Accurate Dereplication of Bioactive Secondary Metabolites from Marine-Derived Fungi by UHPLC-DAD-QTOFMS and a MS/HRMS Library

 ,
,
Abstract
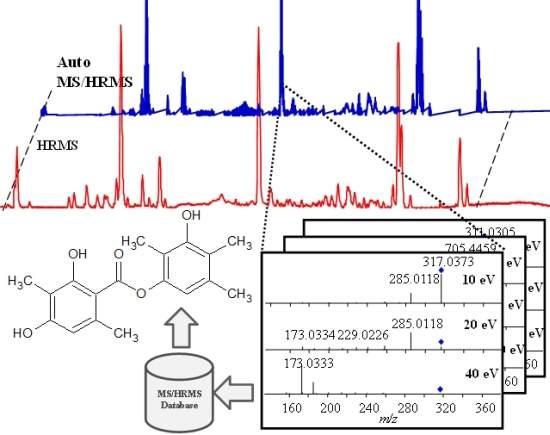
:
1. Introduction
2. Results and Discussion
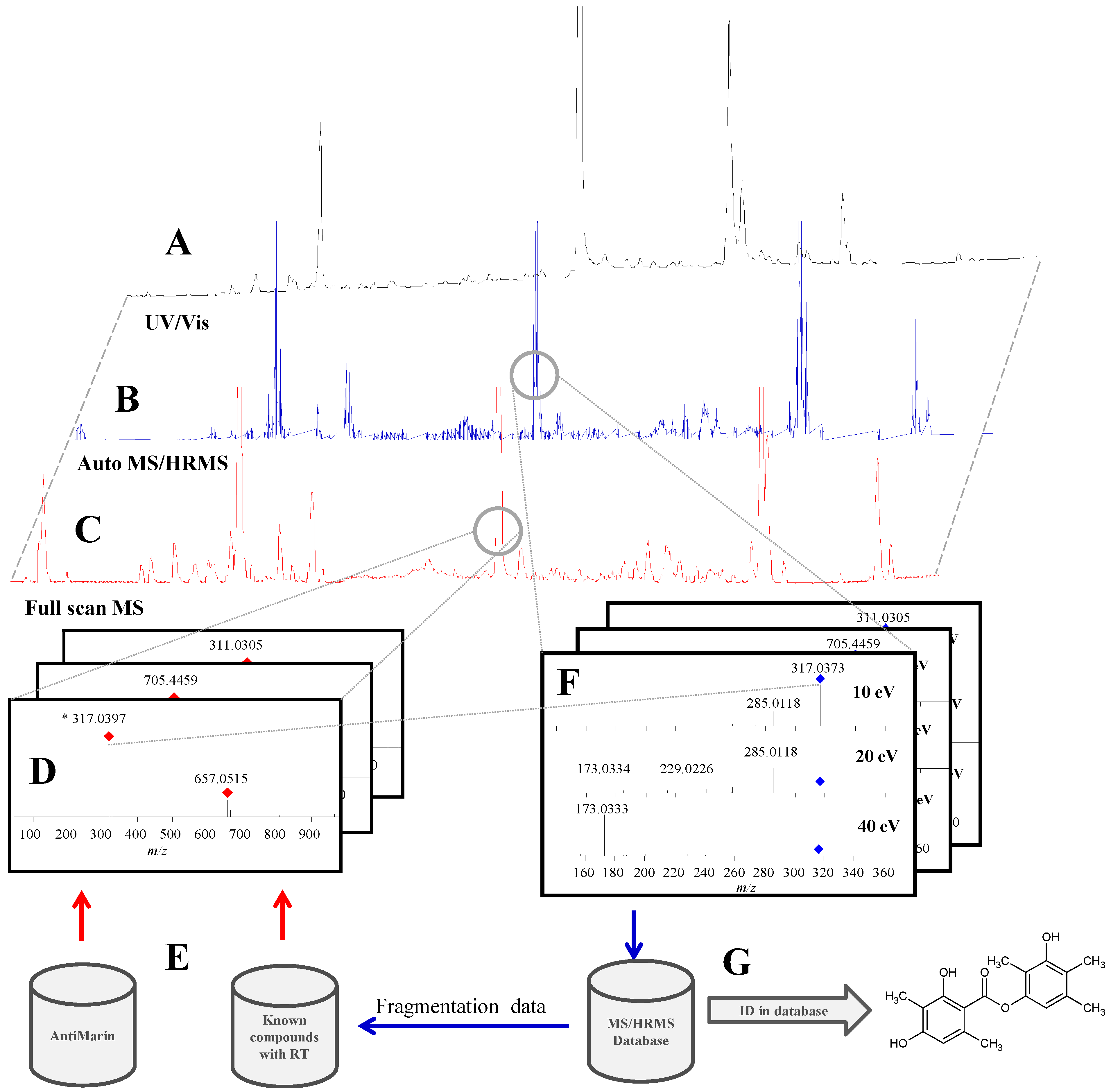
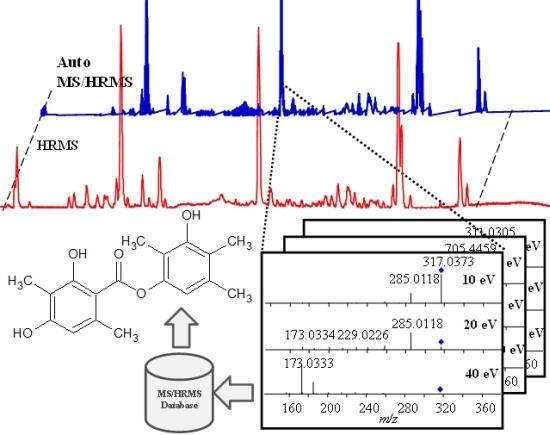
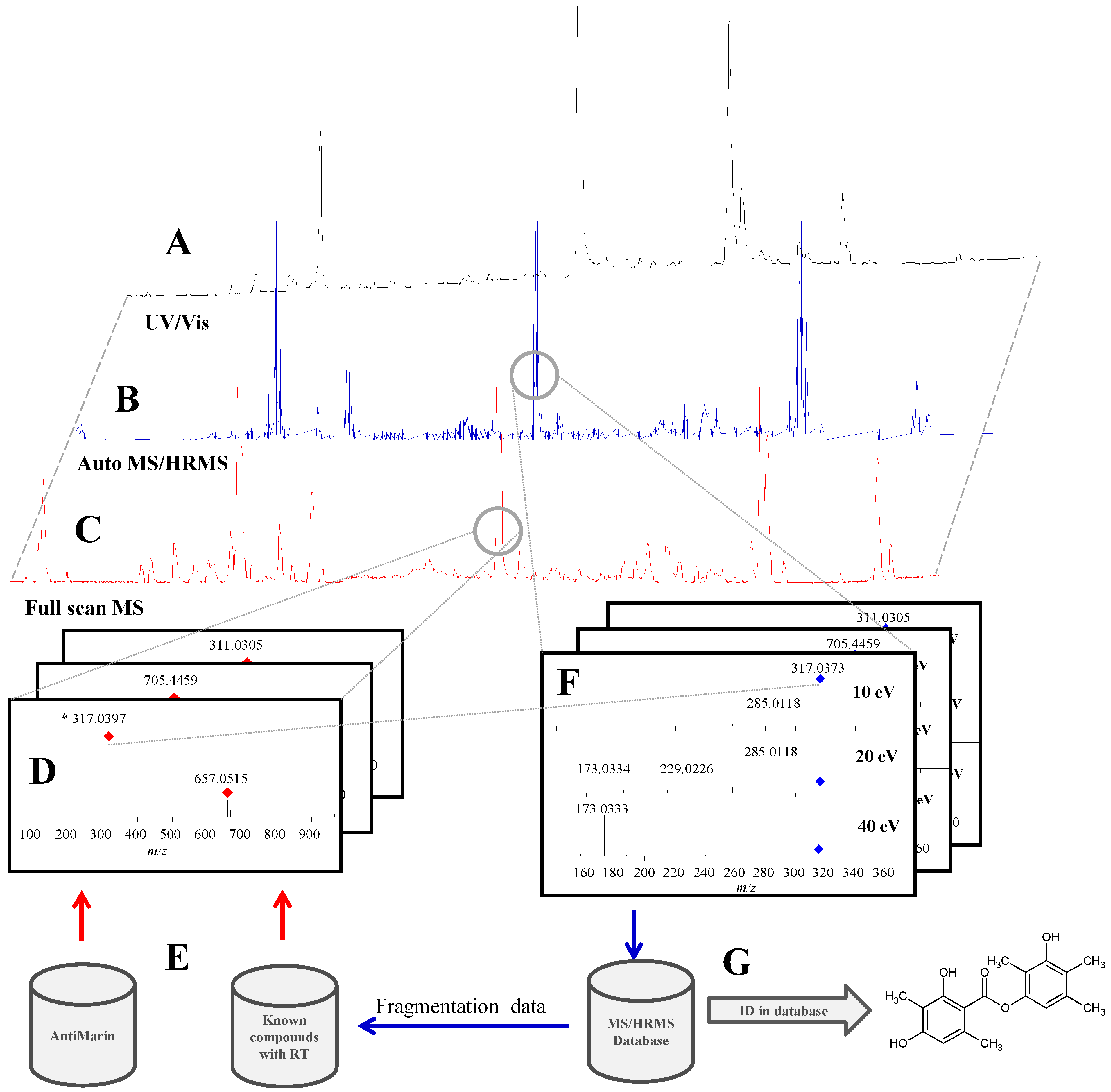
2.1. Data Acquisition and Library Creation
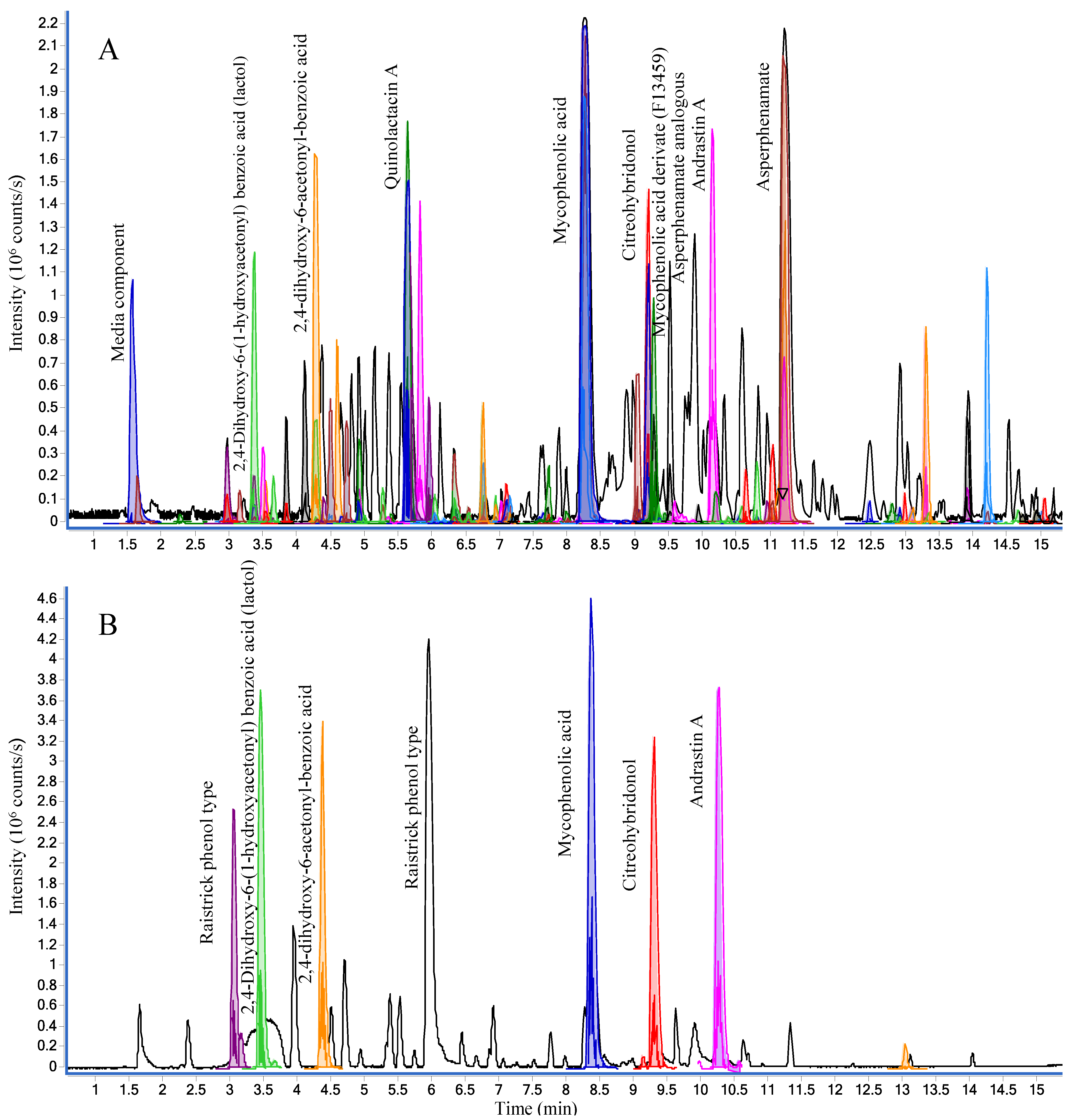
2.1.1. Chromatographic Separation
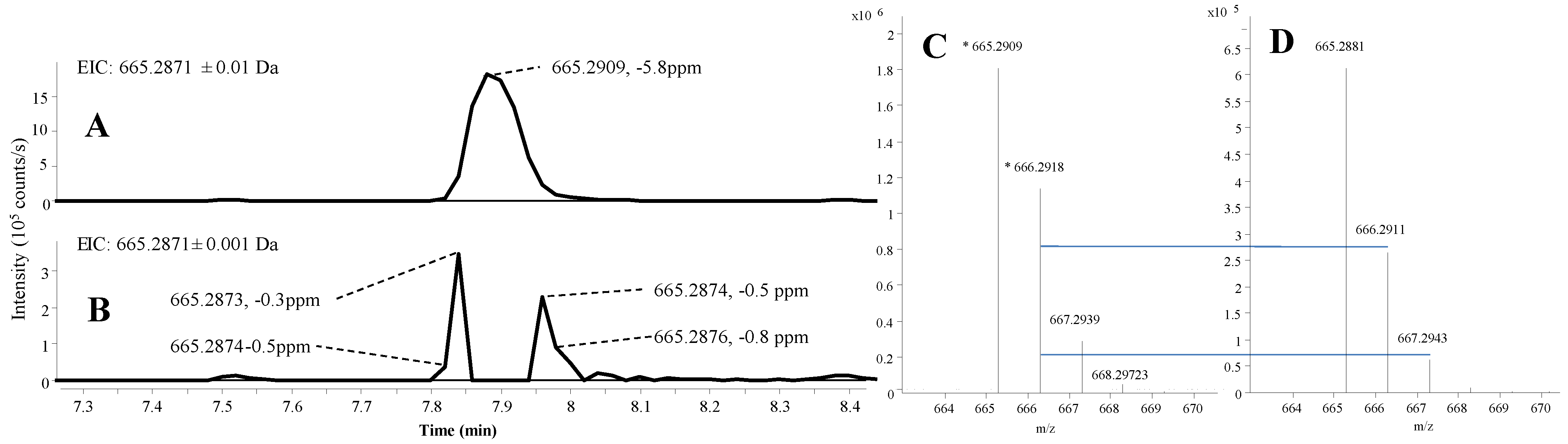
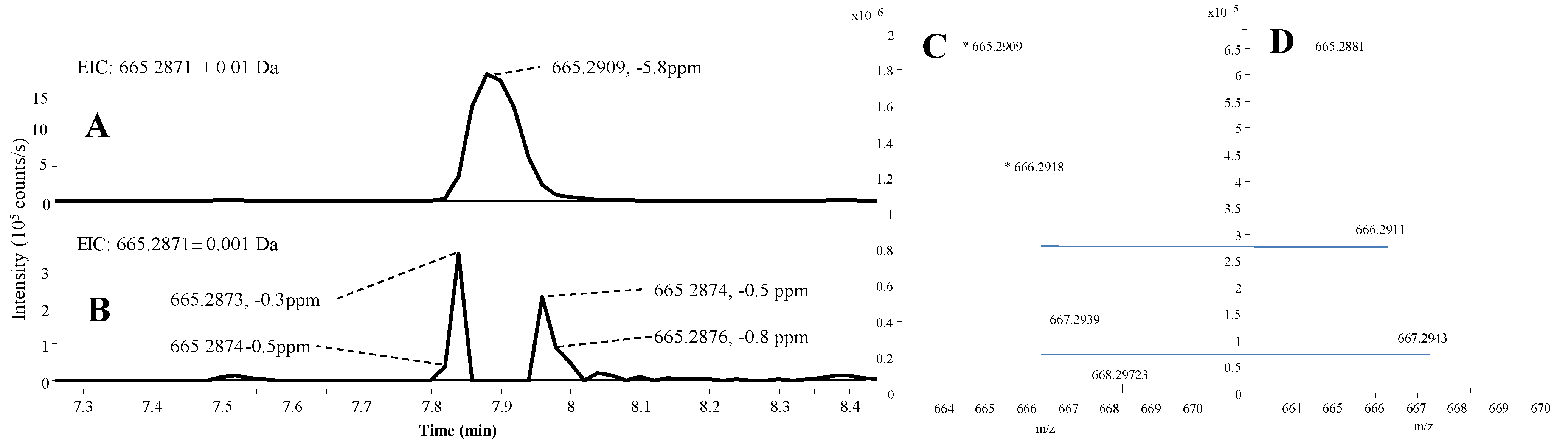
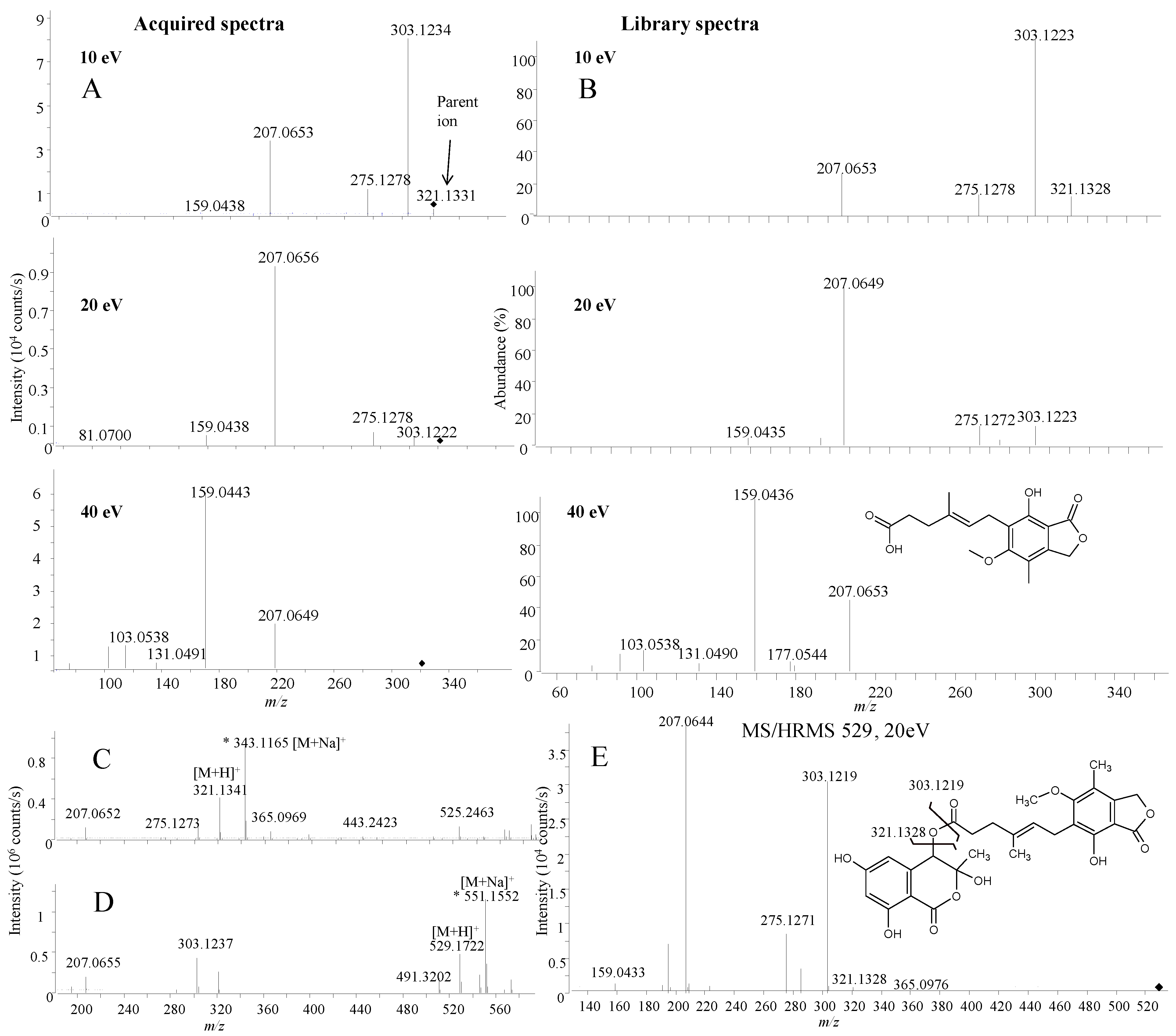
2.1.2. Mass Accuracy and Isotopic Ratio
2.1.3. Precursor Selection
2.1.4. Fragmentation
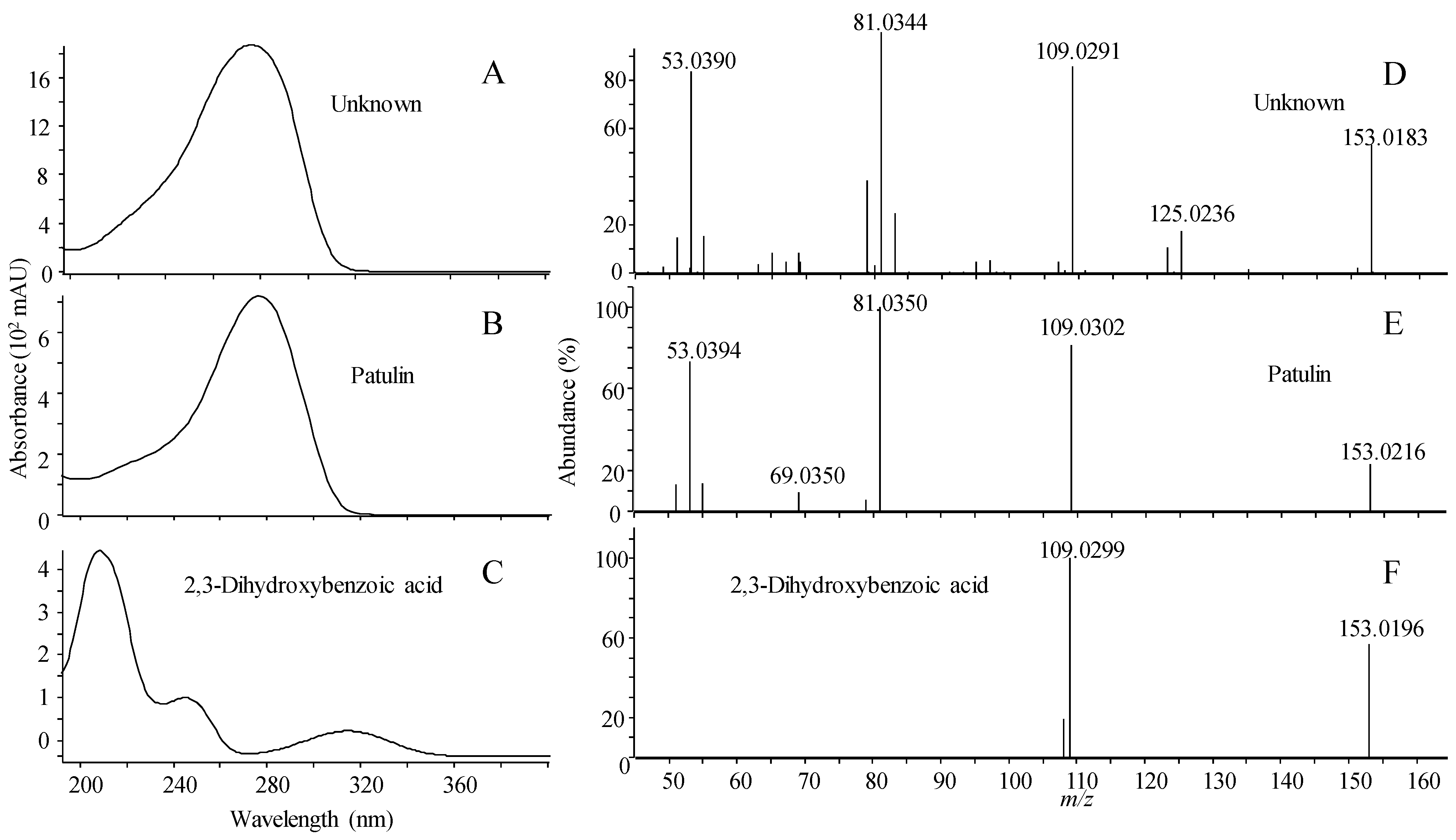
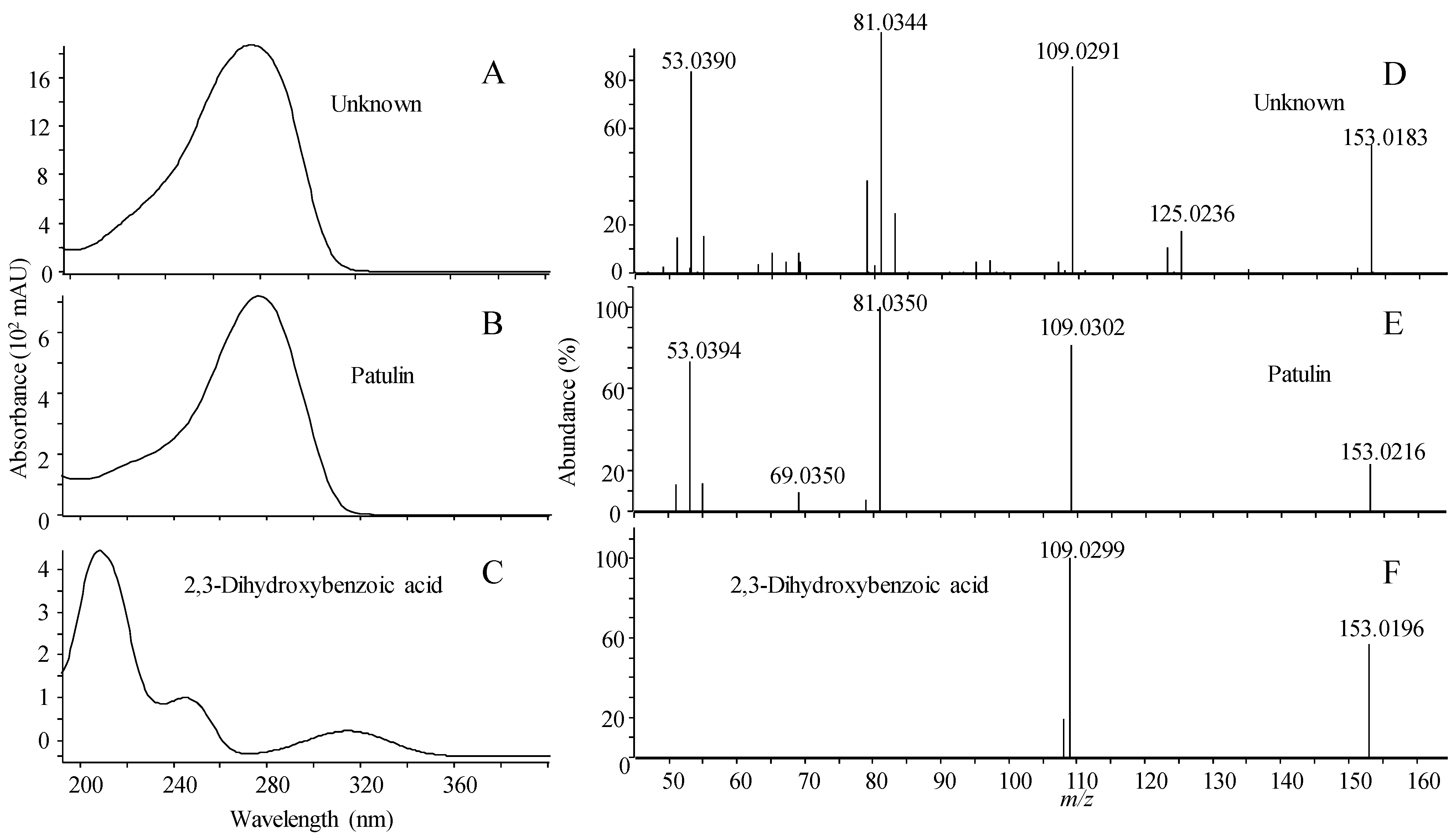
2.1.5. Library Scoring

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | RT (min) | Compound | Forward/Reverse Scoring (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |||
| Patulin | 3.15 | 1 | 100 | 28/50 | 20/62 | 27/65 | 29/90 | 28/87 | 25/52 |
| 2,3-dihydroxybenzoic acid | 3.85 | 2 | 28/32 | 100 | 60/68 | 63/71 | 97/90 | 76/86 | 0/0 |
| 2,4-dihydroxybenzoic acid | 3.74 | 3 | 20/29 | 60/86 | 100 | 86/86 | 55/78 | 88/88 | 6/14 |
| 2,6-dihydroxybenzoic acid | 3.87 | 4 | 21/29 | 63/86 | 86/98 | 100 | 58/79 | 80/92 | 6/14 |
| 3,4-dihydroxybenzoic acid | 2.80 | 5 | 29/33 | 97/97 | 55/61 | 58/64 | 100 | 78/87 | 0/0 |
| 3,5-dihydroxybenzoic acid | 2.63 | 6 | 29/33 | 97/97 | 55/61 | 58/64 | 78/87 | 100 | 0/0 |
| Terreic acid | 3.99 | 7 | 25/44 | 0/0 | 6/39 | 6/38 | 0/0 | 0/0 | 100 |
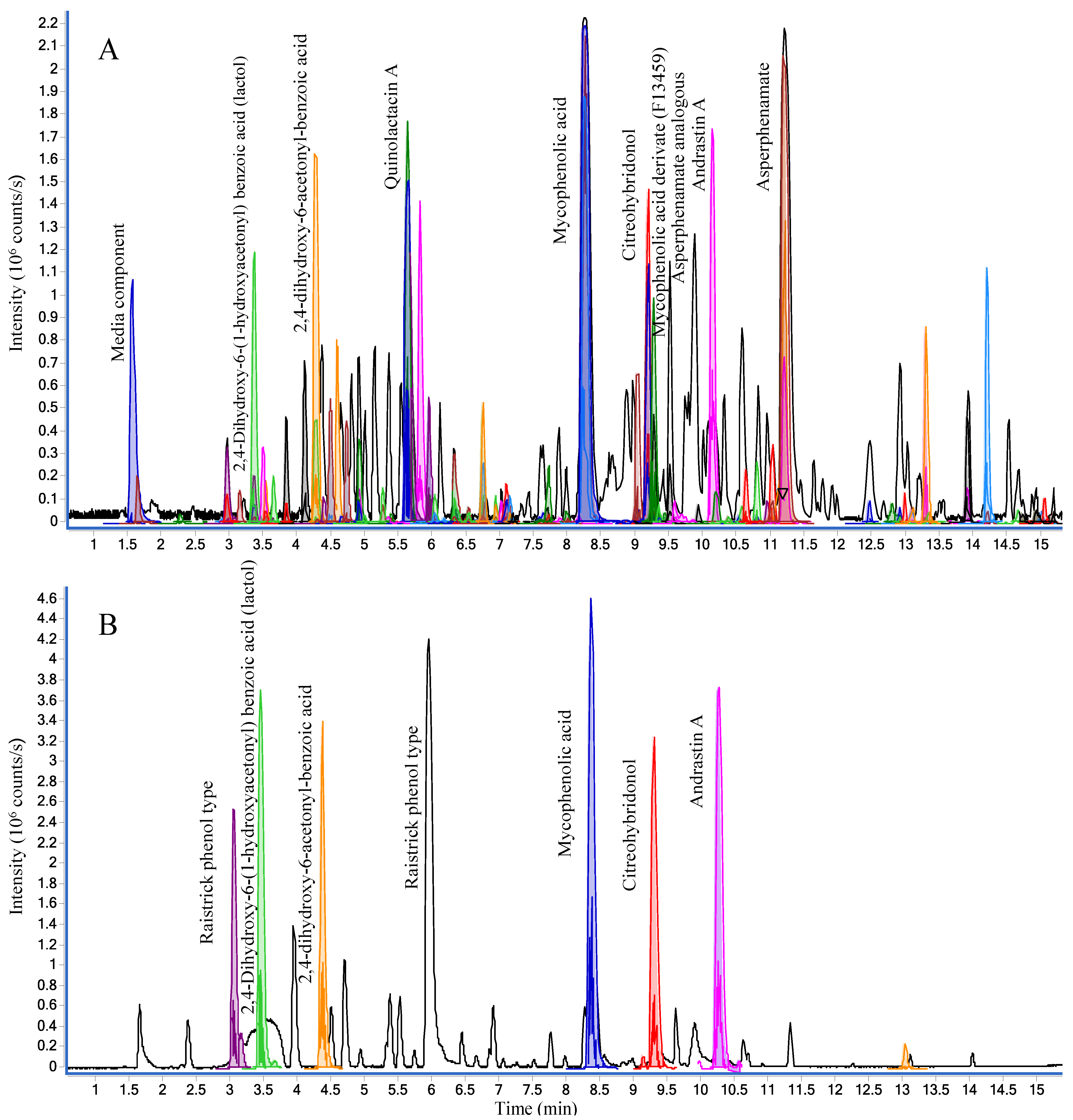
2.2. Dereplication of Marine-Derived Fungi
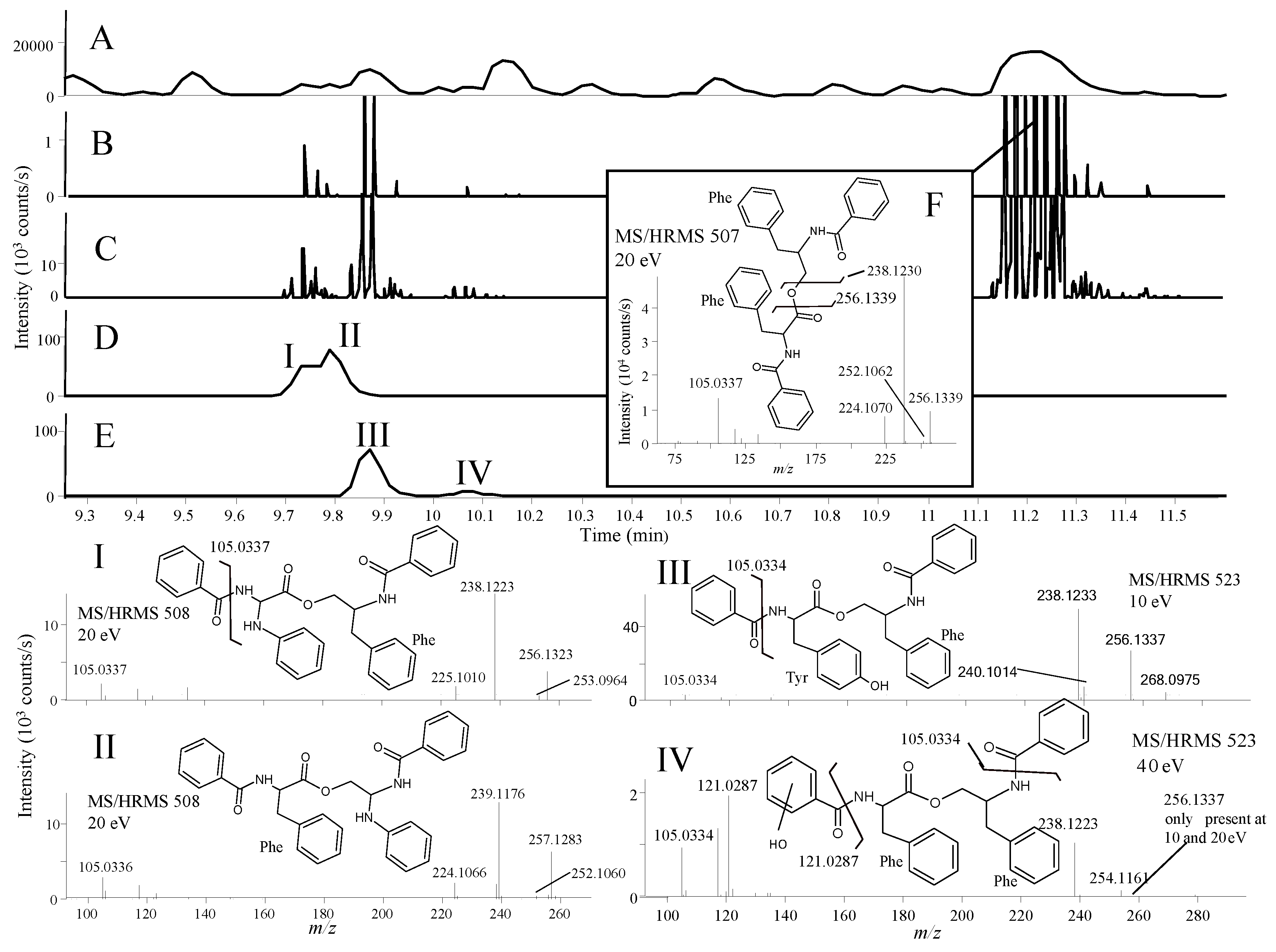
