Four New Jacaranone Analogs from the Fruits of a Beibu Gulf Mangrove Avicennia marina

Abstract
:1. Introduction

2. Results and Discussion

{kind=link}
{kind=link}
| 1 | 2 | |||
|---|---|---|---|---|
| δC, Mult | δH (J in Hz) | δC, Mult | δH (J in Hz) | |
| 1 | 70.2, C | 70.2, C | ||
| 2 | 148.9, CH | 7.16 (d, 2.8) | 148.6, CH | 7.20 (d, 2.8) |
| 3 | 130.5, C | 130.0, C | ||
| 4 | 179.3, C | 179.1, C | ||
| 5 | 125.8, CH | 6.22 (d, 10.0) | 125.4, CH | 6.19 (d, 10.0) |
| 6 | 152.8, CH | 6.99 (dd, 10.0, 2.8) | 153.1, CH | 7.03 (dd, 10.0, 2.8) |
| 1′ | 39.6, CH2 | 2.08 (dd, 11.2, 7.0) | 39.7, CH2 | 2.07 (dd, 11.2, 7.0) |
| 2′α | 63.9, CH2 | 3.94 (ddd, 10.1, 7.0, 2.2) | 64.0, CH2 | 4.00 (dt, 10.4, 7.0, 2.4) |
| β | 3.64 (dd, 10.1, 2.2) | 3.65 (dd, 10.4, 2.4) | ||
| 1ʺ | 102.6, CH | 4.18 (d, 9.2) | 102.8, CH | 4.21 (d, 7.8) |
| 2ʺ | 73.6, CH | 3.13 (9.6, 9.2) | 73.6, CH | 3.14 (dd, 9.6, 7.8) |
| 3ʺ | 76.6, CH | 3.20 (m) | 76.6, CH | 3.12 (m) |
| 4ʺ | 70.1, CH | 3.24 (m) | 69.9, CH | 3.24 (m) |
| 5ʺ | 76.5, CH | 3.29 (m) | 76.6, CH | 3.31 (m) |
| 6ʺα | 61.3, CH2 | 3.81 (dd, 11.9, 2.0) | 61.4, CH2 | 3.84 (d, 11.8) |
| β | 3.62 (m) | 3.65 (m) | ||
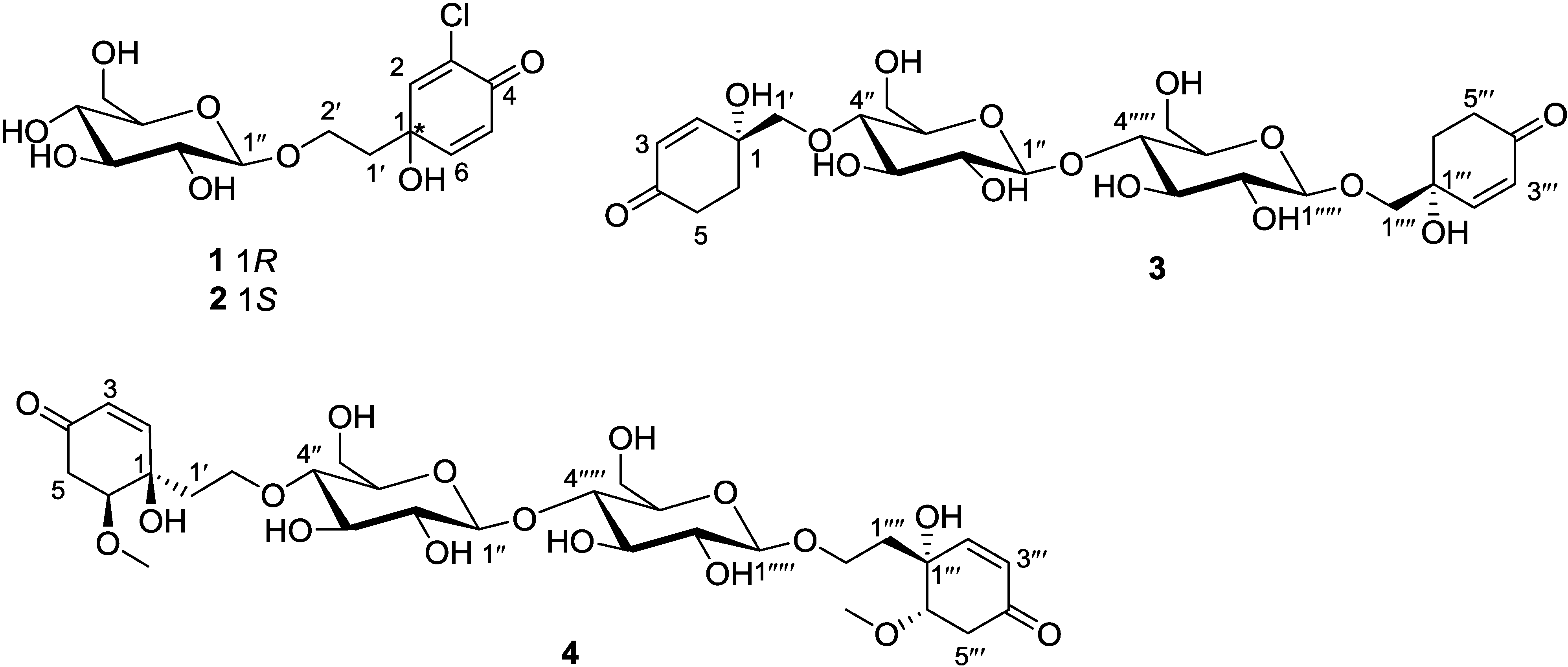
 (MeOH) of −14.7°. The reported rotation value for cornoside is negative −10.5° [13], whose stereochemistry of the aglycone and the β-d-glucosyl residue have been established by enzymatic hydrolysis and other methods [13,14,15]. The reported rotation value after poly-acetylation of cornoside is also negative ( −10.2°) [16]. Moreover, the NMR data of the β-glucosyl residue in compound 1 are in accord with those observed in cornoside [13]. The substitution is simply that of one Cl atom for one H atom, and that many bonds away from the key chiral centre, therefore following those Literature data, we propose that the configuration of C-1 in compound 1 is the same as that found at C-1 in cornoside, namely R. Thus, the structure of 1 is predicted to be as shown in Figure 1. (MeOH) of +10.2°, and the reported value for cornoside ( −10.5°) and poly-acetylated cornoside ( −10.2°) are both negative [13], and the observed value for 1 is −14.7°. Although