Emerging Strategies and Integrated Systems Microbiology Technologies for Biodiscovery of Marine Bioactive Compounds


Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Natural Product or Derivative | Collected Source Organism | Activity | Clinical Status |
|---|---|---|---|---|
| Salinosporamide A (Marizomib) | Natural product | Bacterium | Antitumor | Phase I |
| Plitidepsin (Aplidin) | Natural product | Tunicate | Antitumor | Phase III |
| Bryostatin 1 | Natural product | Bacterium | Antitumor/Anti-Alzheimer | Phase II |
| Cytarabine | Derivative | Sponge | Antitumor | Approved |
| Vidarabine | Derivative | Sponge | Antiviral | Approved |
| Eribulin Mesylate | Derivative | Sponge | Antitumor | Approved |
| Trabectidin (ET-743) | Natural product | Tunicate | Antitumor | EU approved |
| Enzyme | Source | Screening Technologies | Reference |
|---|---|---|---|
| Laccase | Metagenome | Sequence-based | [15] |
| Esterase | Metagenome | Function-based | [16] |
| Fumarase | Metagenome | Sequence-based | [17] |
| α-amilase | Bacillus subtilis S8–18 | Function-based | [18] |
| Glycoside hydrolase | Metagenome | Function-based | [19] |
| Baeyer-Villiger monooxigenase | Stenotrophomonas maltophila PML168 | Function-based | [20] |
