
Cembranoids from a Chinese Collection of the Soft Coral Lobophytum crassum


 and
and
Abstract
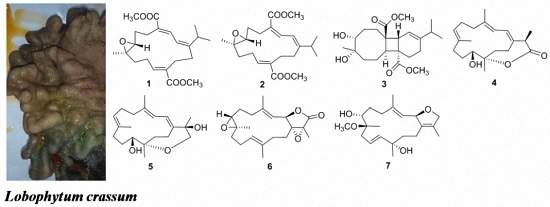
:
1. Introduction
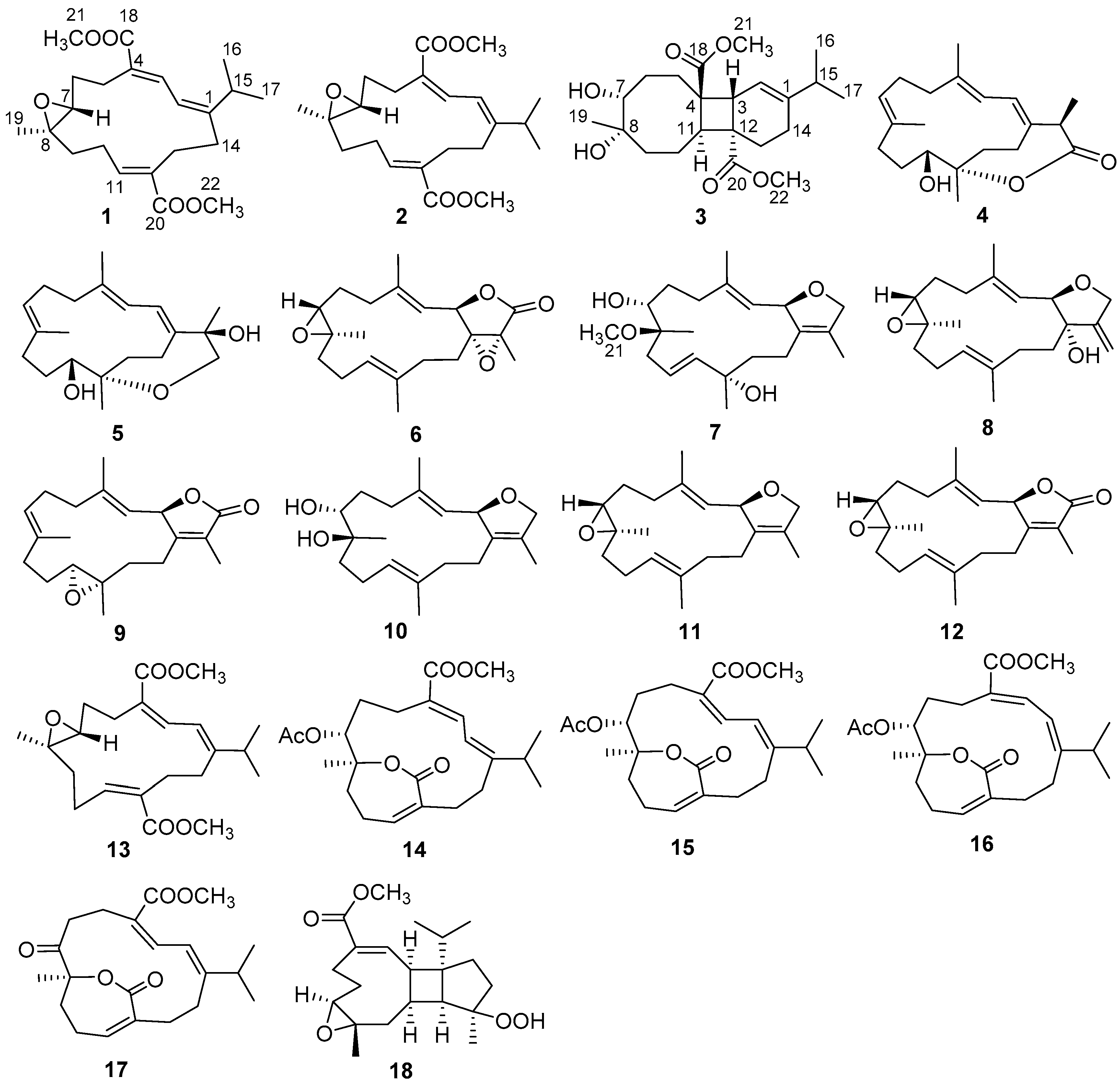
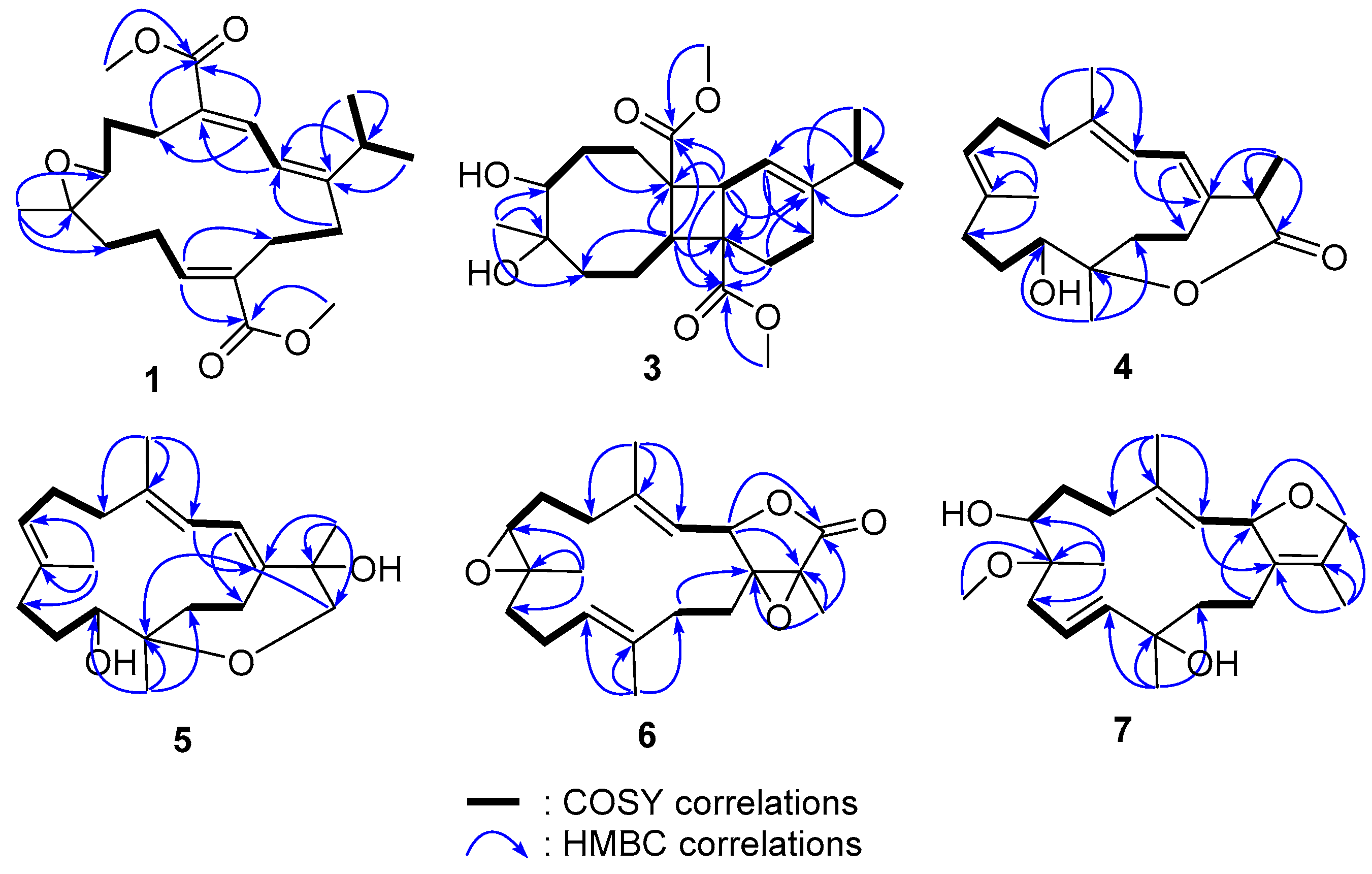
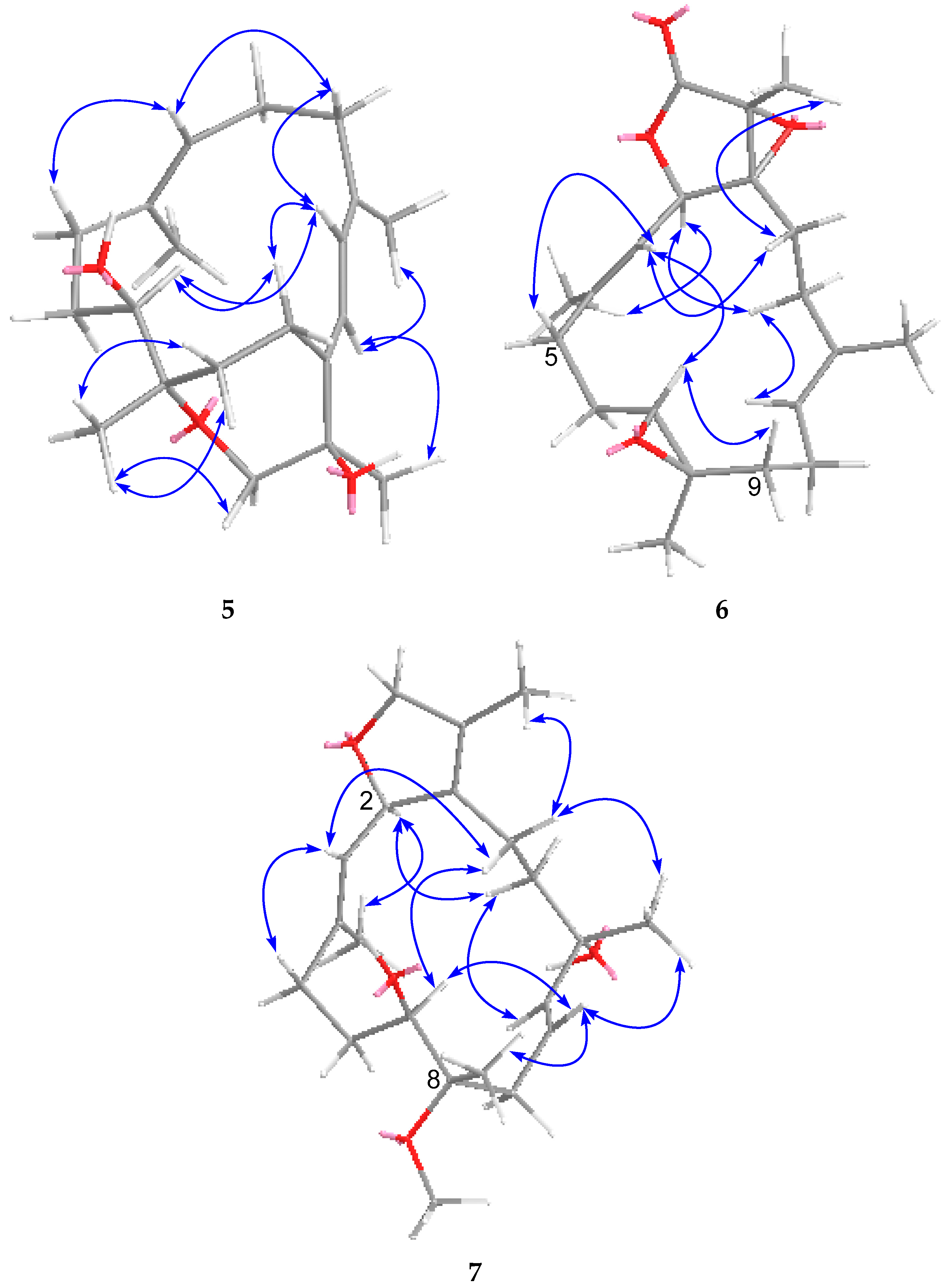
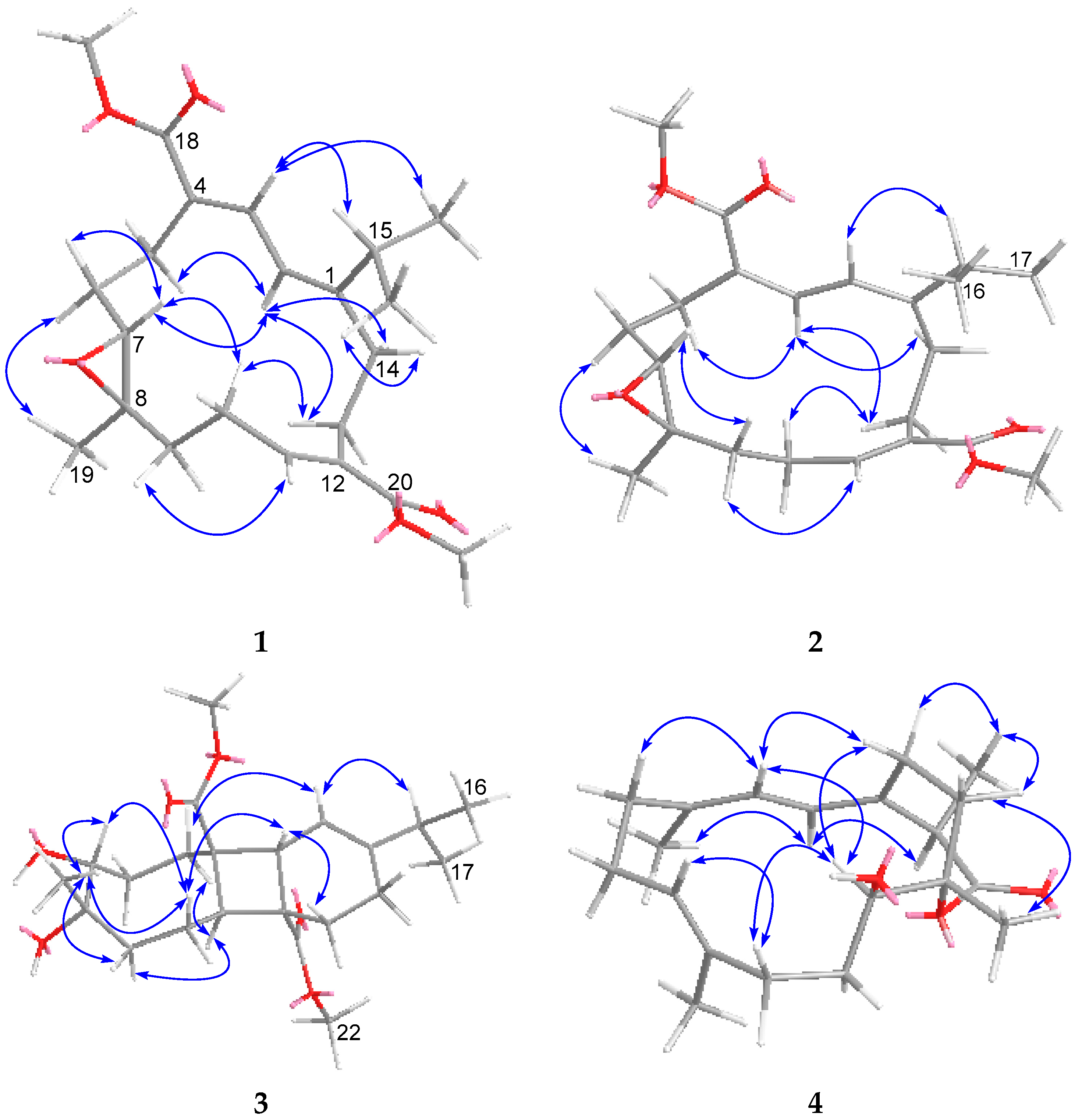
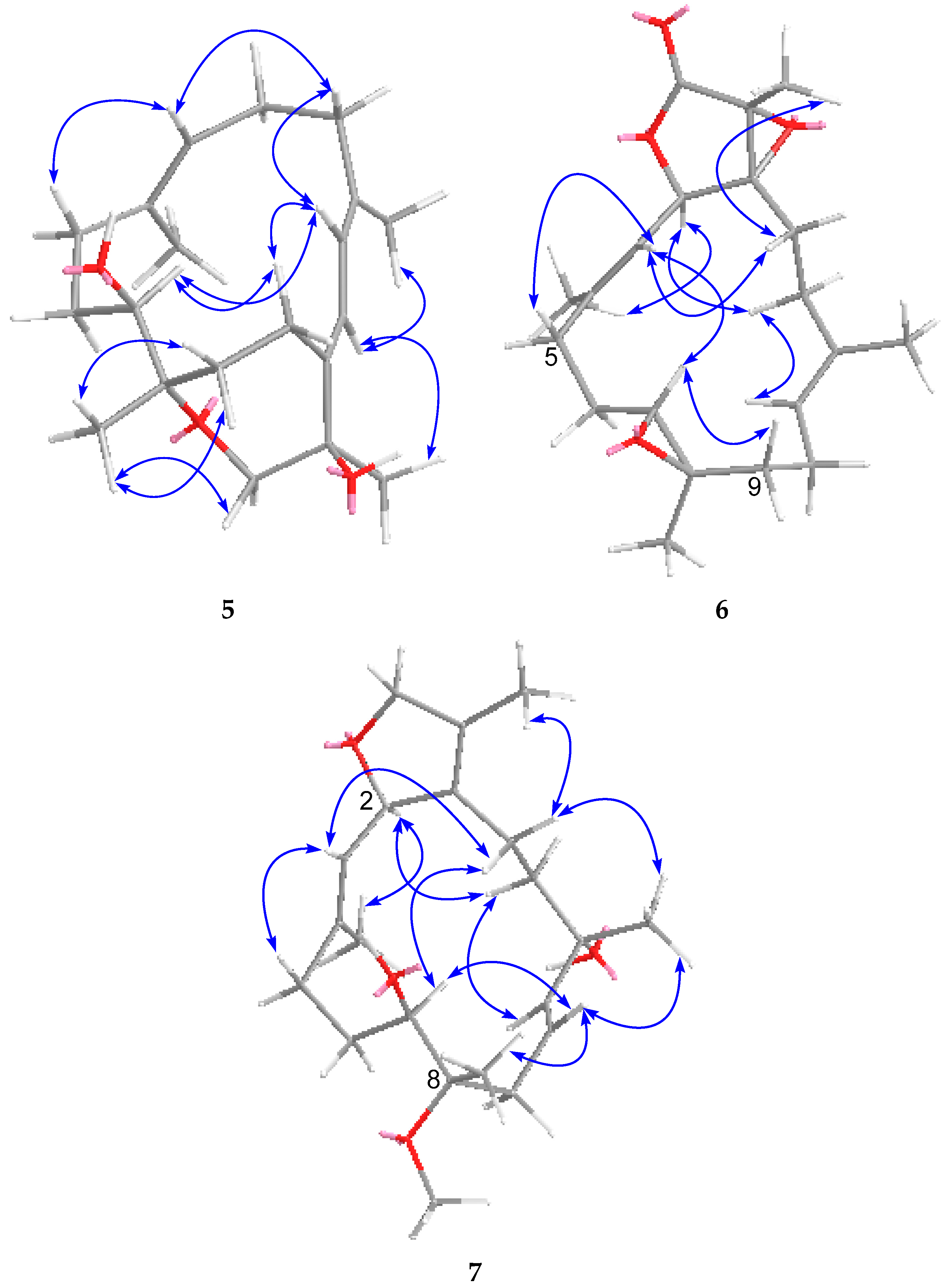
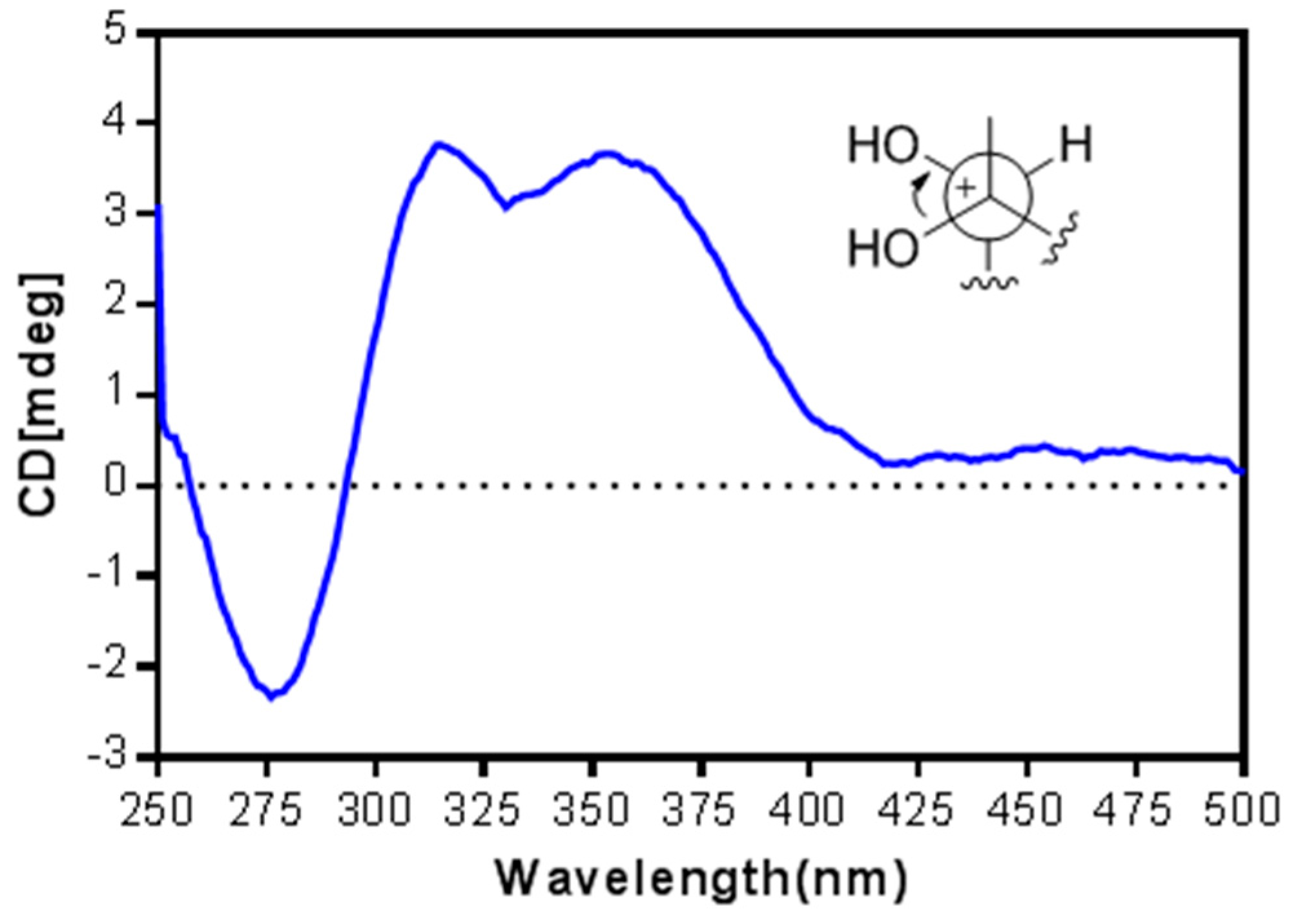
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Animal Material
3.3. Extraction and Isolation
3.4. Assay for Inhibition of Nitric Oxide Production
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Marinlit Database; Department of Chemistry, University of Canterbury. Available online: http://www.chem.canterbury.ac.nz/marinlit/marinlit.shtml (accessed on 3 April 2016).
- Liang, L.-F.; Kurtán, T.; Mándi, A.; Yao, L.-G.; Li, J.; Zhang, W.; Guo, Y.-W. Unprecedented diterpenoids as a PTP1B from the Hainan soft coral Sarcophyton trocheliophorum. Org. Lett. 2013, 15, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Thao, N.P.; Luyen, B.T.T.; Lee, S.H.; Jang, H.D.; Kiem, P.V.; Minh, C.V.; Kim, Y.H. Antiosteoporotic and antioxidant activities of diterpenoids from the Vietnamese soft corals Sinularia maxiama and Lobophytum crassum. Med. Chem. Res. 2015, 24, 3551–3560. [Google Scholar] [CrossRef]
- Cheng, S.-Y.; Wang, S.-K.; Duh, C.-Y. Secocrassumol, a seco-cembranoid from the Dongsha Atoll soft coral Lobophytum crassum. Mar. Drugs 2014, 12, 6028–6037. [Google Scholar] [CrossRef] [PubMed]
- Thao, N.P.; Luyen, B.T.T.; Ngan, N.T.T.; Song, S.B.; Nguyen, X.C.; Nam, N.H.; Kiem, P.V.; Kim, Y.H.; Minh, C.V. New anti-inflammatory cembranoid diterpenoids from the Vietnamese soft coral Lobophytum crassum. Bioorg. Med. Chem. Lett. 2014, 24, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Kao, C.-Y.; Kao, S.-Y.; Chang, C.-H.; Su, J.-H.; Hwang, T.-L.; Kuo, Y.-H.; Wen, Z.-H.; Sung, P.