Abstract
Chitinolytic microorganisms secrete a range of chitin modifying enzymes, which can be exploited for production of chitin derived products or as fungal or pest control agents. Here, we explored the potential of 11 marine bacteria (Pseudoalteromonadaceae, Vibrionaceae) for chitin degradation using in silico and phenotypic assays. Of 10 chitinolytic strains, three strains, Photobacterium galatheae S2753, Pseudoalteromonas piscicida S2040 and S2724, produced large clearing zones on chitin plates. All strains were antifungal, but against different fungal targets. One strain, Pseudoalteromonas piscicida S2040, had a pronounced antifungal activity against all seven fungal strains. There was no correlation between the number of chitin modifying enzymes as found by genome mining and the chitin degrading activity as measured by size of clearing zones on chitin agar. Based on in silico and in vitro analyses, we cloned and expressed two ChiA-like chitinases from the two most potent candidates to exemplify the industrial potential.
1. Introduction
Chitin, the β-1,4-linked homopolymer of N-acetylglucosamine (GlcNAc), is the most abundant polymer in the marine environment, and the second in nature after cellulose. Chitin is the structural basis for exoskeletons of crustaceans and insects, and a component of the fungal cell wall. The global production of chitin is estimated to be 1011 tons per year, however, chitin does not accumulate as it is hydrolyzed by marine microorganisms [1,2]. The hydrolysis is mediated by chitinolytic enzymes and allows the microorganisms to utilize chitin as a carbon and nitrogen source and chitin turnover is important for the biogeochemical C- and N-cycles. Chitin and chitinolytic enzymes are also of biotechnological interest with potential applications in the food, medical and agricultural sectors [3]. Also, chitin in the form of shellfish waste can be considered as a resource potentially used as a carbon-source in microbial fermentations. Shellfish waste constitutes an environmental problem of increasing magnitude [4,5], and the discovery of inexpensive processes, which can degrade chitin into chitooligosaccharides, chitosan and GlcNAc, may address this problem [6].
As mentioned, the cell wall of fungi contains chitin and some chitinolytic microorganisms can inhibit the growth of fungi by chitin degradation [7,8,9]. Fungal plant diseases are of great concern in agriculture and cause large losses at an estimated 5%–10% of the world’s food production [10]. Fungal contamination and mycotoxin production is also a problem in the built environment [11]. Potentially, natural fungicides, such as chitinases, could replace the chemical fungicides in plant biocontrol [12] and toxic chemicals indoors, and since bacterial chitinases can inhibit fungal growth, they are of particular interest for this purpose [7,13].
Chitin is a recalcitrant insoluble polysaccharide and is degraded into soluble oligosaccharides or GlcNAc. Chitinases (EC 3.2.1.14) hydrolyze the β-1,4 glycosidic bonds between the GlcNAc residues to produce chitooligosaccharides. Chitinases are glycosyl hydrolases (GH) and are divided into GH families 18 and 19. Bacterial chitinases usually belong to family 18, although a few belonging to family 19 have been described [14]. The GH18 and GH19 chitinases differ in sequence similarity, three-dimensional structure and catalytic mechanism. Bacteria often secrete many chitinases, and it is believed that they do so in order to efficiently hydrolyze the different forms of chitin they encounter [7,15,16]. The GH19 chitinases are believed to be the primary enzymes involved in breakdown of fungal chitin, but this is also mediated by other types of chitinases [7,17].
Recently, a new extracellular enzyme involved in breakdown of chitin was discovered. Lytic polysaccharide monooxygenases (LPMOs) were first described in 2010 [18] and are metalloenzymes that oxidize the glycosidic bonds in the crystalline surface of chitin and facilitate access of chitinases. LPMOs were first classified as carbohydrate binding module family 33 and they were believed only to be involved in substrate recognition [19]. LPMOs are now reclassified in auxiliary activity group 10 (AA10) in the CAZY-database [20] and their facilitating activity in chitin degradation has become clear [21]. Another facilitating enzyme, which has so far only been described in some marine bacteria, is chitooligosaccharide deacetylase (COD) [22]. COD (EC 3.5.1.105) is secreted in low concentrations and produces a signal molecule, GlcNac-GlcN, which acts as an inducer for chitinase production [23]. Deacetylation by COD is of particular interest for the industrial production of chitosan oligomers. A combination of COD and another chitin deacetylase, NodB, is used for commercial production of defined chitosan oligomers [24].
The purpose of the present work was to determine the potential for chitin degradation in a collection of marine bacteria. The bacteria were isolated due to their production of antibacterial compounds [25] and we have recently shown that chitin influences the production of secondary metabolites, such as antibacterial compounds, in some of these bacteria [26,27]. We therefore rationalized that they likely would have a high potential for chitin degradation. We envision that these bacteria and their chitinolytic activities will be of interest to the biotech and building industries and in agricultural production. We used in silico genome-wide analysis combined with phenotypic testing to unravel the potential and to exemplify the industrial perspective we cloned and heterologously expressed two chitinases.
2. Results
2.1. Chitin Degrading Activity and in Silico Analysis
Chitin degradation by 11 bacterial strains was determined on agar plates containing crystalline chitin, colloidal chitin or chitosan of both shrimp and crab origin. All but one strain, P. fuliginea S3431, degraded chitin. At low temperatures (4 °C and 15 °C), the clearing zones in the chitin containing media were hardly visible. The most pronounced chitin hydrolysis was seen at 35 °C and 25 °C. There was no particular difference between degradation of crab and shrimp chitin. Degradation of crystalline chitin was slower than that of colloidal chitin. None of the strains had chitosanase activity.
The bacterial genomes have previously been mined for chitinase genes and ChiS [28], and we here extend this genome mining including also COD, LPMO and a subdivision of the chitinase genes (Table 1). Two to six putative chitinase genes were found in the 10 chitin degrading strains, and all but one strain, V. galatheae S2757, also encoded one to two putative LPMOs. The gene encoding the ChiS sensor was only present in strains belonging to the Vibrionaceae family, whereas the CdsS/CdsR pair was present in all Pseudoalteromonas strains, including strain S3431 that did not degrade chitin.

Table 1.
Chitinolytic activity and chitinase genes in 11 marine bacteria. Strain S3431 is included as a non-chitin degrading control. Chitinase activity was graded according to clearing zone size (+ is zone size of 0–6.99 mm, ++ is zone size of >7 mm and − is no clearing zone). Activity was evaluated at 25 °C on colloidal shrimp chitin. Chitinases can be grouped into glycosyl hydrolases (GH) families 18 and 19. GH18 chitinases are further sub-grouped into ChiA, ChiD and unspecified (U).