| Cellulase | Marinimicrobium sp. | Function-based | [21] |
| Dehalogenase | Pseudomonas stutzeri DEH130 | Function-based | [22] |
| Aldehyde reductase | Oceanospirillum sp. | Genome-based | [23] |
| Lipase | Bacillus smithii BTMS11 | Function-based | [24] |
| Dehalogenase | Psycromonas ingrahamii | Genome-based | [25] |
| Xylanase | Streptomyces viridochromogenes | Function-based | [26] |
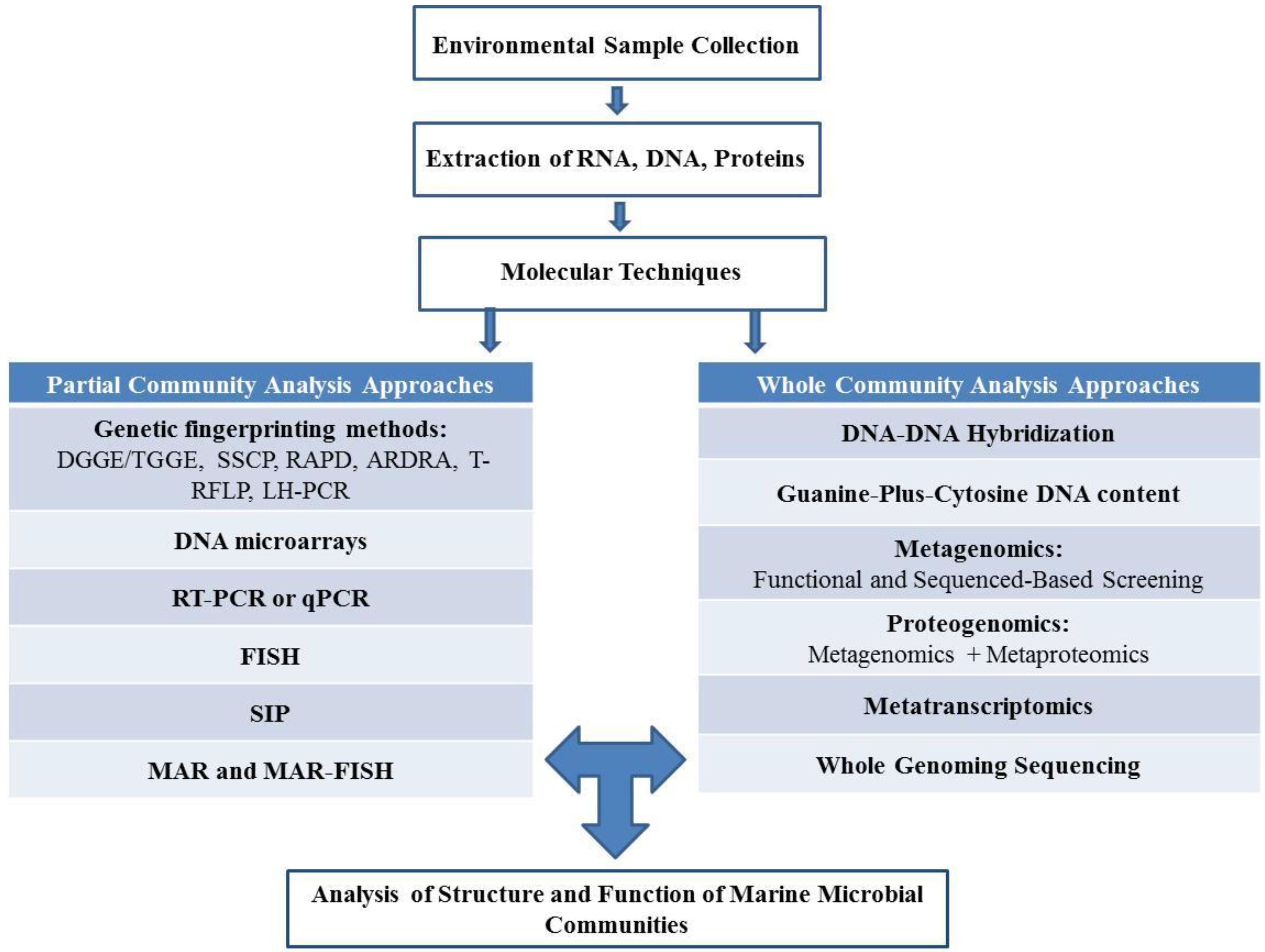
2. Culture Dependent and Independent Isolation of Marine Microorganisms
2.1. Culture Dependent Analysis of Structure and Function of Marine Microbial Communities
2.2. Culture Independent Analysis of Structure and Function of Marine Microbial Communities
2.2.1. Partial Community Analysis Approaches
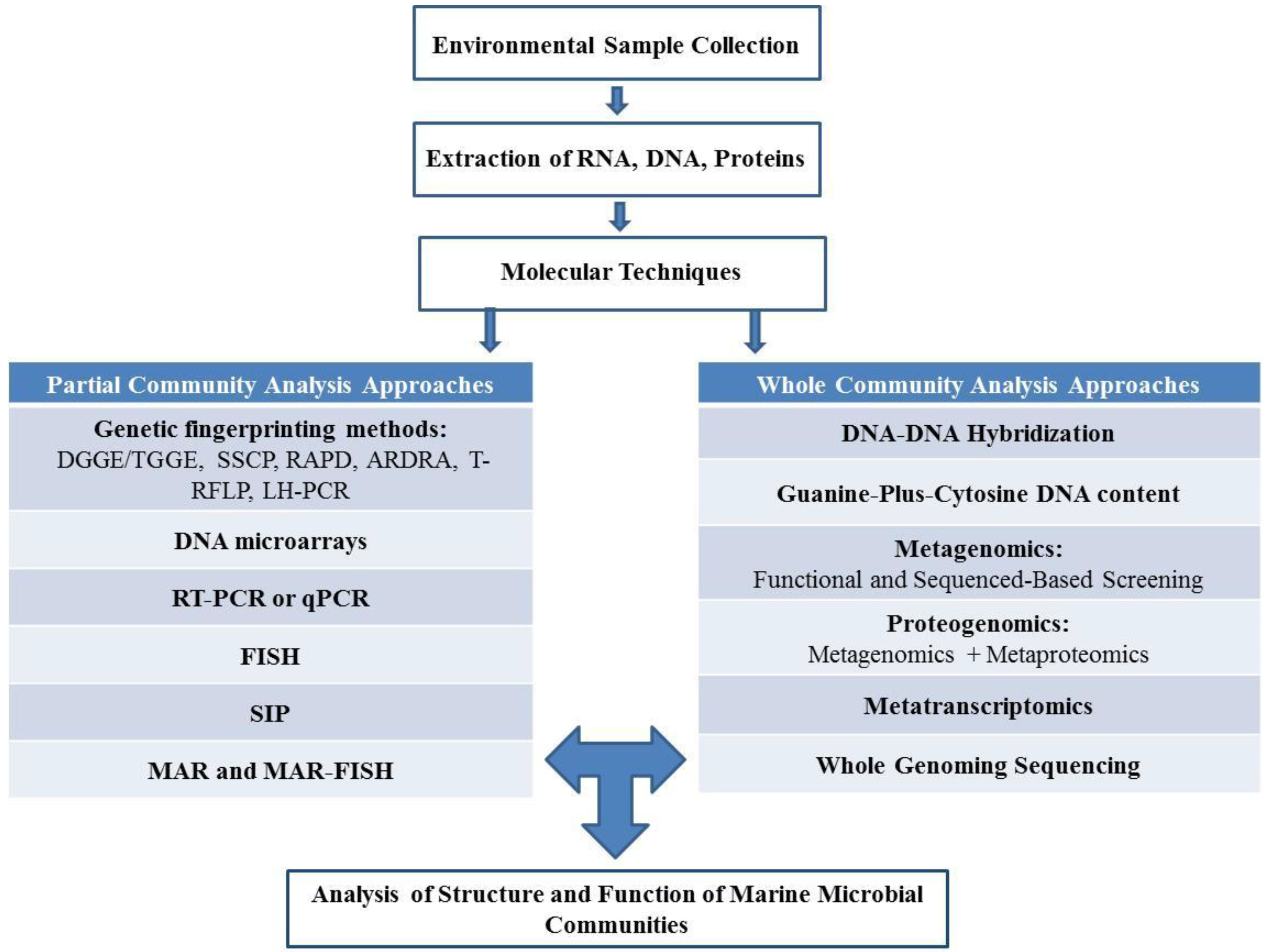
2.2.2. Whole Community Analysis Approaches
2.2.3. Emerging “Omics” Technologies to Analyze the Structure and Function of Marine Microbial Communities
2.3. Culture Dependent and Independent Analysis. Combined or Separately?
3. Strategies for Marine Natural Compounds Discovery from Marine Microbial Communities
3.1. Conventional Screening Methods: Bioactivity-Guided Screening
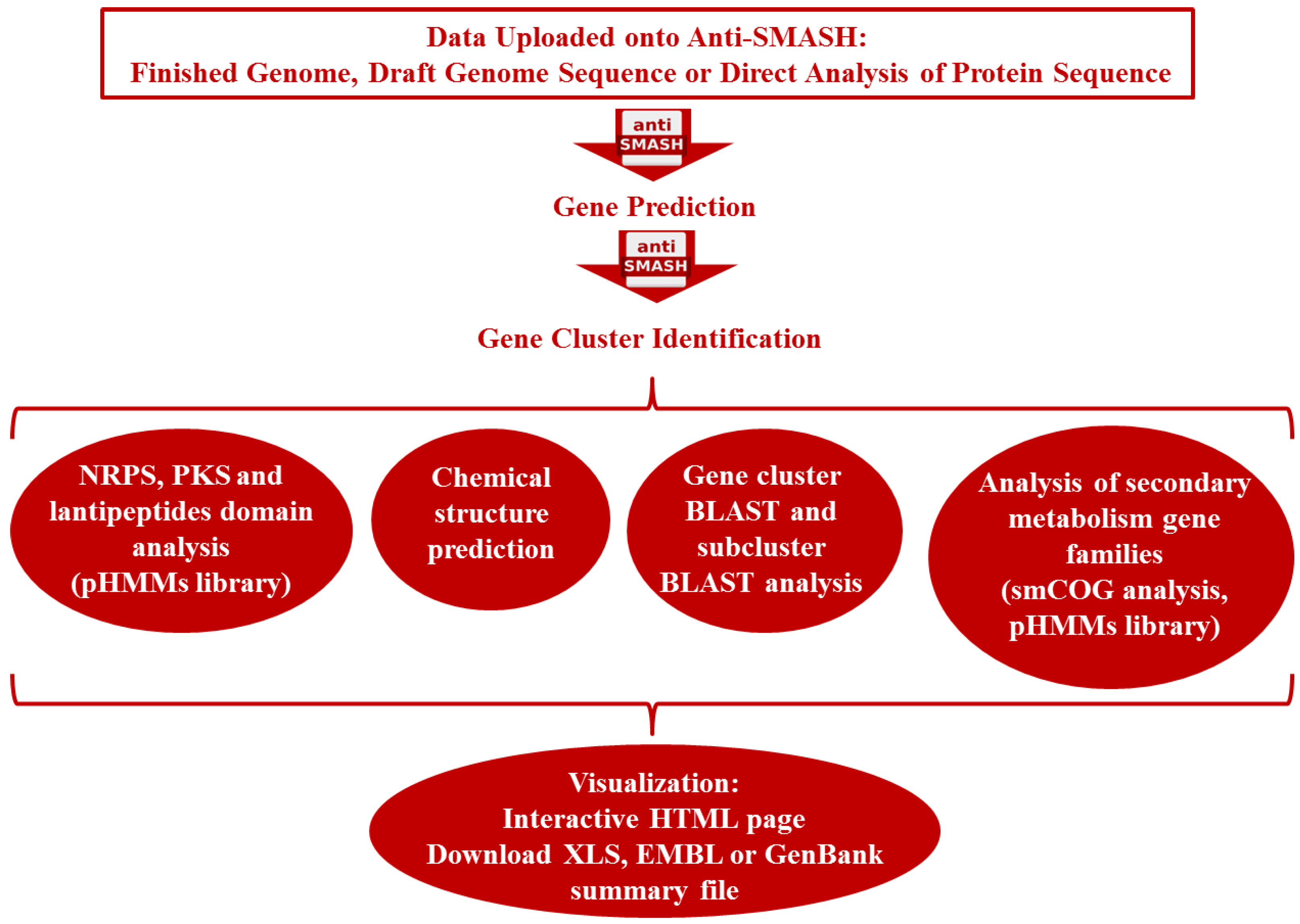
3.2. Genome-Guided Bioprospecting
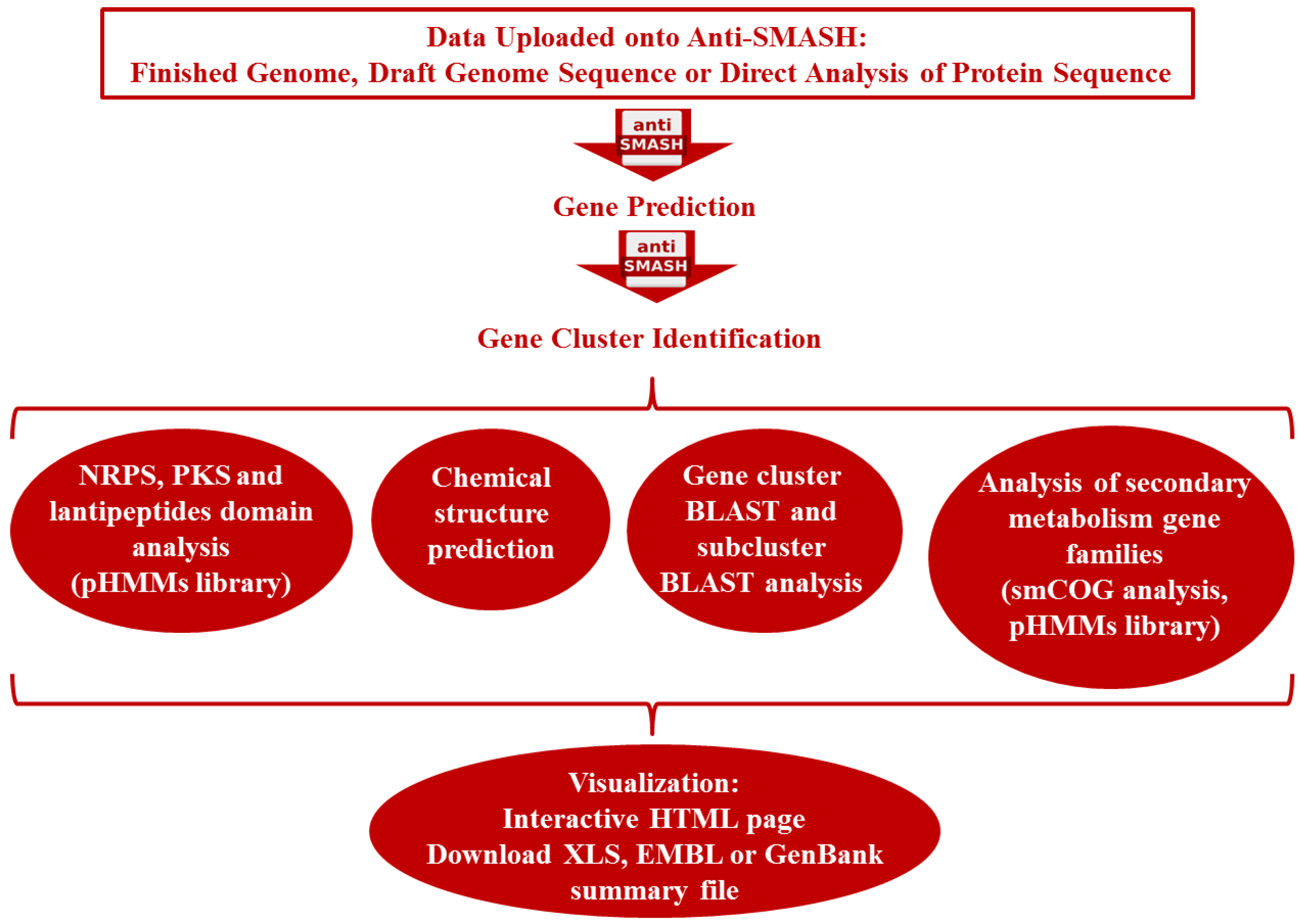
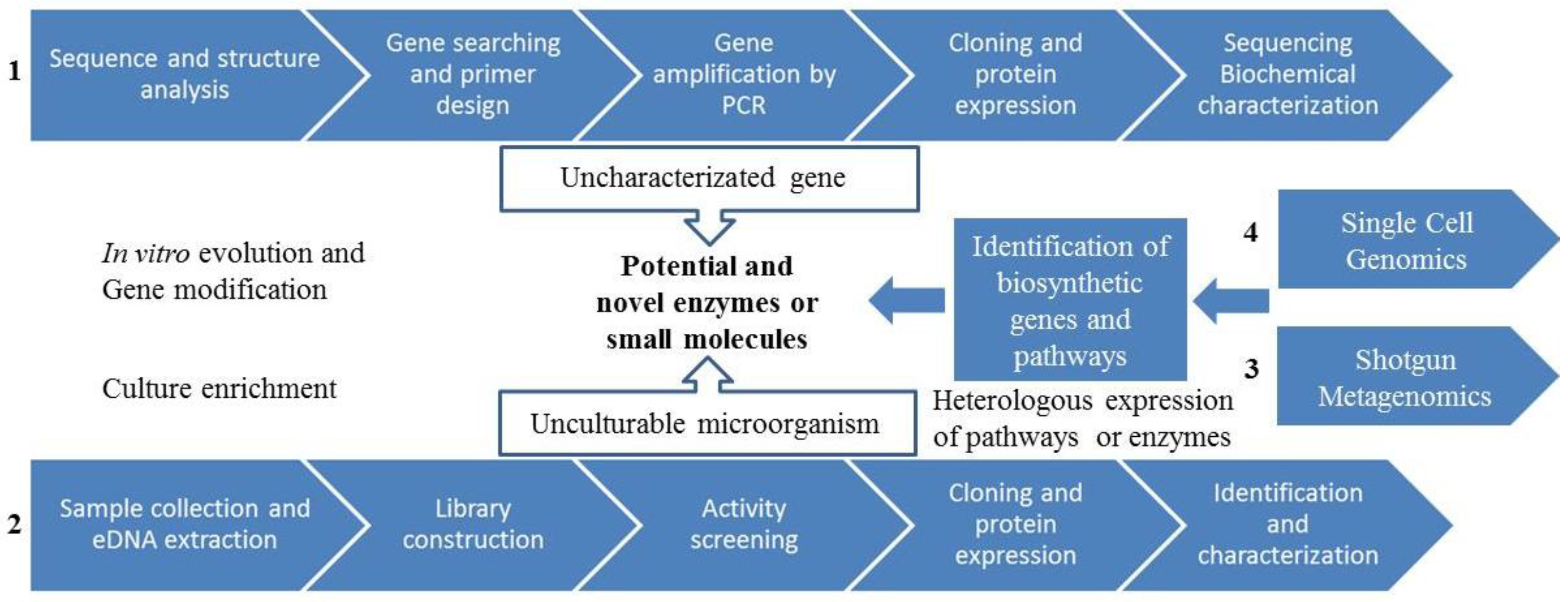