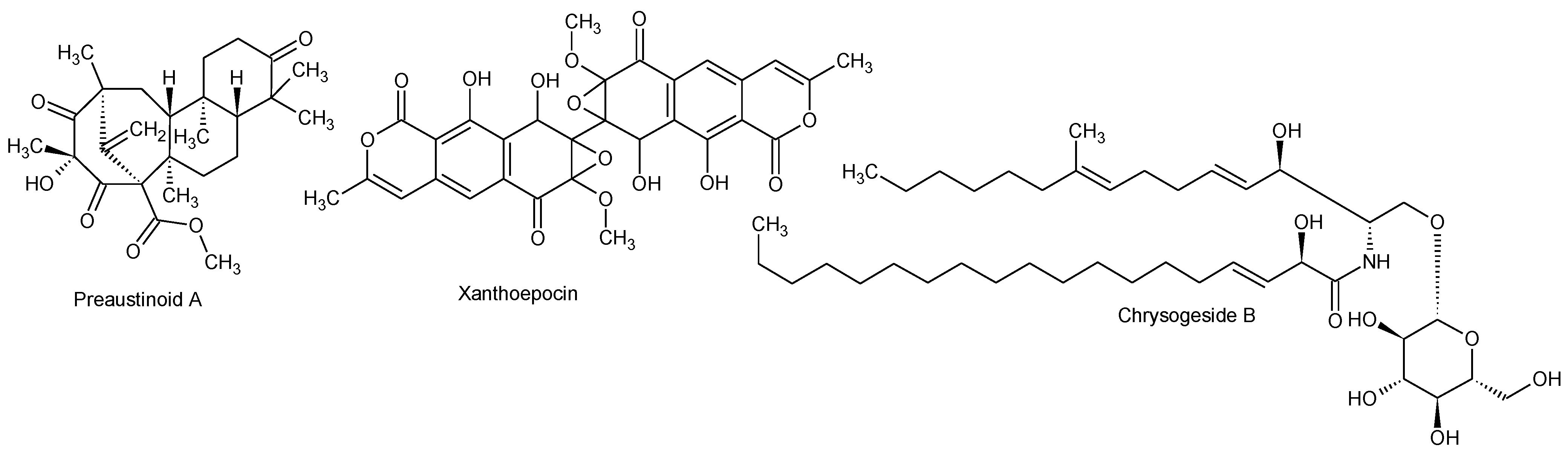
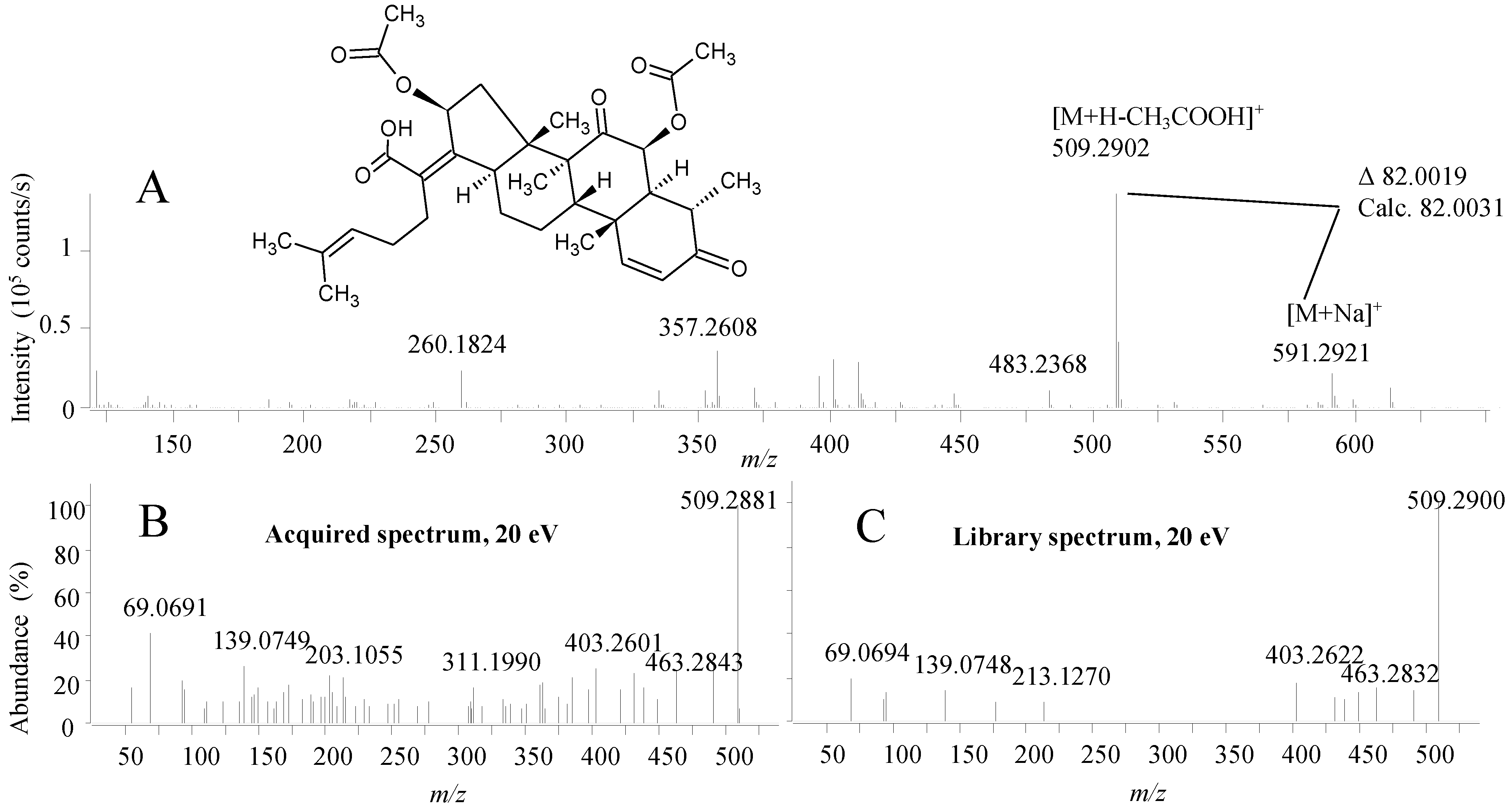
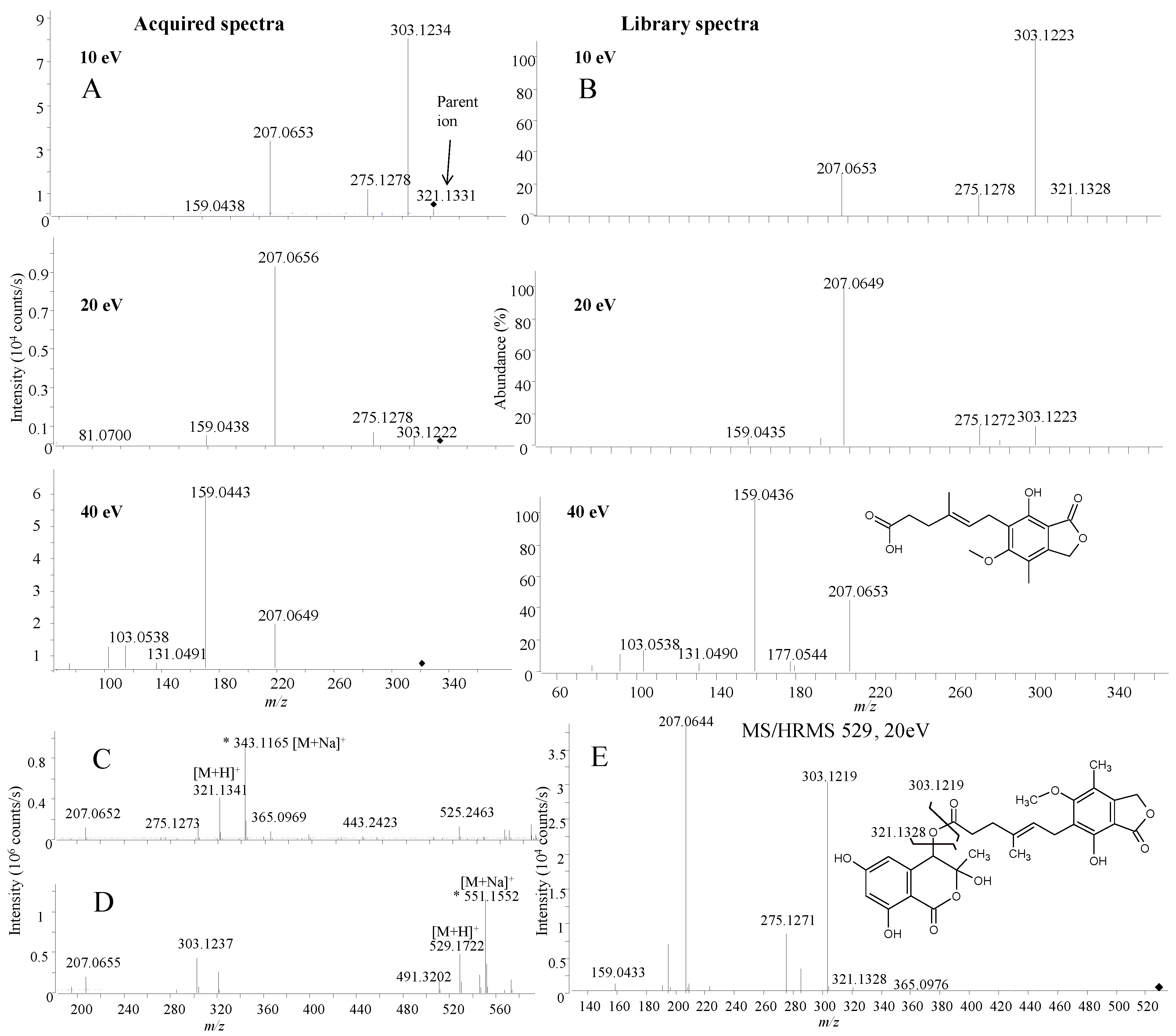
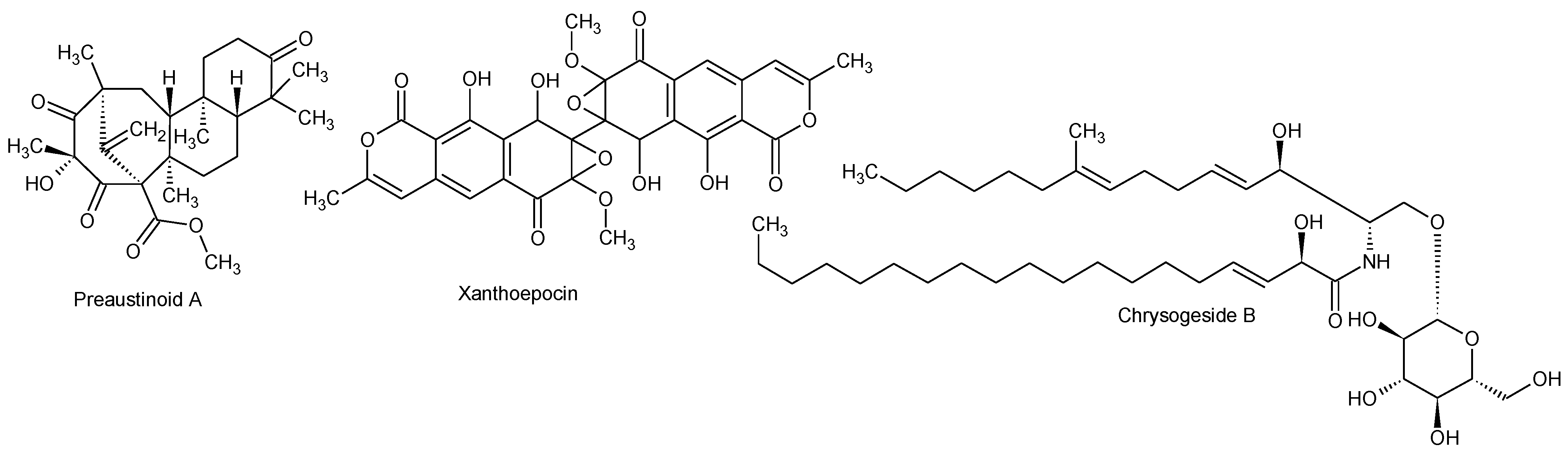
2.2.1. Active Components from a Marine-Derived Penicillium bialowiezense Strain

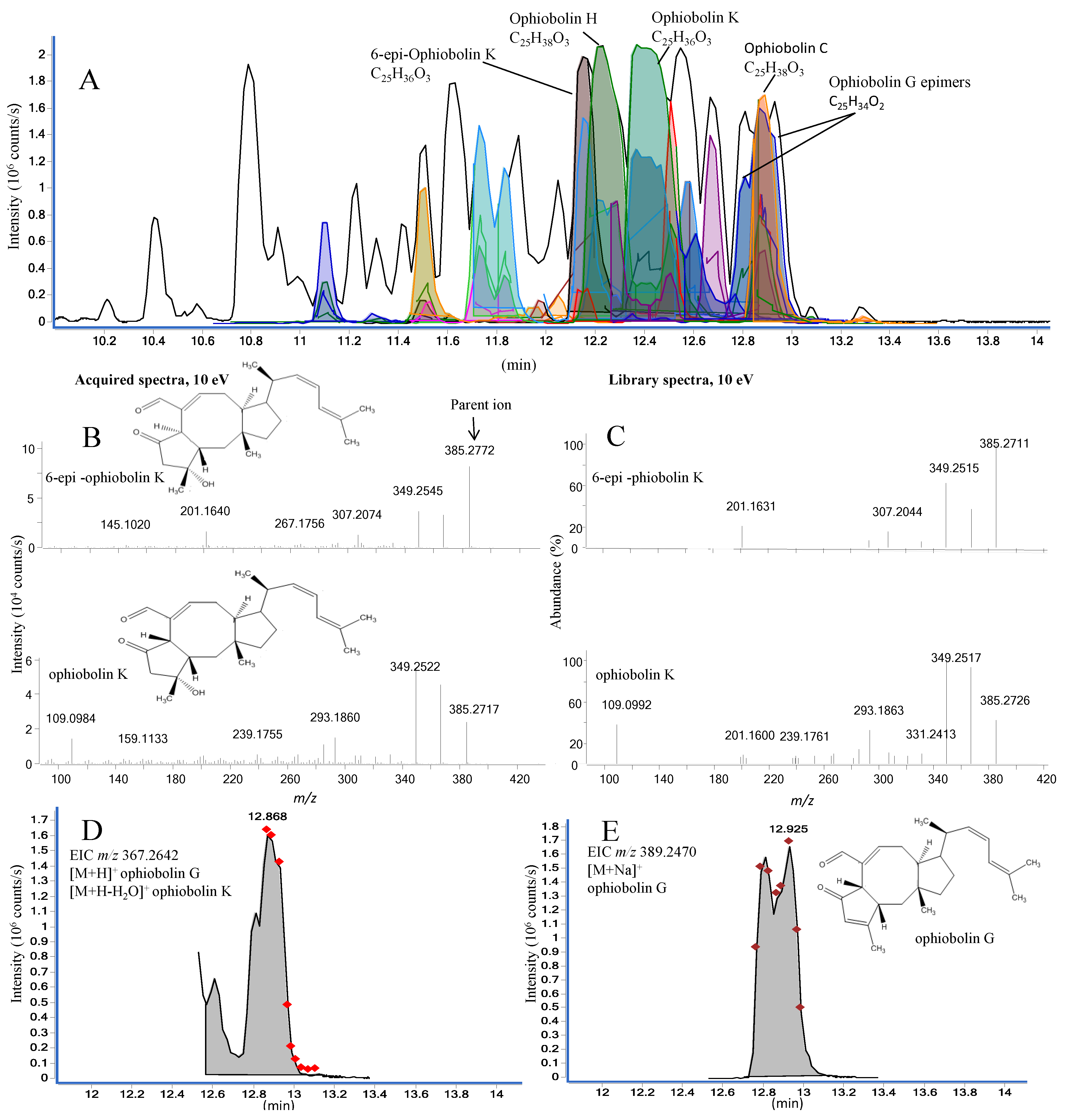
2.2.2. Ophiobolins from a Marine-Derived Aspergillus insuetus

2.2.3. Helvolic Acid as the Anti-Microbial Compound in a Marine-Derived Emericellopsis sp.

3. Experimental Section
3.1. Strains and Cultivation
3.2. Sample Preparation
3.3. Standard Metabolites
3.4. UHPLC-DAD-QTOFMS Analysis
3.5. Library Setup and Auto-MS/MS Data Analysis
3.6. Aggressive Dereplication and Molecular Feature Extraction
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bladt, T.T.; Frisvad, J.C.; Knudsen, P.B.; Larsen, T.O. Anticancer and Antifungal Compounds from Aspergillus, Penicillium and Other Filamentous Fungi. Molecules 2013, 18, 11338–11376. [Google Scholar] [CrossRef]
- El-Elimat, T.; Figueroa, M.; Ehrmann, B.M.; Cech, N.B.; Pearce, C.J.; Oberlies, N.H. High-Resolution MS, MS/MS, and UV Database of Fungal Secondary Metabolites as a Dereplication Protocol for Bioactive Natural Products. J. Nat. Prod. 2013, 76, 1709–1716. [Google Scholar] [CrossRef]
- Mouton, M.; Postma, F.; Wilsenach, J.; Botha, A. Diversity and Characterization of Culturable Fungi from Marine Sediment Collected from St. Helena Bay, South Africa. Microb. Ecol. 2012, 64, 311–319. [Google Scholar] [CrossRef]
- Richards, T.A.; Jones, M.D.; Leonard, G.; Bass, D. Marine Fungi: Their Ecology and Molecular Diversity. Ann. Rev. Mar. Sci. 2012, 4, 495–522. [Google Scholar]