We analysed the chitinases in Pfam, NCBI and SignalP which enabled the classification of chitinases into GH18 and GH19 groups (Table 1). The GH18 chitinases were sub-divided into ChiA, ChiD and an unspecified group. GH18 and, interestingly, also GH19 chitinases were found in all strains, except S3431.
COD genes were found in six strains and in both families. The COD genes were pairwise compared to other known COD genes and chitin deacetylases of bacterial and fungal origin. We included allontoinase genes since BLASTp analysis revealed that the Vibrio COD genes had 99% sequence similarity to allantoinases. Allantionases are enzymes that catalyze the hydrolytic cleavage of the hydantoin ring in allantoin, which is present in purine-derived compounds [29]. Vibrio COD genes had 96%–99% sequence similarity to chitin deacetylases (CDA) and the Pseudoalteromonas COD genes had 98%–99% sequence similarity to polysaccharide deacetylases (Figure 1). The Vibrio COD genes only had a low (10%–14%) similarity to other known COD genes from same genus. However, they share approximately 53% similarity with a chitin deacetylase, which was cloned from a metagenomic sample. The Pseudoalteromonas CODs had low similarity to the known CODs and to the metagenomic chitin deacetylase, 10% and 24%, respectively. The putative COD genes in our study were 10% similar to a Shewanella COD, which had more than 60% similarity to the known COD genes from Vibrio species. Low similarity was also found when comparing to fungal CDA. The Vibrio COD genes had low similarity to allantoinases from Streptomyces coelicolor and Bacillus lichenformis, however approximately 69% similarity to an allantoinase analog (PuuE) from Pseudomonas fluorescens.
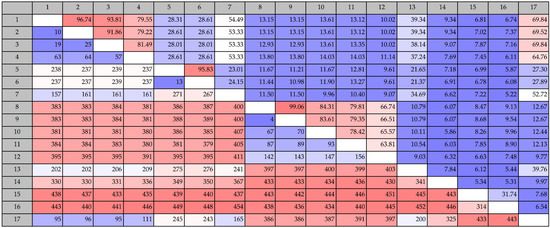
Figure 1.
Pairwise comparison of protein sequence identities of putative COD genes from this study (numbers 1: Vibrio coralliilyticus (KJY71281), 2: Vibrio neptunius S2394 (KJY93856), 3: Vibrio galatheae S2757 (KJY81897), 4: Vibrio nigripulchritudo S2604 (KJY75235), 5: Pseudoalteromonas piscicida S2040 (KJY92988) and 6: Pseudoalteromonas piscicida S2724 (KJY89714)), known COD genes (number 8: Vibrio alginolyticus H-8 (BAB21759), 9: Vibrio parahaemolyticus KN1699 (BAG70715), 10: Vibrio sp. SN84 (BAG82921), 11: Vibrio cholerae O1 (AAF94439) and 12: Shewanella woodyi ATCC 51908 (ACA84860)), chitin deacetylases (number 7: Metagenomic CDA (AEJ31921), 13: Schizosaccharomyces pombe (CAB10114), 14: Colletotrichum Lindemuthianum (2IW0), 15: Steptomyces coelicolor A3(2), (NP_630347) and 16: Bacillus Lichenformis (AAU22686)) and an allantoinase analog PuuE (number 17: Pseudomonas fluorescens, puuE (ACA50280)). Values above the diagonal line refers to percent sequence identity and values below the diagonal refer to number of differences.
2.1.1. Phylogenetic Analysis of Chitinases
A phylogenetic tree comparing all the complete, translated chitinase genes identified in the 10 chitinolytic strains with other known chitinase genes from the GH18 and GH19 families was constructed. The genes clearly clustered within their respective families GH18 ChiA, GH18 ChiD and GH19, and, thus, were correctly annotated (Figure 2). The unclassified GH18 chitinases did not cluster with any known chitinases. The unclassified GH18 chitinase genes clustered together, with the exception of KJZ10112 from P. rubra S2471 that clustered alone, suggesting a new chitinase group.
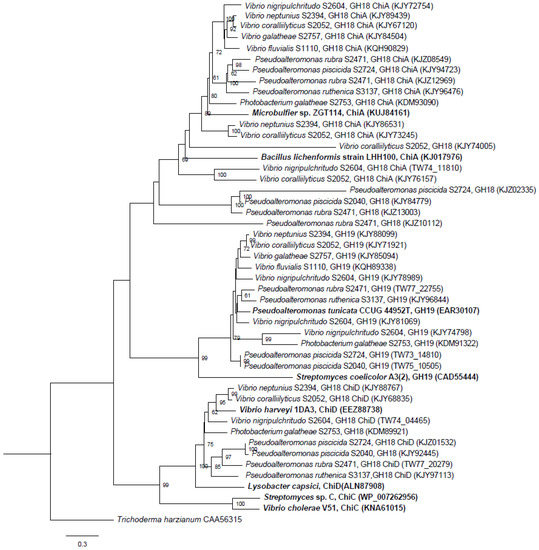
Figure 2.
Phylogenetic relationship of the RAST-annotated chitinases from this study and known chitinases from the NCBI database (marked in bold). Branch support values (bootstrap proportions, with 1000 replicates in the analysis) are associated with nodes indicating that the support was <50%. The bar marker indicates the number of amino acid substitutions. Identifiers include species name and GH family, subfamily and accession numbers.
The unclassified GH18 chitinases contained signal peptides and catalytic GH18 domains (Figure 3). Three of four unclassified chitinases also contained a chitin binding motif (CBM). These domains provide evidence for their classification as chitinases.
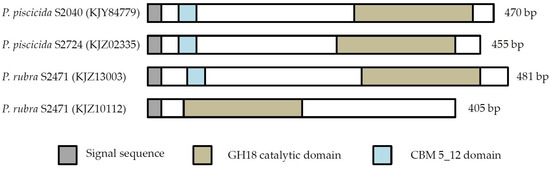
Figure 3.
Domain structure of unclassified GH18 chitinases. Protein domains, as identified in Pfam, are shown. Blank areas in part of the proteins indicate that no match with characterized protein domains were found.
2.1.2. Cloning of Chitinases from S2753 and S2724
Chitinases from two strains with pronounced chitin degradation were cloned into a pBAD_His vector and expressed in an E. coli BL21 (DE3) host (Table 2). Chitinases are extracellular enzymes since a signalP detected signal peptides was found in all chitinases, as seen in the protein domain structure (Figure 4). The GH18 chitinase from S2724 (KJZ02335) was cloned without its signal peptide, as this region was left out from the RAST annotation.
Table 2.
Name, length and expected protein sizes of the cloned chitinases.