3.3. Gene-Guided Bioprospecting
3.4. Metagenomics
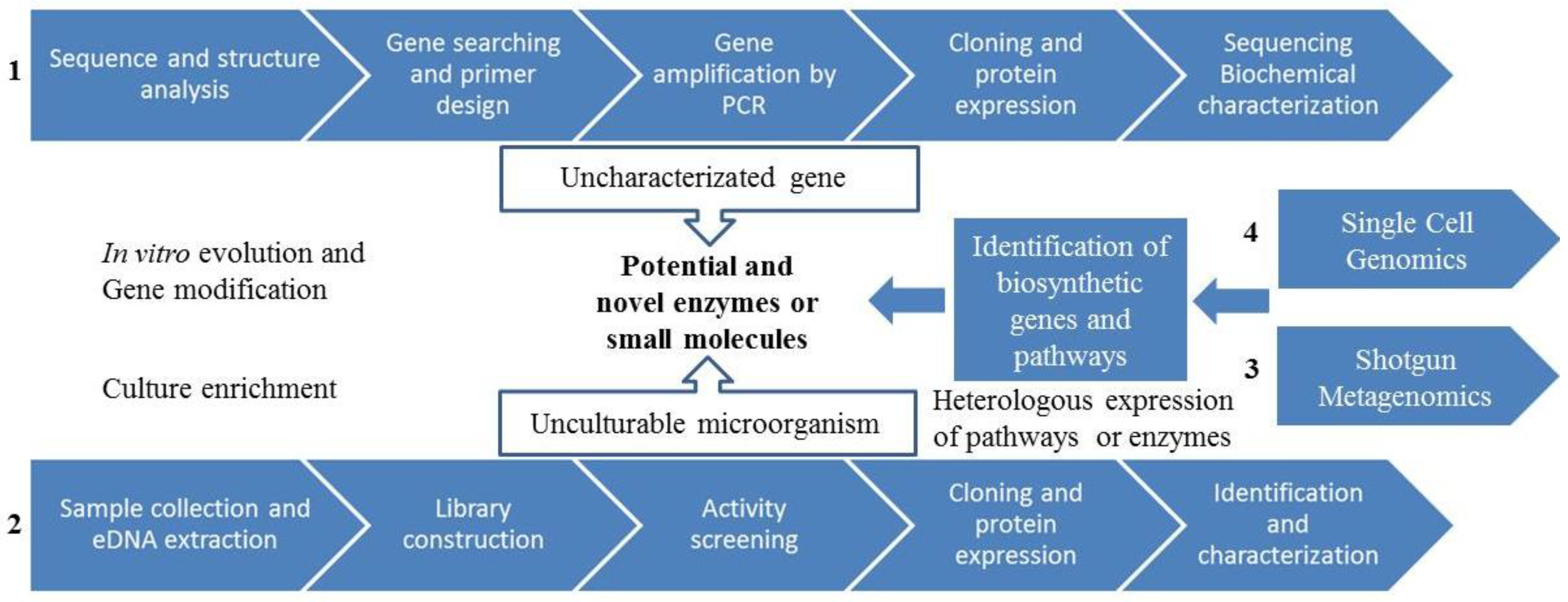
3.4.1. Metagenomics: Functional Screening
3.4.2. Metagenomics: Sequenced-Based Screening
3.4.3. Novel Metagenomics Approaches
3.5. Combinatorial Biosynthesis
3.6. Synthetic Biology
3.7. Heterologous Production of Bioactive Compounds
3.8. “Omic” Integrated Approaches to Characterize Bioactive Compounds Biosynthetic Gene Clusters and Pathways
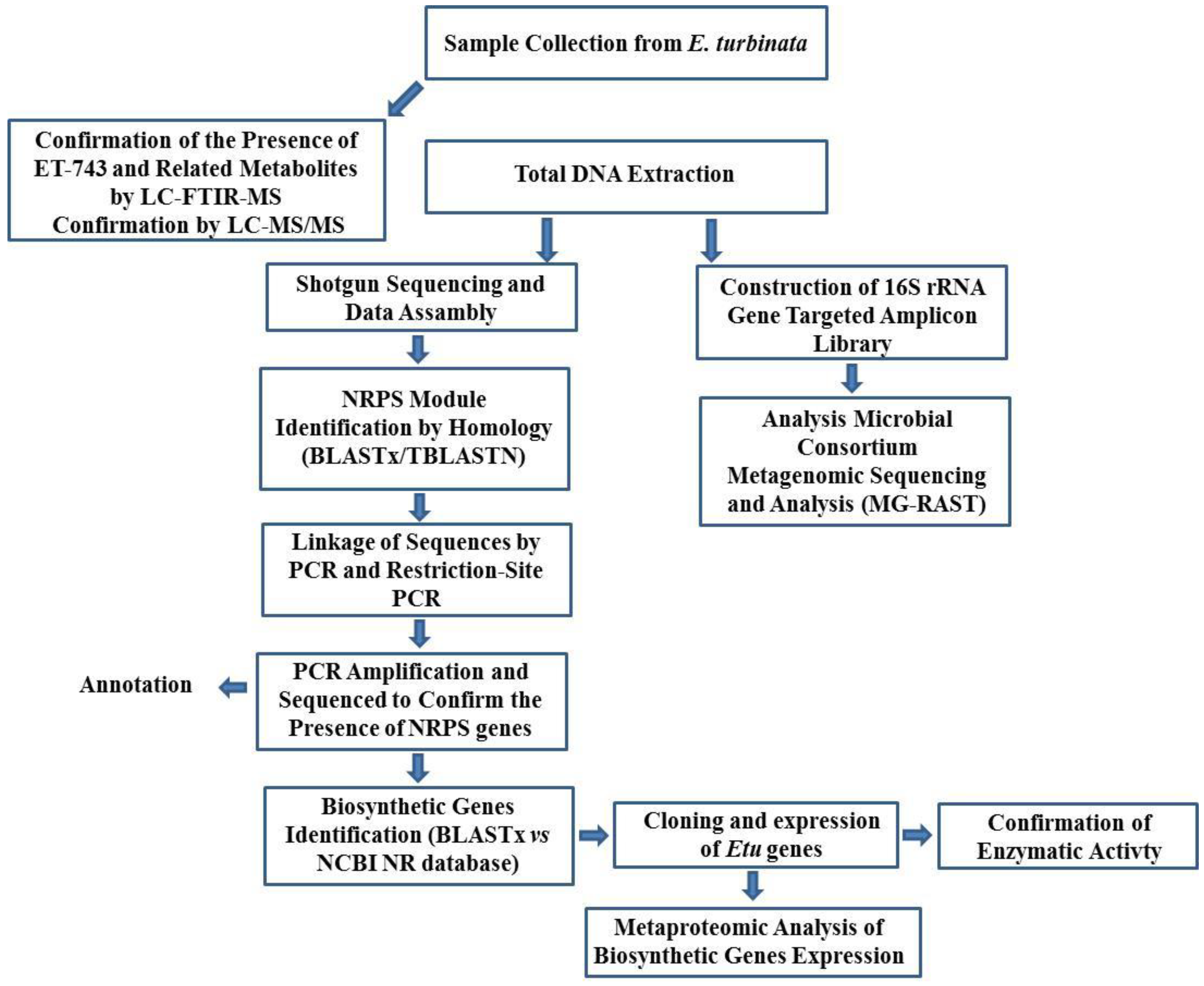
3.8.1. Integrated Approach to Investigate the ET-743 Biosynthetic Pathway
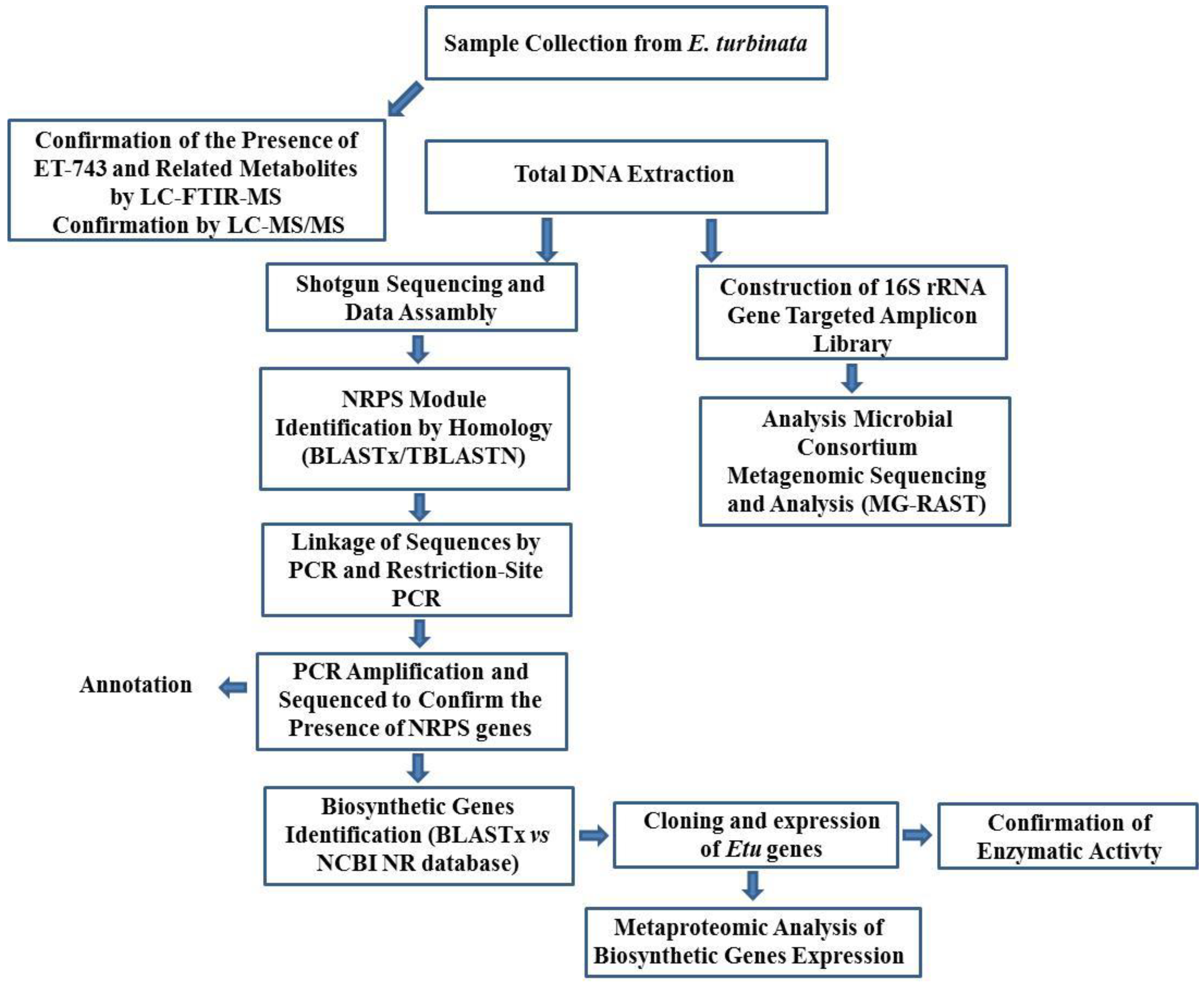
3.8.2. An Integrated Approach in the Discovery a Novel Lantibiotic from B. subtilis Strain Isolated from a Marine Sponge
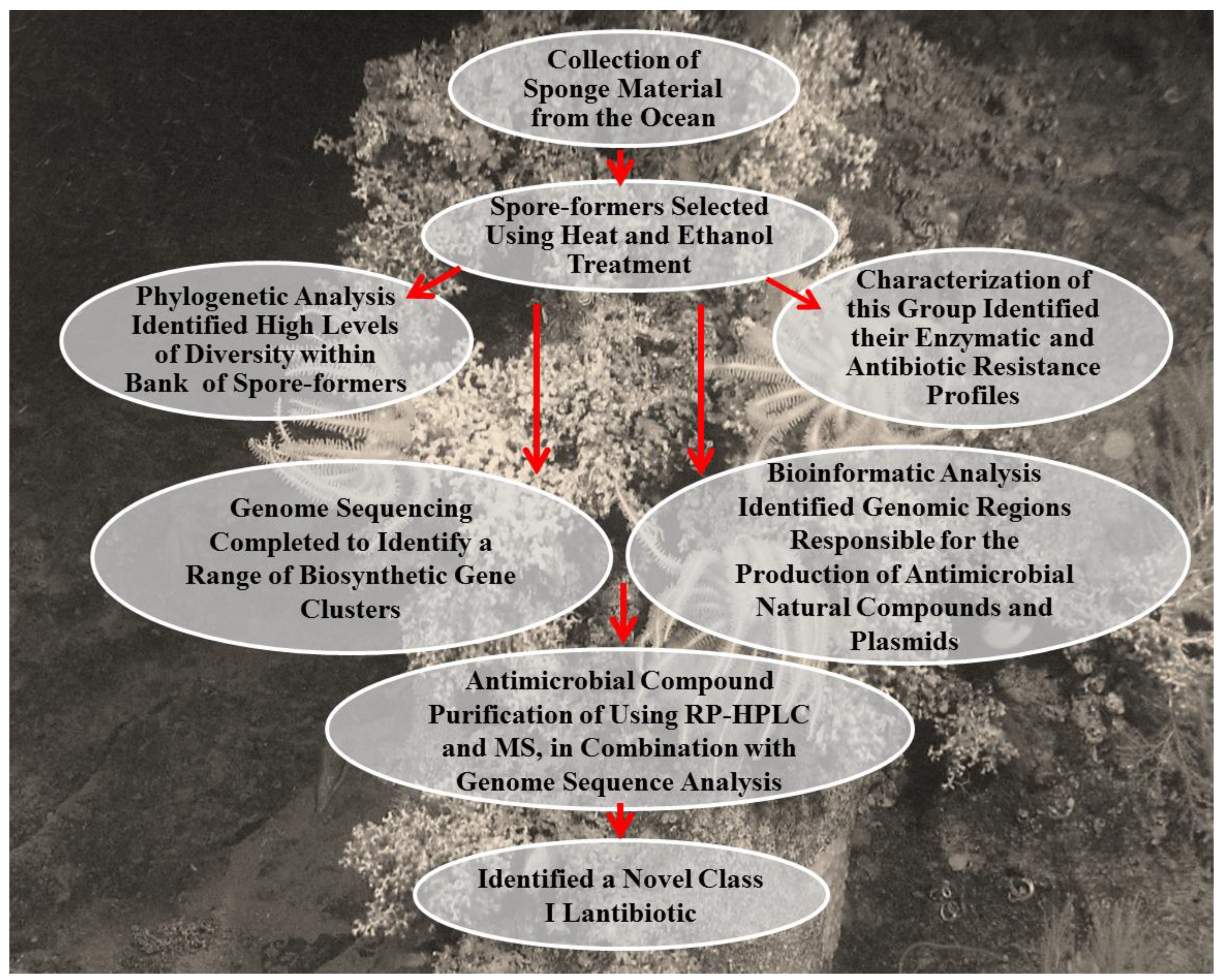
3.8.3. Integrated Approaches in the Investigation of the Biosynthetic Pathways and the Characterization of Glycosylated Bioactive Compounds
3.8.4. Integrated Approaches using Orthogonal Active Site Identification System (OASIS) and Proteomic Interrogation of Secondary Metabolism (PrISM)
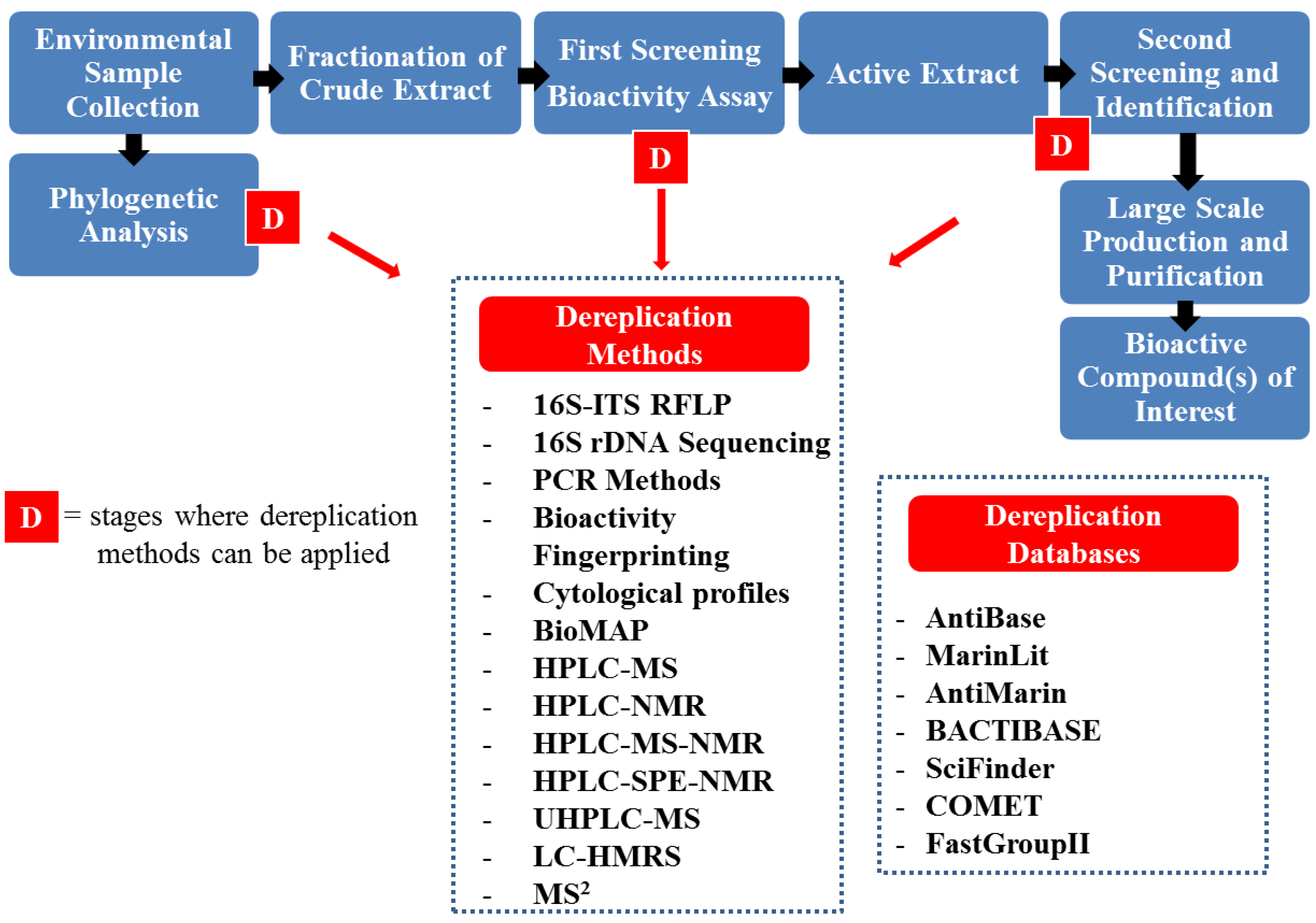
4. Dereplication Strategies
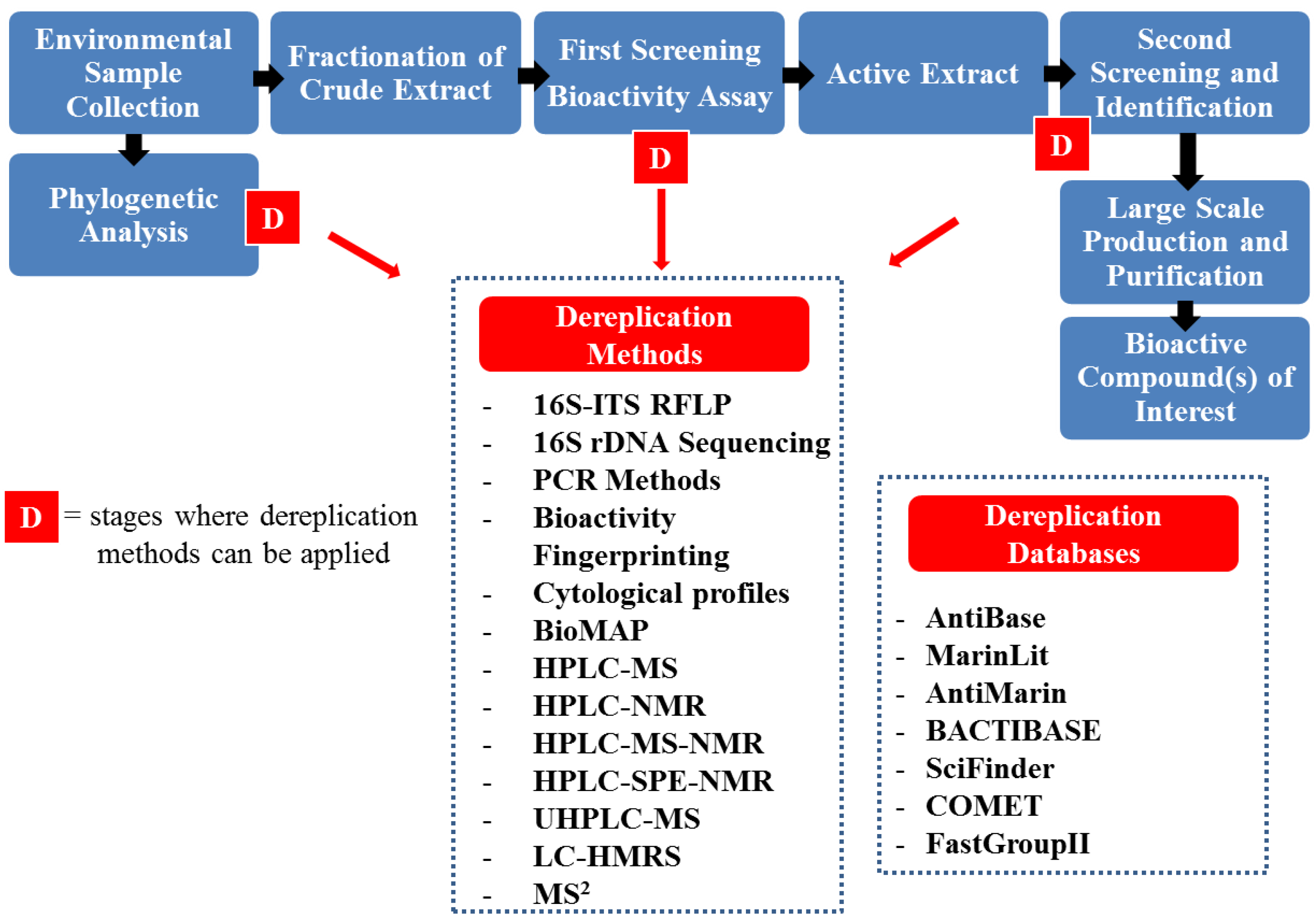
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Houssen, W.; Jaspars, M. Isolation of marine natural products. In Natural Products Isolation: Methods and Protocols, 3rd ed.; Sarker, S.D., Nahar, L., Eds.; Humana Press: Clifton, NJ, USA, 2012; Volume 864, pp. 367–392. [Google Scholar]
