
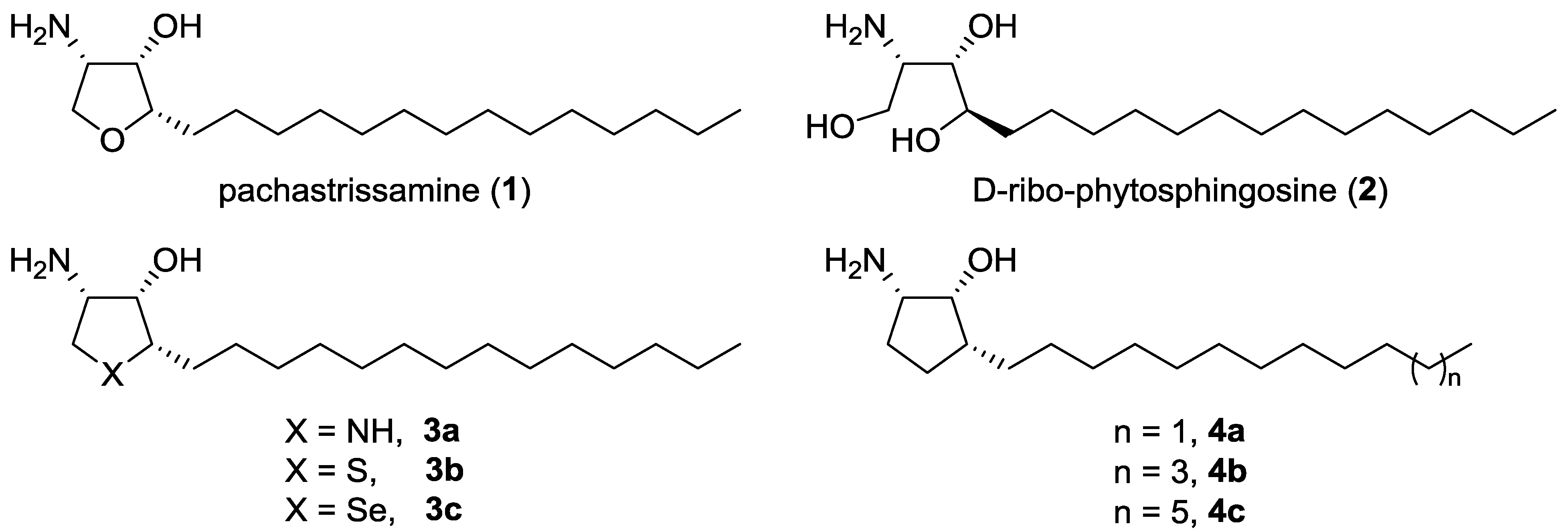
Synthesis and Biological Evaluation of Carbocyclic Analogues of Pachastrissamine

Abstract
:
1. Introduction
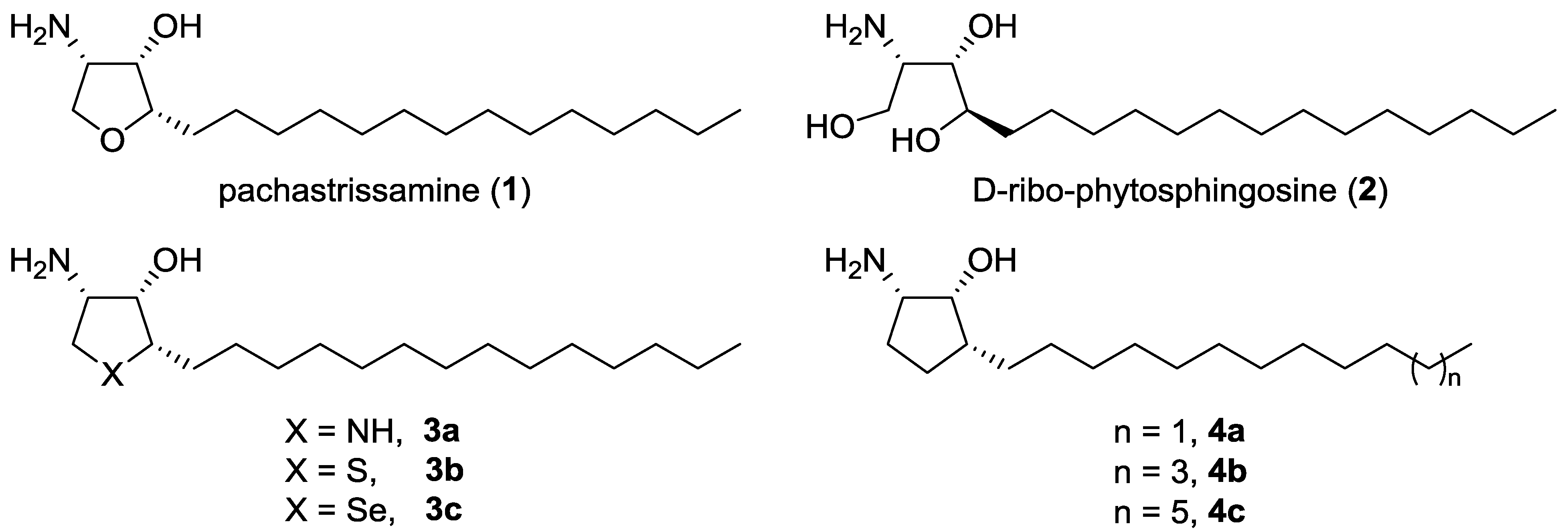
2. Results and Discussion
2.1. Chemistry
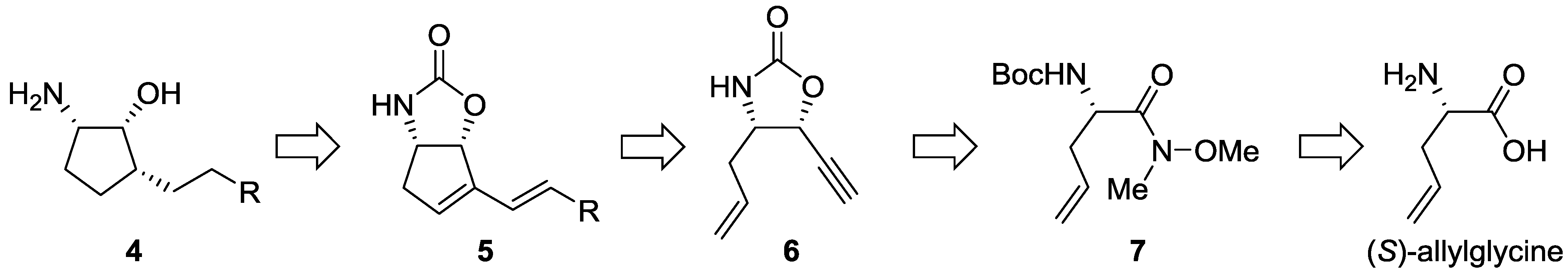
2.1.1. Retrosynthetic Analysis
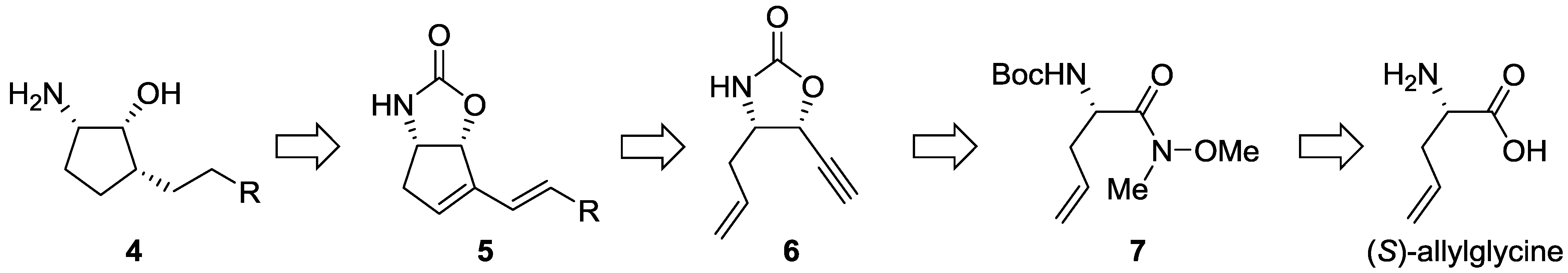
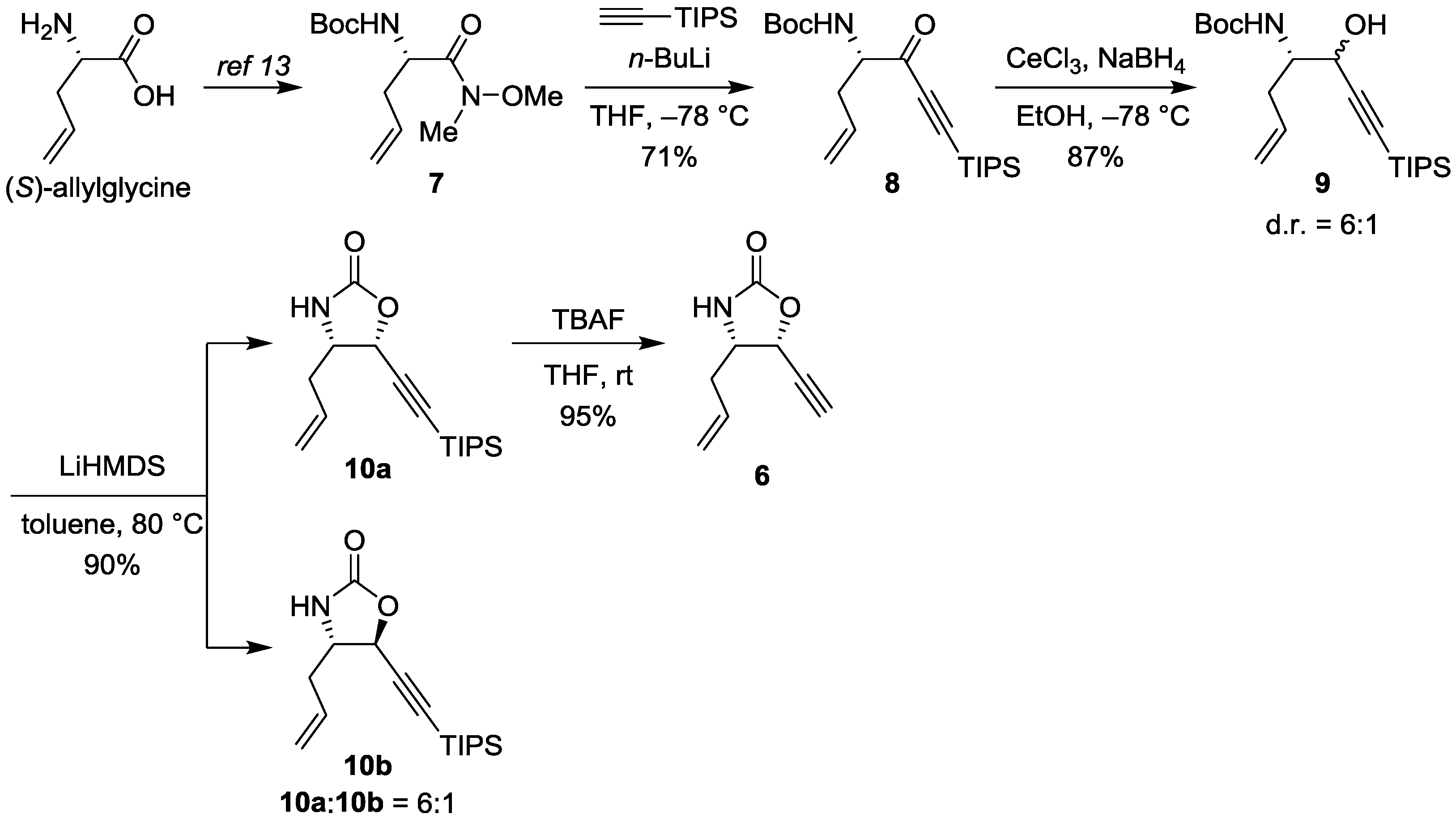
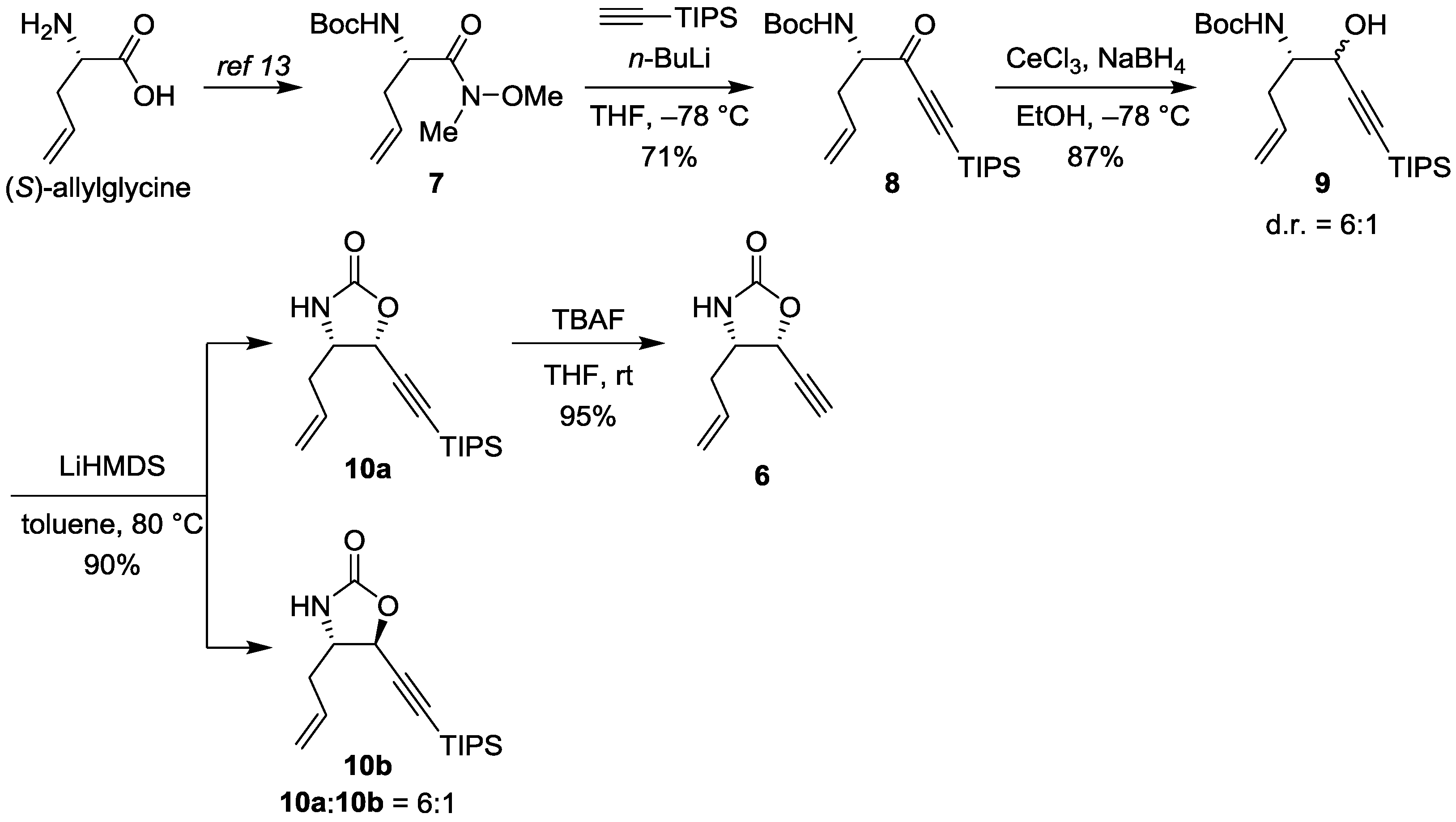
2.1.2. Synthesis

2.2. Biological Evaluation
2.2.1. Cell Viability

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) a | ||||
|---|---|---|---|---|---|
| HCT-116 b | SNU-638 c | MDA-MB-231 d | PC-3 e | Caki-1 f | |
| 1 | 1.0 | 1.2 | 0.7 | 0.7 | 1.7 |
| 4a | 9.7 | 12.8 | 6.1 | 7.6 | 12.1 |
| 4b | 1.0 | 1.7 | 1.8 | 1.0 | 3.2 |
| 4c | 3.2 | 4.4 | 4.1 | 3.1 | 6.6 |
2.2.2. Inhibitory Activity against Sphingosine Kinases
| Compound | IC50 (μM) a | |
|---|---|---|
| SphK1 | SphK2 | |
| 1 | 12.0 | 41.8 |
| 4a | 58.2 | >100 |
| 4b | 7.5 | 20.1 |
| 4c | 41.3 | >100 |
| DMS b | 6.6 | 19.9 |
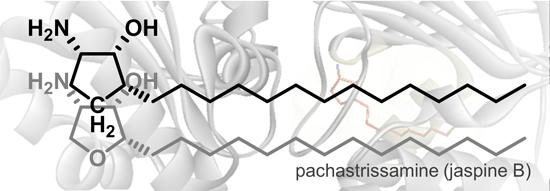
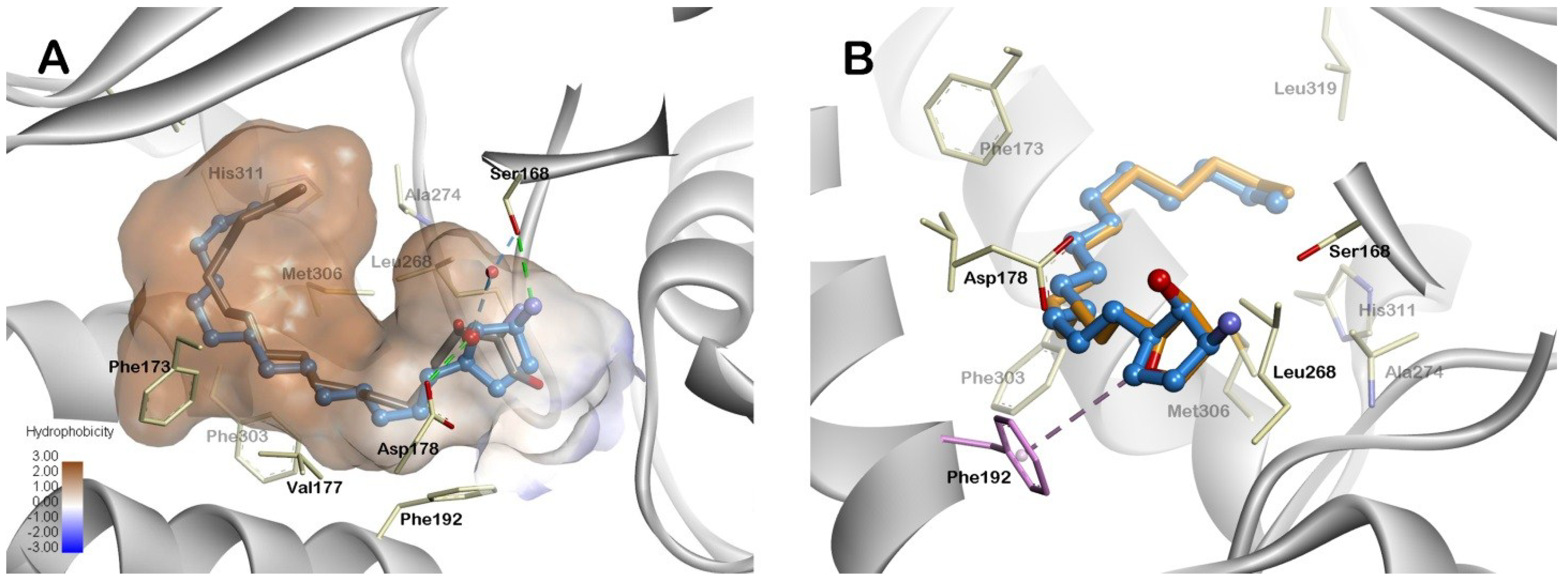
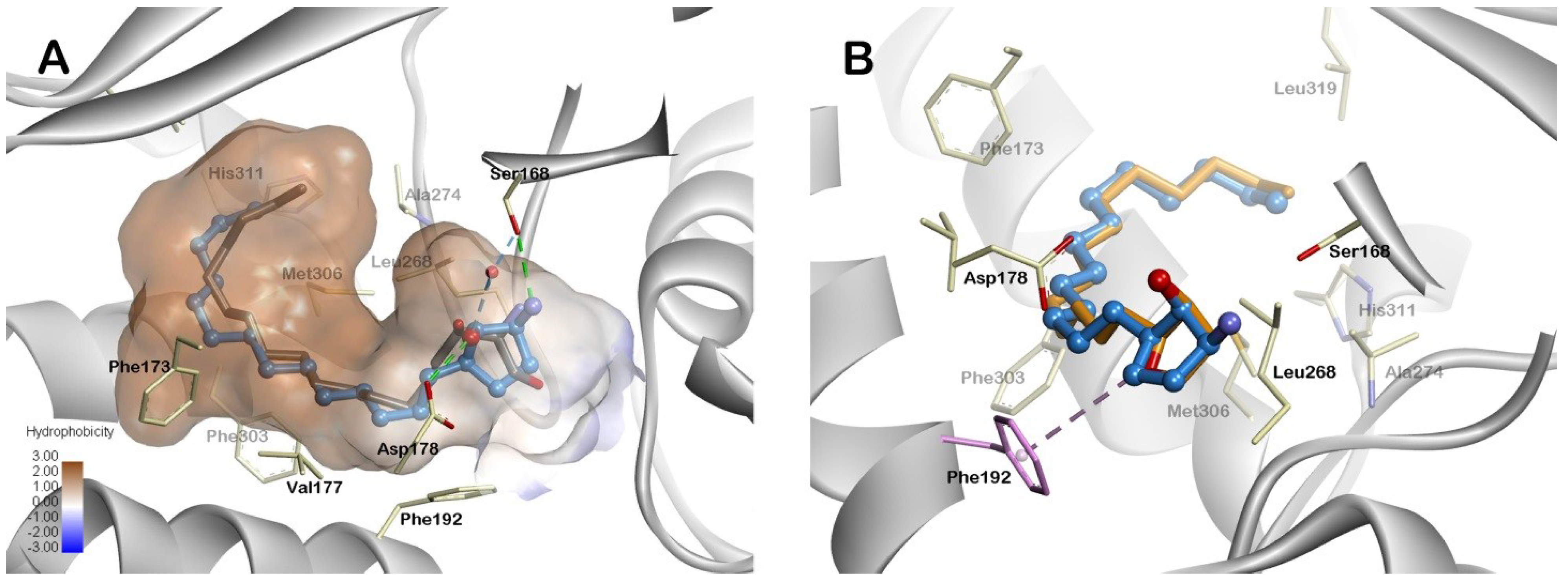
2.3. Molecular Modeling
3. Experimental Section
3.1. Chemistry
3.1.1. General
3.1.2. tert-Butyl (S)-(5-oxo-7-(triisopropylsilyl)hept-1-en-6-yn-4-yl)carbamate (8)
3.1.3. tert-Butyl ((4S)-5-hydroxy-7-(triisopropylsilyl)hept-1-en-6-yn-4-yl)carbamate (9)