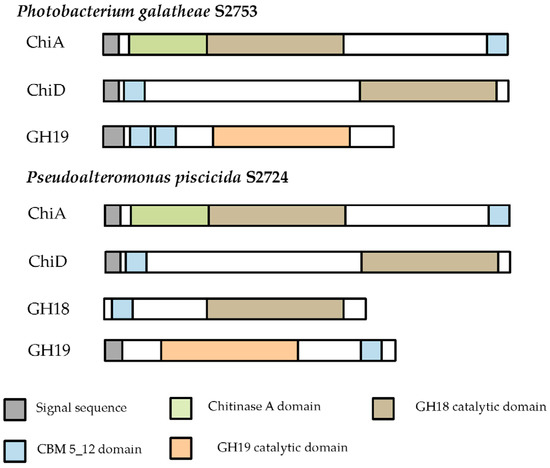
Figure 4.
Domain structure of the cloned chitinases. Protein domains, as identified in Pfam, are shown. Blank areas in part of the proteins indicate that no match with characterized protein domains were found.
SDS-page analysis was used to determine if the chitinases were secreted or accumulated inside the cells. PG_ChiA and PP_ChiA were the only enzymes secreted (Figure 5).
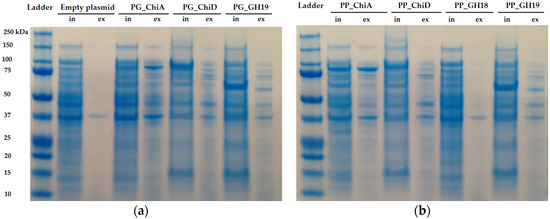
Figure 5.
SDS-PAGE of induced E. coli BL21 harboring the cloned chitinases from (a) P. galatheae S2753 and (b) P. piscicida S2724 including both intracellular proteins (in) and extracellular protein extracts (ex). Expected protein sizes are PG_ChiA: 88 kDa, PG_ChiD: 88 kDa, PG_GH19: 59 kDa, PP_ChiA: 88 kDa, PP_ChiD: 90 kDa, PP_GH18: 49 kDa and PP_GH19: 53 kDa.
To test the substrate specificities of the chitinases, we tested the intracellular and extracellular extracts as well as the induced actively growing E. coli clones on crystalline and colloidal chitin and chitosan. Only ChiA clones and extracts degraded colloidal chitin but not crystalline chitin and not chitosan (Figure 6). Since we tested clone extracts, the protein concentrations of the chitinases were not determined, as other proteins are also present in the extracts.
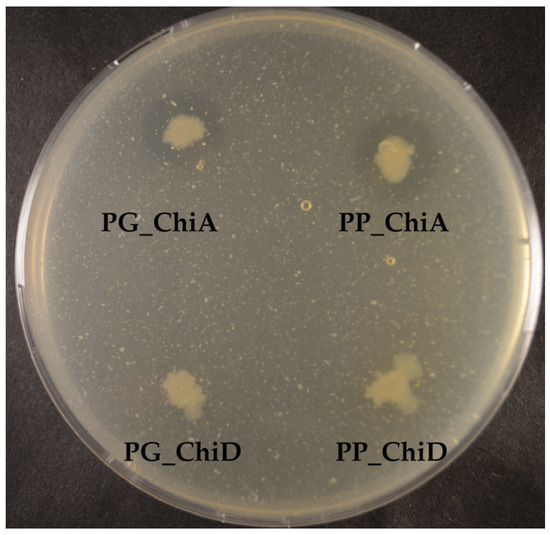
Figure 6.
Colloidal chitin degradation by E. coli BL21 (DE3) expressing chitinase genes. The two ChiA-type chitinases are secreted, whereas the two ChiD-type chitinases accumulate inside the cells.
2.1.3. Antifungal Activity
Some chitinases have antifungal activity, and we tested the extracts from the chitinase clones against seven different fungi. None of the extracts were antifungal. We also tested the wild-type marine bacteria in two settings: one where the bacteria were spotted after fungal inoculation and one where the bacteria were spotted prior to fungal inoculation (Figure 7).
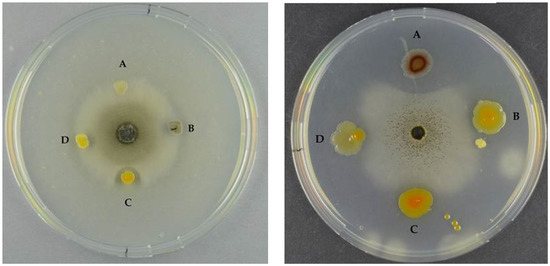
Figure 7.
Left panel: Antifungal assay of S. chartarum by (A) S3137 (no inhibition); (B) S3431 (inhibition); (C) S2040 (inhibition); and (D) S2724 (inhibition). Right panel: A. niger and (A) S2471 (inhibition); (B) S2724 (inhibition); (C) S2040 (inhibition); and (D) S3137 (inhibition). Left panel bacteria were spotted after fungal inoculation, right panel bacteria were spotted prior to fungal inoculation.
In the first setting, one strain, S2040, had a pronounced antifungal effect against all seven fungi (Table 3, columns A). The remaining 10 strains were antifungal against A. niger and B. cinerea, however, they were not capable of retaining the antifungal effect over time. In the second setting (where the bacteria were spotted prior to the fungi), the same scenario as described above occurred. In addition, the two fungi with the slowest growth rates, S. chartarum and B. cinerea were inhibited by the bacterial presence (Table 3, columns P). In this setting, two additional bacteria, S3137 and S2471, retained their antifungal activity over time against P. chrysogenum and A. niger, respectively.
Table 3.
Inhibition of fungi by 11 marine bacteria. Bacteria were spotted after (A) or prior (P) to inoculation of fungi. (+) describes an initial antifungal effect which was not retained after 14 days and (++) describes an antifungal effect which was retained after 14 days. (−) no antifungal effect, (sg) sparse growth of the fungi, (nt) not tested.
3. Discussion
Chitin degradation is an important process in both marine and terrestrial environments. Chitin is also an important resource in different industrial and medical processes, and chitin degrading enzymes or microorganisms are of interest, e.g., as antifungal agents or as bio-insecticides. Hence, there is a growing demand for new enzymes with chitin-modifying properties. We analyzed 11 marine bacteria with antibacterial activity and found a remarkable potential for chitin degradation. In silico, we identified genes involved in the chitin degradation process using an iterative strategy. We found a total of 50 putative chitinase genes of which 11 were considered to be wrongly annotated. The strains contained from two to six putative chitinase genes, and the number of chitinase genes per strain did not correlate with phenotypic chitin degradation ability. The three strains causing the largest clearing zones in chitin agar, S2753, S2724 and S2040, harbored three, three and four chitinase genes, respectively, whereas the poorer chitin degraders, S2050 and S2604, both had six chitinase genes. The chitinases were grouped in different categories of ChiA, ChiD and unclassified belonging to GH18 and GH19 chitinases.