-J. Terpenoids from the Octocorals Menella sp. (Plexauridae) and Lobophytum crassum (Alcyonacea). Mar. Drugs 2012, 10, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-T.; Wang, S.-K.; Duh, C.-Y. Cembranoids from the Dongsha Atoll soft coral Lobophytum crassum. Mar. Drugs 2011, 9, 2705–2716. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.-L.; Su, J.-H. Tetrahydrofuran cembranoids from the cultured soft coral Lobophytum crassum. Mar. Drugs 2011, 9, 2526–2536. [Google Scholar] [CrossRef] [PubMed]
- Duh, C.-Y.; Wang, S.-K.; Huang, B.-T.; Dai, C.F. Cytotoxic cembrenolide diterpenes from the Formosan soft coral Lobophytum crassum. J. Nat. Prod. 2000, 63, 884–885. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.-H.; Wen, Z.-H.; Wu, Y.-C.; Yeh, H.-C.; Sheu, J.H. Cytotoxic and anti-inflammatory cembranoids from the soft coral Lobophytum crassum. J. Nat. Prod. 2008, 71, 1819–1824. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.-J.; Wen, Z.-H.; Hsu, C.-H.; Dai, C.-F.; Sheu, J.H. Bioactive cembranoids from the Dongsha Atoll soft coral Lobophytum crassum. Bull. Chem. Soc. Jpn. 2011, 84, 1102–1106. [Google Scholar] [CrossRef]
- Kao, C.-Y.; Su, J.-H.; Lu, M.-C.; Hwang, T.-L.; Wang, W.-H.; Chen, J.-J.; Sheu, J.-H.; Kuo, Y.-H.; Weng, C.-F.; Fang, L.-S.; et al. Lobocrassins A–E, new cembrane-type diterpenoids from the soft coral Lobophytum crassum. Mar. Drugs 2011, 9, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.-J.; Su, H.-J.; Shyue, Y.-C.; Wen, Z.-H.; Sheu, J.H.; Su, J.H. Two new cembranoids from the soft coral Lobophytum crassum. Bull. Chem. Soc. Jpn. 2011, 84, 653–655. [Google Scholar] [CrossRef]
- Wanzola, M.; Furuta, T.; Kohno, Y.; Fukumitsu, S.; Yasukochi, S.; Watari, K.; Tanaka, C.; Higuchi, R.; Miyamoto, T. Four new cembrane diterpenes isolated from an Okinawan soft coral Lobophytum crassum with inhibitory effects on nitric oxide production. Chem. Pharm. Bull. 2010, 58, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-T.; Wang, S.-K.; Cheng, S.-Y.; Duh, C.-Y. Lobocrasol, a new diterpenoid from the soft coral Lobophytum crassum. Org. Lett. 2009, 11, 3012–3014. [Google Scholar] [CrossRef] [PubMed]