- Debbab, A.; Aly, A.H.; Lin, W.H.; Proksch, P. Bioactive Compounds from Marine Bacteria and Fungi. Microb. Biotechnol. 2010, 3, 544–563. [Google Scholar] [CrossRef]
- Jensen, P.A.; Mincer, T.J.; Williams, P.G.; Fenical, W. Marine actinomycete diversity and natural product discovery. Antonie Van Leeuwenhoek 2005, 87, 43–48. [Google Scholar] [CrossRef]
- Holmstrom, C.; Kjelleberg, S. Marine Pseudoalteromonas species are associated with higher organisms and produce biologically active extracellular agents. FEMS Microbiol. Ecol. 1999, 30, 285–293. [Google Scholar] [CrossRef]
- Bowman, J.P. Bioactive compound synthetic capacity and ecological significance of marine bacterial genus Pseudoalteromonas. Mar. Drugs 2007, 5, 220–241. [Google Scholar] [CrossRef]
- Mansson, M.; Gram, L.; Larsen, T.O. Production of Bioactive Secondary Metabolites by Marine Vibrionaceae. Mar. Drugs 2011, 9, 1440–1468. [Google Scholar] [CrossRef]
- Jones, E. Are there more marine fungi to be described? Bot. Mar. 2011, 54, 343–354. [Google Scholar] [CrossRef]
- Jones, E. Fifty years of marine mycology. Fungal Divers 2011, 50, 73–112. [Google Scholar] [CrossRef]
- Burgaud, G.; Woehlke, S.; Redou, V.; Orsi, W.; Beaudoin, D.; Barbier, G.; Biddle, J.F.; Edgcomb, V.P. Deciphering the presence and activity of fungal communities in marine sediments using a model estuarine system. Aquat. Microb. Ecol. 2013, 70, 45–62. [Google Scholar] [CrossRef]
- Bugni, T.S.; Janso, J.E.; Williamson, R.T.; Feng, X.; Bernan, V.S.; Greenstein, M.; Carter, G.T.; Maiese, W.M.; Ireland, C.M. Dictyosphaeric Acids A and B: New Decalactones from an Undescribed Penicillium sp. Obtained from the Alga Dictyosphaeria versluyii. J. Nat. Prod. 2004, 67, 1396–1399. [Google Scholar] [CrossRef]
