Dereplication Strategies for Targeted Isolation of New Antitrypanosomal Actinosporins A and B from a Marine Sponge Associated-Actinokineospora sp. EG49

 ,
,
Abstract
:
1. Introduction
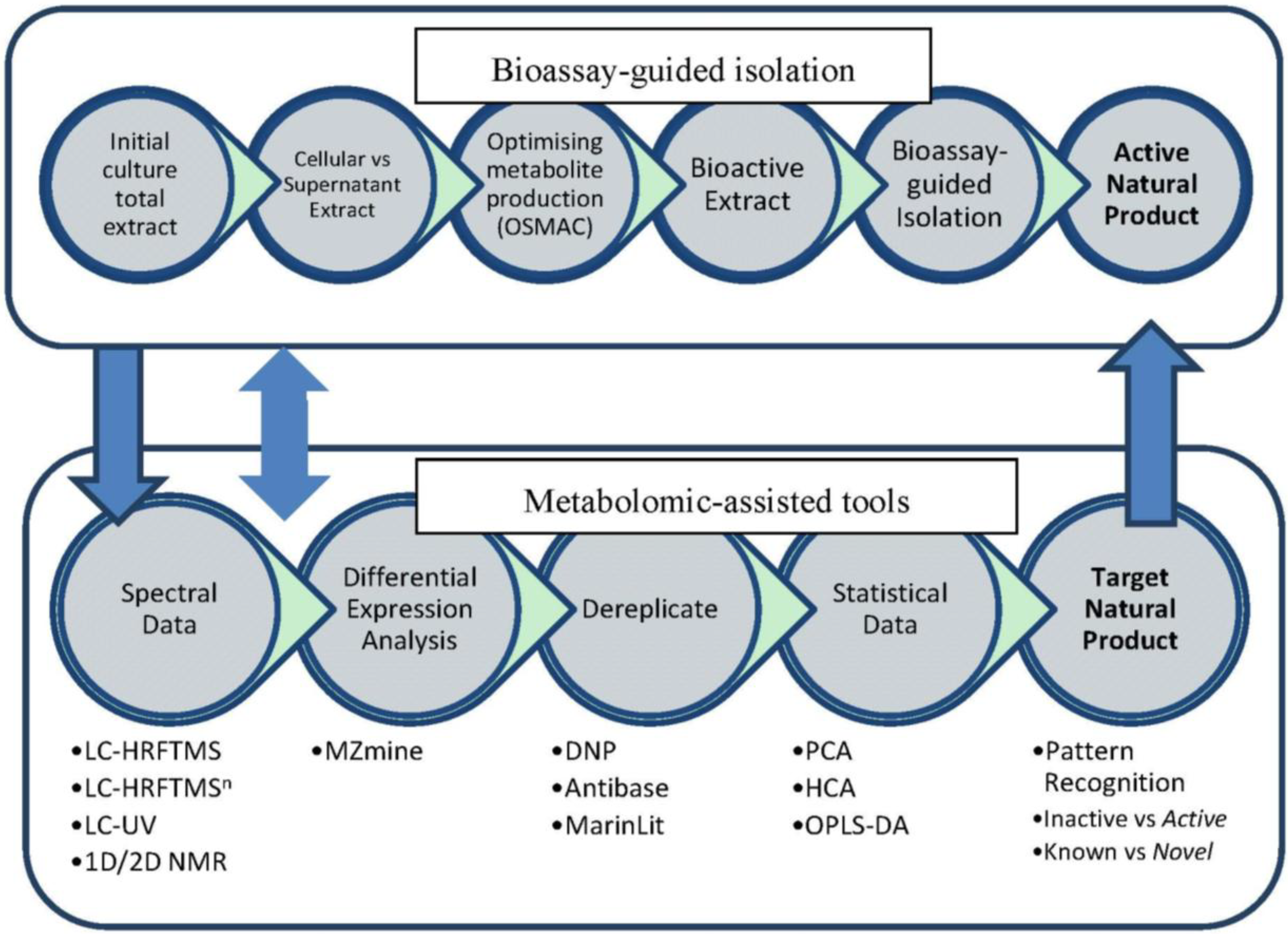
2. Results and Discussion
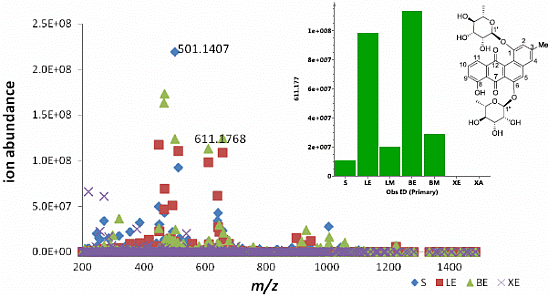
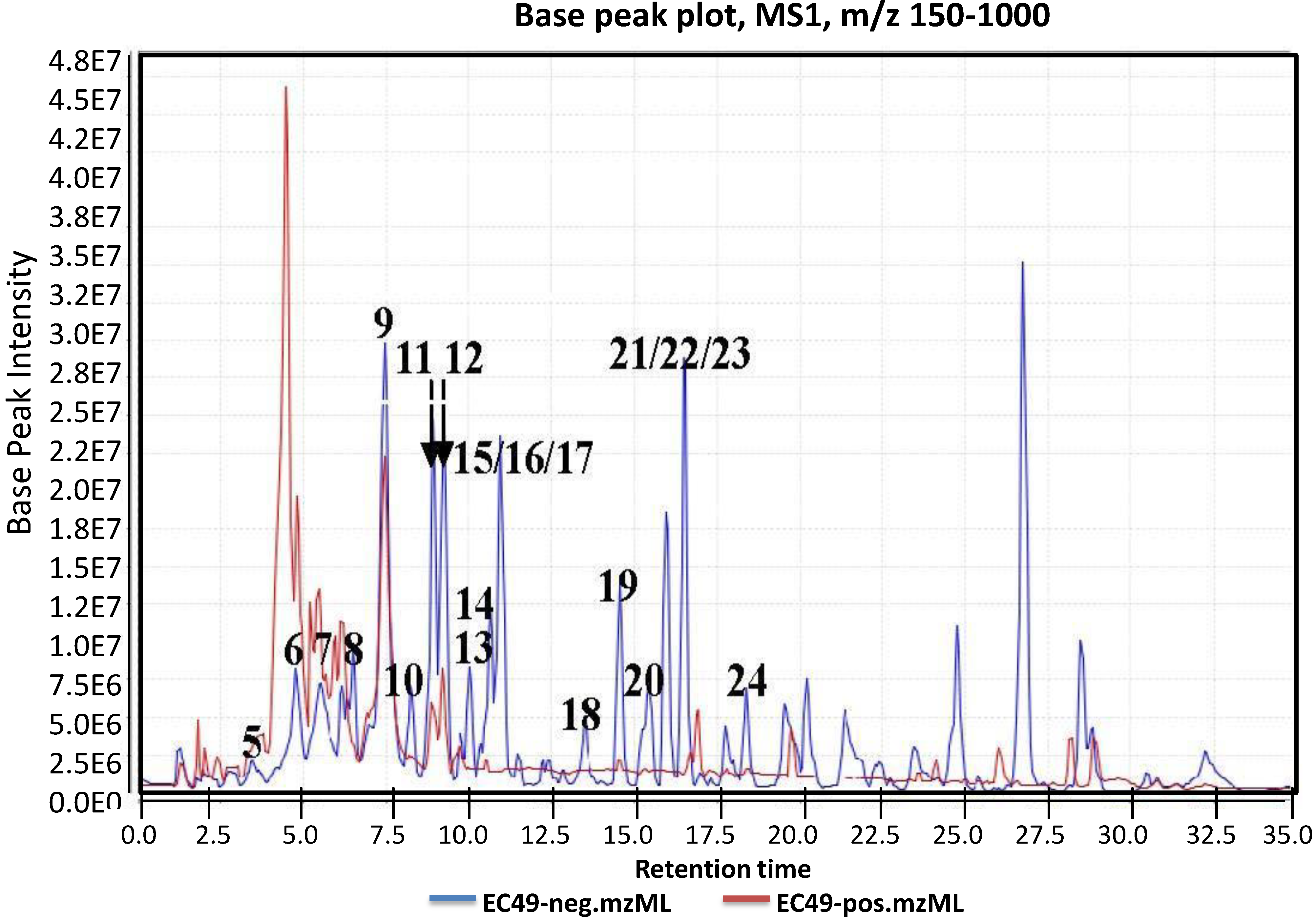
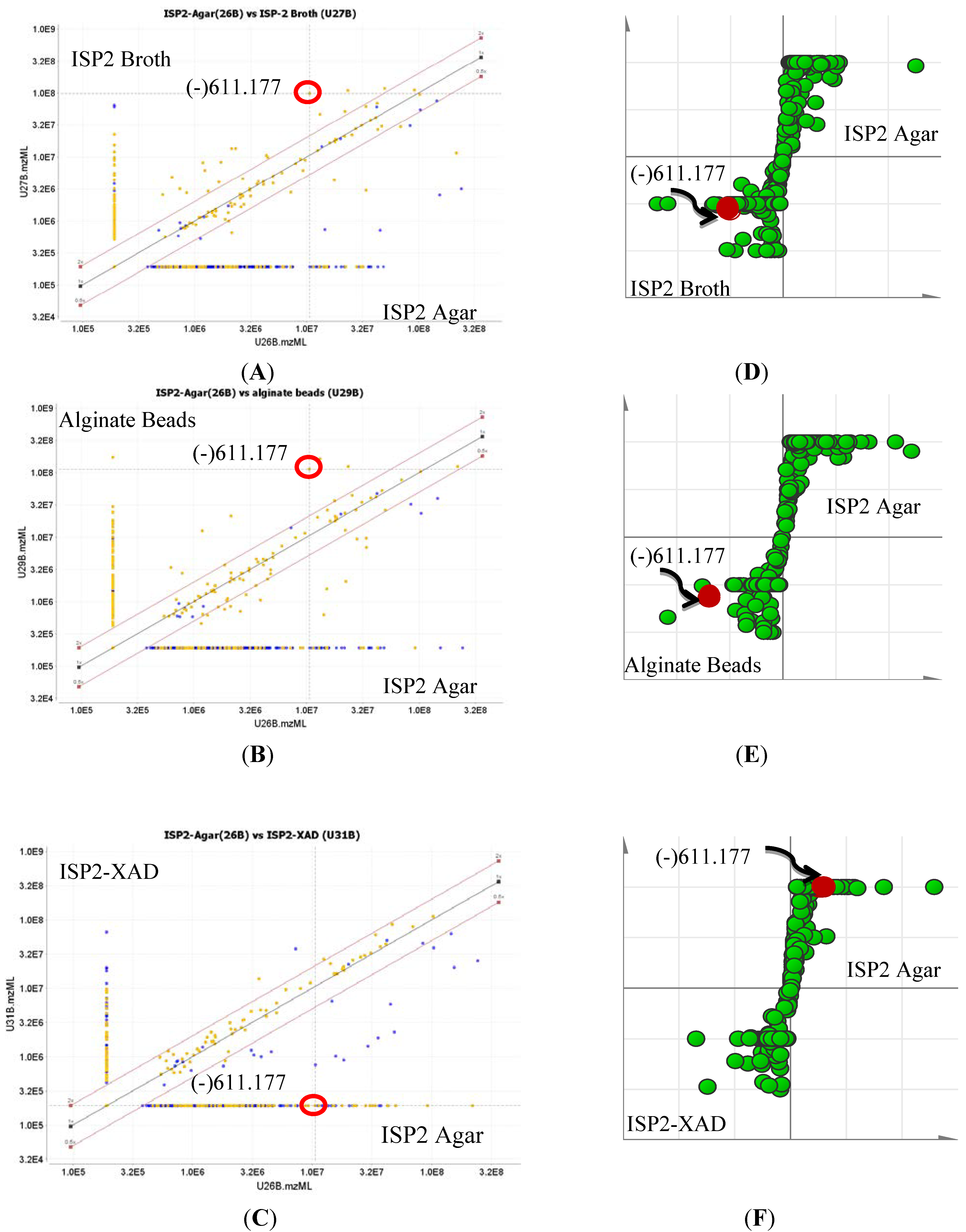
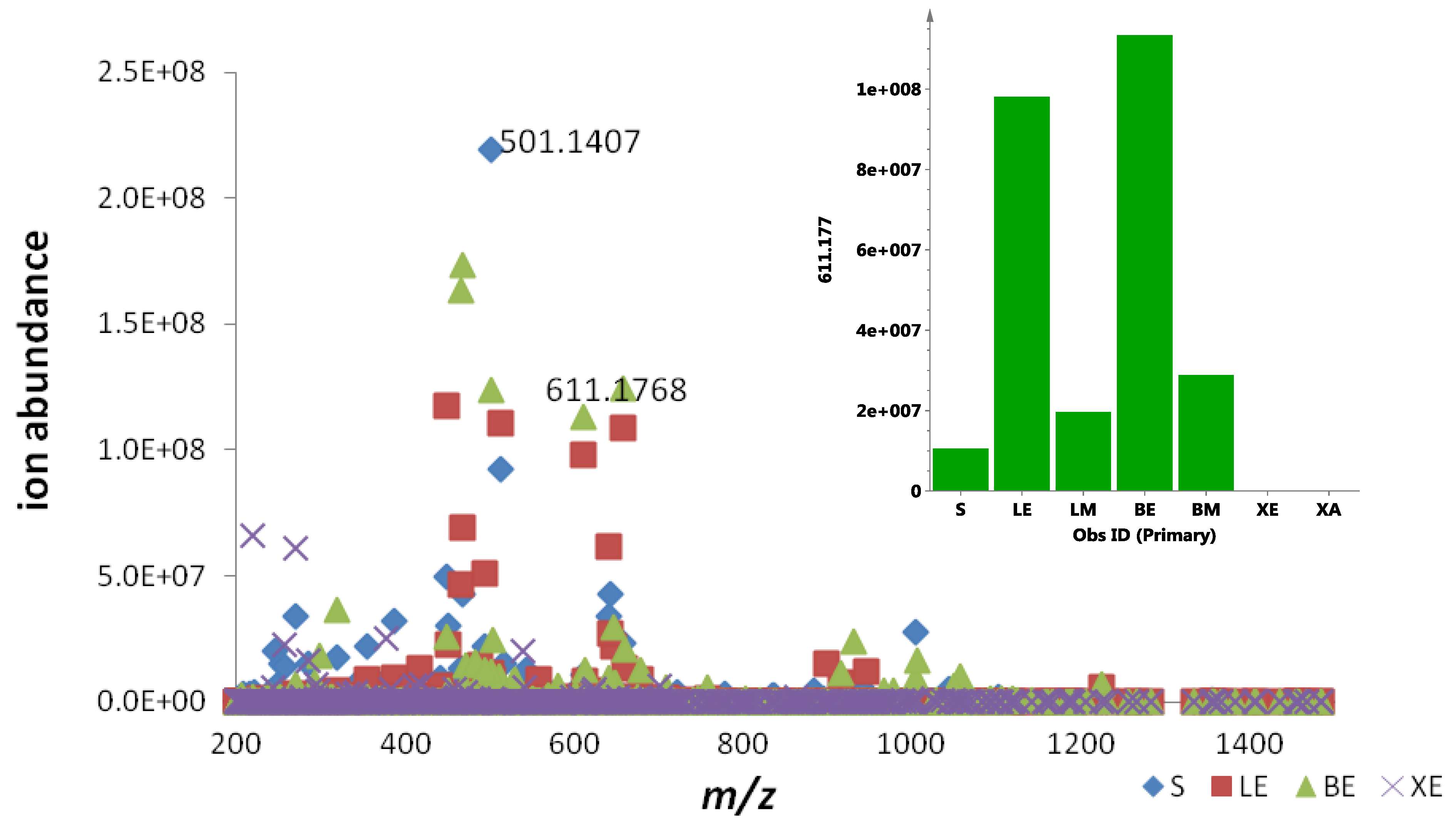
2.1. Metabolomic Profiling of the Crude Ethyl Acetate Extract of ISP2 Agar Culture of EG49
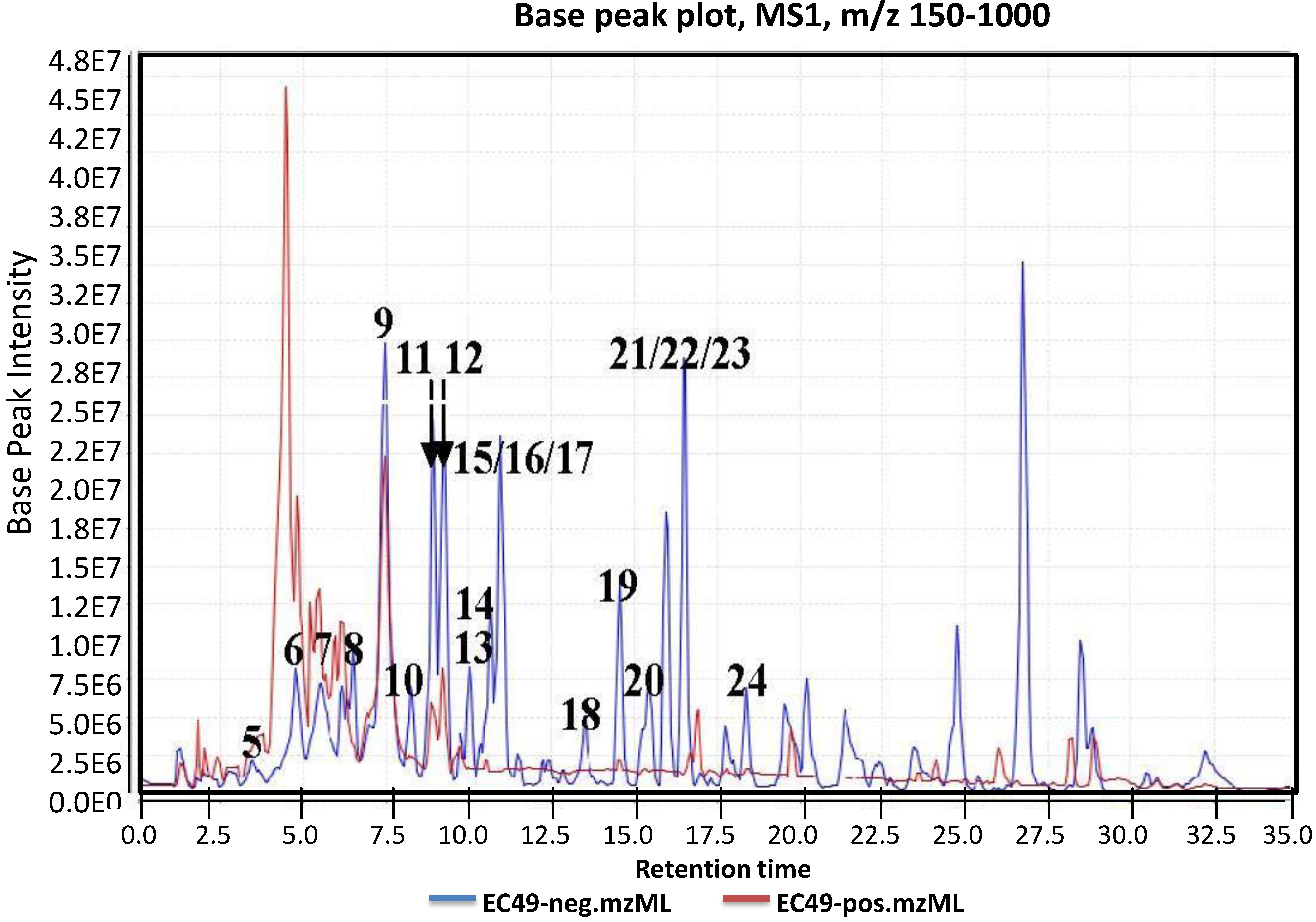

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RT min | [M − H]− | Predicted MF [M − H]− | [M − ADDUCT]− | MS2 | MS3 |
|---|---|---|---|---|---|
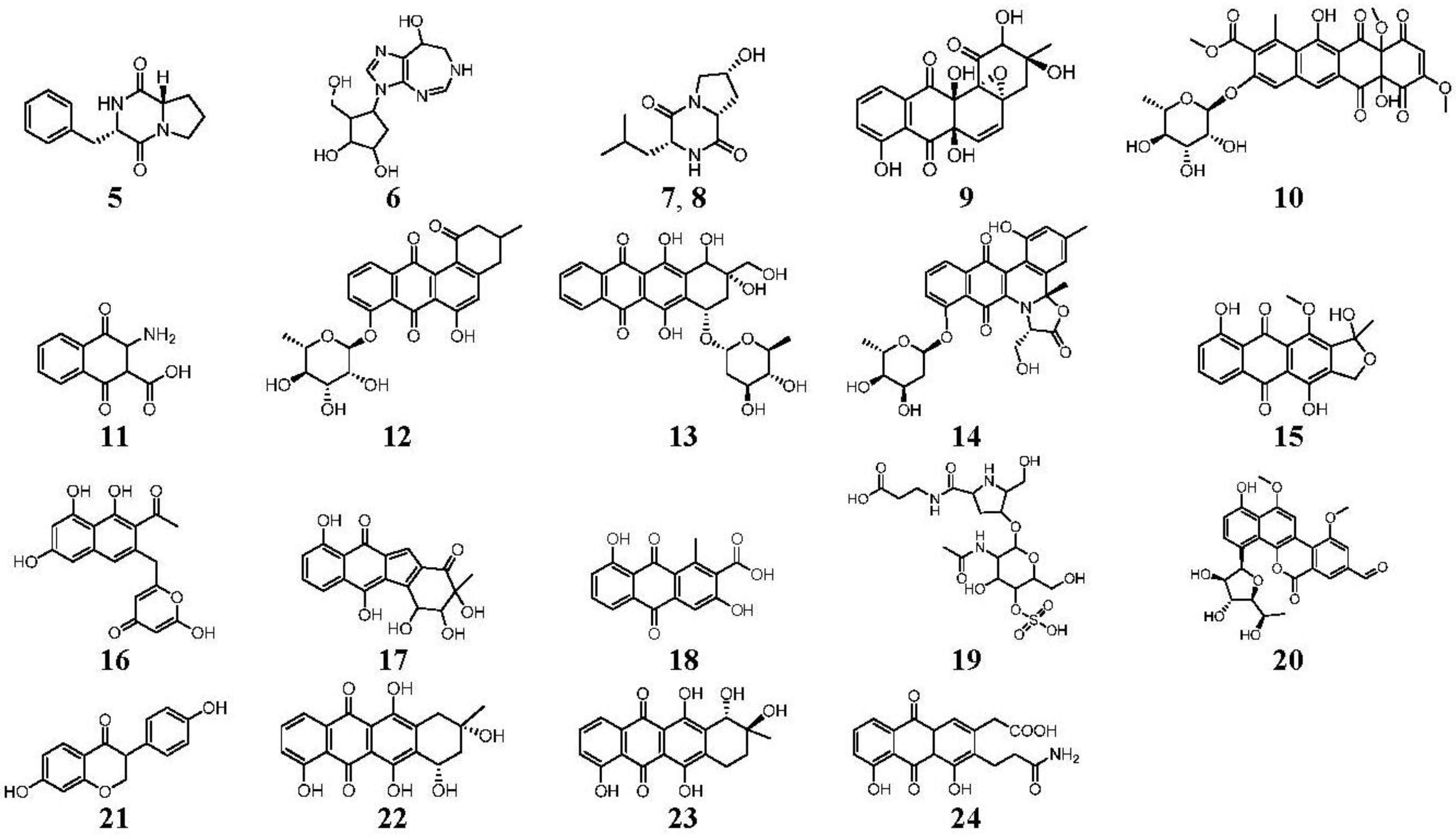
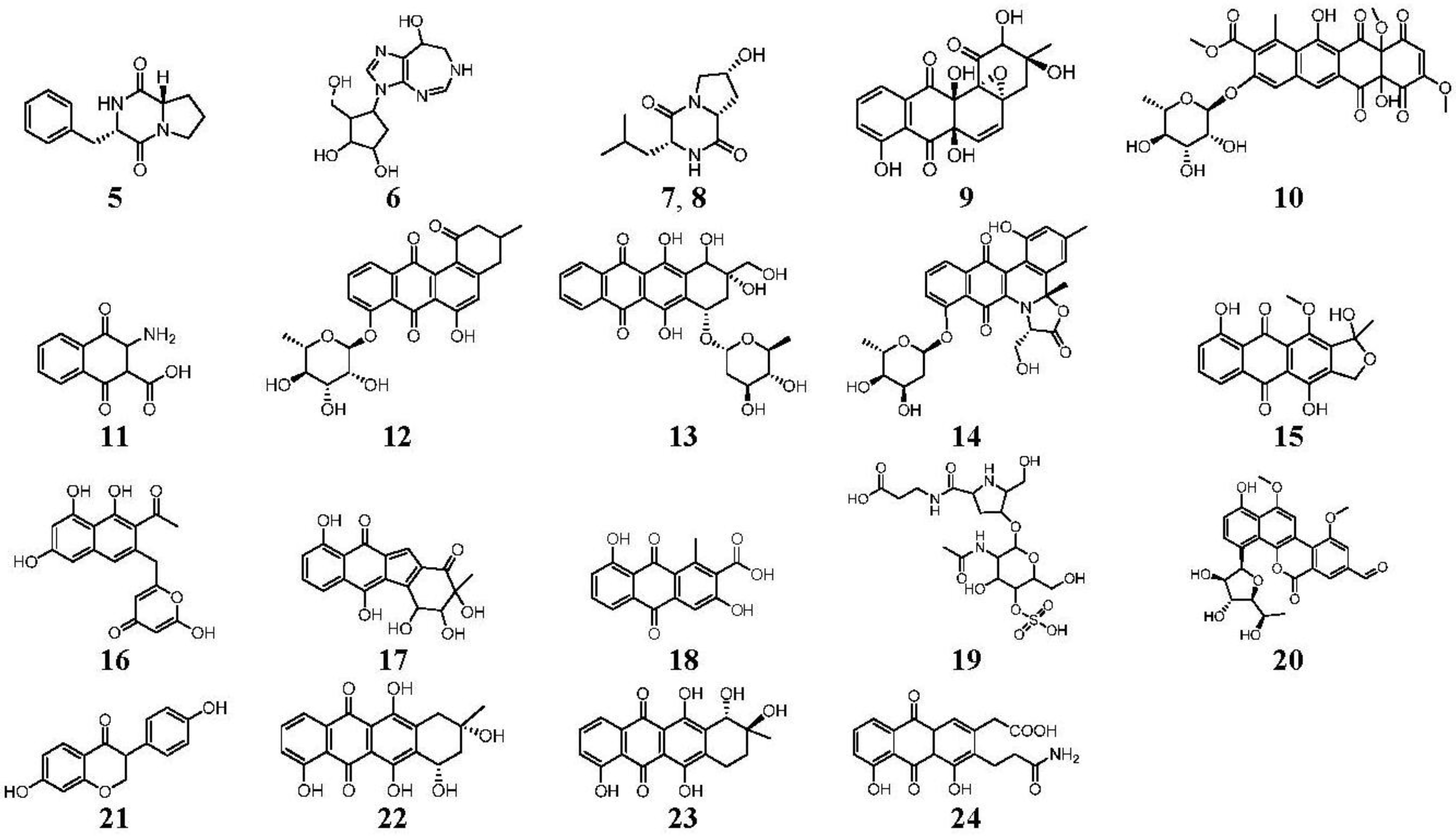