3.1.4. (4S)-4-Allyl-5-((triisopropylsilyl)ethynyl)oxazolidin-2-one (10)
3.1.5. (4S,5R)-4-Allyl-5-ethynyloxazolidin-2-one (6)
3.1.6. General Procedure for the Preparation of 5
3.1.7. (3aS,6aR)-6-((E)-Dodec-1-en-1-yl)-3,3a,4,6a-tetrahydro-2H-cyclopenta[d]oxazol-2-one (5a)
3.1.8. (3aS,6aR)-6-((E)-Tetradec-1-en-1-yl)-3,3a,4,6a-tetrahydro-2H-cyclopenta[d]oxazol-2-one (5b)
3.1.9. (3aS,6aR)-6-((E)-Hexadec-1-en-1-yl)-3,3a,4,6a-tetrahydro-2H-cyclopenta[d]oxazol-2-one (5c)
3.1.10. General Procedure for the Preparation of 11
3.1.11. (3aS,6R,6aR)-6-Dodecylhexahydro-2H-cyclopenta[d]oxazol-2-one (11a)
3.1.12. (3aS,6R,6aR)-6-Tetradecylhexahydro-2H-cyclopenta[d]oxazol-2-one (11b)
3.1.13. (3aS,6R,6aR)-6-Hexadecylhexahydro-2H-cyclopenta[d]oxazol-2-one (11c)
3.1.14. General Procedure for the Preparation of 4
3.1.15. (1R,2S,5R)-2-Amino-5-dodecylcyclopentan-1-ol (4a)
3.1.16. (1R,2S,5R)-2-Amino-5-tetradecylcyclopentan-1-ol (4b)
3.1.17. (1R,2S,5R)-2-Amino-5-hexadecylcyclopentan-1-ol (4c)
3.2. Biological Evaluation
3.2.1. Sulforhodamine B (SRB) Assay
3.2.2. Sphingosine Kinase Inhibition Assay
3.3. Molecular Modeling
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kuroda, I.; Musman, M.; Ohtani, I.I.; Ichiba, T.; Tanaka, J.; Gravalos, D.G.; Higa, T. Pachastrissamine, a cytotoxic anhydrophytosphingosine from a marine sponge, Pachastrissa sp. J. Nat. Prod. 2002, 65, 1505–1506. [Google Scholar] [CrossRef] [PubMed]