- Abdel-Wahab, M.; Gareth Jones, E.B. Three new marine ascomycetes from driftwood in Australia sand dunes. Mycoscience 2000, 41, 379–388. [Google Scholar] [CrossRef]
- Loque, C.P.; Medeiros, A.O.; Pellizzari, F.M.; Oliveira, E.C.; Rosa, C.A.; Rosa, L.H. Fungal community associated with marine macroalgae from Antarctica. Polar Biol. 2010, 33, 641–648. [Google Scholar] [CrossRef]
- Burgaud, G.; Le Calvez, T.; Arzur, D.; Vandenkoornhuyse, P.; Barbier, G. Diversity of culturable marine filamentous fungi from deep-sea hydrothermal vents. Environ. Microbiol. 2009, 11, 1588–1600. [Google Scholar] [CrossRef]
- Khudyakova, Y.V.; Pivkin, M.V.; Kuznetsova, T.A.; Svetashev, V.I. Fungi in sediments of the sea of Japan and their biologically active metabolites. Microbiology 2000, 69, 608–611. [Google Scholar] [CrossRef]
- Alker, A.P.; Smith, G.W.; Kim, K. Characterization of Aspergillus sydowii (Thom et Church), a fungal pathogen of Caribbean sea fan corals. Hydrobiologia 2001, 460, 105–111. [Google Scholar] [CrossRef]
- Roy, K.; Mukhopadhyay, T.; Reddy, G.C.S.; Desikan, K.R.; Ganguli, B.N. Mulundocandin, A New Lipopeptide Antibiotic. 1. Taxonomy, Fermentation, Isolation and Characterization. J. Antibiot. 1987, 40, 275–280. [Google Scholar] [CrossRef]
- Klitgaard, A.; Iversen, A.; Andersen, M.R.; Larsen, T.O.; Frisvad, J.C.; Nielsen, K.F. Aggressive dereplication using UHPLC-DAD-QTOF—Screening extracts for up to 3000 fungal secondary metabolites. Anal. Bioanal. Chem. 2014, 406, 1933–1943. [Google Scholar] [CrossRef] [Green Version]
- Murray, K.K. Glossary of terms for separations coupled to mass spectrometry. J. Chromatogr. A 2010, 1217, 3922–3928. [Google Scholar]