we do not have an explanation for the differences in the absolute values of the optical rotations of cornoside, poly-acetylated of cornoside, and compound 1, and the NMR data of the β-glucosyl residue in compound 2 are greatly similar to those of the β-glucosyl residue in cornoside and compound 1, and also compound 2 showed a positive Cotton effect at 220 nm (Δε +5.97), whereas the observed value for 1 was −4.28, the opposite optical rotation and Cotton effect indicate that 1 and 2 are diastereoisomers. Indeed, we propose that they are enantiomers of the aglycone each with β-glucosyl residues.
(MeOH) of −14.7°. The reported rotation value for cornoside is negative −10.5° [13], whose stereochemistry of the aglycone and the β-d-glucosyl residue have been established by enzymatic hydrolysis and other methods [13,14,15]. The reported rotation value after poly-acetylation of cornoside is also negative ( −10.2°) [16]. Moreover, the NMR data of the β-glucosyl residue in compound 1 are in accord with those observed in cornoside [13]. The substitution is simply that of one Cl atom for one H atom, and that many bonds away from the key chiral centre, therefore following those Literature data, we propose that the configuration of C-1 in compound 1 is the same as that found at C-1 in cornoside, namely R. Thus, the structure of 1 is predicted to be as shown in Figure 1. (MeOH) of +10.2°, and the reported value for cornoside ( −10.5°) and poly-acetylated cornoside ( −10.2°) are both negative [13], and the observed value for 1 is −14.7°. Although we do not have an explanation for the differences in the absolute values of the optical rotations of cornoside, poly-acetylated of cornoside, and compound 1, and the NMR data of the β-glucosyl residue in compound 2 are greatly similar to those of the β-glucosyl residue in cornoside and compound 1, and also compound 2 showed a positive Cotton effect at 220 nm (Δε +5.97), whereas the observed value for 1 was −4.28, the opposite optical rotation and Cotton effect indicate that 1 and 2 are diastereoisomers. Indeed, we propose that they are enantiomers of the aglycone each with β-glucosyl residues.