- Matthée, G.F.; König, G.M.; Wright, A.D. Three new diterpenes from the marine soft coral Lobophytum crassum. J. Nat. Prod. 1998, 61, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Kinamoni, Z.; Groweiss, A.; Carmely, S.; Kashman, Y.; Loya, Y. Several new cembranoid diterpenes from three soft corals of the Red Sea. Tetrahedron 1983, 39, 1643–1648. [Google Scholar] [CrossRef]
- Kashman, Y.; Carmely, S.; Groweiss, A. Further cembranoid derivatives from Red Sea soft corals Alcyonium flaccidum and Lobophytum crassum. J. Org. Chem. 1981, 46, 3592–3596. [Google Scholar] [CrossRef]
- Tursch, B.; Braekman, J.C.; Daloze, D.; Dedeurwaerder, H.; Karlsson, R. Chemical studies of marine invertebrates. XXXI. Crassolide, a highly oxygenated diterpene from the soft coral Lobophytum crassum (Coelenterata, Octocorallia, Alcyonacea). Bull. Soc. Chim. Belg. 1978, 87, 75–81. [Google Scholar] [CrossRef]
- Bowden, B.F.; Brittle, J.A.; Coll, J.C.; Liyanage, N.; Mitchell, S.J.; Stokie, G.J. Studies of Australian soft corals. VI. A new cembranolide diterpene from the soft coral Lobophytum crassum (Coelenterata, Anthozoa, Octocorallia, Alcyonacea). Tetrahedron Lett. 1977, 18, 3661–3662. [Google Scholar] [CrossRef]
- Cuong, N.X.; Thao, N.P.; Luyen, B.T.T.; Ngan, N.T.T.; Thuy, D.T.T.; Song, S.B.; Nam, N.H.; Kiem, P.V.; Kim, Y.H.; Minh, C.V. Cembranoid diterpenes from the soft coral Lobophytum crassum and their anti-inflammatory activities. Chem. Pharm. Bull. 2014, 62, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Krohn, K.; Ding, J.; Miao, Z.-H.; Zhou, X.-H.; Chen, S.-H.; Pescitelli, G.; Salvadori, P.; Kurtan, T.; Guo, Y.-W. Structural and stereochemical studies of α-methylene-γ-lactone-bearing cembrane diterpenoids from a South China Sea soft coral Lobophytum crassum. J. Nat. Prod. 2008, 71, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.W.; Shi, Y.P.; Li, X.M.; Wang, B.G. A new cembranoid diterpene and other related metabolites from the South-China-Sea soft coral Lobophytum crassum. Helv. Chim. Acta 2006, 89, 567–572. [Google Scholar] [CrossRef]