- Schymanski, E.L.; Jeon, J.; Gulde, R.; Fenner, K.; Ruff, M.; Singer, H.P.; Hollender, J. Identifying Small Molecules via High Resolution Mass Spectrometry: Communicating Confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. [Google Scholar]
- Hu, Q.; Noll, R.J.; Li, H.; Makarov, A.; Hardmac, M.; Cooksa, R.G. The Orbitrap: A new mass spectrometer. J. Mass Spectrom. 2005, 40, 430–443. [Google Scholar] [CrossRef]
- Lehner, S.M.; Neumann, N.K.N.; Sulyok, M.; Lemmens, M.; Krska, R.; Schuhmacher, R. Evaluation of LC-high-resolution FT-Orbitrap MS for the quantification of selected mycotoxins and the simultaneous screening of fungal metabolites in food. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2011, 28, 1457–1468. [Google Scholar] [CrossRef]
- Konishi, Y.; Kiyota, T.; Draghici, C.; Gao, J.M.; Yeboah, F.; Acoca, S.; Jarussophon, S.; Purisima, E. Molecular formula analysis by an MS/MS/MS technique to expedite dereplication of natural products. Anal. Chem. 2007, 79, 1187–1197. [Google Scholar] [CrossRef]
- Broecker, S.; Herre, S.; Wust, B.; Zweigenbaum, J.; Pragst, F. Development and practical application of a library of CID accurate mass spectra of more than 2500 toxic compounds for systematic toxicological analysis by LC-QTOF-MS with data-dependent acquisition. Anal. Bioanal. Chem. 2011, 400, 101–117. [Google Scholar] [CrossRef]
- Hill, A.W.; Mortishire-Smith, R.J. Automated assignment of high-resolution collisionally activated dissociation mass spectra using a systematic bond disconnection approach. Rapid Commun. Mass Spectrom. 2005, 19, 3111–3118. [Google Scholar] [CrossRef]