| 3 | 4 | |||
|---|---|---|---|---|
| δC, Mult | δH (J in Hz) | δC, Mult | δH (J in Hz) | |
| 1 | 70.9, C | 72.4, C | ||
| 2 | 152.8, CH | 6.93 (d, 15.1) | 154.7, CH | 6.95 (d, 10.0) |
| 3 | 128.1, CH | 5.93 (dd, 15.1, 2.6) | 127.1, CH | 5.87 (d, 10.0) |
| 4 | 199.1, C | 198.6, C | ||
| 5α | 42.1, CH2 | 2.68 (m) | 38.9, CH2 | 2.82 (dd, 11.1, 3.6) |
| β | 2.49 (m) | 2.49 (dd, 11.1, 7.0) | ||
| 6α | 37.3, CH2 | 2.09 (m) | 82.8, CH | 3.67 (dd, 7.0, 3.6) |
| β | 2.03 (m) | |||
| 1ʹα | 61.3, CH2 | 3.83 (d, 10.1) | 34.4, CH2 | 2.18 (dd, 11.1, 5.3) |
| β | 3.85 (d, 10.1) | 2.00 (dd, 11.1, 6.1) | ||
| 2ʹα | 61.3, CH2 | 3.86 (d, 10.1) | ||
| β | 3.79 (d, 10.1) | |||
| 1ʺ | 102.9, CH | 4.27 (d, 7.8) | 103.0, CH | 4.28 (d, 7.5) |
| 2ʺ | 73.6, CH | 3.13 (dd, 9.4, 7.8) | 73.6, CH | 3.14 (dd, 9.4, 7.8) |
| 3ʺ | 70.9, CH | 4.08 (m) | 70.2, CH | 3.26 (m) |
| 4ʺ | 76.6, CH | 3.32 (m) | 76.7, CH | 3.33 (m) |
| 5ʺ | 76.6, CH | 3.25 (m) | 76.7, CH | 3.26 (m) |
| 6ʺα | 64.9, CH2 | 4.13 (dt, 9.5, 5.0) | 65.1, CH2 | 4.16 (dt, 9.5, 5.0) |
| β | 4.09 (dt, 9.5, 5.0) | 4.14 (dt, 9.5, 5.0) | ||
| 1‴ | 70.7, C | 72.4, C | ||
| 2‴ | 152.5, CH | 6.90 (d, 15.1) | 154.0, CH | 6.93 (d, 10.0) |
| 3‴ | 127.8, CH | 5.92 (dd, 15.1, 2.6) | 127.0, CH | 5.85 (d, 10.0) |
| 4‴ | 199.0, C | 198.6, C | ||
| 5‴α | 42.0, CH2 | 2.67 (m) | 38.9, CH2 | 2.82 (dd, 11.1, 3.6) |
| β | 2.49 (dd, 11.1, 7.0) | |||
| 6‴α | 37.2, CH2 | 2.08 (m)2.01 (m) | 82.7, CH | 3.64 (dd, 7.0, 3.6) |
| β | 2.01 (m) | |||
| 1ʺʺα | 61.3, CH2 | 3.63 (d, 10.1) | 34.3, CH2 | 2.17 (dd, 11.1, 5.3) |
| β | 3.65(d, 10.1) | 1.98 (dd, 11.1, 6.1) | ||
| 2ʺʺα | 61.3, CH2 | 3.84 (d, 10.1) | ||
| β | 3.64(d, 10.1) | |||
| 1ʺ‴ | 102.8, CH | 4.25 (d, 7.8) | 102.8, CH | 4.27 (d, 7.5) |
| 2ʺ‴ | 73.6, CH | 3.13 (dd, 9.4, 7.8) | 73.6, CH | 3.14 (dd, 9.4, 7.8) |
| 3ʺ‴ | 70.2, CH | 3.25 (m) | 70.2, CH | 3.24 (m) |
| 4ʺ‴ | 76.6, CH | 3.31 (m) | 76.6, CH | 3.31 (m) |
| 5ʺ‴ | 76.6, CH | 3.31 (m) | 76.6, CH | 3.24 (m) |
| 6ʺ‴α | 64.9, CH2 | 3.78 (dt, 9.8, 5.5) | 64.8, CH2 | 3.82 (dt, 9.8, 5.5) |
| β | 3.78 (dt, 9.8, 5.5) | 3.80 (dt, 9.8, 5.5) | ||
| OCH3 | 56.9, CH3 | 3.43 (s) | ||
| OCH3 | 56.9, CH3 | 3.43 (s) | ||