| 8.10 | 513.1398 14 RDBE | C26 H25 O11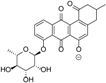 Atramycin A (12) | 467.13 [M − HCOOH] C25 H23 O9 | 321.08 [−C6 H10 O4] C19 H13 O5 13 RDBE 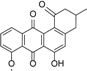 | |
| 8.97 | 657.1822 16 RDBE | C32 H33 O15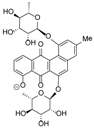 Actinosporin A (1) | 611.18 [M − HCOOH] C31 H31 O13 | 465.12 [−C6 H10 O4] C25 H21 O9 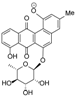 | 319.06 [−C6 H10 O4] C19 H11 O5 14 RDBE 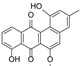 |
| 9.85 | 641.1875 16 RDBE | C32 H33 O14 Unknown | 595 [M − HCOOH] C31 H31 O12 | 451.14 [−C6 H10 O4] C25 H23 O8 14 RDBE | 305.08 [−C6 H10 O4] C19 H13 O4 13 RDBE |
| 10.60 | 657.1826 16 RDBE | C32 H33 O15 Elloramycin E * 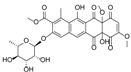 * Elloramycin E not possible due to loss of two sugar units. | 611.18 C31 H31 O13 Unknown | 465.12 [−C6 H10 O4] C25 H21 O9 15 RDBE | 319.06 [−C6 H10 O4] C19 H11 O5 14 RDBE |