- Hufsky, F.; Scheubert, K.; Bocker, S. Computational mass spectrometry for small-molecule fragmentation. TrAC Trends Anal. Chem. 2014, 53, 41–48. [Google Scholar] [CrossRef]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef]
- Smith, C.A.; O’Maille, G.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. METLIN: A metabolite mass spectral database. Ther. Drug Monit. 2005, 27, 747–751. [Google Scholar] [CrossRef]
- Global Natural Products Research and Mass spectrometry. Available online: http://gnps.ucsd.edu/ (accessed on 17 June 2014).
- Champarnaud, E.; Hopley, C. Evaluation of the comparability of spectra generated using a tuning point protocol on twelve electrospray ionisation tandem-in-space mass spectrometers. Rapid Commun. Mass Spectrom. 2011, 25, 1001–1007. [Google Scholar] [CrossRef]
- Yates, J.R.; Cociorva, D.; Liao, L.J.; Zabrouskov, V. Performance of a linear ion trap-orbitrap hybrid for peptide analysis. Anal. Chem. 2006, 78, 493–500. [Google Scholar] [CrossRef]
- Kind, T.; Liu, K.H.; Lee, D.Y.; DeFelice, B.; Meissen, J.K.; Fiehn, O. LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat. Methods 2013, 10, 755–758. [Google Scholar] [CrossRef]
- Fredenhagen, A.; Derrien, C.; Gassmann, E. An MS/MS Library on an Ion-Trap Instrument for Efficient Dereplication of Natural Products. Different Fragmentation Patterns for [M + H]+ and [M + Na]+ Ions. J. Nat. Prod. 2005, 68, 385–391. [Google Scholar] [CrossRef]
- Yang, J.Y.; Sanchez, L.M.; Rath, C.M.; Liu, X.; Boudreau, P.D.; Bruns, N.; Glukhov, E.; Wodtke, A.; de Felicio, R.; Fenner, A.; et al. Molecular Networking as a Dereplication Strategy. J. Nat. Prod. 2013, 76, 1686–1699. [Google Scholar] [CrossRef]