- Yin, S.W.; Shi, Y.P.; Li, X.M.; Wang, B.G. A novel hydroperoxyl-substituted cembranolide diterpene from marine soft coral Lobophytum crassum. Chin. Chem. Lett. 2005, 16, 1489–1491. [Google Scholar]
- Zhao, M.; Yin, J.; Jiang, W.; Ma, M.; Lei, X.; Xiang, Z.; Dong, J.; Huang, K.; Yan, P. Cytotoxic and antibacterial cembranoids from a South China Sea soft coral, Lobophytum sp. Mar. Drugs 2013, 11, 1162–1172. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Zhao, M.; Ma, M.; Xu, Y.; Xiang, Z.; Cai, Y.; Dong, J.; Lei, X.; Huang, K.; Yan, P. New casbane diterpenoids from a South China Sea soft coral, Sinularia sp. Mar. Drugs 2013, 11, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Li, X.; Zhao, F.; Cheng, S.; Xiang, Z.; Dong, J.; Huang, K.; Yan, P. Four new 7,8-epoxycembranoids from a Chinese soft coral Lobophytum sp. Chem. Pharm. Bull. 2013, 61, 1323–1328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, J.; Su, J.; Liang, Y.; Yang, X.; Zheng, K.; Zeng, L. Cytotoxic diterpenoids from the soft coral Sarcophyton crassocaule. J. Nat. Prod. 2006, 69, 1476–1480. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Guo, Y.-W.; Mollo, E.; Cimino, G. Sarcophytonolides A–D, four new cembranolides from the Hainan soft coral Sarcophyton sp. Helv. Chim. Acta 2005, 88, 1028–1033. [Google Scholar] [CrossRef]
- Liang, L.-F.; Gao, L.-X.; Li, J.; Taglialatela-Scafati, O.; Guo, Y.-W. Cembrane diterpenoids from the soft coral Sarcophyton trocheliophorum Marenzeller as a new class of PTP1B inhibitors. Bioorg. Med. Chem. 2013, 21, 5076–5080. [Google Scholar] [CrossRef] [PubMed]
- Frelek, J.; Klimek, A.; Ruskowska, P. Dinuclear transition metal complexes as auxiliary chromophores in chiroptical studies on bioactive compounds. Curr. Org. Chem. 2003, 7, 1081–1104. [Google Scholar] [CrossRef]