- Ledroit, V.; Debitus, C.; Lavaud, C.; Massiot, G. Jaspines A and B: Two new cytotoxic sphingosine derivatives from the marine sponge Jaspis sp. Tetrahedron Lett. 2003, 44, 225–228. [Google Scholar] [CrossRef]
- Liu, J.; Du, Y.; Dong, X.; Meng, S.; Xiao, J.; Cheng, L. Stereoselective synthesis of jaspine B from d-xylose. Carbohydr. Res. 2006, 341, 2653–2657. [Google Scholar] [CrossRef] [PubMed]
- Salma, Y.; Lafont, E.; Therville, N.; Carpentier, S.; Bonnafé, M.-J.; Levade, T.; Génisson, Y.; Andrieu-Abadie, N. The natural marine anhydrophytosphingosine, jaspine B, induces apoptosis in melanoma cells by interfering with ceramide metabolism. Biochem. Pharmacol. 2009, 78, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Yoshimitsu, Y.; Oishi, S.; Miyagaki, J.; Inuki, S.; Ohno, H.; Fujii, N. Pachastrissamine (jaspine B) and its stereoisomers inhibit sphingosine kinases and atypical protein kinase C. Biorg. Med. Chem. 2011, 19, 5402–5408. [Google Scholar] [CrossRef] [Green Version]
- Inuki, S.; Yoshimitsu, Y.; Oishi, S.; Fujii, N.; Ohno, H. Ring-construction/stereoselective functionalization cascade: Total synthesis of pachastrissamine (jaspine B) through palladium-catalyzed bis-cyclization of propargyl chlorides and carbonates. J. Org. Chem. 2010, 75, 3831–3842. [Google Scholar] [CrossRef] [PubMed]