- Sipkema, D.; Franssen, M.R.; Osinga, R.; Tramper, J.; Wijffels, R. Marine sponges as pharmacy. Mar. Biotechnol. 2005, 7, 142–162. [Google Scholar] [CrossRef]
- Bharate, S.B.; Sawant, S.D.; Singh, P.P.; Vishwakarma, R.A. Kinase inhibitors of marine origin. Chem. Rev. 2013, 113, 6761–6815. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2012, 29, 144–222. [Google Scholar]
- Whitman, W.B.; Coleman, D.C.; Wiebe, W.J. Prokaryotes: The unseen majority. Proc. Natl. Acad. Sci. USA 1998, 95, 6578–6583. [Google Scholar] [CrossRef]
- Bhatnagar, I.; Kim, S.-K. Immense essence of excellence: Marine microbial bioactive compounds. Mar. Drugs 2010, 8, 2673–2701. [Google Scholar] [CrossRef]
- Trincone, A. Marine biocatalysts: Enzymatic features and applications. Mar. Drugs 2011, 9, 478–499. [Google Scholar] [CrossRef]
- Giddings, L.-A.; Newman, D. Microbial natural products: Molecular blueprints for antitumor drugs. J. Ind. Microbiol. Biotechnol. 2013, 40, 1181–1210. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Service of the U.S. National Institutes of Health. Available online: http://clinicaltrials.gov/ct2/home (accessed on 21 January 2014).