- Chen, D.; Chen, W.; Liu, D.; van Ofwegen, L.; Proksch, P.; Lin, W. Asteriscane-type sesquiterpenoids from the soft coral Sinularia capillosa. J. Nat. Prod. 2013, 76, 1753–1763. [Google Scholar] [CrossRef] [PubMed]
- Coates, R.M.; Ley, D.A.; Cavender, P.L. Synthesis and carbon-13 nuclear magnetic resonance spectra of all-trans-geranylgeraniol and its nor analogs. J. Org. Chem. 1978, 43, 4915–4922. [Google Scholar] [CrossRef]
- Couperus, P.A.; Clague, A.D.; van Dongen, J.P. 13C chemical shifts of some model olefins. Org. Magn. Chem. 1976, 8, 426–431. [Google Scholar] [CrossRef]
- Bowden, B.F.; Coll, J.C.; Mitchell, S.J. Studies of Australian soft corals. XVIII. Further cembranoid diterpenes from soft corals of the genus Sarcophyton. Aust. J. Chem. 1980, 33, 879–884. [Google Scholar] [CrossRef]
- Quang, T.H.; Ha, T.T.; Minh, C.V.; Kiem, P.V.; Huong, H.T.; Ngan, N.T.T.; Nhiem, N.X.; Tung, N.H.; Tai, B.H.; Thuy, D.T.T.; et al. Cytotoxic and anti-inflammatory cembranoids from the Vietnamese soft coral Lobophytum laevigatum. Bioorg. Med. Chem. 2011, 19, 2625–2632. [Google Scholar] [CrossRef] [PubMed]
- Kusumi, T.; Yamada, K.; Ishitsuka, M.O.; Fujita, Y.; Kakisawa, H. New cembranoids from the Okinawan soft coral Sinularia mayi. Chem. Lett. 1990, 19, 1315–1318. [Google Scholar] [CrossRef]
- Kobayashi, M.; Hirase, T. Marine terpenes and terpenoids. XI. Structures of new dihydrofuranocembranoids isolated from a Sarcophyton sp. soft coral of Okinawa. Chem. Pharm. Bull. 1990, 38, 2442–2445. [Google Scholar] [CrossRef]
- Bowden, B.F.; Coll, J.C.; Heaton, A.; König, G.; Bruck, M.A.; Cramer, R.E.; Klein, D.M.; Scheuer, P.J. The structures of four isomeric dihydrofuran-containing cembranoid diterpenes from several species of soft coral. J. Nat. Prod. 1987, 50, 650–659. [Google Scholar] [CrossRef]
- Liang, L.-F.; Lan, L.-F.; Taglialatela-Scafati, O.; Guo, Y.-W. Sartrolides A–G and bissartrolide, new cembranolides from the South China Sea soft coral Sarcophyton trocheliophorum Marenzeller. Tetrahedron 2013, 69, 7381–7386. [Google Scholar] [CrossRef]