was observed, which was in accord with that observed in cornoside ( −10.5°), and analysis of the 13C NMR data of C-1 and C-1‴ in 3 indicated that they were greatly similar to that of C-1 (δC 68.9) in 4-[2-(β-d-glucopyranosyloxy)ethyl]-4-hydroxy-2-cyclohexen-1-one obtained from Millingtonia hortensis[19]. Consequently, the structure of 3 was determined as showed in Figure 1. in MeOH of −26.4°, which is in accord with that observed in 1,6-dihydroxy-4-oxo-2-cyclohexene-1-acetic acid ethyl ester ( −12.5°) [17]. From the aforementioned analyses, the configurations of 4 were assumed to be 1R, 6S, 1‴R and 6‴S. On the basis of this cumulative analysis, the structure of 4 was thus determined as shown in Figure 1.3. Experimental Section
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
−14.7° (c 0.18, MeOH); CD (MeOH) Δε220nm −4.28, IR (KBr) νmax 3425, 1720 and 1682 cm−1. 1H (CD3OD, 600 MHz) and 13C (CD3OD, 150 MHz) NMR data, see Table 1; HRESIMS: m/z 351.0835 (calcd. for C14H19ClO8 + H, 351.0841). +10.2° (c 0.21, MeOH); CD (MeOH) Δε220nm +5.97; IR (KBr) νmax 3424, 1711 and 1684 cm−1. 1H (CD3OD, 600 MHz) and 13C (CD3OD, 150 MHz) NMR data, see Table 1; HRESIMS: m/z 351.0837 (calcd. for C14H19ClO8 + H, 351.0841). −15.8° (c 0.25, MeOH); CD (MeOH) Δε220nm −6.32; IR (KBr) νmax 3452, 1701 and 1675 cm−1. 1H (CD3OD, 600 MHz) and 13C (CD3OD, 150 MHz) NMR data, see Table 1; HRESIMS: m/z 591.2281 (calcd. for C26H38lO15 + H, 591.2283). −26.4° (c 0.31, MeOH); CD (MeOH) Δε220nm −5.42; IR (KBr) νmax 3447, 1705 and 1679 cm−1. 1H (CD3OD, 600 MHz) and 13C (CD3OD, 150 MHz) NMR data, see Table 1; HRESIMS: m/z 701.2630 (calcd. for C30H46O17 + Na, 701.2633).3.4. Cellular Antioxidant Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sun, Y.; Ouyang, J.; Deng, Z.W.; Li, Q.S.; Lin, W.H. Structure elucidation of five new iridoid glucosides from the leaves of Avicennia marina. Magn. Reson. Chem. 2008, 46, 638–642. [Google Scholar] [CrossRef]
- Fauvel, M.T.; Taoubi, K.; Gleye, J.; Fourasté, I. Phenylpropanoid glycosides from Avicennia marina. Planta Med. 1993, 59, 387. [Google Scholar]
- Konig, G.; Rimpler, H. Iridoid glucosides in Avicennia marina. Phytochemistry 1985, 24, 1245–1248. [Google Scholar] [CrossRef]
- Feng, Y.; Li, X.M.; Duan, X.J.; Wang, B.G. Iridoid glucosides and flavones from the aerial parts of Avicennia marina. Chem. Biodivers. 2006, 3, 799–806. [Google Scholar] [CrossRef]
- Fauvel, M.T.; Bousquetmelou, A.; Moulis, C.; Gleye, J.; Jensen, S.R. Iridoid glucosides from Avicennia germinans. Phytochemistry 1995, 38, 893–894. [Google Scholar]