| 15.37 | 467.1353 14 RDBE | C25 H23 O9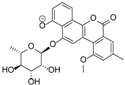 BE-12406-A | 320.07 [−C6 H10 O4] C19 H12 O5 14 RDBE 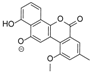 | 291.07 [−OCH3] C18 H11 O4 13 RDBE 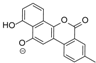 | |
| 15.98 | 465.1191 15 RDBE | C25 H21 O9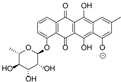 Galtamycinone | 449.12 [−O] C25 H21 O8 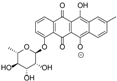 | 319.06 [−C6 H10 O4] C19 H11 O5 14 RDBE 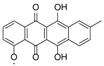 | 303.07 [−OH] C19 H11 O4 14 RDB 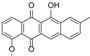 |
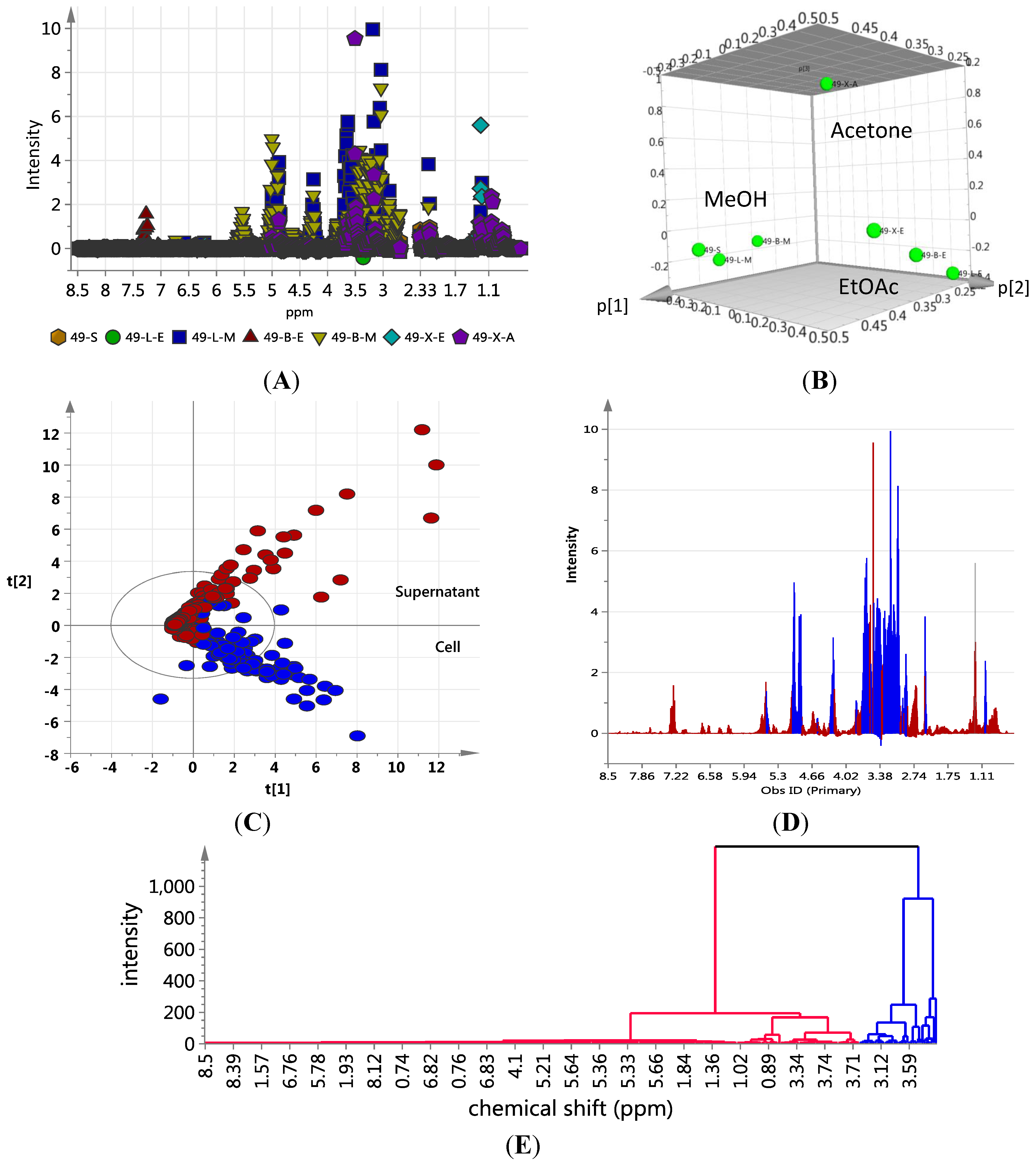
2.2. Metabolomic Profiling of Cellular and Supernatant Extracts from Liquid Broth Cultures of EG49
| Solvent | Ionisation Mode | MS m/z | Rt (min) | Chemical Formula | Name |
|---|---|---|---|---|---|