- Watrous, J.; Roach, P.; Alexandrov, T.; Heath, B.S.; Yang, J.Y.; Kersten, R.D.; van der Voort, M.; Pogliano, K.; Gross, H.; Raaijmakers, J.M.; et al. Mass spectral molecular networking of living microbial colonies. Proc. Natl. Acad. Sci. USA 2012, 109, E1743–E1752. [Google Scholar] [CrossRef]
- PharmaSea. Available online: http://www.pharma-sea.eu/ (accessed on 13 June 2014).
- Hopfgartner, G.; Vilbois, F. The impact of accurate mass measurements using quadrupole/time-of-flight mass spectrometry on the characterisation and screening of drug metabolites. Analusis 2000, 28, 906–914. [Google Scholar] [CrossRef]
- Colombo, M.; Sirtori, F.R.; Rizzo, V. A fully automated method for accurate mass determination using high-performance liquid chromatography with a quadrupole/orthogonal acceleration time-of-flight mass spectrometer. Rapid Commun. Mass Spectrom. 2004, 18, 511–517. [Google Scholar] [CrossRef]
- Kind, T.; Fiehn, O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinform. 2007, 8, 105. [Google Scholar] [CrossRef]
- Oberacher, H.; Pavlic, M.; Libiseller, K.; Schubert, B.; Sulyok, M.; Schuhmacher, R.; Csaszar, E.; Kofeler, H.C. On the inter-instrument and inter-laboratory transferability of a tandem mass spectral reference library: 1. Results of an Austrian multicenter study. J. Mass Spectrom. 2009, 44, 485–493. [Google Scholar] [CrossRef]
- Nielsen, K.F.; Månsson, M.; Rank, C.; Frisvad, J.C.; Larsen, T.O. Dereplication of microbial natural products by LC-DAD-TOFMS. J. Nat. Prod. 2011, 74, 2338–2348. [Google Scholar] [CrossRef]
- DTU Mycotoxin-Fungal Secondary Metabolite MS/HRMS library. Available online: http://www.bio.dtu.dk/english/Research/Platforms/Metabolom/MSMSLib (accessed on 13 June 2014).