- Toth, J.A.; Burreson, B.J.; Scheuer, P.J.; Finer-Moore, J.; Clardy, J. Emblide, a new polyfunctional cembranolide from the soft coral Sarcophyton glaucum. Tetrahedron 1980, 36, 1307–1309. [Google Scholar] [CrossRef]
- Uchio, Y.; Nitta, M.; Nakayama, M.; Iwagawa, T.; Hase, T. Ketoemblide and sarcophytolide, two new cembranolides with ε-lactone function from the soft coral Sarcophyta elegans. Chem. Lett. 1983, 12, 613–616. [Google Scholar] [CrossRef]
- Yan, P.; Deng, Z.; van Ofwegen, L.; Proksch, P.; Lin, W. Lobophytones O–T, new biscembranoids and cembranoid from soft coral Lobophytum pauciflorum. Mar. Drugs 2010, 8, 2837–2848. [Google Scholar] [CrossRef] [PubMed]

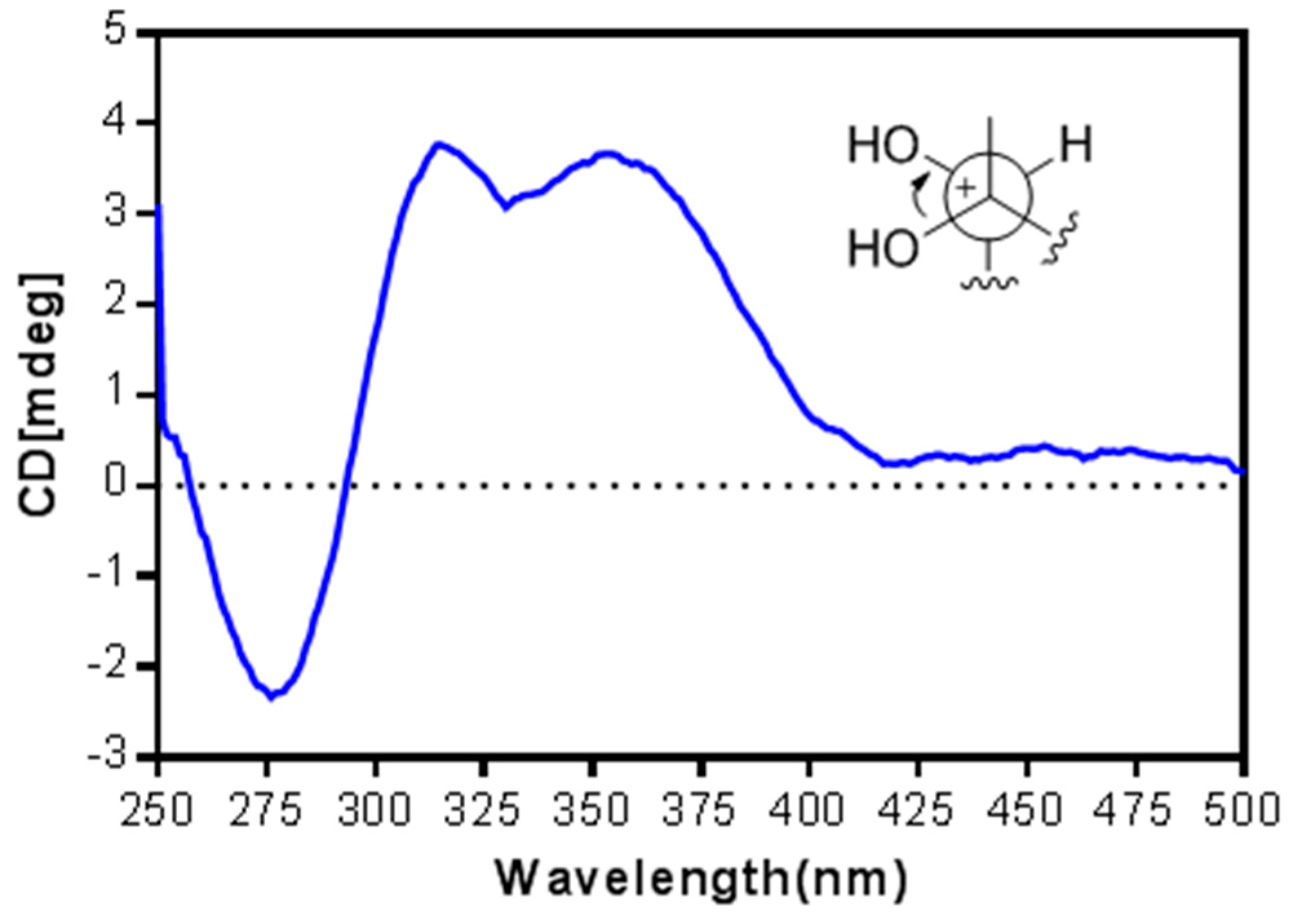

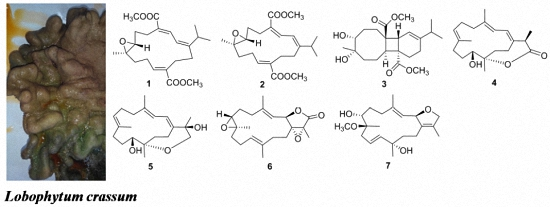{kind=link}
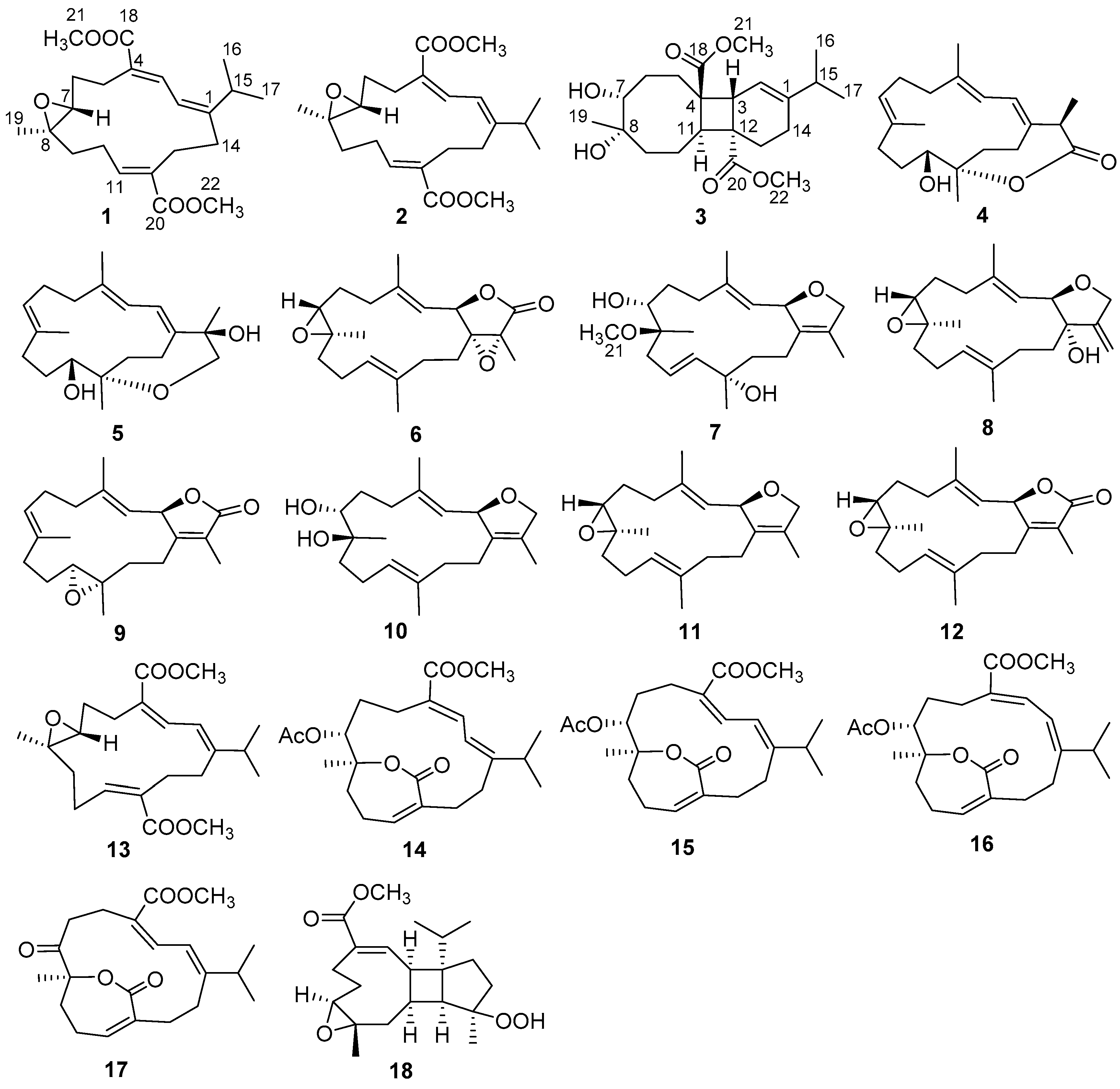{kind=link}
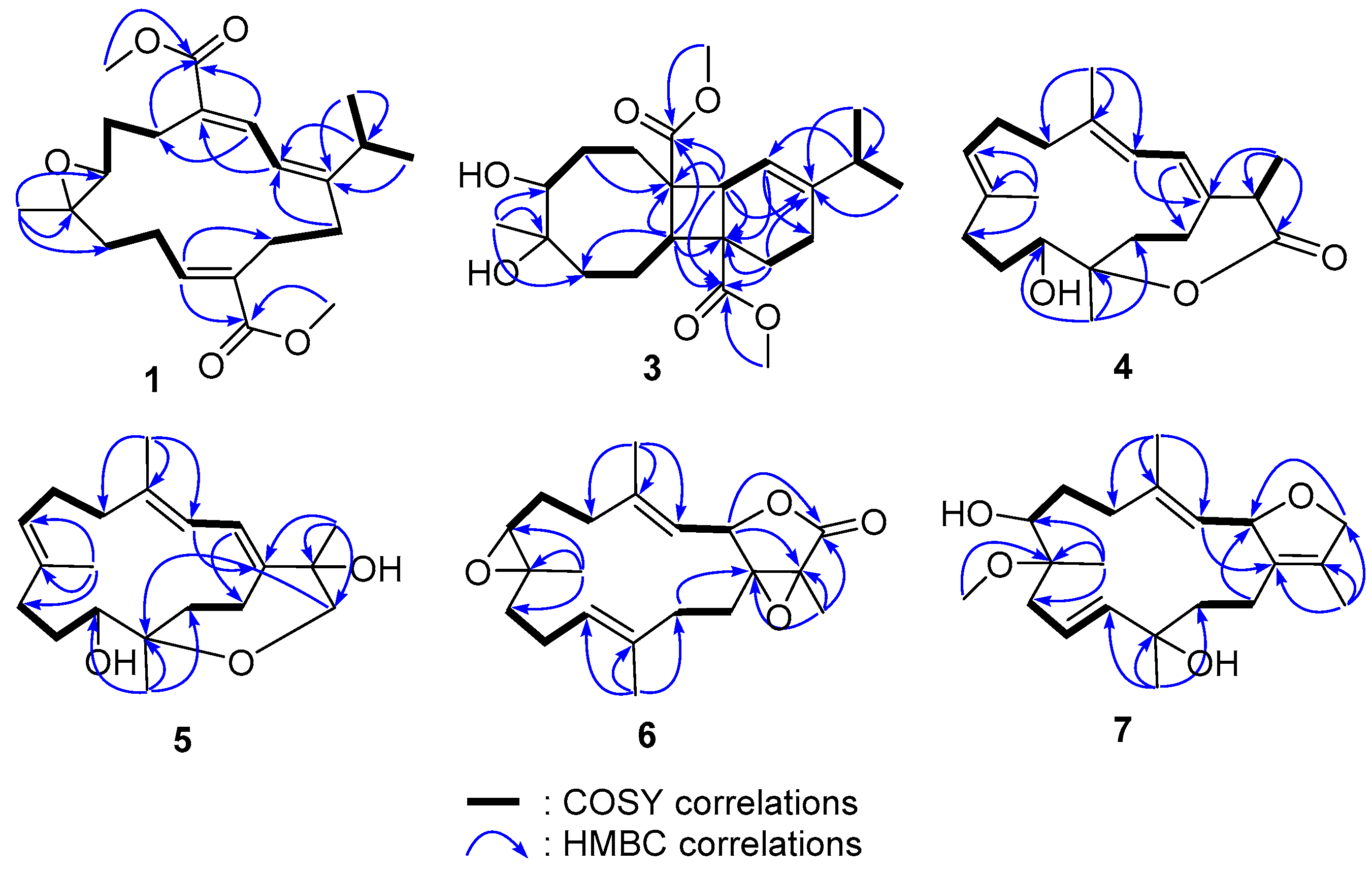{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| 2 | 6.24, d (12.0) | 6.23, d (12.0) | 5.31, d (3.0) | 6.02, dd (11.4, 1.8) | 6.06, dd (11.4, 1.8) | 5.29, d (10.8) | 5.44, br d (9.6) |
| 3 | 7.70, d (12.0) | 7.62, d (12.0) | 3.17, d (3.0) | 5.87, d (11.4) | 5.96, d (11.4) | 5.19, d (10.8) | 5.08, d (9.6) |
| 5 | 2.63, m | 2.66, m | 1.92, m | 2.28, m | 2.26, m | 2.40, m | 2.32, m |
| 2.55, m | 2.63, m | 1.75, m | 1.98, m | 2.01, m | 2.37, m | 2.15, m | |
| 6 | 2.07, m | 1.91, m | 1.74, m | 2.27, m | 2.28, m | 1.95, m | 1.86, td (13.8, 3.0) |
| 1.57, m | 1.71, m | 1.57, m | 2.04, m | 2.03, m | 1.92, m | ||
| 1.35, m | |||||||
| 7 | 2.87, dd (9.6, 3.0) | 2.64, m | 3.58, d (10.8) | 5.11, dd (10.2, 5.4) | 5.09, dd (10.2, 4.8) | 2.64, br t (3.6) | 3.34, d (10.8) |
| 9 | 1.82, m | 2.05, m | 1.97, m | 2.25, m | 2.20, m | 2.15, m | 2.31, m |
| 1.49, m | 1.19, m | 1.66, m | 2.15, m | 2.10, m | 0.95, t (13.2) | ||
| 10 | 2.18, m | 2.12, m | 1.87, m | 1.82, m | 1.82, m | 2.28, m | 5.53, m |