- Ito, C.; Katsuno, S.; Kondo, Y.; Tan, H.T.W.; Furukawa, H. Chemical constituents of Avicennia alba. Isolation and structural elucidation of new naphthoquinones and their analogues. Chem. Pharm. Bull. 2000, 48, 339–343. [Google Scholar] [CrossRef]
- Han, L.; Huang, X.S.; Dahse, H.M.; Moellmann, U.; Fu, H.Z.; Grabley, S.; Sattler, I.; Lin, W.H. Unusual naphthoquinone derivatives from the twigs of Avicennia marina. J. Nat. Prod. 2007, 70, 923–927. [Google Scholar] [CrossRef]
- Sharaf, M.; El-Ansari, M.A.; Saleh, N.A.M. New flavonoids from Avicennia marina. Fitoterapia 2000, 71, 274–277. [Google Scholar] [CrossRef]
- Han, L.; Huang, X.S.; Dahse, H.M.; Moellmann, U.; Grabley, S.; Lin, W.H.; Sattler, I. New abietane diterpenoids from the mangrove Avicennia marina. Planta Med. 2008, 74, 432–437. [Google Scholar] [CrossRef]
- Parker, K.A.; Kang, S.K. Regiospecific nucleophilic aromatic-substitution—Conjugate addition of active methylene-compounds to quinone monoacetals and aromatization of the adducts. J. Org. Chem. 1980, 45, 1218–1224. [Google Scholar] [CrossRef]
- Hollenst, R.; Philipsb, W.V. Carbon magnetic-resonance spectra of α, β, γ, σ- and α, β, α′, β′-unsaturated-ketones. Helv. Chim. Acta 1972, 55, 2030–2044. [Google Scholar] [CrossRef]
- Breitmaier, E.; Voelter, W. Carbon-13 NMR Spectroscopy. VCH Publishers: New York, NY, USA, 1987; pp. 380–394. [Google Scholar]
- Endo, K.; Hikino, H. Structures of rengyol, rengyoxide, and rengyolone, new cyclohexylethane derivatives from Forsythia suspensa fruits. Can. J. Chem. 1984, 62, 2011–2014. [Google Scholar] [CrossRef]
- Bianco, A.; Scalzo, R.L.; Scarpati, M.L. Isolation of cornoside from Olea europaea and its transformation into halleridone. Phytochemistry 1993, 32, 445–457. [Google Scholar] [CrossRef]
- Jensen, S.R.; Kjaer, A.; Nielsen, B.J. A quinol glucoside isolated from Cornus species. Acta Chem. Scand. 1973, 27, 367–369. [Google Scholar] [CrossRef]
- Sasaki, H.; Taguchi, H.; Endo, T.; Yosioka, I. The glycosides of Martynia louisiana Mill. A new phenylpropanoid glycoside, martynoside. Chem. Pharm. Bull. 1978, 26, 2111–2121. [Google Scholar] [CrossRef]
- Tian, X.Y.; Wang, Y.H.; Yang, Q.Y.; Yu, S.S.; Fang, W.S. Jacaranone analogs from Senecio scandens. J. Asian Nat. Prod. Res. 2009, 11, 63–68. [Google Scholar] [CrossRef]
- Seo, S.; Tomita, Y.; Tori, K.; Yoshimura, Y. Determination of the absolute configuration of a secondary hydroxy group in a chiral secondary alcohol using glycosidation shifts in carbon-13 nuclear magnetic resonance sepctroscopy. J. Am. Chem. Soc. 1978, 100, 3331–3339. [Google Scholar] [CrossRef]