- Passiniemi, M.; Koskinen, A.M.P. Asymmetric synthesis of pachastrissamine (jaspine B) and its diastereomers via η3-allylpalladium intermediates. Org. Biomol. Chem. 2011, 9, 1774–1783. [Google Scholar] [CrossRef] [PubMed]
- Srinivas Rao, G.; Venkateswara Rao, B. A common strategy for the stereoselective synthesis of anhydrophytosphingosine pachastrissamine (jaspine B) and N,O,O,O-tetra-acetyl d-lyxo-phytosphingosine. Tetrahedron Lett. 2011, 52, 6076–6079. [Google Scholar]
- Llaveria, J.; Díaz, Y.; Matheu, M.I.; Castillón, S. Enantioselective synthesis of jaspine B (pachastrissamine) and its C-2 and/or C-3 epimers. Eur. J. Org. Chem. 2011, 1514–1519. [Google Scholar]
- Zhao, M.-L.; Zhang, E.; Gao, J.; Zhang, Z.; Zhao, Y.-T.; Qu, W.; Liu, H.-M. An efficient and convenient formal synthesis of jaspine B from d-xylose. Carbohydr. Res. 2012, 351, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Schmiedel, V.M.; Stefani, S.; Reissig, H.-U. Stereodivergent synthesis of jaspine B and its isomers using a carbohydrate-derived alkoxyallene as C3-building block. Beilstein J. Org. Chem. 2013, 9, 2564–2569. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, P.; Ajay, S.; Meena, S.; Sinha, S.; Shaw, A.K. Stereoselective total synthesis of jaspine B (pachastrissamine) utilizing iodocyclization and an investigation of its cytotoxic activity. Tetrahedron Asymmetry 2013, 24, 903–908. [Google Scholar] [CrossRef]
- Jana, A.K.; Panda, G. Stereoselective synthesis of jaspine B and its C2 epimer from garner aldehyde. RSC Adv. 2013, 3, 16795–16801. [Google Scholar] [CrossRef]