Chitinolytic bacteria harbor different chitinases, most likely due to a specificity for different substrates [16,30,31,32]. Here, all but one bacterial strain contained at least one chitinase belonging to GH18 ChiA, whereas GH18 ChiD and unclassified GH18 chitinases were only present in some strains. This is not surprising, since ChiA-type chitinases are the most dominant chitinases in bacteria and play a key role in chitin degradation [16,33,34]. Also, this was the dominant chitinase group found in un-cultured bacteria [35]. P. piscicida S2040 does not encode for a ChiA-like chitinase, and yet was one of the most potent chitin degraders. The genome of S2040 contained one chitinase of the ChiD-like type, one belonging to GH19 and one unclassified GH18 chitinase, and chitinases of the ChiA-like type are not responsible for the pronounced chitin degradation by this strain. Four chitinases from strains S2724, S2040 and S2471 did not cluster into any existing chitinase subgroups, suggesting they belong to a new subgroup of chitinases. The domain structure provides further evidence that these are indeed chitinases, as they contain the main characteristics of a chitinase (signal peptide, CBM and catalytic domain). However, one chitinase (KJZ10112) does not contain a CBM. This does not disqualify its classification as a chitinase. CBMs are important for the overall performance of the enzyme, but chitinases do not lose function without CBMs, they merely display weaker binding [36,37,38]. These four chitinases were only identified in the Pseudoalteromonadaceae family, and it is interesting to note that two of the chitinases, KJZ02335 and KJY84779, originate from two of the three most potent chitin degrading strains. These unknown chitinases may play a significant role in chitin degradation by Pseudoalteromonas species.
All strains had at least one gene encoding chitinases belonging to the GH19 family, which for many years were considered to be unique to higher plants. The first bacterial GH19 chitinase was found in 1996 [39] and the majority of GH19 chitinases have been found in Streptomyces species [14]. However, GH19 chitinases from Vibrio, Aeromonas, Pseudoalteromonas, Chitiniphilus, Nocardiopsis and Burkholderia species have also been described [8,40,41,42,43,44]. The presence of the GH19 chitinase in all 10 chitinolytic strains could indicate that this gene is more widespread in the marine environment than hitherto believed.
Since the discovery of the function of LPMOs as facilitators of chitin degradation, the interest in these particular enzymes has increased. Our in silico analysis revealed that LPMOs are present in nine of the 10 genomes from chitinolytic bacteria. Recombinant LPMOs have not yet been characterized in marine bacteria and this potential should be further explored. These LPMOs could be of interest in enzyme cocktails as shown in a recent study where a LPMO from a Streptomyces griseus increased the chitin solubilization yields by up to 30-fold when combined with a Serratia marcescens GH18 chitinase [45].
COD genes have so far only been identified in Vibrio species and in Shewanella woodyi ATCC51908 [46,47]. Here, we found six putative COD genes, however, they only had little homology to known CODs. The Vibrio COD genes were similar to the allantoinase analog PuuE indicating a wrong annotation. Additional evidence for the wrong annotation is the observation that the neighboring genes to the putative Vibrio CODs are involved in purine degradation. PuuE type allantoinases have high similarity to polysaccharide deacetylases, which is also the observation in this study, where the Vibrio COD genes had approximately 50% identity to a CDA from a metagenomic sample [48]. The putative COD genes from the two Pseudoalteromonas strains were only 23%–27% identical to the metagenomic CDA and the PuuE allantoinase. PuuE proteins can be distinguished from polysaccharide deacetylases by two highly conserved segments, which are only present in PuuE proteins. These conserved segments are present in the Vibrio genes (data not shown), but not in the Pseudoalteromonas genes, which provide further evidence for the classification of Vibrio CODs as PuuE type allantoinases. In contrast to the Vibrio COD genes, the Pseudoalteromonas COD genes have signal peptides. Thus, these CODs are likely secreted, and they may therefore potentially act as CDAs. CDAs of bacterial origin are of particular industrial interest, as they catalyze the conversion of chitin to chitosan, a highly coveted polymer [49]. Irrespective of the real function of the so-called Pseudoalteromonas COD, the enzymatic deacetylation of polymers has a high industrial potential.
Two of the seven cloned chitinases were secreted whereas the remaining five accumulated inside the cells. The signal peptide of ChiA type chitinases can be recognized by the secretion system in E. coli, and hence it is not surprising that ChiA is secreted by E. coli. [50,51]. However, cloned ChiA type chitinases are not always secreted [52], so the secretion does not seem to be specific to the chitinase type, but the secretion of the two ChiA-type chitinases from this study allows for an easy purification and exemplifies the potential industrial use of these enzymes. Extracellular and intracellular extracts were tested on different chitinous media, but only the extracts of the two ChiA enzymes degraded colloidal chitin, which may be due to substrate specificity and/or need for synergy between chitinases, as already mentioned.
We tested the extracts against seven fungi, covering both indoor contaminants and plant pathogens. GH19 chitinases are thought to be the main antifungal chitinases, but ChiA type chitinases have also displayed antifungal activity, like StmChiA from Stenotrophomonas maltophilia which was antifungal against F. oxysporum [52]. However, extracts from ChiA clones were not antifungal, nor were any of the other extracts. Since chitinase concentration in the extracts was unknown, an up-concentration could potentially result in a measurable effect.
All 11 bacterial strains, including the non-chitin degrader, were antifungal when tested as live cultures, however, with different fungal targets. Allowing pre-growth of the potential producer prior to fungal inoculation increased the antifungal activity of some of the bacteria, which is in agreement with the study by Giubergia and co-workers [27], in which bioactivity of a collection of Vibrionaceae increased 3-fold when the producer was allowed a 2-day pre-growth period. P. piscicida S2040 displayed pronounced antifungal effect towards all fungi, independently of the time of spotting, and hence would serve as a candidate for a broad-range antifungal bio-pesticide.
In summary, three of the ten strains were of interest due to their remarkable chitin degrading abilities and their antifungal activities. These strains could have potential in biodegradation of chitin-waste, and in biocontrol of unwanted fungal growth in agriculture and the building industry.
4. Materials and Methods
4.1. Strains and Plasmids
The bacterial strains used in this study (Table 4).
Table 4.
Marine strains used in this study.
The bacteria were isolated during the Galathea 3 expedition [25] and they have been whole-genome sequenced [28]. Genomes were assembled using CLC Genomics Workbench 7 (CLC bio, Aarhus, Denmark) and contig-based draft genomes were obtained. Gene annotation was performed using Rapid Annotation using Subsystem Technology (RAST), [53,54,55]. The genomes are available at the National Center for Biotechnology Information (NCBI) [56]. To compare annotations, the genomes were also downloaded from NCBI containing the NCBI-annotated genes. Escherichia coli Top10 was used for cloning and propagation of plasmids, and E. coli BL21 (DE3) was used for expression of chitinase genes from P. galatheae S2753 and P. piscicida S2724. The cloning and expression vector was pBAD_Myc_HisA. Plasmids were isolated using the QIAprep® Spin Miniprep kit (Qiagen, 27106, Hilden, Germany), and genomic DNA was extracted using the NucleoSpin® Tissue kit (Machery-Nagel, 740952, Düren, Germany). Strains and plasmids used for cloning can be seen in Table 5.