- Hase, T.; Kawamoto, Y.; Ohtani, K.; Kasai, R.; Yamasaki, K.; Picheansoonthon, C. Cyclohexylethanoids and related glucosides from Millingtonia hortensis. Phytochemistry 1995, 39, 235–241. [Google Scholar] [CrossRef]
- Huang, R.M.; Ma, W.; Dong, J.D.; Zhou, X.F.; Xu, T.H.; Lee, K.J.; Yang, X.W.; Xu, S.H.; Liu, Y.H. A new 1,4-diazepine from South China Sea marine sponge Callyspongia species. Molecules 2010, 15, 871–877. [Google Scholar] [CrossRef]
- Wang, Y.L.; Li, Z.L.; Zhang, H.L.; Sha, Y.; Pei, Y.H.; Hua, H.M. New germacrane sequiterpenes from Salvia chinensis. Chem. Pharm. Bull. 2008, 61, 843–846. [Google Scholar]
- Faller, A.L.K.; Fialho, E.; Liu, R.H. Cellular antioxidant activity of Feijoada whole meal coupled with an in vitro digestion. J. Agric. Food Chem. 2012, 60, 4826–4832. [Google Scholar] [CrossRef]
- Song, W.; Derito, C.M.; Liu, M.K.; He, X.J.; Dong, M.; Liu, R.H. Cellular antioxidant activity of common vegetables. J. Agric. Food Chem. 2010, 58, 6621–6629. [Google Scholar] [CrossRef]
- Wolfe, K.L.; Kang, X.M.; He, X.J.; Dong, M.; Zhang, Q.Y.; Liu, R.H. Cellular antioxidant activity of common fruits. J. Agric. Food Chem. 2008, 56, 8418–8426. [Google Scholar]
- Wolfe, K.L.; Liu, R.H. Cellular antioxidant activity (CAA) assay for assessing antioxidants, foods, and dietary supplements. J. Agric. Food Chem. 2007, 55, 8896–8907. [Google Scholar] [CrossRef]
- Felice, D.L.; Sun, J.; Liu, R.H. A modified methylene blue assay for accurate cell counting. J. Funct. Foods 2009, 1, 109–118. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yi, X.-X.; Chen, Y.; Xie, W.-P.; Xu, M.-B.; Chen, Y.-N.; Gao, C.-H.; Huang, R.-M. Four New Jacaranone Analogs from the Fruits of a Beibu Gulf Mangrove Avicennia marina. Mar. Drugs 2014, 12, 2515-2525. https://doi.org/10.3390/md12052515
Yi X-X, Chen Y, Xie W-P, Xu M-B, Chen Y-N, Gao C-H, Huang R-M. Four New Jacaranone Analogs from the Fruits of a Beibu Gulf Mangrove Avicennia marina. Marine Drugs. 2014; 12(5):2515-2525. https://doi.org/10.3390/md12052515
Chicago/Turabian StyleYi, Xiang-Xi, Yong Chen, Wen-Pei Xie, Ming-Ben Xu, Yin-Ning Chen, Cheng-Hai Gao, and Ri-Ming Huang. 2014. "Four New Jacaranone Analogs from the Fruits of a Beibu Gulf Mangrove Avicennia marina" Marine Drugs 12, no. 5: 2515-2525. https://doi.org/10.3390/md12052515