| EtOAc | N | 467.1350 | 8.27 | C25H24O9 | Atramycin A (12) |
| EtOAc | P | 469.1494 | 8.28 | C25H24O9 | Atramycin A (12) |
| EtOAc | N | 513.1404 | 8.28 | C26H25O11 | Unknown |
| EtOAc | N | 611.1768 | 8.98 | C31H32O13 (new) * | Actinosporin A (1) |
| EtOAc | N | 657.1828 | 10.50 | C32H34O15 | Elloramycin E (10) |
| EtOAc | N | 501.1407 | 10.76 | C25H26O11 (new) * | Actinosporin B (2) |
| EtOAc | N | 467.1350 | 15.13 | C25H24O9 | BE-12406-A |
| EtOAc | N | 449.1247 | 15.97 | C25H22O8 | Galtamycinone |
| MeOH | N | 377.0860 | 1.38 | C15H9O3N10 | Unknown |
| MeOH | N | 452.2787 | 21.50 | C25H40O7 | Unknown |
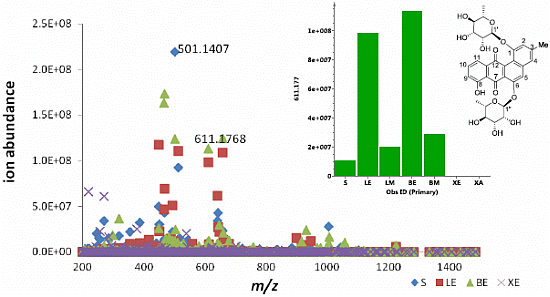
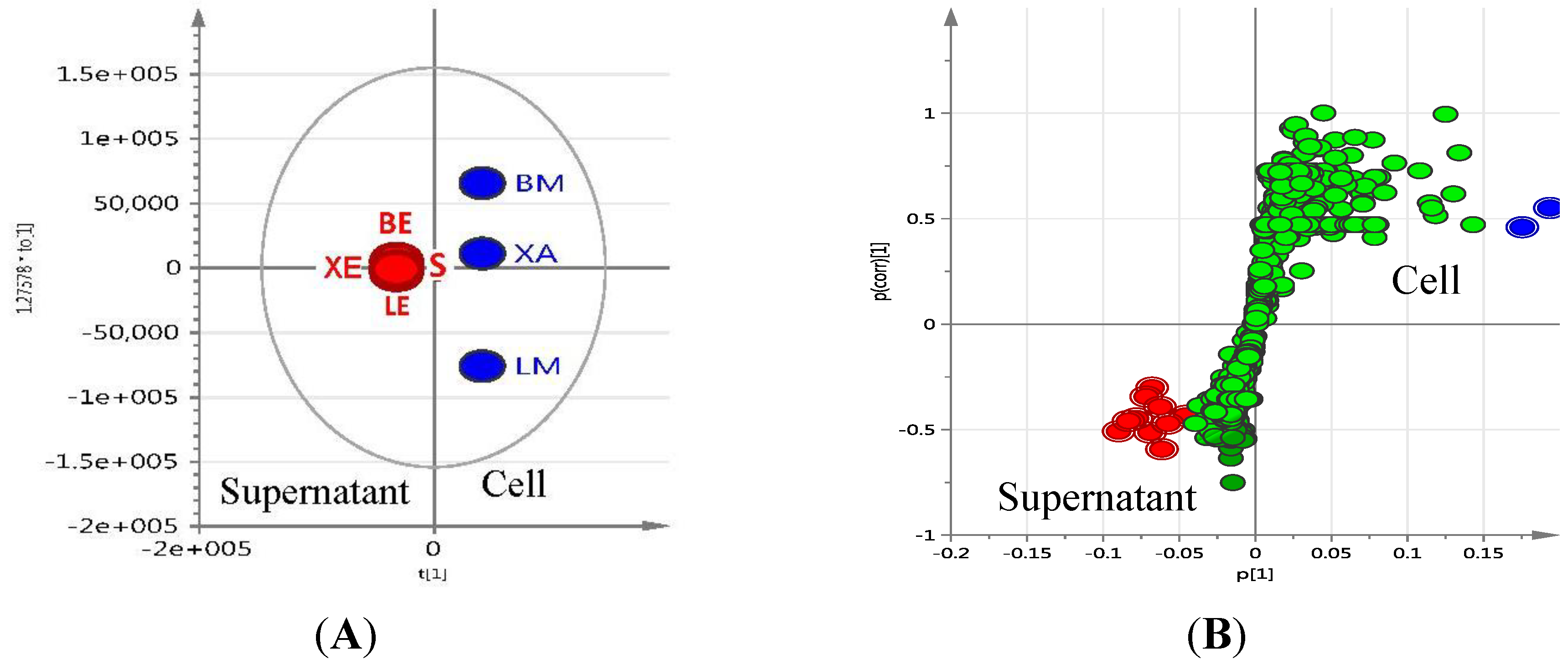
 S; ●LE; ■LM; ▲BE; ▼BM; ♦XE;
S; ●LE; ■LM; ▲BE; ▼BM; ♦XE;  XA); (B) 3D Principal component analysis (PCA)-score plot; (C) PCA-loading plot of cellular (blue) vs. supernatant extracts (red); (D) Line plots obtained from the hierarchical clustering analysis of the PCA results as shown in (E). The primary observed ID (obs ID) is synonymous to the chemical shift in ppm.
S; ●LE; ■LM; ▲BE; ▼BM; ♦XE; XA); (B) 3D Principal component analysis (PCA)-score plot; (C) PCA-loading plot of cellular (blue) vs. supernatant extracts (red); (D) Line plots obtained from the hierarchical clustering analysis of the PCA results as shown in (E). The primary observed ID (obs ID) is synonymous to the chemical shift in ppm.
XA); (B) 3D Principal component analysis (PCA)-score plot; (C) PCA-loading plot of cellular (blue) vs. supernatant extracts (red); (D) Line plots obtained from the hierarchical clustering analysis of the PCA results as shown in (E). The primary observed ID (obs ID) is synonymous to the chemical shift in ppm.