| 2.15, m | 2.00, m | 1.69, m | 1.35, m | 1.20, m | 1.90, m | ||
| 11 | 6.80, t (7.2) | 6.60, dd (7.2, 4.2) | 3.24, dd (7.2, 5.4) | 3.79, br d (10.8) | 3.44, br d (10.8) | 5.12, dd (9.0, 6.6) | 5.52, d (18.6) |
| 13 | 2.63, m | 2.69, m | 2.08, m | 2.26, m | 2.08, m | 2.29, m | 1.67, m |
| 2.42, m | 2.43, m | 1.78, m | 1.94, m | 1.95, ddd (15.0, 6.0, 2.4) | 1.99, m | 1.53, m | |
| 14 | 2.42, m | 2.83, m | 1.94, m | 2.98, m | 2.84, ddd (15.0, 11.4, 6.0) | 1.91, m | 2.14, m |
| 2.28, m | 2.37, m | 1.72, m | 2.17, m | 1.71, m | 1.69, m | ||
| 2.26, m | |||||||
| 15 | 3.20, m | 2.17, m | 2.22, m | 3.43, q (7.2) | |||
| 16 | 1.00, d (6.6) | 1.00, d (6.6) | 1.00, d (6.6) | 1.47,d (7.2) | 1.49, s | 4.51, d (11.4) | |
| 4.46, d (11.4) | |||||||
| 17 | 0.95, d (6.6) | 1.08, d (6.6) | 1.01, d (6.6) | 3.82, d (12.6) | 1.55, s | 1.65, s | |
| 3.44, d (12.6) | |||||||
| 18 | 1.79, s | 1.79, s | 1.88, s | 1.74, s | |||
| 19 | 1.18, s | 1.12, s | 1.14, s | 1.30, s | 1.34, s | 1.28, s | 1.15, s |
| 20 | 1.28, s | 0.99, s | 1.58, s | 1.33, s | |||
| 21 | 3.77, s | 3.77, s | 3.76, s | 3.23, s | |||
| 22 | 3.75, s | 3.76, s | 3.68, s |
| No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| 1 | 158.8, C | 158.7, C | 148.3, C | 133.7, C | 140.9, C | 71.7, C | 132.4, C |
| 2 | 120.4, CH | 118.8, CH | 113.8, CH | 125.7, CH | 120.4, CH | 78.0, CH | 84.0, CH |
| 3 | 134.3, CH | 136.6, CH | 42.0, CH | 121.7, CH | 122.8, CH | 119.8, CH | 126.8, CH |
| 4 | 127.9, C | 127.7, C | 50.0, C | 138.2, C | 137.3, C | 144.4, C | 139.6, C |
| 5 | 23.2, CH2 | 23.6, CH2 | 24.4, CH2 | 40.4, CH2 | 40.5, CH2 | 37.7, CH2 | 35.6, CH2 |
| 6 | 27.2, CH2 | 26.8, CH2 | 29.9, CH2 | 26.4, CH2 | 26.3, CH2 | 25.3, CH2 | 26.2, CH2 |
| 7 | 60.7, CH | 62.7, CH | 73.7, CH | 125.0, CH | 124.9, CH | 61.7, CH | 71.2, CH |
| 8 | 60.9, C | 61.1, C | 74.9, C | 136.3, C | 136.5, C | 59.7, C | 78.4, C |
| 9 | 36.2, CH2 | 36.3, CH2 | 39.3, CH2 | 35.5, CH2 | 36.3, CH2 | 40.0, CH2 | 36.7, CH2 |
| 10 | 24.3, CH2 | 24.0, CH2 | 20.4, CH2 | 26.3, CH2 | 27.0, CH2 | 23.7, CH2 | 124.4, CH |