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2014, 31, 160–258. [Google Scholar] [CrossRef]
- Hu, G.-P.; Yuan, J.; Sun, L.; She, Z.-G.; Wu, J.-H.; Lan, X.-J.; Zhu, X.; Lin, Y.-C.; Chen, S.-P. Statistical research on marine natural products based on data obtained between 1985 and 2008. Mar. Drugs 2011, 9, 514–525. [Google Scholar] [CrossRef]
- Taylor, M.W.; Radax, R.; Steger, D.; Wagner, M. Sponge-associated microorganisms: Evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 2007, 71, 295–347. [Google Scholar] [CrossRef]
- Webster, N.S.; Taylor, M.W. Marine sponges and their microbial symbionts: Love and other relationships. Environ. Microbiol. 2012, 14, 335–346. [Google Scholar] [CrossRef]
- Li, J.W.-H.; Vederas, J.C. Drug discovery and natural products: End of an era or an endless frontier? Science 2009, 325, 161–165. [Google Scholar] [CrossRef]
- Fang, Z.; Li, T.; Wang, Q.; Zhang, X.; Peng, H.; Fang, W.; Hong, Y.; Ge, H.; Xiao, Y. A bacterial laccase from marine microbial metagenome exhibiting chloride tolerance and dye decolorization ability. Appl. Microbiol. Biotechnol. 2011, 89, 1103–1110. [Google Scholar] [CrossRef]
- Fu, C.; Hu, Y.; Xie, F.; Guo, H.; Ashforth, E.J.; Polyak, S.W.; Zhu, B.; Zhang, L. Molecular cloning and characterization of a new cold-active esterase from a deep-sea metagenomic library. Appl. Microbiol. Biotechnol. 2011, 90, 961–970. [Google Scholar] [CrossRef]
- Jiang, C.; Wu, L.-L.; Zhao, G.-C.; Shen, P.-H.; Jin, K.; Hao, Z.-Y.; Li, S.-X.; Ma, G.-F.; Luo, F.-F.; Hu, G.-Q.; et al. Identification and characterization of a novel fumarase gene by metagenome expression cloning from marine microorganisms. Microb. Cell Fact. 2010, 9, 1–9. [Google Scholar] [CrossRef]
- Kalpana, B.J.; Sindhulakshmi, M.; Pandian, S.K. Amylase enzyme from Bacillus subtilis S8-18: A potential desizing agent from the marine environment. Biotechnol. Appl. Biochem. 2014, 61, 134–144. [Google Scholar] [CrossRef]
- Wierzbicka-Wos, A.; Bartasun, P.; Cieslinski, H.; Kur, J. Cloning and characterization of a novel cold-active glycoside hydrolase family 1 enzyme with beta-glucosidase, beta-fucosidase and beta-galactosidase activities. BMC Biotechnol. 2013, 13, 22. [Google Scholar] [CrossRef]
- Willetts, A.; Joint, I.; Gilbert, J.A.; Trimble, W.; Mühling, M. Isolation and initial characterization of a novel type of Baeyer-Villiger monooxygenase activity from a marine microorganism. Microb. Biotechnol. 2012, 5, 549–559. [Google Scholar] [CrossRef]
- Zhao, K.; Guo, L.Z.; Lu, W.D. Extracellular production of novel halotolerant, thermostable, and alkali-stable carboxymethyl cellulase by marine bacterium Marinimicrobium sp. LS-A18. Appl. Biochem. Biotechnol. 2012, 168, 550–567. [Google Scholar] [CrossRef]
- Zhang, J.; Cao, X.; Xin, Y.; Xue, S.; Zhang, W. Purification and characterization of a dehalogenase from Pseudomonas stutzeri DEH130 isolated from the marine sponge Hymeniacidon perlevis. World J. Microb. Biotechnol. 2013, 29, 1791–1799. [Google Scholar] [CrossRef]
- Li, G.; Ren, J.; Wu, Q.; Feng, J.; Zhu, D.; Ma, Y. Identification of a marine NADPH-dependent aldehyde reductase for chemoselective reduction of aldehydes. J. Mol. Catal. B-Enzym. 2013, 90, 17–22. [Google Scholar] [CrossRef]
- Lailaja, V.P.; Chandrasekaran, M. Detergent compatible alkaline lipase produced by marine Bacillus smithii BTMS 11. World J. Microb. Biotechnol. 2013, 29, 1349–1360. [Google Scholar] [CrossRef]
- Novak, H.; Sayer, C.; Panning, J.; Littlechild, J. Characterisation of an l-haloacid dehalogenase from the marine psychrophile Psychromonas ingrahamii with potential industrial application. Mar. Biotechnol. 2013, 15, 695–705. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, X.; Bai, F. Production of xylanase by an alkaline-tolerant marine-derived Streptomyces viridochromogenes strain and improvement by ribosome engineering. Appl. Microbiol. Biotechnol. 2013, 97, 4361–4368. [Google Scholar] [CrossRef]
- Bernard, L.; Schäfer, H.; Joux, F.; Courties, C.; Muyzer, G.; Lebaron, P. Genetic diversity of total, active and culturable marine bacteria in coastal seawater. Aquat. Microb. Ecol. 2000, 23, 1–11. [Google Scholar] [CrossRef]
- Hugenholtz, P. Exploring prokaryotic diversity in the genomic era. Genome Biol. 2002, 3, 0003.0001–0003.0008. [Google Scholar] [CrossRef]
- Ferrari, B.C.; Winsley, T.; Gillings, M.; Binnerup, S. Cultivating previously uncultured soil bacteria using a soil substrate membrane system. Nat. Protoc. 2008, 3, 1261–1269. [Google Scholar] [CrossRef]
- Schut, F.; de Vries, E.J.; Gottschal, J.C.; Robertson, B.R.; Harder, W.; Prins, R.A.; Button, D.K. Isolation of typical marine bacteria by dilution culture: Growth, maintenance, and characteristics of isolates under laboratory conditions. Appl. Environ. Microbiol. 1993, 59, 2150–2160. [Google Scholar]
- Shigematsu, T.; Hayashi, M.; Kikuchi, I.; Ueno, S.; Masaki, H.; Fujii, T. A culture-dependent bacterial community structure analysis based on liquid cultivation and its application to a marine environment. FEMS Microbiol. Lett. 2009, 293, 240–247. [Google Scholar] [CrossRef]
- Simu, K.; Holmfeldt, K.; Zweifel, U.L.; Hagström, Å. Culturability and coexistence of colony-forming and single-cell marine bacterioplankton. Appl. Environ. Microbiol. 2005, 71, 4793–4800. [Google Scholar] [CrossRef]
- Rappé, M.S.; Connon, S.A.; Vergin, K.L.; Giovannoni, S.J. Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature 2002, 418, 630–633. [Google Scholar] [CrossRef]
- Connon, S.A.; Giovannoni, S.J. High-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Appl. Environ. Microbiol. 2002, 68, 3878–3885. [Google Scholar] [CrossRef]
- Vartoukian, S.R.; Palmer, R.M.; Wade, W.G. Strategies for culture of “unculturable” bacteria. FEMS Microbiol. Lett. 2010, 309, 1–7. [Google Scholar]
- Stewart, E.J. Growing unculturable bacteria. J. Bacteriol. 2012, 194, 4151–4160. [Google Scholar] [CrossRef]
- Kaeberlein, T.; Lewis, K.; Epstein, S.S. Isolating “uncultivable” microorganisms in pure culture in a simulated natural environment. Science 2002, 296, 1127–1129. [Google Scholar] [CrossRef]
- Zengler, K.; Toledo, G.; Rappé, M.; Elkins, J.; Mathur, E.J.; Short, J.M.; Keller, M. Cultivating the uncultured. Proc. Natl. Acad. Sci. USA 2002, 99, 15681–15686. [Google Scholar]
- Rastogi, G.; Sani, R. Molecular techniques to assess microbial community structure, function, and dynamics in the environment. In Microbes and Microbial Technology; Ahmad, I., Ahmad, F., Pichtel, J., Eds.; Springer: New York, NY, USA, 2011; pp. 29–57. [Google Scholar]
- DeSantis, T.; Brodie, E.; Moberg, J.; Zubieta, I.; Piceno, Y.; Andersen, G. High-density universal 16S rRNA microarray analysis reveals broader diversity than typical clone library when sampling the environment. Microb. Ecol. 2007, 53, 371–383. [Google Scholar] [CrossRef]
- Cottrell, M.T.; Kirchman, D.L. Community composition of marine bacterioplankton determined by 16S rRNA gene clone libraries and fluorescence in situ hybridization. Appl. Environ. Microbiol. 2000, 66, 5116–5122. [Google Scholar] [CrossRef]
- Alex, A.; Silva, V.; Vasconcelos, V.; Antunes, A. Evidence of unique and generalist microbes in distantly related sympatric intertidal marine sponges (Porifera: Demospongiae). PLoS One 2013, 8, e80653. [Google Scholar]
- Urakawa, H.; Kita-Tsukamoto, K.; Ohwada, K. Microbial diversity in marine sediments from Sagami Bay and Tokyo Bay, Japan, as determined by 16S rRNA gene analysis. Microbiology 1999, 145, 3305–3315. [Google Scholar]
- Harrington, C.; Del Casale, A.; Kennedy, J.; Neve, H.; Picton, B.E.; Mooij, M.J.; O’Gara, F.; Kulakov, L.A.; Larkin, M.J.; Dobson, A.D.W. Evidence of bacteriophage-mediated horizontal transfer of bacterial 16S rRNA genes in the viral metagenome of the marine sponge Hymeniacidon perlevis. Microbiology 2012, 158, 2789–2795. [Google Scholar] [CrossRef]
- Nübel, U.; Garcia-Pichel, F.; Kühl, M.; Muyzer, G. Quantifying microbial diversity: Morphotypes, 16S rRNA genes, and carotenoids of oxygenic phototrophs in microbial mats. Appl. Environ. Microbiol. 1999, 65, 422–430. [Google Scholar]
- Muyzer, G.; Smalla, K. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie van Leeuwenhoek 1998, 73, 127–141. [Google Scholar] [CrossRef]
- Neilan, B.A. Identification and phylogenetic analysis of toxigenic cyanobacteria by multiplex randomly amplified polymorphic DNA PCR. Appl. Environ. Microbiol. 1995, 61, 2286–2291. [Google Scholar]
- Roberts, M.A.; Crawford, D.L. Use of randomly amplified polymorphic DNA as a means of developing genus- and strain-specific Streptomyces DNA probes. Appl. Environ. Microbiol. 2000, 66, 2555–2564. [Google Scholar] [CrossRef]
- Heyndrickx, M.; Vauterin, L.; Vandamme, P.; Kersters, K.; de Vos, P. Applicability of combined amplified ribosomal DNA restriction analysis (ARDRA) patterns in bacterial phylogeny and taxonomy. J. Microbiol. Methods 1996, 26, 247–259. [Google Scholar] [CrossRef]
- Moeseneder, M.M.; Arrieta, J.M.; Muyzer, G.; Winter, C.; Herndl, G.J. Optimization of terminal-restriction fragment length polymorphism analysis for complex marine bacterioplankton communities and comparison with denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 1999, 65, 3518–3525. [Google Scholar]
- Fuhrman, J.A.; Steele, J.A.; Hewson, I.; Schwalbach, M.S.; Brown, M.V.; Green, J.L.; Brown, J.H. A latitudinal diversity gradient in planktonic marine bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 7774–7778. [Google Scholar]
- Brown, M.V.; Schwalbach, M.S.; Hewson, I.; Fuhrman, J.A. Coupling 16S-ITS rDNA clone libraries and automated ribosomal intergenic spacer analysis to show marine microbial diversity: Development and application to a time series. Environ. Microbiol. 2005, 7, 1466–1479. [Google Scholar] [CrossRef]
- Peplies, J.; Lau, S.C.K.; Pernthaler, J.; Amann, R.; Glöckner, F.O. Application and validation of DNA microarrays for the 16S rRNA-based analysis of marine bacterioplankton. Environ. Microbiol. 2004, 6, 638–645. [Google Scholar] [CrossRef]
- Zhou, J. Microarrays for bacterial detection and microbial community analysis. Curr. Opin. Microbiol. 2003, 6, 288–294. [Google Scholar] [CrossRef]
- Wu, L.; Kellogg, L.; Devol, A.H.; Tiedje, J.M.; Zhou, J. Microarray-based characterization of microbial community functional structure and heterogeneity in marine sediments from the Gulf of Mexico. Appl. Environ. Microbiol. 2008, 74, 4516–4529. [Google Scholar]
- Smith, C.J.; Osborn, A.M. Advantages and limitations of quantitative PCR (Q-PCR)-based approaches in microbial ecology. FEMS Microbiol. Ecol. 2009, 67, 6–20. [Google Scholar] [CrossRef]
- Cassler, M.; Peterson, C.L.; Ledger, A.; Pomponi, S.A.; Wright, A.E.; Winegar, R.; McCarthy, P.J.; Lopez, J.V. Use of real-time qPCR to quantify members of the unculturable heterotrophic bacterial community in a deep sea marine sponge, Vetulina sp. Microb. Ecol. 2008, 55, 384–394. [Google Scholar] [CrossRef]
- Glöckner, F.O.; Fuchs, B.M.; Amann, R. Bacterioplankton compositions of lakes and oceans: A first comparison based on fluorescence in situ hybridization. Appl. Environ. Microbiol. 1999, 65, 3721–3726. [Google Scholar]
- Moter, A.; Göbel, U.B. Fluorescence in situ hybridization (FISH) for direct visualization of microorganisms. J. Microbiol. Methods 2000, 41, 85–112. [Google Scholar] [CrossRef]
- Kindaichi, T.; Ito, T.; Okabe, S. Ecophysiological interaction between nitrifying bacteria and heterotrophic bacteria in autotrophic nitrifying biofilms as determined by microautoradiography-fluorescence in situ hybridization. Appl. Environ. Microbiol. 2004, 70, 1641–1650. [Google Scholar] [CrossRef]
- Konstantinidis, K.T.; Tiedje, J.M. Prokaryotic taxonomy and phylogeny in the genomic era: Advancements and challenges ahead. Curr. Opin. Microbiol. 2007, 10, 504–509. [Google Scholar] [CrossRef]
- Colwell, R.R. Viable but nonculturable bacteria: A survival strategy. J. Infect. Chemother. 2000, 6, 121–125. [Google Scholar] [CrossRef]
- Torsvik, V.; Øvreås, L. Microbial diversity and function in soil: From genes to ecosystems. Curr. Opin. Microbiol. 2002, 5, 240–245. [Google Scholar] [CrossRef]
- Wellington, E.M.H.; Berry, A.; Krsek, M. Resolving functional diversity in relation to microbial community structure in soil: Exploiting genomics and stable isotope probing. Curr. Opin. Microbiol. 2003, 6, 295–301. [Google Scholar] [CrossRef]
- Webster, G.; Watt, L.C.; Rinna, J.; Fry, J.C.; Evershed, R.P.; Parkes, R.J.; Weightman, A.J. A comparison of stable-isotope probing of DNA and phospholipid fatty acids to study prokaryotic functional diversity in sulfate-reducing marine sediment enrichment slurries. Environ. Microbiol. 2006, 8, 1575–1589. [Google Scholar] [CrossRef]
- Radajewski, S.; McDonald, I.R.; Murrell, J.C. Stable-isotope probing of nucleic acids: A window to the function of uncultured microorganisms. Curr. Opin. Biotechnol. 2003, 14, 296–302. [Google Scholar] [CrossRef]
- Adamczyk, J.; Hesselsoe, M.; Iversen, N.; Horn, M.; Lehner, A.; Nielsen, P.H.; Schloter, M.; Roslev, P.; Wagner, M. The isotope array, a new tool that employs substrate-mediated labeling of rRNA for determination of microbial community structure and function. Appl. Environ. Microbiol. 2003, 69, 6875–6887. [Google Scholar] [CrossRef]
- Nielsen, J.L.; Nielsen, P.H. Combined microautoradiography and fluorescence in situ hybridization (MAR-FISH) for the identification of metabolically active microorganisms. In Handbook of Hydrocarbon and Lipid Microbiology; Timmis, K., Ed.; Springer Berlin Heidelberg: Berlin, Germany, 2010; pp. 4093–4102. [Google Scholar]
- Pinhassi, J.; Zweifel, U.L.; Hagström, A. Dominant marine bacterioplankton species found among colony-forming bacteria. Appl. Environ. Microbiol. 1997, 63, 3359–3366. [Google Scholar]
- Nüsslein, K.; Tiedje, J.M. Characterization of the dominant and rare members of a young Hawaiian soil bacterial community with small-subunit ribosomal DNA amplified from DNA fractionated on the basis of its guanine and cytosine composition. Appl. Environ. Microbiol. 1998, 64, 1283–1289. [Google Scholar]
- Holben, W.E.; Feris, K.P.; Kettunen, A.; Apajalahti, J.H.A. GC fractionation enhances microbial community diversity assessment and detection of minority populations of bacteria by denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 2004, 70, 2263–2270. [Google Scholar]
- Riesenfeld, C.S.; Schloss, P.D.; Handelsman, J. Metagenomics: Genomic analysis of microbial communities. Annu. Rev. Genet. 2004, 38, 525–552. [Google Scholar] [CrossRef]
- Kennedy, J.; Codling, C.E.; Jones, B.V.; Dobson, A.D.W.; Marchesi, J.R. Diversity of microbes associated with the marine sponge, Haliclona simulans, isolated from Irish waters and identification of polyketide synthase genes from the sponge metagenome. Environ. Microbiol. 2008, 10, 1888–1902. [Google Scholar] [CrossRef]
- Kisand, V.; Valente, A.; Lahm, A.; Tanet, G.; Lettieri, T. Phylogenetic and functional metagenomic profiling for assessing microbial biodiversity in environmental monitoring. PLoS One 2012, 7, e43630. [Google Scholar]
- Webster, N.S.; Taylor, M.W.; Behnam, F.; Lücker, S.; Rattei, T.; Whalan, S.; Horn, M.; Wagner, M. Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ. Microbiol. 2010, 12, 2070–2082. [Google Scholar]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Welch, D.M.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar] [CrossRef]
- Kennedy, J.; Flemer, B.; Jackson, S.A.; Lejon, D.P.H.; Morrissey, J.P.; O’Gara, F.; Dobson, A.D.W. Marine metagenomics: New tools for the study and exploitation of marine microbial metabolism. Mar. Drugs 2010, 8, 608–628. [Google Scholar] [CrossRef]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef]
- Aziz, R.; Bartels, D.; Best, A.; DeJongh, M.; Disz, T.; Edwards, R.; Formsma, K.; Gerdes, S.; Glass, E.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genomics 2008, 9, 75. [Google Scholar] [CrossRef]
- Rapid Annotation using Subsystem Technology. Available online: http://rast.nmpdr.org/ (accessed on 21 January 2014).
- Horton, M.; Bodenhausen, N.; Bergelson, J. MARTA: A suite of Java-based tools for assigning taxonomic status to DNA sequences. Bioinformatics 2010, 26, 568–569. [Google Scholar]
- Metagenomic And rDNA Taxonomy Assigner. Available online: http://bergelson.uchicago.edu/software/marta (accessed on 21 January 2014).
- Yu, F.; Sun, Y.; Liu, L.; Farmerie, W. GSTaxClassifier: A genomic signature based taxonomic classifier for metagenomic data analysis. Bioinformation 2010, 4, 46–49. [Google Scholar]
- Genomic Signature based Taxonomic Classifier. Available online: http://helix2.biotech.ufl.edu:26878/metagenomics/ (accessed on 21 January 2014).
- Markowitz, V.M.; Chen, I.-M.A.; Palaniappan, K.; Chu, K.; Szeto, E.; Pillay, M.; Ratner, A.; Huang, J.; Woyke, T.; Huntemann, M.; et al. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 2014, 42, D560–D567. [Google Scholar] [CrossRef]
- Integrated Microbial Genomes with Microbiome Samples. Available online: https://img.jgi.doe.gov/cgi-bin/m/main.cgi (accessed on 21 January 2014).
- Seshadri, R.; Kravitz, S.A.; Smarr, L.; Gilna, P.; Frazier, M. CAMERA: A community resource for metagenomics. PLoS Biol. 2007, 5, e75. [Google Scholar] [CrossRef]
- Community CyberInfrastructure for Advanced Marine Microbial Ecology Research and Analysis. Available online: http://camera.calit2.net/ (accessed on 21 January 2014).
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [Green Version]
- MG-RAST: Metagenomics Analysis Server. Available online: http://metagenomics.anl.gov/ (accessed on 22 January 2014).
- NCBI: National Center for Biotechnology Information. Available online: http://www.ncbi.nlm.nih.gov/genome (accessed on 22 January 2014).
- Bernal, A.; Ear, U.; Kyrpides, N. Genomes OnLine Database (GOLD): A monitor of genome projects world-wide. Nucleic Acids Res. 2001, 29, 126–127. [Google Scholar] [CrossRef]
- GOLD: Genomes Online Database. Available online: http://www.genomesonline.org (accessed on 22 February 2014).