S; ●LE; ■LM; ▲BE; ▼BM; ♦XE; XA); (B) 3D Principal component analysis (PCA)-score plot; (C) PCA-loading plot of cellular (blue) vs. supernatant extracts (red); (D) Line plots obtained from the hierarchical clustering analysis of the PCA results as shown in (E). The primary observed ID (obs ID) is synonymous to the chemical shift in ppm.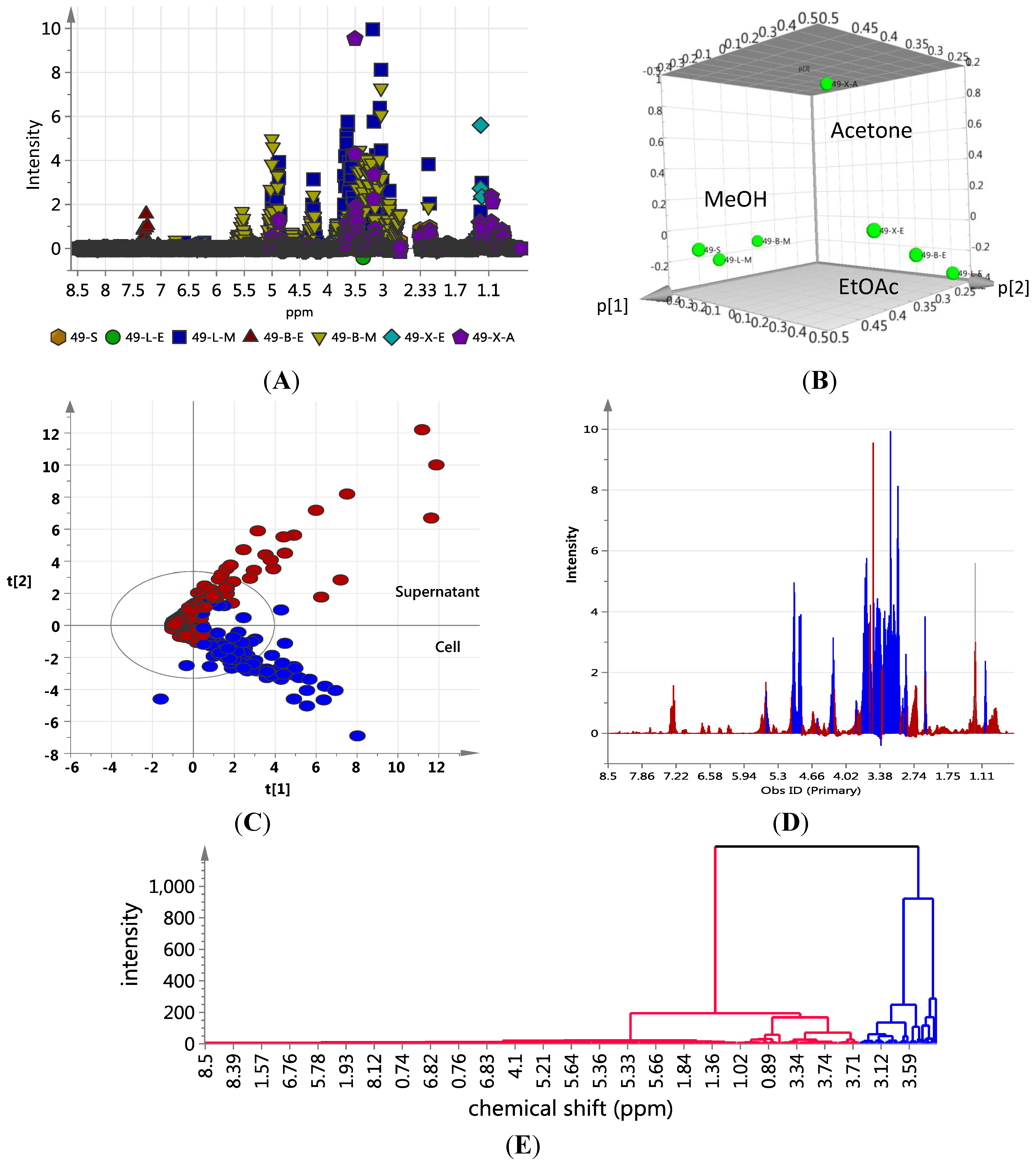
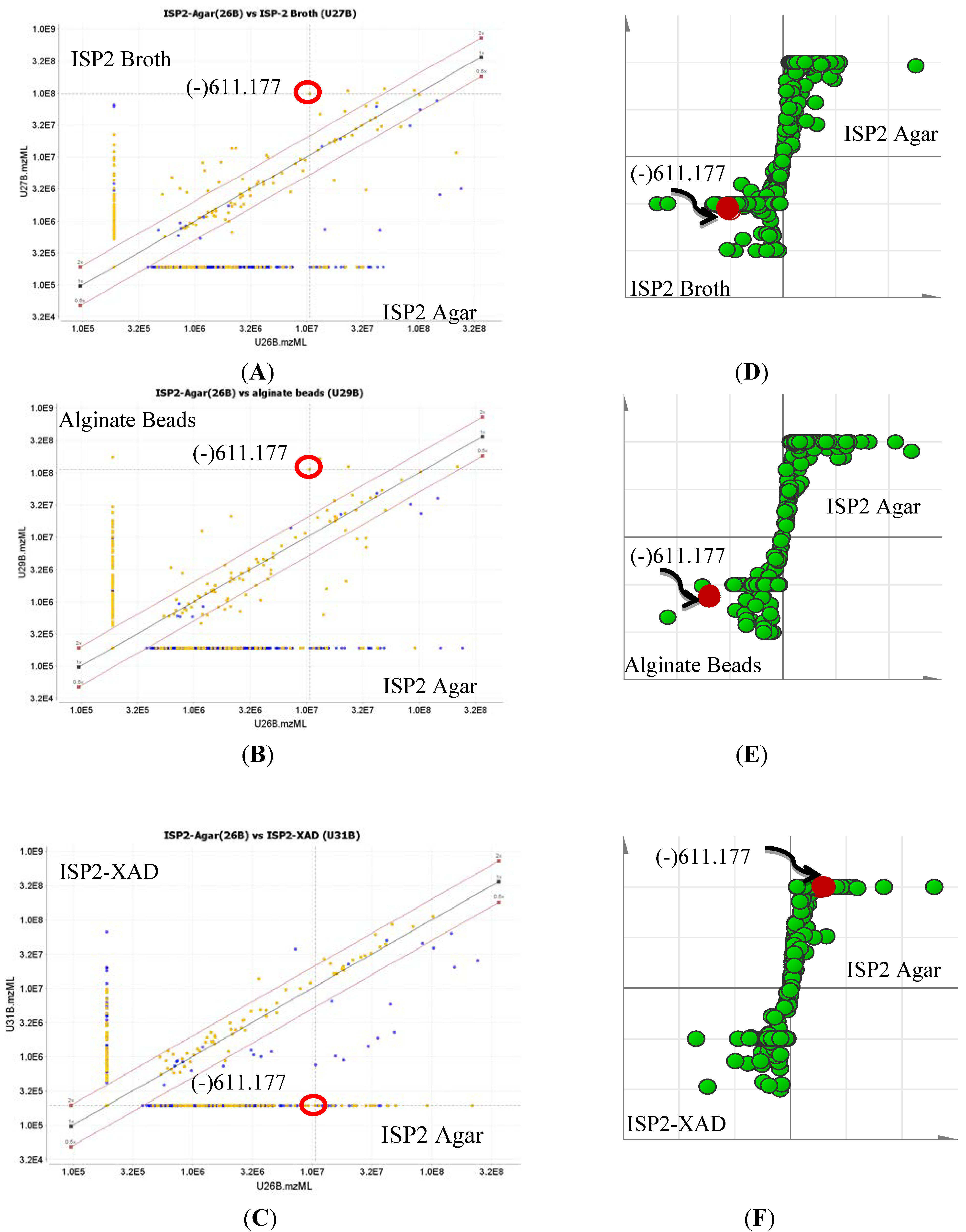
2.3. Quantification of Actinosporin A in Four Different Culture Media
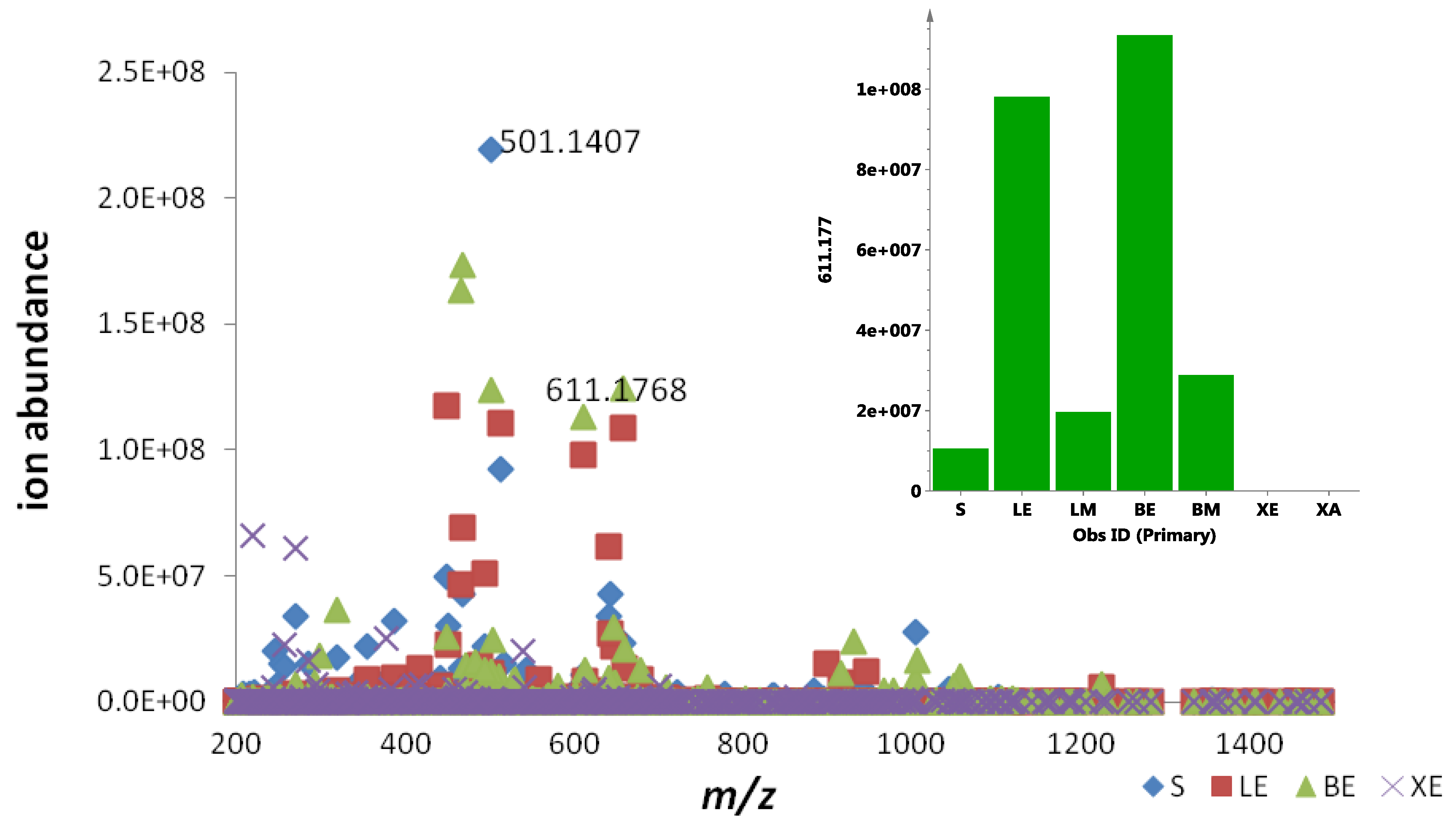
2.4. Bioassay-Guided Isolation and Structure Elucidation of the New Actinosporins A and B
| Position C/H No. | δH, Mult (J in Hz) | COSY | δC | Mult | HMBC (δH to δC) | ROESY |
|---|---|---|---|---|---|---|
| 1 | 153.9 | C | ||||
| 2 | 7.19, s | H-3-CH3 | 112.2 | CH | C-1, C-3, C-3-CH3, C-4, C-12a, C-12b | H-3-CH3, H-1A |
| 3 | 142.0 | C | ||||
| 3-CH3 | 2.47, s | H-2, H-4 | 20.8 | CH3 | C-2, C-3, C-4 | |
| 4 | 7.26, s | H-3-CH3 | 119.6 | CH | C-2, C-3-CH3, C-4a, C-12b | H-3-CH3, H-5 |
| 4a | 138.9 | C | ||||
| 5 | 7.69, s | 116.9 | CH | C-4, C-6, C-6a, C-12b | H-4, H-1B, H-5B | |
| 6 | 152.3 | C | ||||
| 6a | 124.0 | C | ||||
| 7 | 186.2 | C | ||||
| 7a | 115.7 | C | ||||
| 8 | 161.2 | C | ||||
| 9 | 7.21, d (7.3) | H-10, H-11 | 122.6 | CH | C-7a, C-11, C-10 | |
| 10 | 7.66, t (7.3) | H-9, H-11 | 136.1 | CH | C-8, C-11, C-11a | |
| 11 | 7.52, d (7.3) | H-9, H-10 | 117.1 | CH | C-7, C-7a, C-9, C-10, C-12 | |
| 11a | 135.5 | C | ||||
| 12 | 187.5 | C | ||||
| 12a | 140.0 | C | ||||
| 12b | 116.1 | C | ||||
| 1′ | 5.48, brs | H-2′ | 99.6 | CH | C-1, C-2′, C-5′ | H-2 |
| 2′ | 4.05, s | H-1′, H-3′ | 70.4 | CH | C-3′, C-4′ | |
| 3′ | 3.80, dd (9.4, 3.4) | H-2′, H-4′ | 70.9 | CH | C-4′, C-5′ | |
| 4′ | 3.48, t (9.4) | H-3′, H-5′ | 72.4 | CH | C-3′, C-5′, C-6′ | H-6′ |
| 5′ | 3.76, m | H-6′ | 69.6 | CH | C-6′ | H-3′ |
| 6′ | 1.25, d (6.3) | H-5′ | 16.5 | CH3 | C-5′, C-4′ | |