- Dhand, V.; Chang, S.; Britton, R. Total synthesis of the cytotoxic anhydrophytosphingosine pachastrissamine (jaspine B). J. Org. Chem. 2013, 78, 8208–8213. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-W.; Liu, S.-W.; Hou, D.-R. Formation of tetrahydrofurans via a 5-endo-tet cyclization of aziridines—Synthesis of (−)-pachastrissamine. Org. Biomol. Chem. 2013, 11, 5292–5299. [Google Scholar] [CrossRef] [PubMed]
- Martinková, M.; Mezeiová, E.; Gonda, J.; Jacková, D.; Pomikalová, K. Total synthesis of (−)-jaspine B and its 4-epi-analogue from d-xylose. Tetrahedron Asymmetry 2014, 25, 750–766. [Google Scholar] [CrossRef]
- Génisson, Y.; Lamandé, L.; Salma, Y.; Andrieu-Abadie, N.; André, C.; Baltas, M. Enantioselective access to a versatile 4-oxazolidinonecarbaldehyde and application to the synthesis of a cytotoxic jaspine B truncated analogue. Tetrahedron Asymmetry 2007, 18, 857–864. [Google Scholar] [CrossRef]
- Salma, Y.; Ballereau, S.; Maaliki, C.; Ladeira, S.; Andrieu-Abadie, N.; Génisson, Y. Flexible and enantioselective access to jaspine B and biologically active chain-modified analogues thereof. Org. Biomol. Chem. 2010, 8, 3227–3243. [Google Scholar] [CrossRef] [PubMed]
- Canals, D.; Mormeneo, D.; Fabriàs, G.; Llebaria, A.; Casas, J.; Delgado, A. Synthesis and biological properties of pachastrissamine (jaspine B) and diastereoisomeric jaspines. Biorg. Med. Chem. 2009, 17, 235–241. [Google Scholar] [CrossRef]