- Liu, B.-H.; Yu, F.; Wu, T.-S.; Li, W. Evaluation of genotoxic risk and oxidative DNA damage in mammalian cells exposed to mycotoxins, patulin and citrinin. Toxicol. Appl. Pharmacol. 2003, 191, 255–263. [Google Scholar] [CrossRef]
- Vansteelandt, M.; Kerzaon, I.; Blanchet, E.; Tankoua, O.F.; du Pont, T.R.; Joubert, Y.; Monteau, F.; Le Bizec, B.; Frisvad, J.C.; Pouchus, Y.F.; et al. Patulin and secondary metabolite production by marine-derived Penicillium strains. Fungal Biol. 2012, 116, 954–961. [Google Scholar] [CrossRef]
- Rasmussen, T.B.; Skindersoe, M.E.; Bjarnsholt, T.; Phipps, R.K.; Christensen, K.B.; Jensen, P.O.; Andersen, J.B.; Koch, B.; Larsen, T.O.; Hentzer, M.; et al. Identity and effects of quorum-sensing inhibitors produced by Penicillium species. Microbiology 2005, 151, 1325–1340. [Google Scholar] [CrossRef]
- Gram, L.; Melchiorsen, J.; Bruhn, J.B. Antibacterial Activity of Marine Culturable Bacteria Collected from a Global Sampling of Ocean Surface Waters and Surface Swabs of Marine Organisms. Mar. Biotechnol. 2010, 12, 439–451. [Google Scholar] [CrossRef]
- Lind, K.F.; Hansen, E.; Osterud, B.; Eilertsen, K.E.; Bayer, A.; Engqvist, M.; Leszczak, K.; Jorgensen, T.O.; Andersen, J.H. Antioxidant and Anti-Inflammatory Activities of Barettin. Mar. Drugs 2013, 11, 2655–2666. [Google Scholar] [CrossRef]
- Bohni, N.; Lorena Cordero-Maldonado, M.; Maes, J.; Siverio-Mota, D.; Marcourt, L.; Munck, S.; Kamuhabwa, A.R.; Moshi, M.J.; Esguerra, C.V.; de Witte, P.A.; et al. Integration of Microfractionation, qNMR and Zebrafish Screening for the In Vivo Bioassay-Guided Isolation and Quantitative Bioactivity Analysis of Natural Products. PLoS One 2013, 8, e64006. [Google Scholar] [Green Version]
- Buenafe, O.E.; Orellana-Paucar, A.; Maes, J.; Huang, H.; Ying, X.; de Borggraeve, W.; Crawford, A.D.; Luyten, W.; Esguerra, C.V.; de Witte, P. Tanshinone IIA Exhibits Anticonvulsant Activity in Zebrafish and Mouse Seizure Models. ACS Chem. Neurosci. 2013, 4, 1479–1487. [Google Scholar] [CrossRef]
- Frisvad, J.C.; Samson, R.A. Polyphasic taxonomy of Penicillium subgenus Penicillium. A guide to identification of the food and air-borne terverticillate Penicillia and their mycotoxins. Stud. Mycol. 2004, 49, 1–173. [Google Scholar]
- Overy, D.P.; Frisvad, J.C. Mycotoxin production and postharvest storage rot of ginger (Zingiber officinale) by Penicillium brevicompactum. J. Food Prot. 2005, 68, 607–609. [Google Scholar]
- Andersen, B. Consistent production of phenolic compounds by Penicillium brevicompactum from chemotaxonomic characterization. Antonie Van Leeuwenhoek 1991, 60, 115–123. [Google Scholar] [CrossRef]
- Frisvad, J.C.; Smedsgaard, J.; Larsen, T.O.; Samson, R.A. Mycotoxins, drugs and other extrolites produced by species in Penicillium subgenus Penicillium. Stud. Mycol. 2004, 49, 201–241. [Google Scholar]
- Bentley, R. Mycophenolic acid: A one hundred year Odyssey from antibiotic to immunosuppressant. Chem. Rev. 2000, 100, 3801–3825. [Google Scholar] [CrossRef]
- Williams, R.H.; Lively, D.H.; Delong, D.C.; Cline, J.C.; Sweeney, M.J.; Poore, G.A.; Larsen, S.H. Mycophenolic Acid—Antiviral and Antitumor Properties. J. Antibiot. 1968, 21, 463–464. [Google Scholar] [CrossRef]
- Kern, I.; Xu, R.; Julien, S.; Suter, D.; Preynat-Seauve, O.; Baquie, M.; Poncet, A.; Combescure, C.; Stoppini, L.; Thriel, C.V.; et al. Embryonic Stem Cell-Based Screen for Small Molecules: Cluster Analysis Reveals Four Response Patterns in Developing Neural Cells. Curr. Med. Chem. 2013, 20, 710–723. [Google Scholar] [CrossRef]
- Koshino, H.; Muroi, M.; Tajika, T.; Kimura, Y.; Takatsuki, A. F13459, a new derivative of mycophenolic acid: II. Physico-chemical properties and structural elucidation. J. Antibiot. 2001, 54, 494–500. [Google Scholar] [CrossRef]
- Muroi, M.; Sano, K.; Okada, G.; Koshino, H.; Oku, T.; Takatsuki, A. F13459, a new derivative of mycophenolic acid: I. Taxonomy, isolation, and biological properties. J. Antibiot. 2001, 54, 489–493. [Google Scholar] [CrossRef]
- Yuan, L.; Li, Y.; Zou, C.; Wang, C.; Gao, J.; Miao, C.; Ma, E.; Sun, T. Synthesis and in vitro antitumor activity of asperphenamate derivatives as autophagy inducer. Bioorg. Med. Lett. 2012, 22, 2216–2220. [Google Scholar] [CrossRef]
- Hu, Y.M.; Yu, Z.L.; Yang, Z.J.; Zhu, G.Y.; Fong, W.F. Comprehensive chemical analysis of Venenum Bufonis by using liquid chromatography/electrospray ionization tandem mass spectrometry. J. Pharm. Biomed. Anal. 2011, 56, 210–220. [Google Scholar] [CrossRef]
- Peng, X.; Wang, Y.; Sun, K.; Liu, P.; Yin, X.; Zhu, W. Cerebrosides and 2-Pyridone Alkaloids from the Halotolerant Fungus Penicillium chrysogenum Grown in a Hypersaline Medium. J. Nat. Prod. 2011, 74, 1298–1302. [Google Scholar] [CrossRef]
- Ando, T.; Igarashi, Y.; Kuwamori, Y.; Takagi, K.; Ando, T.; Fudou, R.; Furumai, T.; Oki, T. Xanthoepocin, a new antibiotic fom Penicillium simplicissimum IFO5762. J. Antibiot. 2000, 53, 928–933. [Google Scholar] [CrossRef]
- Bladt, T.T.; Duerr, C.; Knudsen, P.B.; Kildgaard, S.; Frisvad, J.C.; Gotfredsen, C.H.; Seiffert, M.; Larsen, T.O. Bio-Activity and Dereplication-Based Discovery of Ophiobolins and Other Fungal Secondary Metabolites Targeting Leukemia Cells. Molecules 2013, 18, 14629–14650. [Google Scholar] [CrossRef]
- Zuccaro, A.; Summerbell, R.C.; Gams, W.; Schroers, H.J.; Mitchell, J.I. A new Acremonium species associated with Fucus spp., and its affinity with a phylogenetically distinct marine Emericellopsis clade. Stud. Mycol. 2004, 50, 283–297. [Google Scholar]
- Graca, A.P.; Bondoso, J.; Gaspar, H.; Xavier, J.R.; Monteiro, M.C.; de la Cruz, M.; Oves-Costales, D.; Vicente, F.; Lage, O.M. Antimicrobial Activity of Heterotrophic Bacterial Communities from the Marine Sponge Erylus discophorus (Astrophorida, Geodiidae). PLoS One 2013, 8, e78992. [Google Scholar] [CrossRef]
- Ratnaweera, P.B.; Williams, D.E.; de Silva, E.D.; Wijesundera, R.L.C.; Dalisay, D.S.; Andersen, R.J. Helvolic acid, an antibacterial nortriterpenoid from a fungal endophyte, Xylaria sp. of orchid Anoectochilus setaceus endemic to Sri Lanka. Mycology 2014, 5, 23–28. [Google Scholar] [CrossRef]
- Qin, L.; Li, B.; Guan, J.; Zhang, G. In vitro synergistic antibacterial activities of helvolic acid on multi-drug resistant Staphylococcus aureus. Nat. Prod. Res. 2009, 23, 309–318. [Google Scholar] [CrossRef]
- Pinheiro, A.; Dethoup, T.; Bessa, J.; Silva, A.M.; Kijjoa, A. A new bicyclic sesquiterpene from the marine sponge associated fungus Emericellopsis minima. Phytochem. Lett. 2012, 5, 68–70. [Google Scholar] [CrossRef]
- Bills, G.F.; Platas, G.; Gams, W. Conspecificity of the cerulenin and helvolic acid producing ‘Cephalosporium caerulens’, and the hypocrealean fungus Sarocladium oryzae. Mycol. Res. 2004, 108, 1291–1300. [Google Scholar] [CrossRef]
- Thirumalachar, M.J. Antiamoebin Anti Parasit A New Anti Protozoal Anti Helminthic Antibiotic I Production and Biological Studies Emericellopsis-Poonensis Emericellopsis-Synnematicola Cephalosporium-Pimprina. Hindustan Antibiot. Bull. 1968, 10, 287–289. [Google Scholar]
- Stoppacher, N.; Neumann, N.K.N.; Burgstaller, L.; Zeilinger, S.; Degenkolb, T.; Bruckner, H.; Schuhmacher, R. The Comprehensive Peptaibiotics Database. Chem. Biodiv. 2013, 10, 734–743. [Google Scholar] [CrossRef]
- Monteiro, M.C.; de la Cruz, M.; Cantizani, J.; Moreno, C.; Tormo, J.R.; Mellado, E.; de Lucas, J.R.; Asensio, F.; Valiante, V.; Brakhage, A.A.; et al. A New Approach to Drug Discovery: High-Throughput Screening of Microbial Natural Extracts against Aspergillus fumigatus Using Resazurin. J. Biomol. Screen. 2012, 17, 542–549. [Google Scholar] [CrossRef]
- De la Cruz, M.; Martin, J.; Gonzalez-Menendez, V.; Perez-Victoria, I.; Moreno, C.; Tormo, J.R.; El Aouad, N.; Guarro, J.; Vicente, F.; Reyes, F.; et al. Chemical and Physical Modulation of Antibiotic Activity in Emericella Species. Chem. Biodiv. 2012, 9, 1095–1113. [Google Scholar] [CrossRef]
- Frisvad, J.C.; Thrane, U. Standardised High-Performance Liquid Chromatography of 182 mycotoxins and other fungal metabolites based on alkylphenone retention indices and UV-VIS spectra (Diode Array Detection). J. Chromatogr. 1987, 404, 195–214. [Google Scholar] [CrossRef]
- Zhu, Z.J.; Schultz, A.W.; Wang, J.H.; Johnson, C.H.; Yannone, S.M.; Patti, G.J.; Siuzdak, G. Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the METLIN database. Nat. Protoc. 2013, 8, 451–460. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kildgaard, S.; Mansson, M.; Dosen, I.; Klitgaard, A.; Frisvad, J.C.; Larsen, T.O.; Nielsen, K.F. Accurate Dereplication of Bioactive Secondary Metabolites from Marine-Derived Fungi by UHPLC-DAD-QTOFMS and a MS/HRMS Library. Mar. Drugs 2014, 12, 3681-3705. https://doi.org/10.3390/md12063681
Kildgaard S, Mansson M, Dosen I, Klitgaard A, Frisvad JC, Larsen TO, Nielsen KF. Accurate Dereplication of Bioactive Secondary Metabolites from Marine-Derived Fungi by UHPLC-DAD-QTOFMS and a MS/HRMS Library. Marine Drugs. 2014; 12(6):3681-3705. https://doi.org/10.3390/md12063681
Chicago/Turabian StyleKildgaard, Sara, Maria Mansson, Ina Dosen, Andreas Klitgaard, Jens C. Frisvad, Thomas O. Larsen, and Kristian F. Nielsen. 2014. "Accurate Dereplication of Bioactive Secondary Metabolites from Marine-Derived Fungi by UHPLC-DAD-QTOFMS and a MS/HRMS Library" Marine Drugs 12, no. 6: 3681-3705. https://doi.org/10.3390/md12063681