| 1″ | 5.66, brs | H-2″ | 99.0 | CH | C-8, C-5″, C-4″ | H-5 |
| 2″ | 4.22, d (3.4) | H-1″, H-3″ | 70.6 | CH | C-3″, C-4″ | |
| 3″ | 4.25, s | H-2″, H-4″ | 70.7 | CH | C-4″ | H-5″ |
| 4″ | 3.53, t (9.2) | H-3″, H-5″ | 72.5 | CH | C-2″ C-3″, C-6″ | |
| 5″ | 3.74, m | H-6″ | 69.9 | CH | C-3″, C-4″, C-6″ | |
| 6″ | 1.27, d (6.2) | H-5″ | 16.6 | CH3 | C-4″, C-5″ |
3. Experimental Section
3.1. Fermentation Approaches
3.1.1. Solid Culture
3.1.2. Liquid Culture
3.1.3. Liquid Culture with XAD
3.1.4. Liquid Culture with Calcium Alginate Beads
3.2. General Procedure for Dereplication Study Using MS and NMR Data
3.3. Purification of Actinosporins A and B
3.4. Compound Characterisation
 +180 (c 0.002, MeOH); UV (MeOH): λmax (log ε) = 227 (4.41), 312 (4.06), 408 (3.61) nm; IR: 3389, 3206, 1677, 1644, 1625, 1364 cm−1; 1H (CD3OD, 600 MHz), and 13C NMR (CD3OD, 125 MHz) data see Table 3; HR-ESIMS: found at m/z 613.1915 [M + H]+, calcd for C31H33O13, (613.1916), and at m/z 611.1768 [M − H]−, calcd for C31H31O13 (611.1770). +18.0 (c 0.05, MeOH); UV (MeOH): λmax (log ε) = 254 (4.41), 379 (4.06), 408 (3.61) nm; 1H NMR (CD3OD, 600 MHz) δH 7.96 (1H, d, J11,10 7.4, H-11), 7.87 (1H, dd, J10,11 7.4, J10,9 8.2, H-10), 7.79 (1H, d, J5,6 7.8, H-5), 7.70 (1H, d, J9,10 8.2, H-9), 7.62 (1H, d, J6,5 7.8, H-6), 5.64 (1H, br s, H-1′), 4.93 (1H, bs, H-1), 4.83 (1H, d, J3A,3B 11.6, H-13A), 4.01 (2H, m, H-2 and H-2′), 3.51 (1H, m, H-5′) 3.33–3.53 (underneath HOD peak, H-13B, H-3′, H-4′), 3.01 (1H, d, J4A,4B 13.2, H-4A), 2.90 (d, J4B,4A 13.2, H-4B), 1.10 (3H, d, J6′,5′ 6.3, H-6′); 13C NMR (151 MHz, DMSO) δ = 181.0 (C-7), 156.9 (C-8), 140.7 (C-4a), 135.6 (C-11a), 133.27 (C-6a), 133.31 (C-12a), 123.9 (C-7a), 122.0 (C-11), 116.0 (C-12b), 72.5 (C-13), 71.7 (C-3), 71.5 (C-4′), 71.4 (C-3′), 70.5 (C-2′), 70.4 (C-2), 64.0 (C-5′), and 42.7 (C-4); HR-ESIMS: [M − H]− found at m/z 501.1402, calcd for C25H25O11 (501.1402); [M + H]+ found at m/z 503.1554, calcd for C25H27O11 (503.1548).