- Xu, J.-M.; Zhang, E.; Shi, X.-J.; Wang, Y.-C.; Yu, B.; Jiao, W.-W.; Guo, Y.-Z.; Liu, H.-M. Synthesis and preliminary biological evaluation of 1,2,3-triazole-jaspine B hybrids as potential cytotoxic agents. Eur. J. Med. Chem. 2014, 80, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Rives, A.; Ladeira, S.; Levade, T.; Andrieu-Abadie, N.; Génisson, Y. Synthesis of cytotoxic aza analogues of jaspine B. J. Org. Chem. 2010, 75, 7920–7923. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.; Bae, H.; Baek, D.J.; Kwak, Y.-S.; Kim, D.; Kim, S. Syntheses of sulfur and selenium analogues of pachastrissamine via double displacements of cyclic sulfate. Org. Biomol. Chem. 2011, 9, 7237–7242. [Google Scholar] [CrossRef] [PubMed]
- Patani, G.A.; LaVoie, E.J. Bioisosterism: A rational approach in drug design. Chem. Rev. 1996, 96, 3147–3176. [Google Scholar] [CrossRef] [PubMed]
- Arjona, O.; Gómez, A.M.; López, J.C.; Plumet, J. Synthesis and conformational and biological aspects of carbasugars. Chem. Rev. 2007, 107, 1919–2036. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, M.; Gotor, V. Biocatalytic selective modifications of conventional nucleosides, carbocyclic nucleosides, and C-nucleosides. Chem. Rev. 2000, 100, 4319–4348. [Google Scholar] [CrossRef] [PubMed]