Table 5.
Cloning and expression hosts and plasmids used in this study.
4.2. Preparation of Colloidal Chitin
Colloidal chitin was prepared from shrimp shell chitin (Sigma, C7170, Deisenhofen, Germany) or crab shell (Sigma, C9752, Deisenhofen, Germany) chitin as follows: 10 g chitin was hydrolyzed in 400 mL ice-cold 37% HCl for 6 h at 4 °C with stirring. The solution was transferred to 4 L cold dH2O over night for settlement of chitin. The solution was neutralized using NaOH and adjusted to pH 7. Colloidal chitin was collected by centrifugation at 4000× g for 5 min and resuspended in dH2O for a final concentration of 2%. The chitin solution was autoclaved at 121 °C for 15 min.
4.3. Chitinase and Chitosanase Activity Screening
The strains were tested for chitinase and chitosanase activity on plates containing different chitinous substrates. The basic media consisted of 2% Sea Salt (Sigma, S9883, Deisenhofen, Germany), 1,5% agar, 0.3% casamino acids and was supplemented with either 0.2% colloidal chitin from shrimp or crab, 0.2% crystalline shrimp chitin or 0.2% shrimp chitosan (Sigma, 50494, Deisenhofen, Germany) or crab chitosan (Sigma, 48165, Deisenhofen, Germany). Plates were spotted with one single colony from a streaked culture from freeze-stock on marine agar plates (Difco 2216). The plates were incubated at 4, 15, 25 and 35 °C for 11 days for colloidal chitin and chitosan and 35 days for crystalline chitin. The natural turbidity of the media allows for visual evaluation of chitin/chitosan degradation appearing as a clearing zone around the spotted bacteria. A qualitative grading of chitinase activity (clearing zone) was given, determined from the edge of the bacterial colony to the edge of the clearing zone, where a zone of 0–6.99 mm was graded one plus, +, and >7 mm was graded two plusses, ++.
4.4. In Silico Analysis
An annotation based search for chosen genes involved in chitin degradation was conducted using CLC Main Workbench 7 and included genes putatively encoding chitinases, chitin sensors (ChiS or CdsS), COD and LPMOs. 50 chitinase-encoding genes were found using RAST. The complete genes were translated to protein, and divided into GH18 and GH19 families by analysis using the Pfam protein family database [57,58] and further divided into subfamilies by a protein blast in the non-redundant database in NCBI [59]. Since chitinases are extracellular proteins they were checked for signal peptides using SignalP 4.1 [60]. 11 RAST-annotated chitinases were eliminated from the analysis, as they did not contain either GH18 or GH19 domains, and two others were deleted as they did not contain signal peptides or CBMs. All included and eliminated chitinases can be seen in Supplementary Materials Table S1. Since the COD proteins showed little homology to other known CODs, they were blasted against the non-redundant protein database to find proteins of high sequence similarity. The COD encoding genes were compared to already known CODs, chitin deacetylases and an allantoinase analog by pairwise comparison in CLC.
4.5. Phylogenetic Analysis of Chitinases
A phylogenetic tree was created consisting of the identified chitinase genes from the ten chitinolytic strains. To indicate correct groupings and potentially group the unidentified chitinase genes, known chitinase genes belonging to the GH18 subfamilies A, C and D and GH19 chitinases from the NCBI database were included. The phylogenetic tree was created from a multiple alignment of the translated chitinase proteins with a neighbor-joining construction method and a bootstrap analysis with 1000 replicates was included. The tree was visualized using FigTree [61]. A fungal chitinase from Trichoderma harzianum (accession number CAA56315) was used as root.
4.6. Construction of Plasmids for Chitinase Expression
Expression plasmids for the seven chitinase genes from P. galatheae S2753 and P. piscicida S2724 were constructed via USER cloning, as preciously described [62], using pBAD-Myc_HisA as the vector plasmid. Plasmid and chitinase genes were amplified by PCR using PfuX7 polymerase [63] and primers (Table 6) using following settings: initial denaturation at 98 °C for 2 min, 30 cycles of 98 °C for 20 s, 57 °C for 20 s, 72 °C for 1:30 min and a final extension at 72 °C of 2:30 min. For PG_GH19, PP_GH18 and PP_GH19 extension time was reduced to 50 s and final extension time reduced to 1:45 min. For plasmid DNA, extension time was prolonged to 2:30 min and final extension prolonged to 4:30 min. Each PCR reaction (50 μL) consisted of 5 μL Pfu buffer (200 mM Tris-HCl pH 8.8, 100 mM KCl, 60 mM (NH4)2SO4, 20 mM MgSO4, 1 mg/mL BSA (in nuclease-free water) and 1% Triton X-100), 5 μL dNTPs (2 mM), 1.2 μL MgCl2 (50 mM), 5 μL forward primer (5 μM), 5 μL reverse primer (5 μM), 1 μL PfuX7 polymerase and 0.5–1 μL DNA.
Table 6.
Primers used for chitinase gene amplification.
In short, the chitinase containing plasmids were constructed in a reaction of 10 μL, consisting of 100 ng of each purified PCR product, 1 μL T4 DNA ligase buffer (New England BioLabs, #B0202S, Ipswich, MA, USA ) and 1 μL USER™ enzyme (New England BioLabs, #M5505S, Ipswich, MA, USA). The reaction was incubated at 37 °C for 15 min, followed by 26 °C for 15 min and 10 °C for 10 min. 3 μL USER reaction was mixed with 40 μL chemically competent E. coli Top10. The mixture was incubated on ice for 30 min, followed by a 60 s heat shock at 42 °C, and 2 min incubation on ice. Cells were recovered in 1 mL LB for 1 h and subsequently harvested at 1600× g for 2 min. The pellet was plated on LB agar containing 100 μg/mL ampicillin and incubated at 37 °C overnight, and the following day colonies were grown in LB media with antibiotics and stored as glycerol stocks. Plasmids were purified and confirmed by sequencing (Macrogen, Amsterdam, The Netherlands).