| 11 | 142.5, CH | 144.4, CH | 43.5, CH | 67.9, CH | 70.8, CH | 124.5, CH | 138.9, CH |
| 12 | 132.4, C | 130.1, C | 45.2, C | 87.0, C | 80.2, C | 135.2, C | 73.0, C |
| 13 | 27.7, CH2 | 27.0, CH2 | 25.3, CH2 | 32.9, CH2 | 30.9, CH2 | 34.8, CH2 | 41.3, CH2 |
| 14 | 30.0, CH2 | 28.9, CH2 | 23.2, CH2 | 22.1, CH2 | 22.6, CH2 | 27.0, CH2 | 21.8, CH2 |
| 15 | 29.8, CH | 36.8, CH | 35.6, CH | 51.0, CH | 77.6, C | 60.7, C | 127.9, C |
| 16 | 21.2, CH3 | 22.3, CH3 | 21.4, CH3 | 16.3, CH3 | 28.0, CH3 | 172.8, C | 78.5, CH2 |
| 17 | 20.4, CH3 | 20.8, CH3 | 20.7, CH3 | 177.8, C | 73.6, CH2 | 9.9, CH3 | 10.1, CH3 |
| 18 | 168.8, C | 168.5, C | 177.4, C | 16.5, CH3 | 16.3, CH3 | 16.2, CH3 | 15.5, CH3 |
| 19 | 17.9, CH3 | 16.9, CH3 | 21.1, CH3 | 14.8, CH3 | 14.9, CH3 | 16.7, CH3 | 17.8, CH3 |
| 20 | 168.0, C | 167.8, C | 176.7, C | 23.1, CH3 | 19.0, CH3 | 14.8, CH3 | 26.9, CH3 |
| 21 | 51.8, CH3 | 51.7, CH3 | 52.1, CH3 | 49.2, CH3 | |||
| 22 | 51.8, CH3 | 51.8, CH3 | 52.0, CH3 |
| Compound | IC50 (μM) | CC50 b (μM) |
|---|---|---|
| 1 | 17 ± 3 | >60.0 |
| 7 | 13 ± 2 | >60.0 |
| 12 | 24 ± 2 | >60.0 |
| 13 | 8 ± 1 | >60.0 |
| 17 | 12 ± 2 | >60.0 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, M.; Cheng, S.; Yuan, W.; Xi, Y.; Li, X.; Dong, J.; Huang, K.; Gustafson, K.R.; Yan, P. Cembranoids from a Chinese Collection of the Soft Coral Lobophytum crassum. Mar. Drugs 2016, 14, 111. https://doi.org/10.3390/md14060111
Zhao M, Cheng S, Yuan W, Xi Y, Li X, Dong J, Huang K, Gustafson KR, Yan P. Cembranoids from a Chinese Collection of the Soft Coral Lobophytum crassum. Marine Drugs. 2016; 14(6):111. https://doi.org/10.3390/md14060111
Chicago/Turabian StyleZhao, Min, Shimiao Cheng, Weiping Yuan, Yiyuan Xi, Xiubao Li, Jianyong Dong, Kexin Huang, Kirk R. Gustafson, and Pengcheng Yan. 2016. "Cembranoids from a Chinese Collection of the Soft Coral Lobophytum crassum" Marine Drugs 14, no. 6: 111. https://doi.org/10.3390/md14060111