- Villar, H.; Frings, M.; Bolm, C. Ring closing enyne metathesis: A powerful tool for the synthesis of heterocycles. Chem. Soc. Rev. 2007, 36, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Grubbs, R.H. Olefin metathesis. Tetrahedron 2004, 60, 7117–7140. [Google Scholar] [CrossRef]
- Royer, F.; Vilain, C.; Elkaïm, L.; Grimaud, L. Selective domino ring-closing metathesis–cross-metathesis reactions between enynes and electron-deficient alkenes. Org. Lett. 2003, 5, 2007–2009. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Y.; Kim, H.Y.; Tae, H.; Kim, B.G.; Lee, J. One-pot three-component tandem metathesis/Diels−alder reaction. Org. Lett. 2003, 5, 3439–3442. [Google Scholar] [CrossRef] [PubMed]
- Borzilleri, R.M.; Zheng, X.; Schmidt, R.J.; Johnson, J.A.; Kim, S.-H.; DiMarco, J.D.; Fairchild, C.R.; Gougoutas, J.Z.; Lee, F.Y.F.; Long, B.H.; et al. A novel application of a Pd(0)-catalyzed nucleophilic substitution reaction to the regio- and stereo-selective synthesis of lactam analogues of the epothilone natural products. J. Am. Chem. Soc. 2000, 122, 8890–8897. [Google Scholar] [CrossRef]
- Lee, S.K.; Nam, K.-A.; Heo, Y.-H. Cytotoxic activity and G2/M cell cycle arrest mediated by antofine, a phenanthroindolizidine alkaloid isolated from cynanchum paniculatum. Planta Med. 2003, 69, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Yatomi, Y.; Ruan, F.; Megidish, T.; Toyokuni, T.; Hakomori, S.