- Wilmes, P.; Bond, P.L. Metaproteomics: Studying functional gene expression in microbial ecosystems. Trends Microbiol. 2006, 14, 92–97. [Google Scholar] [CrossRef]
- Slattery, M.; Ankisetty, S.; Corrales, J.; Marsh-Hunkin, K.E.; Gochfeld, D.J.; Willett, K.L.; Rimoldi, J.M. Marine proteomics: A critical assessment of an emerging technology. J. Nat. Prod. 2012, 75, 1833–1877. [Google Scholar] [CrossRef]
- Sowell, S.M.; Wilhelm, L.J.; Norbeck, A.D.; Lipton, M.S.; Nicora, C.D.; Barofsky, D.F.; Carlson, C.A.; Smith, R.D.; Giovanonni, S.J. Transport functions dominate the SAR11 metaproteome at low-nutrient extremes in the Sargasso Sea. ISME J. 2009, 3, 93–105. [Google Scholar] [CrossRef]
- Morris, R.M.; Nunn, B.L.; Frazar, C.; Goodlett, D.R.; Ting, Y.S.; Rocap, G. Comparative metaproteomics reveals ocean-scale shifts in microbial nutrient utilization and energy transduction. ISME J. 2010, 4, 673–685. [Google Scholar] [CrossRef]
- Williams, T.J.; Long, E.; Evans, F.; Demaere, M.Z.; Lauro, F.M.; Raftery, M.J.; Ducklow, H.; Grzymski, J.J.; Murray, A.E.; Cavicchioli, R. A metaproteomic assessment of winter and summer bacterioplankton from Antarctic Peninsula coastal surface waters. ISME J. 2012, 6, 1883–1900. [Google Scholar] [CrossRef]
- Stokke, R.; Roalkvam, I.; Lanzen, A.; Haflidason, H.; Steen, I.H. Integrated metagenomic and metaproteomic analyses of an ANME-1-dominated community in marine cold seep sediments. Environ. Microbiol. 2012, 14, 1333–1346. [Google Scholar] [CrossRef]
- Banfield, J.F.; Verberkmoes, N.C.; Hettich, R.L.; Thelen, M.P. Proteogenomic approaches for the molecular characterization of natural microbial communities. OMICS 2005, 9, 301–333. [Google Scholar] [CrossRef]
- Christie-Oleza, J.A.; Miotello, G.; Armengaud, J. Proteogenomic definition of biomarkers for the large Roseobacter clade and application for a quick screening of new environmental isolates. J. Proteome Res. 2013, 12, 5331–5339. [Google Scholar] [CrossRef]
- D’Haeseleer, P.; Gladden, J.M.; Allgaier, M.; Chain, P.S.G.; Tringe, S.G.; Malfatti, S.A.; Aldrich, J.T.; Nicora, C.D.; Robinson, E.W.; Paša-Tolić, L.; et al. Proteogenomic analysis of a thermophilic bacterial consortium adapted to deconstruct switchgrass. PLoS One 2013, 8, e68465. [Google Scholar] [CrossRef]
- Ogura, A.; Lin, M.; Shigenobu, Y.; Fujiwara, A.; Ikeo, K.; Nagai, S. Effective gene collection from the metatranscriptome of marine microorganisms. BMC Genomics 2011, 12, S15. [Google Scholar]
- Coll-Lladó, M.; Acinas, S.G.; Pujades, C.; Pedrós-Alió, C. Transcriptome fingerprinting analysis: An approach to explore gene expression patterns in marine microbial communities. PLoS One 2011, 6, e22950. [Google Scholar]
- Shi, Y.; McCarren, J.; DeLong, E.F. Transcriptional responses of surface water marine microbial assemblages to deep-sea water amendment. Environ. Microbiol. 2012, 14, 191–206. [Google Scholar] [CrossRef]
- Martinez, A.; Ventouras, L.A.; Wilson, S.T.; Karl, D.M.; DeLong, E.F. Metatranscriptomic and functional metagenomic analysis of methylphosphonate utilization by marine bacteria. Front. Microbiol. 2013, 4, 340. [Google Scholar]
- Wu, J.; Gao, W.; Johnson, R.; Zhang, W.; Meldrum, D. Integrated metagenomic and metatranscriptomic analyses of microbial communities in the meso- and bathypelagic realm of North Pacific Ocean. Mar. Drugs 2013, 11, 3777–3801. [Google Scholar] [CrossRef]
- Al-Awadhi, H.; Dashti, N.; Khanafer, M.; Al-Mailem, D.; Ali, N.; Radwan, S. Bias problems in culture-independent analysis of environmental bacterial communities: A representative study on hydrocarbonoclastic bacteria. SpringerPlus 2013, 2, 369. [Google Scholar] [CrossRef]
- Öztürk, B.; De Jaeger, L.; Smidt, H.; Sipkema, D. Culture-dependent and independent approaches for identifying novel halogenases encoded by Crambe crambe (marine sponge) microbiota. Sci. Rep. 2013, 3, 2780. [Google Scholar]
- Zeng, Y.; Zou, Y.; Grebmeier, J.; He, J.; Zheng, T. Culture-independent and -dependent methods to investigate the diversity of planktonic bacteria in the northern Bering Sea. Polar Biol. 2012, 35, 117–129. [Google Scholar] [CrossRef]
- Hirayama, H.; Sunamura, M.; Takai, K.; Nunoura, T.; Noguchi, T.; Oida, H.; Furushima, Y.; Yamamoto, H.; Oomori, T.; Horikoshi, K. Culture-dependent and -independent characterization of microbial communities associated with a shallow submarine hydrothermal system occurring within a coral reef off Taketomi Island, Japan. Appl. Environ. Microbiol. 2007, 73, 7642–7656. [Google Scholar] [CrossRef]
- Besaury, L.; Marty, F.; Buquet, S.; Mesnage, V.; Muyzer, G.; Quillet, L. Culture-dependent and independent studies of microbial diversity in highly copper-contaminated chilean marine sediments. Microb. Ecol. 2013, 65, 311–324. [Google Scholar] [CrossRef]
- Al-Awadhi, H.; Al-Mailem, D.; Dashti, N.; Khanafer, M.; Radwan, S. Indigenous hydrocarbon-utilizing bacterioflora in oil-polluted habitats in Kuwait, two decades after the greatest man-made oil spill. Arch. Microbiol. 2012, 194, 689–705. [Google Scholar] [CrossRef]
- Tripp, H.J.; Kitner, J.B.; Schwalbach, M.S.; Dacey, J.W.H.; Wilhelm, L.J.; Giovannoni, S.J. SAR11 marine bacteria require exogenous reduced sulphur for growth. Nature 2008, 452, 741–744. [Google Scholar] [CrossRef]
- Tong, J.; Trapido-Rosenthal, H.; Wang, J.; Wang, Y.; Li, Q.X.; Lu, Y. Antiviral activities and putative identification of compounds in microbial extracts from the Hawaiian coastal waters. Mar. Drugs 2012, 10, 521–538. [Google Scholar] [CrossRef]
- Cheng, Z.-B.; Xiao, H.; Fan, C.-Q.; Lu, Y.-N.; Zhang, G.; Yin, S. Bioactive polyhydroxylated sterols from the marine sponge Haliclona crassiloba. Steroids 2013, 78, 1353–1358. [Google Scholar] [CrossRef]
- Engelhardt, K.; Degnes, K.F.; Kemmler, M.; Bredholt, H.; Fjærvik, E.; Klinkenberg, G.; Sletta, H.; Ellingsen, T.E.; Zotchev, S.B. Production of a new thiopeptide antibiotic, TP-1161, by a marine Nocardiopsis species. Appl. Environ. Microbiol. 2010, 76, 4969–4976. [Google Scholar] [CrossRef] [Green Version]
- Graça, A.P.; Bondoso, J.; Gaspar, H.; Xavier, J.R.; Monteiro, M.C.; de la Cruz, M.; Oves-Costales, D.; Vicente, F.; Lage, O.M. Antimicrobial activity of heterotrophic bacterial communities from the marine sponge Erylus discophorus (Astrophorida, Geodiidae). PLoS One 2013, 8, e78992. [Google Scholar] [CrossRef]
- Sanchez, L.M.; Wong, W.R.; Riener, R.M.; Schulze, C.J.; Linington, R.G. Examining the fish microbiome: Vertebrate-derived bacteria as an environmental niche for the discovery of unique marine natural products. PLoS One 2012, 7, e35398. [Google Scholar]
- Dalisay, D.S.; Williams, D.E.; Wang, X.L.; Centko, R.; Chen, J.; Andersen, R.J. Marine sediment-derived Streptomyces bacteria from British Columbia, Canada are a promising microbiota resource for the discovery of antimicrobial natural products. PLoS One 2013, 8, e77078. [Google Scholar]
- Liu, X.; Bolla, K.; Ashforth, E.; Zhuo, Y.; Gao, H.; Huang, P.; Stanley, S.; Hung, D.; Zhang, L. Systematics-guided bioprospecting for bioactive microbial natural products. Antonie van Leeuwenhoek 2012, 101, 55–66. [Google Scholar] [CrossRef]
- CAMERA: Community CyberInfrastructure for Advanced Marine Microbial Ecology Research and Analysis. Available online: https://portal.camera.calit2.net/gridsphere/gridsphere?cid=microgenome (accessed on 22 January 2014).
- Ohnishi, Y.; Ishikawa, J.; Hara, H.; Suzuki, H.; Ikenoya, M.; Ikeda, H.; Yamashita, A.; Hattori, M.; Horinouchi, S. Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J. Bacteriol. 2008, 190, 4050–4060. [Google Scholar] [CrossRef]
- Medema, M.H.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef]
- Blin, K.; Medema, M.H.; Kazempour, D.; Fischbach, M.A.; Breitling, R.; Takano, E.; Weber, T. antiSMASH 2.0—A versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013, 41, W204–W212. [Google Scholar] [CrossRef]
- van Heel, A.J.; de Jong, A.; Montalbán-López, M.; Kok, J.; Kuipers, O.P. BAGEL3: Automated identification of genes encoding bacteriocins and (non-)bactericidal posttranslationally modified peptides. Nucleic Acids Res. 2013, 41, W448–W453. [Google Scholar]
- Bacteriocin Genome Mining Tool 3. Available online: http://bagel2.molgenrug.nl/index.php/bagel3 (accessed on 22 February 2014).
- Meier, J.L.; Burkart, M.D. The chemical biology of modular biosynthetic enzymes. Chem. Soc. Rev. 2009, 38, 2012–2045. [Google Scholar] [CrossRef]
- Li, M.; Ung, P.; Zajkowski, J.; Garneau-Tsodikova, S.; Sherman, D. Automated genome mining for natural products. BMC Bioinform. 2009, 10, 185. [Google Scholar] [CrossRef]
- NP searcher. Available online: http://dna.sherman.lsi.umich.edu/ (accessed on 22 January 2014).
- Mallika, V.; Sivakumar, K.C.; Jaichand, S.; Soniya, E.V. Kernel based machine learning algorithm for the efficient prediction of type III polyketide synthase family of proteins. J. Integr. Bioinform. 2010, 7, 143:1–143:8. [Google Scholar]
- PSIPRED Protein Sequence Analysis Workbench. Available online: http://bioinf.cs.ucl.ac.uk/psipred/ (accessed on 22 February 2014).
- Anand, S.; Prasad, M.V.R.; Yadav, G.; Kumar, N.; Shehara, J.; Ansari, M.Z.; Mohanty, D. SBSPKS: Structure based sequence analysis of polyketide synthases. Nucleic Acids Res. 2010, 38, W487–W496. [Google Scholar] [CrossRef]
- Structure-Based Sequence Analysis of PKS (SBSPKS). Available online: http://www.nii.ac.in/~pksdb/sbspks/master.html (accessed on 22 January 2014).
- Hertweck, C. The biosynthetic logic of polyketide diversity. Angew. Chem. Int. Ed. 2009, 48, 4688–4716. [Google Scholar] [CrossRef]
- Fischbach, M.A.; Walsh, C.T. Assembly-line enzymology for nolyketide and nonribosomal peptide antibiotics: Logic, machinery, and mechanisms. Chem. Rev. 2006, 106, 3468–3496. [Google Scholar] [CrossRef]
- Udwary, D.W.; Zeigler, L.; Asolkar, R.N.; Singan, V.; Lapidus, A.; Fenical, W.; Jensen, P.R.; Moore, B.S. Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinispora tropica. Proc. Natl. Acad. Sci. USA 2007, 104, 10376–10381. [Google Scholar] [CrossRef]
- Lai, J.R.; Koglin, A.; Walsh, C.T. Carrier protein structure and recognition in polyketide and nonribosomal peptide biosynthesis. Biochemistry 2006, 45, 14869–14879. [Google Scholar] [CrossRef]
- Walsh, C.T. Polyketide and nonribosomal peptide antibiotics: Modularity and versatility. Science 2004, 303, 1805–1810. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, H.; Wang, Y.; Cui, H.; Xie, Z.; Pu, Y.; Pei, S.; Li, F.; Qin, S. Genomic sequence-based discovery of novel angucyclinone antibiotics from marine Streptomyces sp. W007. FEMS Microbiol. Lett. 2012, 332, 105–112. [Google Scholar] [CrossRef]
- Zazopoulos, E.; Huang, K.; Staffa, A.; Liu, W.; Bachmann, B.O.; Nonaka, K.; Ahlert, J.; Thorson, J.S.; Shen, B.; Farnet, C.M. A genomics-guided approach for discovering and expressing cryptic metabolic pathways. Nat. Biotechnol. 2003, 21, 187–190. [Google Scholar] [CrossRef]
- Kalan, L.; Gessner, A.; Thaker, M.N.; Waglechner, N.; Zhu, X.; Szawiola, A.; Bechthold, A.; Wright, G.D.; Zechel, D.L. A cryptic polyene biosynthetic gene cluster in Streptomyces calvus is expressed upon complementation with a functional blda gene. Chem. Biol. 2013, 20, 1214–1224. [Google Scholar] [CrossRef]
- Prieto, M.L.; O’Sullivan, L.; Tan, S.P.; McLoughlin, P.; Hughes, H.; O’Connor, P.M.; Cotter, P.D.; Lawlor, P.G.; Gardiner, G.E. Assessment of the bacteriocinogenic potential of marine bacteria reveals lichenicidin production by seaweed-derived Bacillus spp. Mar. Drugs 2012, 10, 2280–2299. [Google Scholar] [CrossRef]
- Wu, C.; Tan, Y.; Gan, M.; Wang, Y.; Guan, Y.; Hu, X.; Zhou, H.; Shang, X.; You, X.; Yang, Z.; et al. Identification of elaiophylin derivatives from the marine-derived actinomycete Streptomyces sp. 7-145 using PCR-based screening. J. Nat. Prod. 2013, 76, 2153–2157. [Google Scholar]