4.7. Protein Expression and SDS-Page Analysis
Protein expression was initiated by transformation of the purified plasmids into electrocompetent E. coli BL21 (DE3) cells. Transformants were incubated at 37 °C overnight and one colony was used to incubate 10 mL LB media supplemented with 100 μg/mL ampicillin and incubated at 37 °C, 250 rpm, overnight. ON culture was used to incubate fresh LB media supplemented with ampicillin. At OD600 between 0.5 and 0.8 the cultures were induced with arabinose (total concentration 0.02%) and the cultures were further incubated at 37 °C for 4 h. 2 mL cell suspension was centrifuged for 1 min at 12,000× g. The supernatant was separated from the pellet, and the pellet was resuspended in 1 mL lysis buffer (50 mM potassium phosphate, pH 7.4, 500 mM sodium acetate, 0.1 mM EDTA and 20% glycerol). The lysis mixtures were kept on ice and lysed by sonication (Soniprep, amplitude 10 μm) for 2 × 30 s, and hereafter centrifuged for 1 min at 12,000× g. For SDS-page, protein concentration was estimated using the method of Bradford [64] with BSA as standard. Protein concentration of extracellular extracts ranged from 0.1 to 0.3 mg/mL and intracellular were diluted to a total concentration of 0.5 mg/mL. Fifty μL of protein extract were incubated with 10 μL loading dye (300 mM Tris HCl, pH 6.8, 0.01% bromophenol blue, 15% v/v glycerol and 6% SDS) at 95 °C for 5 min and 20 μL of each solution was loaded on a precast 4%–12% Bis-Tris gel (NuPAGE™ Novex™, Thermo Scientific, NP0321, Waltham, MA, USA). The gel was run for 1.5 h at 90 V and stained with Coomassie Brilliant Blue G-250. To identify the proteins on the gel, the sizes were estimated using the compute PI webtool [65].
4.8. Antifungal Activity
Seven different fungi were used for antifungal activity testing of both wildtype bacteria as well as the cloned chitinase enzyme extracts. The following fungi were chosen to cover indoor mold and plant-pathogenic fungi: Penicillium chrysogenum (IBT 33843), Stachybotrys chartarum (IBT 7709), Chaetomium globosum (IBT 7029), Neosartorya hiratsukae (IBT 28630), Aspergillus niger (IBT 32191), Fusarium oxysporum (IBT 41964) and Botrytis cinerea (IBT 41856) and were from the IBT Culture Collection at Department of Biotechnology and Biomedicine, Technical University of Denmark. The antifungal activity of wildtype bacterial strains was tested by adding a 20 μL spore suspension to a puncture well on the center of a MA plate. Due to different growth rates, the fungi were allowed to grow for 4–8 days, after which colony mass of each wildtype bacteria was spotted approximately 2 mm from the edge of the fungal colony. The plates were left for 4 days and antifungal activity was observed. We also tested the antifungal activity of the wild-type strains in a setting where the bacteria were spotted 2 days prior to the inoculating the fungi. The bacteria were spotted 2 cm from center of the plates. In both the above settings, plates were checked again after approximately 14 days to see if the bacteria retained their inhibitive effect. Antifungal effect was graded qualitatively, where one plus (+) describes antifungal effect which did not retain the effect after 14 days, and two pluses (++) describes the antifungal effect which was retained after 14 days.
For the chitinase extracts, the fungi were inoculated as described above and after 3–4 days of growth, holes were punched 2 mm from the edge of the fungal colony and 50 μL of each enzyme solution, prepared as in Section 4.7, was added to the wells. Plates were incubated for 2–4 days and checked for antifungal activity. All experiments were conducted at room temperature.
Supplementary Materials
The following are available online at http://www.mdpi.com/1660-3397/14/12/230/s1, Table S1: List of included and excluded chitinases.
Acknowledgments
H.M. was supported by a Ph.D. grant from the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme FP7-People-2012-ITN, under grant agreement No. 317058, ‘BACTORY’. B.A. was supported by a grant from the VILLUM Foundation. S.S.P. received funding from the Commission on Health, Food and Welfare under the Danish Council for Strategic Research and the MaCuMBA Project under the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement No. 311975. The publication reflects the views only of the author, and the European Union cannot be held responsible for any use which may be made of the information contained therein. The present work was carried out as part of the Galathea 3 expedition under the auspices of the Danish Expedition Foundation. This is Galathea 3 contribution No. P122.
Author Contributions
H.M., S.S.P. and L.G. conceived and designed the experiments; S.S.P. performed the experiments with assistance from H.M. and B.A.; S.S.P. and H.M. analyzed the data; S.S.P. wrote the paper with support from H.M., L.G. and B.A.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Zobell, C.E.; Rittenberg, S.C. The Occurrence and Characteristics of Chitinoclastic Bacteria in the Sea. J Bacteriol. 1938, 35, 275–287. [Google Scholar] [PubMed]
- Gooday, G.W. Physiology of microbial degradation of chitin and chitosan. Biodegradation 1990, 1, 177–190. [Google Scholar] [CrossRef]
- Hayes, M.; Carney, B.; Slater, J.; Brück, W. Mining marine shellfish wastes for bioactive molecules: Chitin and chitosan—Part B: Applications. Biotechnol. J. 2008, 3, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Xi, C. Don’t waste seafood waste. Nature 2015, 524, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.; Sotelo, C.G.; Chapela, M.J.; Pérez-Martin, R.I. Towards sustainable and efficient use of fishery resources: Present and future trends. Trends Food Sci. Technol. 2007, 18, 29–36. [Google Scholar] [CrossRef]
- Kim, S.; Mendis, E. Bioactive compounds from marine processing byproducts—A review. J. Food Res. 2006, 39, 383–393. [Google Scholar] [CrossRef]
- Kawase, T.; Yokokawa, S.; Saito, A.; Fujii, T.; Miyashita, K.; Watanabe, T. Comparison of Enzymatic and Antifungal Properties between Family 18 and 19 Chitinases from S. coelicolor A3(2). Biosci. Biotechnol. Biochem. 2014, 8451, 988–998. [Google Scholar]
- García-Fraga, B.; Da Silva, A.F.; López-Seijas, J.; Sieiro, C. A novel family 19 chitinase from the marine-derived Pseudoalteromonas tunicata CCUG 44952T: Heterologous expression, characterization and antifungal activity. Biochem. Eng. J. 2015, 93, 84–93. [Google Scholar] [CrossRef]
