide NH and the N=C groups forms a rigid, planar 5-membered ring and places the OH group anti to the H-bonded donor electron pair on the imine nitrogen (viz. X-ray structures of bastadin-4 [1] and bastadin-10, Molinski, T. F.; Olmstead, M. M. unpublished results.). Both E/Z oxime isomers have been detected in related α-ketoximines from marine sponges and theE geometry has been correlated with an upfield 13C-NMR signal for the benzylic CH2 group (δ ~28 ppm, c.f. Z- δ ~35 ppm. Arabshahi, L.; Schmitz, F. J. Brominated Tyrosine Metabolites from an Unidentified Sponge. J. Org. Chem 1987, 52, 3584. [Google Scholar]). Compounds 2, 3 and 5 have E oximes.
- Ragan, M. A. Phenol Sulfate Esters: Ultraviolet, Infrared, Proton and Carbon-13 Nuclear Magnetic Resonance Spectroscopic Investigation. Can. J. Chem 1978, 56, 2681–2685. [Google Scholar]
- Gulavita, N. K.; Wright, A. E.; McCarthy, P. J.; Pomponi, S. A.; Kelly-Borges, M.; Chin, M.; Sills, M. A. Isolation and Structure Elucidation of 34-Sulfatobastadin 13, An Inhibitor of the Endothelin A Receptor, from a Marine Sponge of the Genus Ianthella. J. Nat. Prod 1993, 56, 1613–1617. [Google Scholar]
- This is also consistent with the C1’ 13C-NMR assignment (δ 154.0, s) of the parent alcohol, hemibastadin-1 [6b].
- Jaspars, M.; Rali, T.; Laney, M.; Schatzman, R. C.; Diaz, M. C.; Schmitz, F. J.; Pordesimo, E. O.; Crews, P. The Search for Inosine 5′-Phosphate Dehydrogenase (IMPDH) Inhibitors from Marine Sponges. Evaluation of the Bastadin Alkaloids. Tetrahedron 1994, 50, 7367–7374. [Google Scholar]
- Searle, P. A.; Richter, R. K.; Molinski, T. F. Bengazoles C-G From the Sponge Jaspis sp. Synthesis of the Side Chain and Determination of Absolute Configuration. J. Org. Chem 1996, 61, 4073–4079. [Google Scholar]
- Saito, A.; Seiler, S.; Chu, A.; Fleischer, S. Preparation and morphology of sarcoplasmic reticulum terminal cisternae from rabbit skeletal muscle. J. Cell Biol 1984, 99, 875–885. [Google Scholar]
- Wong, P. W.; Pessah, I. N. Ortho-substituted polychlorinated biphenyls alter calcium regulation by a ryanodine receptor-mediated mechanism: structural specificity toward skeletal-and cardiac-type microsomal calcium release channels. Mol. Pharmacol 1996, 49, 740–751. [Google Scholar]Wong, P. W.; Pessah, I. N. Noncoplanar PCB 95 alters microsomal calcium transport by an immunophilin FKBP12-dependent mechanism. Mol. Pharmacol 1997, 51, 693–702. [Google Scholar]
© 2004 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
Masuno, M.N.; Hoepker, A.C.; Pessah, I.N.; Molinski, T.F. 1-O-Sulfatobastadins-1 and -2 from Ianthella basta (Pallas).Antagonists of the yR1-FKBP12 Ca2+ Channel. Mar. Drugs 2004, 2, 176-184. https://doi.org/10.3390/md204176
Masuno MN, Hoepker AC, Pessah IN, Molinski TF. 1-O-Sulfatobastadins-1 and -2 from Ianthella basta (Pallas).Antagonists of the yR1-FKBP12 Ca2+ Channel. Marine Drugs. 2004; 2(4):176-184. https://doi.org/10.3390/md204176
Chicago/Turabian StyleMasuno, Makoto N., Alexander C. Hoepker, Isaac N. Pessah, and Tadeusz F. Molinski. 2004. "1-O-Sulfatobastadins-1 and -2 from Ianthella basta (Pallas).Antagonists of the yR1-FKBP12 Ca2+ Channel" Marine Drugs 2, no. 4: 176-184. https://doi.org/10.3390/md204176
APA StyleMasuno, M. N., Hoepker, A. C., Pessah, I. N., & Molinski, T. F. (2004). 1-O-Sulfatobastadins-1 and -2 from Ianthella basta (Pallas).Antagonists of the yR1-FKBP12 Ca2+ Channel. Marine Drugs, 2(4), 176-184. https://doi.org/10.3390/md204176