- Wang, H.X.; Chen, Y.Y.; Ge, L.; Fang, T.T.; Meng, J.; Liu, Z.; Fang, X.Y.; Ni, S.; Lin, C.; Wu, Y.Y.; et al. PCR screening reveals considerable unexploited biosynthetic potential of ansamycins and a mysterious family of AHBA-containing natural products in actinomycetes. J. Appl. Microbiol. 2013, 115, 77–85. [Google Scholar]
- Wang, H.; Liu, N.; Xi, L.; Rong, X.; Ruan, J.; Huang, Y. Genetic screening strategy for rapid access to polyether ionophore producers and products in actinomycetes. Appl. Environ. Microbiol. 2011, 77, 3433–3442. [Google Scholar] [CrossRef]
- Hornung, A.; Bertazzo, M.; Dziarnowski, A.; Schneider, K.; Welzel, K.; Wohlert, S.-E.; Holzenkämpfer, M.; Nicholson, G.J.; Bechthold, A.; Süssmuth, R.D.; et al. A genomic screening approach to the structure-guided identification of drug candidates from natural sources. ChemBioChem 2007, 8, 757–766. [Google Scholar]
- Pearson, W.R.; Lipman, D.J. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 1988, 85, 2444–2448. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Noguchi, H.; Park, J.; Takagi, T. MetaGene: Prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 2006, 34, 5623–5630. [Google Scholar] [CrossRef]
- Lukashin, A.V.; Borodovsky, M. GeneMark: New solutions for gene finding. Nucleic Acids Res. 1998, 26, 1107–1115. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- van Loo, B.; Kingma, J.; Arand, M.; Wubbolts, M.G.; Janssen, D.B. Diversity and biocatalytic potential of epoxide hydrolases identified by genome analysis. Appl. Environ. Microbiol. 2006, 72, 2905–2917. [Google Scholar] [CrossRef]
- Kwon, Y.-C.; Lee, K.-H.; Kim, H.-C.; Han, K.; Seo, J.-H.; Kim, B.-G.; Kim, D.-M. Cloning-independent expression and analysis of ω-transaminases by use of a cell-free protein synthesis system. Appl. Environ. Microbiol. 2010, 76, 6295–6298. [Google Scholar] [CrossRef]
- Vergne-Vaxelaire, C.; Bordier, F.; Fossey, A.; Besnard-Gonnet, M.; Debard, A.; Mariage, A.; Pellouin, V.; Perret, A.; Petit, J.-L.; Stam, M.; et al. Nitrilase activity screening on structurally diverse substrates: Providing biocatalytic tools for organic synthesis. Adv. Synth. Catal. 2013, 355, 1763–1779. [Google Scholar] [CrossRef]
- Mondol, M.; Shin, H.; Islam, M. Diversity of secondary metabolites from marine Bacillus species: Chemistry and biological activity. Mar. Drugs 2013, 11, 2846–2872. [Google Scholar] [CrossRef]
- Zhang, W.; Li, Z.; Miao, X.; Zhang, F. The screening of antimicrobial bacteria with diverse novel nonribosomal peptide synthetase (NRPS) genes from South China Sea sponges. Mar. Biotechnol. 2009, 11, 346–355. [Google Scholar] [CrossRef]
- Handelsman, J. Metagenomics: Application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 2004, 68, 669–685. [Google Scholar] [CrossRef]
- Lee, H.S.; Kwon, K.K.; Kang, S.G.; Cha, S.-S.; Kim, S.-J.; Lee, J.-H. Approaches for novel enzyme discovery from marine environments. Curr. Opin. Biotechnol. 2010, 21, 353–357. [Google Scholar] [CrossRef]
- Shokralla, S.; Spall, J.L.; Gibson, J.F.; Hajibabaei, M. Next-generation sequencing technologies for environmental DNA research. Mol. Ecol. 2012, 21, 1794–1805. [Google Scholar] [CrossRef]
- Schofield, M.M.; Sherman, D.H. Meta-omic characterization of prokaryotic gene clusters for natural product biosynthesis. Curr. Opin. Biotechnol. 2013, 24, 1151–1158. [Google Scholar] [CrossRef]
- Biver, S.; Portetelle, D.; Vandenbol, M. Characterization of a new oxidant-stable serine protease isolated by functional metagenomics. SpringerPlus 2013, 2, 410. [Google Scholar] [CrossRef]
- Waschkowitz, T.; Rockstroh, S.; Daniel, R. Isolation and characterization of metalloproteases with a novel domain structure by construction and screening of metagenomic libraries. Appl. Environ. Microbiol. 2009, 75, 2506–2516. [Google Scholar] [CrossRef]
- Selvin, J.; Kennedy, J.; Lejon, D.P.H.; Kiran, G.S.; Dobson, A.D.W. Isolation identification and biochemical characterization of a novel halo-tolerant lipase from the metagenome of the marine sponge Haliclona simulans. Microb. Cell Fact. 2012, 11, 72. [Google Scholar] [CrossRef]
- Hårdeman, F.; Sjöling, S. Metagenomic approach for the isolation of a novel low-temperature-active lipase from uncultured bacteria of marine sediment. FEMS Microbiol. Ecol. 2007, 59, 524–534. [Google Scholar] [CrossRef]
- Jiang, X.; Xu, X.; Huo, Y.; Wu, Y.; Zhu, X.; Zhang, X.; Wu, M. Identification and characterization of novel esterases from a deep-sea sediment metagenome. Arch. Microbiol. 2012, 194, 207–214. [Google Scholar] [CrossRef]
- Jeon, J.H.; Lee, H.S.; Kim, J.T.; Kim, S.J.; Choi, S.H.; Kang, S.G.; Lee, J.H. Identification of a new subfamily of salt-tolerant esterases from a metagenomic library of tidal flat sediment. Appl. Microbiol. Biotechnol. 2012, 93, 623–631. [Google Scholar] [CrossRef]
- Chu, X.; He, H.; Guo, C.; Sun, B. Identification of two novel esterases from a marine metagenomic library derived from South China Sea. Appl. Microbiol. Biotechnol. 2008, 80, 615–625. [Google Scholar] [CrossRef]
- Gillespie, D.E.; Brady, S.F.; Bettermann, A.D.; Cianciotto, N.P.; Liles, M.R.; Rondon, M.R.; Clardy, J.; Goodman, R.M.; Handelsman, J. Isolation of antibiotics turbomycin A and B from a metagenomic library of soil microbial DNA. Appl. Environ. Microbiol. 2002, 68, 4301–4306. [Google Scholar] [CrossRef]
- Wang, G.-Y.-S.; Graziani, E.; Waters, B.; Pan, W.; Li, X.; McDermott, J.; Meurer, G.; Saxena, G.; Andersen, R.J.; Davies, J. Novel natural products from soil DNA libraries in a streptomycete host. Org. Lett. 2000, 2, 2401–2404. [Google Scholar] [CrossRef]
- Brady, S.F.; Clardy, J. Synthesis of long-chain fatty acid enol esters isolated from an environmental DNA clone. Org. Lett. 2002, 5, 121–124. [Google Scholar]
- Feng, Z.; Kallifidas, D.; Brady, S.F. Functional analysis of environmental DNA-derived type II polyketide synthases reveals structurally diverse secondary metabolites. Proc. Natl. Acad. Sci. USA 2011, 108, 12629–12634. [Google Scholar] [CrossRef]
- Brady, S.F.; Simmons, L.; Kim, J.H.; Schmidt, E.W. Metagenomic approaches to natural products from free-living and symbiotic organisms. Nat. Prod. Rep. 2009, 26, 1488–1503. [Google Scholar] [CrossRef]
- Banik, J.J.; Brady, S.F. Recent application of metagenomic approaches toward the discovery of antimicrobials and other bioactive small molecules. Curr. Opin. Microbiol. 2010, 13, 603–609. [Google Scholar] [CrossRef]
- Robertson, D.E.; Chaplin, J.A.; DeSantis, G.; Podar, M.; Madden, M.; Chi, E.; Richardson, T.; Milan, A.; Miller, M.; Weiner, D.P.; et al. Exploring nitrilase sequence space for enantioselective catalysis. Appl. Environ. Microbiol. 2004, 70, 2429–2436. [Google Scholar] [CrossRef]
- Bayer, S.; Birkemeyer, C.; Ballschmiter, M. A nitrilase from a metagenomic library acts regioselectively on aliphatic dinitriles. Appl. Microbiol. Biotechnol. 2011, 89, 91–98. [Google Scholar] [CrossRef]
- Johnston, C.; Ibrahim, A.; Magarvey, N. Informatic strategies for the discovery of polyketides and nonribosomal peptides. MedChemComm 2012, 3, 932–937. [Google Scholar] [CrossRef]
- Cottrell, M.T.; Yu, L.; Kirchman, D.L. Sequence and expression analyses of Cytophaga-like hydrolases in a Western Arctic metagenomic library and the Sargasso Sea. Appl. Environ. Microbiol. 2005, 71, 8506–8513. [Google Scholar] [CrossRef]
- Xu, M.; Xiao, X.; Wang, F. Isolation and characterization of alkane hydroxylases from a metagenomic library of Pacific deep-sea sediment. Extremophiles 2008, 12, 255–262. [Google Scholar] [CrossRef]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental Genome Shotgun Sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef]
- Bayer, K.; Scheuermayer, M.; Fieseler, L.; Hentschel, U. Genomic mining for novel FADH2-dependent halogenases in marine sponge-associated microbial consortia. Mar. Biotechnol. 2013, 15, 63–72. [Google Scholar] [CrossRef]
- Bayer, T.S.; Widmaier, D.M.; Temme, K.; Mirsky, E.A.; Santi, D.V.; Voigt, C.A. Synthesis of methyl halides from biomass using engineered microbes. J. Am. Chem. Soc. 2009, 131, 6508–6515. [Google Scholar]
- Stepanauskas, R.; Sieracki, M.E. Matching phylogeny and metabolism in the uncultured marine bacteria, one cell at a time. Proc. Natl. Acad. Sci. USA 2007, 104, 9052–9057. [Google Scholar] [CrossRef]
- Siegl, A.; Kamke, J.; Hochmuth, T.; Piel, J.; Richter, M.; Liang, C.; Dandekar, T.; Hentschel, U. Single-cell genomics reveals the lifestyle of Poribacteria, a candidate phylum symbiotically associated with marine sponges. ISME J. 2011, 5, 61–70. [Google Scholar] [CrossRef]
- Williamson, L.L.; Borlee, B.R.; Schloss, P.D.; Guan, C.; Allen, H.K.; Handelsman, J. Intracellular screen to identify metagenomic clones that induce or inhibit a quorum-sensing biosensor. Appl. Environ. Microbiol. 2005, 71, 6335–6344. [Google Scholar] [CrossRef]
- Uchiyama, T.; Miyazaki, K. Substrate-induced gene expression screening: A method for high-throughput screening of metagenome libraries. In Metagenomics: Methods and Protocols; Humana Press: Clifton, NJ, USA, 2010; Volume 668, pp. 153–168. [Google Scholar]
- Uchiyama, T.; Watanabe, K. Substrate-induced gene expression (SIGEX) screening of metagenome libraries. Nat. Protoc. 2008, 3, 1202–1212. [Google Scholar] [CrossRef]
- Meiring, T.; Mulako, I.; Tuffin, M.; Meyer, Q.; Cowan, D. Retrieval of full-length functional genes using subtractive hybridization magnetic bead capture. In Metagenomics: Methods and Protocols; Streit, W.R., Daniel, R., Eds.; Humana Press: Clifton, NJ, USA, 2010; Volume 668, pp. 287–297. [Google Scholar]
- Meyer, Q.C.; Burton, S.G.; Cowan, D.A. Subtractive hybridization magnetic bead capture: A new technique for the recovery of full-length ORFs from the metagenome. Biotechnol. J. 2007, 2, 36–40. [Google Scholar]
- Margassery, L.M.; Kennedy, J.; O’Gara, F.; Dobson, A.D.; Morrissey, J.P. A high-throughput screen to identify novel calcineurin inhibitors. J. Microbiol. Methods 2012, 88, 63–66. [Google Scholar] [CrossRef]
- Menzella, H.G.; Reeves, C.D. Combinatorial biosynthesis for drug development. Curr. Opin. Microbiol. 2007, 10, 238–245. [Google Scholar] [CrossRef]
- Wong, F.T.; Khosla, C. Combinatorial biosynthesis of polyketides-a perspective. Curr. Opin. Chem. Biol. 2012, 16, 117–123. [Google Scholar] [CrossRef]
- Fisch, K.M. Biosynthesis of natural products by microbial iterative hybrid PKS-NRPS. RSC Adv. 2013, 3, 18228–18247. [Google Scholar] [CrossRef]
- Kim, E.J.; Lee, J.H.; Choi, H.; Pereira, A.R.; Ban, Y.H.; Yoo, Y.J.; Kim, E.; Park, J.W.; Sherman, D.H.; Gerwick, W.H.; et al. Heterologous production of 4-O-demethylbarbamide, a marine cyanobacterial natural product. Org. Lett. 2012, 14, 5824–5827. [Google Scholar] [CrossRef]
- Doekel, S.; Coëffet-Le Gal, M.-F.; Gu, J.-Q.; Chu, M.; Baltz, R.H.; Brian, P. Non-ribosomal peptide synthetase module fusions to produce derivatives of daptomycin in Streptomyces roseosporus. Microbiology 2008, 154, 2872–2880. [Google Scholar] [CrossRef]
- Seipke, R.F.; Hutchings, M.I. The regulation and biosynthesis of antimycins. Beilstein J. Org. Chem. 2013, 9, 2556–2563. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Chen, J.; Zhang, L.; Zheng, Q.; Han, Y.; Zhang, H.; Zhang, D.; Awakawa, T.; Abe, I.; Liu, W. Multiplexing of combinatorial chemistry in antimycin biosynthesis: Expansion of molecular diversity and utility. Angew. Chem. Int. Ed. 2013, 52, 12308–12312. [Google Scholar] [CrossRef]
- Eustáquio, A.S.; O’Hagan, D.; Moore, B.S. Engineering fluorometabolite production: Fluorinase expression in Salinispora tropica yields fluorosalinosporamide. J. Nat. Prod. 2010, 73, 378–382. [Google Scholar] [CrossRef]