- Hoster, F.; Schmitz, J.E.; Daniel, R. Enrichment of chitinolytic microorganisms: Isolation and characterization of a chitinase exhibiting antifungal activity against phytopathogenic fungi from a novel Streptomyces strain. Appl. Microbiol. Biotechnol. 2005, 66, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.I.; Hocking, A.D. Introduction. In Fungi and Food Spoilage; Springer Science: New York, NY, USA, 2009; p. 1. [Google Scholar]
- Andersen, B.; Dosen, I.; Lewinska, A.M.; Nielsen, K.F. Pre-contamination of new gypsum wallboard with potentially harmful fungal species. Indoor Air 2016. [Google Scholar] [CrossRef] [PubMed]
- Brzezinska, M.S.; Jankiewicz, U.; Burkowska, A.; Walczak, M. Chitinolytic microorganisms and their possible application in environmental protection. Curr. Microbiol. 2014, 68, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Cai, J.; Xie, C.; Liu, C.; Chen, Y. Purification and partial characterization of a 36-kDa chitinase from Bacillus thuringiensis subsp. colmeri, and its biocontrol potential. Enzyme Microb. Technol. 2010, 46, 252–256. [Google Scholar] [CrossRef]
- Beier, S.; Bertilsson, S. Bacterial chitin degradation-mechanisms and ecophysiological strategies. Front. Microbiol. 2013, 4, 149. [Google Scholar] [CrossRef] [PubMed]
- Svitil, A.; Chadhain, S.; Moore, J.; Kirchman, D. Chitin Degradation Proteins Produced by the Marine Bacterium Vibrio harveyi Growing on Different Forms of Chitin. Appl. Environ. Microbiol. 1997, 63, 408–413. [Google Scholar] [PubMed]
- Orikoshi, H.; Nakayama, S.; Miyamoto, K.; Hanato, C.; Yasuda, M.; Inamori, Y.; Tsujibo, H. Roles of four chitinases (ChiA, ChiB, ChiC, and ChiD) in the chitin degradation system of marine bacterium Alteromonas sp. strain O-7. Appl. Environ. Microbiol. 2005, 71, 1811–1815. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kanai, R.; Kawase, T.; Tanabe, T.; Mitsutomi, M.; Sakuda, S.; Miyashita, K. Family 19 chitinases of Streptomyces species: Characterization and distribution. Microbiology 1999, 145, 3353–3363. [Google Scholar] [CrossRef] [PubMed]
- Vaaje-Kolstad, G.; Westereng, B.; Horn, S.; Liu, Z.; Zhai, H.; Sørlie, M.; Eisjink, V. An Oxidative Enzyme Boosting the enzymatic conversion of recalcitrant polysaccharides. Science 2010, 330, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Fushinobu, S. Metalloproteins: A new face for biomass breakdown. Nat. Chem. Biol. 2013, 10, 88–89. [Google Scholar] [CrossRef] [PubMed]
- Levasseur, A.; Drula, E.; Lombard, V.; Coutinho, P.M.; Henrissat, B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 2013, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Hamre, A.G.; Eide, K.B.; Wold, H.H.; Sørlie, M. Activation of enzymatic chitin degradation by a lytic polysaccharide monooxygenase. Carbohydr. Res. 2015, 407, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Kadokura, K.; Rokutani, A.; Yamamoto, M.; Ikegami, T.; Sugita, H.; Itoi, S.; Hakamata, W.; Oku, T.; Nishio, T. Purification and characterization of Vibrio parahaemolyticus extracellular chitinase and chitin oligosaccharide deacetylase involved in the production of heterodisaccharide from chitin. Appl. Microbiol. Biotechnol. 2007, 75, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Kadokura, K.; Ikegami, T.; Shigeta, Y.; Kumaki, Y.; Hakamata, W.; Oku, T.; Nishio, T. Heterodisaccharide 4-O-(N-acetyl-beta-d-glucosaminyl)-d-glucosamine is a specific inducer of chitinolytic enzyme production in Vibrios harboring chitin oligosaccharide deacetylase genes. Glycobiology 2009, 19, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Hamer, S.N.; Cord-Landwehr, S.; Planas, A.; Waegeman, H.; Moerschbacher, B.M.; Kolkenbrock, S. Enzymatic production of defined chitosan oligomers with a specific pattern of acetylation using a combination of chitin oligosaccharide deacetylases. Sci. Rep. 2015, 5, 8716. [Google Scholar] [CrossRef] [PubMed]
- Gram, L.; Melchiorsen, J.; Bruhn, J.B. Antibacterial activity of marine culturable bacteria collected from a global sampling of ocean surface waters and surface swabs of marine organisms. Mar. Biotechnol. 2010, 12, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Wietz, M.; Månsson, M.; Gram, L. Chitin stimulates production of the antibiotic andrimid in a Vibrio coralliilyticus strain. Environ. Microbiol. Rep. 2011, 3, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Giubergia, S.; Phippen, C.; Gotfredsen, C.H.; Nielsen, K.F.; Gram, L. Influence of niche-specific nutrients on secondary metabolism in Vibrionaceae. Appl. Environ. Microbiol. 2016, 82, 4035–4044. [Google Scholar] [CrossRef] [PubMed]
- Machado, H.; Sonnenschein, E.C.; Melchiorsen, J.; Gram, L. Genome mining reveals unlocked bioactive potential of marine Gram-negative bacteria. BMC Genom. 2015, 16, 158. [Google Scholar] [CrossRef] [PubMed]
- Ramazzina, I.; Cendron, L.; Folli, C.; Berni, R.; Monteverdi, D.; Zanotti, G.; Percudani, R. Logical Identification of an Allantoinase Analog (puuE) Recruited from Polysaccharide Deacetylases. J. Biol. Chem. 2008, 283, 23295–23304. [Google Scholar] [CrossRef] [PubMed]
- Boer, H.; Munck, N.; Natunen, J.; Wohlfahrt, G.; Söderlund, H.; Renkonen, O.; Koivula, A. Differential recognition of animal type β4-galactosylated and -fucosylated chito-oligosaccharides by two family 18 chitinases from Trichoderma harzianum. Glycobiology 2004, 14, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Vaaje-Kolstad, G.; Bunæs, A.C.; Mathiesen, G.; Eijsink, V.G.H. The chitinolytic system of Lactococcus lactis ssp. lactis comprises a nonprocessive chitinase and a chitin-binding protein that promotes the degradation of α- And β-chitin. FEBS J. 2009, 276, 2402–2415. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Sugawara, N.; Suzuki, M.; Uchiyama, T.; Katouno, F.; Nikaidou, N.; Watanabe, T. Chitinases A, B, and C1 of Serratia marcescens 2170 produced by recombinant Escherichia coli: Enzymatic properties and synergism on chitin degradation. Biosci. Biotechnol. Biochem. 2002, 66, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, T.; Katouno, F.; Nikaidou, N.; Nonaka, T.; Sugiyama, J.; Watanabe, T. Roles of the Exposed Aromatic Residues in Crystalline Chitin Hydrolysis by Chitinase A from Serratia marcescens 2170. J. Biol. Chem. 2001, 276, 41343–41349. [Google Scholar] [CrossRef] [PubMed]