-I.; Igarashi, Y. N,N-dimethylsphingosine inhibition of sphingosine kinase and sphingosine 1-phosphate activity in human platelets. Biochemistry 1996, 35, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Min, X.; Xiao, S.-H.; Johnstone, S.; Romanow, W.; Meininger, D.; Xu, H.; Liu, J.; Dai, J.; An, S.; et al. Molecular basis of sphingosine kinase 1 substrate recognition and catalysis. Structure 2013, 21, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Bissantz, C.; Kuhn, B.; Stahl, M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef] [PubMed]
- Protein Data Bank. Available online: http://www.rcsb.org (accessed on 27 November 2014).
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, Y.; Song, J.; Bae, H.; Kim, W.-J.; Lee, J.-Y.; Han, G.-H.; Lee, S.K.; Kim, S. Synthesis and Biological Evaluation of Carbocyclic Analogues of Pachastrissamine. Mar. Drugs 2015, 13, 824-837. https://doi.org/10.3390/md13020824
Kwon Y, Song J, Bae H, Kim W-J, Lee J-Y, Han G-H, Lee SK, Kim S. Synthesis and Biological Evaluation of Carbocyclic Analogues of Pachastrissamine. Marine Drugs. 2015; 13(2):824-837. https://doi.org/10.3390/md13020824
Chicago/Turabian StyleKwon, Yongseok, Jayoung Song, Hoon Bae, Woo-Jung Kim, Joo-Youn Lee, Geun-Hee Han, Sang Kook Lee, and Sanghee Kim. 2015. "Synthesis and Biological Evaluation of Carbocyclic Analogues of Pachastrissamine" Marine Drugs 13, no. 2: 824-837. https://doi.org/10.3390/md13020824