- Winter, J.M.; Tang, Y. Synthetic biological approaches to natural product biosynthesis. Curr. Opin. Biotechnol. 2012, 23, 736–743. [Google Scholar]
- Isaacs, F.J.; Carr, P.A.; Wang, H.H.; Lajoie, M.J.; Sterling, B.; Kraal, L.; Tolonen, A.C.; Gianoulis, T.A.; Goodman, D.B.; Reppas, N.B.; et al. Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science 2011, 333, 348–353. [Google Scholar] [CrossRef]
- Wang, H.H.; Isaacs, F.J.; Carr, P.A.; Sun, Z.Z.; Xu, G.; Forest, C.R.; Church, G.M. Programming cells by multiplex genome engineering and accelerated evolution. Nature 2009, 460, 894–898. [Google Scholar] [CrossRef]
- Khalil, A.S.; Collins, J.J. Synthetic biology: Applications come of age. Nat. Rev. Genet. 2010, 11, 367–379. [Google Scholar] [CrossRef]
- Keasling, J.D. Manufacturing molecules through metabolic engineering. Science 2010, 330, 1355–1358. [Google Scholar] [CrossRef]
- Park, S.R.; Park, J.W.; Ban, Y.H.; Sohng, J.K.; Yoon, Y.J. 2-Deoxystreptamine-containing aminoglycoside antibiotics: Recent advances in the characterization and manipulation of their biosynthetic pathways. Nat. Prod. Rep. 2013, 30, 11–20. [Google Scholar] [CrossRef]
- Yuzawa, S.; Kim, W.; Katz, L.; Keasling, J.D. Heterologous production of polyketides by modular type I polyketide synthases in Escherichia coli. Curr. Opin. Biotechnol. 2012, 23, 727–735. [Google Scholar] [CrossRef]
- Gao, X.; Wang, P.; Tang, Y. Engineered polyketide biosynthesis and biocatalysis in Escherichia coli. Appl. Microbiol. Biotechnol. 2010, 88, 1233–1242. [Google Scholar] [CrossRef]
- Zakeri, B.; Lu, T.K. Synthetic biology of antimicrobial discovery. ACS Synth. Biol. 2013, 2, 358–372. [Google Scholar] [CrossRef]
- Carbonell, P.; Planson, A.-G.; Faulon, J.-L. Retrosynthetic design of heterologous pathways. In Systems Metabolic Engineering: Methods and Protocols; Alper, H.S., Ed.; Humana Press: Clifton, NJ, USA, 2013; Volume 985, pp. 149–173. [Google Scholar]
- Turner, N.J.; O’Reilly, E. Biocatalytic retrosynthesis. Nat. Chem. Biol. 2013, 9, 285–288. [Google Scholar] [CrossRef]
- Loeschcke, A.; Markert, A.; Wilhelm, S.; Wirtz, A.; Rosenau, F.; Jaeger, K.-E.; Drepper, T. TREX: A universal tool for the transfer and expression of biosynthetic pathways in bacteria. ACS Synth. Biol. 2012, 2, 22–33. [Google Scholar]
- Wang, J.; Xiong, Z.; Meng, H.; Wang, Y.; Wang, Y. Synthetic biology triggers new era of antibiotics development. In Reprogramming Microbial Metabolic Pathways; Wang, X., Chen, J., Quinn, P., Eds.; Springer: Dordrecht, The Netherlands, 2012; Volume 64, pp. 95–114. [Google Scholar]
- Gomez-Escribano, J.; Bibb, M. Heterologous expression of natural product biosynthetic gene clusters in Streptomyces coelicolor: From genome mining to manipulation of biosynthetic pathways. J. Ind. Microbiol. Biotechnol. 2014, 41, 425–431. [Google Scholar] [CrossRef]
- Zhang, H.; Boghigian, B.A.; Armando, J.; Pfeifer, B.A. Methods and options for the heterologous production of complex natural products. Nat. Prod. Rep. 2011, 28, 125–151. [Google Scholar] [CrossRef]
- Pfeifer, B.A.; Admiraal, S.J.; Gramajo, H.; Cane, D.E.; Khosla, C. Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science 2001, 291, 1790–1792. [Google Scholar] [CrossRef]
- Kao, C.; Katz, L.; Khosla, C. Engineered biosynthesis of a complete macrolactone in a heterologous host. Science 1994, 265, 509–512. [Google Scholar]
- Ichinose, K.; Bedford, D.J.; Tornus, D.; Bechthold, A.; Bibb, M.J.; Peter Revill, W.; Floss, H.G.; Hopwood, D.A. The granaticin biosynthetic gene cluster of Streptomyces violaceoruber Tü22: Sequence analysis and expression in a heterologous host. Chem. Biol. 1998, 5, 647–659. [Google Scholar] [CrossRef]
- Ichinose, K.; Ozawa, M.; Itou, K.; Kunieda, K.; Ebizuka, Y. Cloning, sequencing and heterologous expression of the medermycin biosynthetic gene cluster of Streptomyces sp. AM-7161: Towards comparative analysis of the benzoisochromanequinone gene clusters. Microbiology 2003, 149, 1633–1645. [Google Scholar] [CrossRef]
- Tang, L.S. Cloning and heterologous expression of the epothilone gene cluster. Science 2000, 287, 640. [Google Scholar] [CrossRef]
- Bedford, D.J.; Schweizer, E.; Hopwood, D.A.; Khosla, C. Expression of a functional fungal polyketide synthase in the bacterium Streptomyces coelicolor A3(2). J. Bacteriol. 1995, 177, 4544–4548. [Google Scholar]
- Eustáquio, A.S.; Gust, B.; Galm, U.; Li, S.-M.; Chater, K.F.; Heide, L. Heterologous expression of novobiocin and clorobiocin biosynthetic gene clusters. Appl. Environ. Microbiol. 2005, 71, 2452–2459. [Google Scholar] [CrossRef]
- Zhang, W.; Ames, B.D.; Tsai, S.-C.; Tang, Y. Engineered biosynthesis of a novel amidated polyketide, using the malonamyl-specific initiation module from the oxytetracycline polyketide synthase. Appl. Environ. Microbiol. 2006, 72, 2573–2580. [Google Scholar] [CrossRef]
- Winter, J.M.; Moffitt, M.C.; Zazopoulos, E.; McAlpine, J.B.; Dorrestein, P.C.; Moore, B.S. Molecular basis for chloronium-mediated meroterpene cyclization: Cloning, sequencing, and heterologous expression of the napyradiomycin biosynthetic gene cluster. J. Biol. Chem. 2007, 282, 16362–16368. [Google Scholar] [CrossRef]
- Ito, T.; Roongsawang, N.; Shirasaka, N.; Lu, W.; Flatt, P.M.; Kasanah, N.; Miranda, C.; Mahmud, T. Deciphering pactamycin biosynthesis and engineered production of new pactamycin analogues. ChemBioChem 2009, 10, 2253–2265. [Google Scholar] [CrossRef]
- Penn, J.; Li, X.; Whiting, A.; Latif, M.; Gibson, T.; Silva, C.; Brian, P.; Davies, J.; Miao, V.; Wrigley, S.; et al. Heterologous production of daptomycin in Streptomyces lividans. J. Ind. Microbiol. Biotechnol. 2006, 33, 121–128. [Google Scholar] [CrossRef]
- Pfeifer, B.A.; Wang, C.C.C.; Walsh, C.T.; Khosla, C. Biosynthesis of yersiniabactin, a complex polyketide-nonribosomal peptide, using Escherichia coli as a heterologous host. Appl. Environ. Microbiol. 2003, 69, 6698–6702. [Google Scholar] [CrossRef]
- Mutka, S.C.; Carney, J.R.; Liu, Y.; Kennedy, J. Heterologous production of epothilone C and D in Escherichia coli. Biochemistry 2006, 45, 1321–1330. [Google Scholar] [CrossRef]
- Gruenewald, S.; Mootz, H.D.; Stehmeier, P.; Stachelhaus, T. In vivo production of artificial nonribosomal peptide products in the heterologous host Escherichia coli. Appl. Environ. Microbiol. 2004, 70, 3282–3291. [Google Scholar] [CrossRef]
- Watanabe, K.; Hotta, K.; Praseuth, A.P.; Koketsu, K.; Migita, A.; Boddy, C.N.; Wang, C.C.C.; Oguri, H.; Oikawa, H. Total biosynthesis of antitumor nonribosomal peptides in Escherichia coli. Nat. Chem. Biol. 2006, 2, 423–428. [Google Scholar] [CrossRef]
- Fu, J.; Wenzel, S.C.; Perlova, O.; Wang, J.; Gross, F.; Tang, Z.; Yin, Y.; Stewart, A.F.; Müller, R.; Zhang, Y. Efficient transfer of two large secondary metabolite pathway gene clusters into heterologous hosts by transposition. Nucleic Acids Res. 2008, 36, e113. [Google Scholar] [CrossRef]
- Rath, C.M.; Janto, B.; Earl, J.; Ahmed, A.; Hu, F.Z.; Hiller, L.; Dahlgren, M.; Kreft, R.; Yu, F.; Wolff, J.J.; et al. Meta-omic characterization of the marine invertebrate microbial consortium that produces the chemotherapeutic natural product ET-743. ACS Chem. Biol. 2011, 6, 1244–1256. [Google Scholar] [CrossRef]
- Phelan, R.; Barret, M.; Cotter, P.; Connor, P.; Chen, R.; Morrissey, J.; Dobson, A.; Gara, F.; Barbosa, T. Subtilomycin: A new lantibiotic from Bacillus subtilis strain MMA7 isolated from the marine sponge Haliclona simulans. Mar. Drugs 2013, 11, 1878–1898. [Google Scholar] [CrossRef]
- Kersten, R.D.; Ziemert, N.; Gonzalez, D.J.; Duggan, B.M.; Nizet, V.; Dorrestein, P.C.; Moore, B.S. Glycogenomics as a mass spectrometry-guided genome-mining method for microbial glycosylated molecules. Proc. Natl. Acad. Sci. USA 2013, 110, E4407–E4416. [Google Scholar]
- Bumpus, S.B.; Evans, B.S.; Thomas, P.M.; Ntai, I.; Kelleher, N.L. A proteomics approach to discovering natural products and their biosynthetic pathways. Nat. Biotechnol. 2009, 27, 951–956. [Google Scholar] [CrossRef]
- Chen, Y.; McClure, R.A.; Zheng, Y.; Thomson, R.J.; Kelleher, N.L. Proteomics guided discovery of flavopeptins: Anti-proliferative aldehydes synthesized by a reductase domain-containing non-ribosomal peptide synthetase. J. Am. Chem. Soc. 2013, 135, 10449–10456. [Google Scholar] [CrossRef]
- Evans, B.S.; Ntai, I.; Chen, Y.; Robinson, S.J.; Kelleher, N.L. Proteomics-based discovery of koranimine, a cyclic imine natural product. J. Am. Chem. Soc. 2011, 133, 7316–7319. [Google Scholar]
- Meier, J.L.; Burkart, M.D. Proteomic analysis of polyketide and nonribosomal peptide biosynthesis. Curr. Opin. Chem. Biol. 2011, 15, 48–56. [Google Scholar] [CrossRef]
- Meier, J.L.; Niessen, S.; Hoover, H.S.; Foley, T.L.; Cravatt, B.F.; Burkart, M.D. An orthogonal active site identification system (OASIS) for proteomic profiling of natural product biosynthesis. ACS Chem. Biol. 2009, 4, 948–957. [Google Scholar]
- Yang, J.Y.; Sanchez, L.M.; Rath, C.M.; Liu, X.; Boudreau, P.D.; Bruns, N.; Glukhov, E.; Wodtke, A.; de Felicio, R.; Fenner, A.; et al. Molecular networking as a dereplication strategy. J. Nat. Prod. 2013, 76, 1686–1699. [Google Scholar] [CrossRef]
- Forner, D.; Berrué, F.; Correa, H.; Duncan, K.; Kerr, R.G. Chemical dereplication of marine actinomycetes by liquid chromatography–high resolution mass spectrometry profiling and statistical analysis. Anal. Chim. Acta 2013, 805, 70–79. [Google Scholar] [CrossRef]
- FastGroupII. Available online: http://fastgroup.sdsu.edu/ (accessed on 22 February 2014).
- BACTIBASE. Available online: http://bactibase.pfba-lab-tun.org/main.php (accessed on 22 February 2014).
- Laatsch, H. Antibase Version 4.0; Wiley-VCH Verlag GmbH & Co: Weinheim, Germany, 2012. [Google Scholar]
- Hammami, R.; Zouhir, A.; Le Lay, C.; Ben Hamida, J.; Fliss, I. BACTIBASE second release: A database and tool platform for bacteriocin characterization. BMC Microbiol. 2010, 10, 22. [Google Scholar] [CrossRef]
- Yu, Y.; Breitbart, M.; McNairnie, P.; Rohwer, F. FastGroupII: A web-based bioinformatics platform for analyses of large 16S rDNA libraries. BMC Bioinform. 2006, 7, 57. [Google Scholar] [CrossRef]
- Microbial Screening Technologies. Available online: http://www.microbialscreening.com/dereplication.htm (accessed on 23 February 2014).
- Murray, P.M.; Moane, S.; Collins, C.; Beletskaya, T.; Thomas, O.P.; Duarte, A.W.F.; Nobre, F.S.; Owoyemi, I.O.; Pagnocca, F.C.; Sette, L.D.; et al. Sustainable production of biologically active molecules of marine based origin. N. Biotechnol. 2013, 30, 839–850. [Google Scholar] [CrossRef]
- Carr, P.A.; Wang, H.H.; Sterling, B.; Isaacs, F.J.; Lajoie, M.J.; Xu, G.; Church, G.M.; Jacobson, J.M. Enhanced multiplex genome engineering through co-operative oligonucleotide co-selection. Nucleic Acids Res. 2012, 40, e132. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rocha-Martin, J.; Harrington, C.; Dobson, A.D.W.; O'Gara, F. Emerging Strategies and Integrated Systems Microbiology Technologies for Biodiscovery of Marine Bioactive Compounds. Mar. Drugs 2014, 12, 3516-3559. https://doi.org/10.3390/md12063516
Rocha-Martin J, Harrington C, Dobson ADW, O'Gara F. Emerging Strategies and Integrated Systems Microbiology Technologies for Biodiscovery of Marine Bioactive Compounds. Marine Drugs. 2014; 12(6):3516-3559. https://doi.org/10.3390/md12063516
Chicago/Turabian StyleRocha-Martin, Javier, Catriona Harrington, Alan D.W. Dobson, and Fergal O'Gara. 2014. "Emerging Strategies and Integrated Systems Microbiology Technologies for Biodiscovery of Marine Bioactive Compounds" Marine Drugs 12, no. 6: 3516-3559. https://doi.org/10.3390/md12063516