- Kawase, T.; Yokokawa, S.; Saito, A.; Fujii, T.; Nikaidou, N.; Miyashita, K.; Watanabe, T. Comparison of enzymatic and antifungal properties between family 18 and 19 chitinases from S. coelicolor A3(2). Biosci. Biotechnol. Biochem. 2006, 70, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Jones, C.M.; Mohit, V.; Hallin, S.; Bertilsson, S. Global phylogeography of chitinase genes in aquatic metagenomes. Appl. Environ. Microbiol. 2011, 77, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Ito, Y.; Yamada, T.; Hashimoto, M.; Sekine, S.; Tanaka, H. The Roles of the C-Terminal Domain and Type and Type III Domains of Chitinase A1 from Bacillus circulans WL-12 in chitin degradation. J. Bacteriol. 1994, 176, 4465–4472. [Google Scholar] [CrossRef] [PubMed]
- Katouno, F.; Taguchi, M.; Sakurai, K.; Uchiyama, T.; Nikaidou, N.; Nonaka, T.; Sugiyama, J.; Watanabe, T. Importance of Exposed Aromatic Residues in Chitinase B from Serratia marcescens 2170 for Crystalline Chitin Hydrolysis. J. Biochem. 2004, 168, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Limón, M.C.; Lora, J.M.; García, I.; De La Cruz, J.; Llobell, A.; Benítez, T.; Pintor-Toro, J.A. Primary structure and expression pattern of the 33-kDa chitinase gene from the mycoparasitic fungus Trichoderma harzianum. Curr. Genet. 1995, 28, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Armand, S.; Hata, T.; Nikaidou, N.; Henrissat, B.; Mitsutomi, M.; Watanabe, T. A modular family 19 chitinase found in the prokaryotic organism Streptomyces griseus HUT 6037. J. Bacteriol. 1996, 178, 5065–5070. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Taniguchi, H.; Kitaoka, M. A reducing-end-acting chitinase from Vibrio proteolyticus belonging to glycoside hydrolase family 19. Appl. Microbiol. Biotechnol. 2008, 78, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Kojima, M.; Yoshikawa, T.; Mitsuda, N.; Araki, K.; Kawaguchi, T.; Miyatake, K.; Arai, M.; Fukamizo, T. A novel type of family 19 chitinase from Aeromonas sp. No. 10S-24. Eur. J. Biochem. 2003, 270, 2513–2520. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Garbulewska, E.; Sato, K.; Kato, Y.; Nogawa, M.; Taguchi, G.; Shimosaka, M. Isolation of genes coding for chitin-degrading enzymes in the novel chitinolytic bacterium, Chitiniphilus shinanonensis, and characterization of a gene coding for a family 19 chitinase. JBIOSC 2012, 113, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Tsujibo, H.; Orikoshi, H.; Baba, N.; Miyamoto, K.; Yasuda, M.; Miyahara, M. Identification and Characterization of the Gene Cluster Involved in Chitin Degradation in a Marine Bacterium, Alteromonas sp. Appl. Environ. Microbiol. 2002, 68, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Shimosaka, M.; Ando, Y.; Nishiyama, K. Species-specific distribution of a modular family 19 chitinase gene in Burkholderia gladioli. FEMS Microbiol. Ecol. 2001, 37, 135–141. [Google Scholar] [CrossRef]
- Nakagawa, Y.S.; Kudo, M.; Loose, J.S.M.; Ishikawa, T.; Totani, K. A small lytic polysaccharide monooxygenase from Streptomyces griseus targeting alpha and beta chitin. FEBS J. 2015, 282, 1065–1079. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, L.X.; Wang, X.; Roseman, S. The chitin catabolic cascade in the marine bacterium Vibrio cholerae: Characterization of a unique chitin oligosaccharide deacetylase. Glycobiology 2007, 17, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Uehera, R.; Shiraishu, H.; Hakamata, W.; Nishio, T. Chitin Oligosaccharide Deacetylase from Shewanella woodyi ATCC51908. J. Appl. Glycosci. 2015. [Google Scholar] [CrossRef]
- Liu, J.; Jia, Z.; Li, S.; Li, Y.; You, Q.; Zhang, C.; Zheng, X.; Xiong, G.; Zhao, J.; Qi, C.; et al. Identification and characterization of a chitin deacetylase from a metagenomic library of deep-sea sediments of the Arctic Ocean. Gene 2016, 590, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Park, R.D.; Muzzarelli, R.A.A. Chitin deacetylases: Properties and applications. Mar. Drugs 2010, 8, 24–46. [Google Scholar] [CrossRef] [PubMed]
- De Fuente-Salcido, N.M.; Casados-Vázquez, L.E.; García-Pérez, A.P.; Barboza-Pérez, U.E.; Bideshi, D.K.; Salcedo-hernández, R.; Almendarez, B.E.G.; Barboza-Corona, J.E. The endochitinase ChiA Btt of Bacillus thuringiensis 2803 and its potential use to control the phytopathogen Colletotrichum gloeosporioides. Microbiol. Open 2016, 5, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Lobo, M.D.P.; Silva, F.A.D.; de Castro Landim, P.G.; da Cruz, P.R.; de Brito, T.L.; de Medeiros, S.C.; Oliveira, J.T.A.; Vasconcelos, I.M.; Pereira, H.D.; Grangeiro, T.B. Expression and efficient secretion of a functional chitinase from Chromobacterium violaceum in Escherichia coli. BMC Biotechnol. 2013, 13, 46. [Google Scholar] [CrossRef] [PubMed]
- Suma, K.; Podile, A.R. Chitinase A from Stenotrophomonas maltophilia shows transglycosylation and antifungal activities. Bioresour. Technol. 2013, 133, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Rapid Annotation Using Subsystem Technology Server. Available online: http://rast.nmpdr.org/ (accessed on 14 December 2016).
- The National Center for Biotechnology Information. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 14 December 2016).
- Pfam. Available online: http://pfam.xfam.org/ (accessed on 14 December 2016).
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, 279–285. [Google Scholar] [CrossRef] [PubMed]
- BLAST. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 13 December 2016).
- SignalP. Available online: http://www.cbs.dtu.dk/services/SignalP/ (accessed on 13 December 2016).
- FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 13 December 2016).
- Nour-Eldin, H.H.; Hansen, B.G.; Nørholm, M.H.H.; Jensen, J.K.; Halkier, B.A. Advancing uracil-excision based cloning towards an ideal technique for cloning PCR fragments. Nucleic Acids Res. 2006, 34, e122. [Google Scholar] [CrossRef] [PubMed]
- Nørholm, M.H.H. A mutant Pfu DNA polymerase designed for advanced uracil-excision DNA engineering. BMC Biotechnol. 2010, 10, 21. [Google Scholar]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Chem. 1976, 254, 248–254. [Google Scholar] [CrossRef]
- Compute PI. Available online: http://web.expasy.org/compute_pi/ (accessed on 13 December 2016).
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).