+180 (c 0.002, MeOH); UV (MeOH): λmax (log ε) = 227 (4.41), 312 (4.06), 408 (3.61) nm; IR: 3389, 3206, 1677, 1644, 1625, 1364 cm−1; 1H (CD3OD, 600 MHz), and 13C NMR (CD3OD, 125 MHz) data see Table 3; HR-ESIMS: found at m/z 613.1915 [M + H]+, calcd for C31H33O13, (613.1916), and at m/z 611.1768 [M − H]−, calcd for C31H31O13 (611.1770). +18.0 (c 0.05, MeOH); UV (MeOH): λmax (log ε) = 254 (4.41), 379 (4.06), 408 (3.61) nm; 1H NMR (CD3OD, 600 MHz) δH 7.96 (1H, d, J11,10 7.4, H-11), 7.87 (1H, dd, J10,11 7.4, J10,9 8.2, H-10), 7.79 (1H, d, J5,6 7.8, H-5), 7.70 (1H, d, J9,10 8.2, H-9), 7.62 (1H, d, J6,5 7.8, H-6), 5.64 (1H, br s, H-1′), 4.93 (1H, bs, H-1), 4.83 (1H, d, J3A,3B 11.6, H-13A), 4.01 (2H, m, H-2 and H-2′), 3.51 (1H, m, H-5′) 3.33–3.53 (underneath HOD peak, H-13B, H-3′, H-4′), 3.01 (1H, d, J4A,4B 13.2, H-4A), 2.90 (d, J4B,4A 13.2, H-4B), 1.10 (3H, d, J6′,5′ 6.3, H-6′); 13C NMR (151 MHz, DMSO) δ = 181.0 (C-7), 156.9 (C-8), 140.7 (C-4a), 135.6 (C-11a), 133.27 (C-6a), 133.31 (C-12a), 123.9 (C-7a), 122.0 (C-11), 116.0 (C-12b), 72.5 (C-13), 71.7 (C-3), 71.5 (C-4′), 71.4 (C-3′), 70.5 (C-2′), 70.4 (C-2), 64.0 (C-5′), and 42.7 (C-4); HR-ESIMS: [M − H]− found at m/z 501.1402, calcd for C25H25O11 (501.1402); [M + H]+ found at m/z 503.1554, calcd for C25H27O11 (503.1548).3.5. Antitrypanosomal Activity Testing
3.6. Cytotoxicity Assay
4. Conclusions
Supplementary Files
Acknowledgments
Conflicts of Interest
References
- Yuliana, N.D.; Khatib, A.; Choi, Y.H.; Verpoorte, R. Metabolomics for bioactivity assessment of natural products. Phytother. Res. 2011, 25, 157–169. [Google Scholar]
- Tawfike, A.; Viegelmann, C.; Edrada-Ebel, R. Metabolomics and Dereplication Strategies in Natural Products. In Metabolomics Tools for Natural Product Discovery: Methods and Protocols: Methods in Molecular Biology; Roessner, U., Dias, D.A., Eds.; Humana Press: New York, NY, USA, 2013; pp. 227–244. [Google Scholar]
- Kjer, J.; Debbab, A.; Aly, A.H.; Proksch, P. Methods for isolation of marine-derived endophytic fungi and their bioactive secondary products. Nat. Protoc. 2010, 5, 479–490. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2013, 30, 237–323. [Google Scholar] [CrossRef]
- Shin, M.H.; Lee do, Y.; Liu, K.H.; Fiehn, O.; Kim, K.H. Evaluation of sampling and extraction methodologies for the global metabolic profiling of Saccharophagus degradans. Anal. Chem. 2010, 82, 6660–6666. [Google Scholar] [CrossRef]
- Bosque-Sendra, J.M.; Cuadros-Rodriguez, L.; Ruiz-Samblas, C.; de la Mata, A.P. Combining chromatography and chemometrics for the characterization and authentication of fats and oils from triacylglycerol compositional data—A review. Anal. Chim. Acta 2012, 724, 1–11. [Google Scholar] [CrossRef]
- Ali, K.; Iqbal, M.; Korthout, H.A.; Maltese, F.; Fortes, A.M.; Pais, M.S.; Verpoorte, R.; Choi, Y.H. NMR spectroscopy and chemometrics as a tool for anti-TNFalpha activity screening in crude extracts of grapes and other berries. Metabolomics 2012, 8, 1148–1161. [Google Scholar] [CrossRef]
- Zheng, L.; Watson, D.G.; Johnston, B.F.; Clark, R.L.; Edrada-Ebel, R.; Elseheri, W. A chemometric study of chromatograms of tea extracts by correlation optimization warping in conjunction with PCA, support vector machines and random forest data modeling. Anal. Chim. Acta 2009, 642, 257–265. [Google Scholar] [CrossRef]
- Mitova, M.I.; Murphy, A.C.; Lang, G.; Blunt, J.W.; Cole, A.L.; Ellis, G.; Munro, M.H. Evolving trends in the dereplication of natural product extracts. 2. The isolation of chrysaibol, an antibiotic peptaibol from a New Zealand sample of the mycoparasitic fungus Sepedonium chrysospermum. J. Nat. Prod. 2008, 71, 1600–1603. [Google Scholar] [CrossRef]
- Lang, G.; Mayhudin, N.A.; Mitova, M.I.; Sun, L.; van der Sar, S.; Blunt, J.W.; Cole, A.L.; Ellis, G.; Laatsch, H.; Munro, M.H. Evolving trends in the dereplication of natural product extracts: New methodology for rapid, small-scale investigation of natural product extracts. J. Nat. Prod. 2008, 71, 1595–1599. [Google Scholar] [CrossRef]
- Krug, D.; Zurek, G.; Schneider, B.; Garcia, R.; Muller, R. Efficient mining of myxobacterial metabolite profiles enabled by liquid chromatography-electrospray ionisation-time-of-flight mass spectrometry and compound-based principal component analysis. Anal. Chim. Acta 2008, 624, 97–106. [Google Scholar] [CrossRef]
- Mansson, M.; Phipps, R.K.; Gram, L.; Munro, M.H.; Larsen, T.O.; Nielsen, K.F. Explorative solid-phase extraction (E-SPE) for accelerated microbial natural product discovery, dereplication, and purification. J. Nat. Prod. 2010, 73, 1126–1132. [Google Scholar] [CrossRef]
- Hou, Y.; Braun, D.R.; Michel, C.R.; Klassen, J.L.; Adnani, N.; Wyche, T.P.; Bugni, T.S. Microbial strain prioritization using metabolomics tools for the discovery of natural products. Anal. Chem. 2012, 84, 4277–4283. [Google Scholar]
- Ebada, S.S.; Edrada, R.A.; Lin, W.; Proksch, P. Methods for isolation, purification and structural elucidation of bioactive secondary metabolites from marine invertebrates. Nat. Protoc. 2008, 3, 1820–1831. [Google Scholar] [CrossRef]
- Lampert, Y.; Kelman, D.; Nitzan, Y.; Dubinsky, Z.; Behar, A.; Hill, R.T. Phylogenetic diversity of bacteria associated with the mucus of Red Sea corals. FEMS Microbiol. Ecol. 2008, 64, 187–198. [Google Scholar] [CrossRef]
- Prieto-Davo, A.; Villarreal-Gomez, L.J.; Forschner-Dancause, S.; Bull, A.T.; Stach, J.E.; Smith, D.C.; Rowley, D.C.; Jensen, P.R. Targeted search for actinomycetes from near-shore and deep-sea marine sediments. FEMS Microbiol. Ecol. 2013, 84, 510–518. [Google Scholar] [CrossRef]
- Bull, A.T.; Stach, J.E. Marine actinobacteria: New opportunities for natural product search and discovery. Trends Microbiol. 2007, 15, 491–499. [Google Scholar] [CrossRef]
- Vicente, J.; Stewart, A.; Song, B.; Hill, R.; Wright, J. Biodiversity of actinomycetes associated with Caribbean sponges and their potential for natural product discovery. Mar. Biotechnol. 2013, 15, 413–424. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, W.; Jin, Y.; Jin, M.; Yu, X. A comparative study on the phylogenetic diversity of culturable actinobacteria isolated from five marine sponge species. Antonie Van Leeuwenhoek 2008, 93, 241–248. [Google Scholar] [CrossRef]
- Subramani, R.; Aalbersberg, W. Marine actinomycetes: An ongoing source of novel bioactive metabolites. Microbiol. Res. 2012, 167, 571–580. [Google Scholar] [CrossRef]
- Jiao, R.H.; Xu, H.; Cui, J.T.; Ge, H.M.; Tan, R.X. Neuraminidase inhibitors from marine-derived actinomycete Streptomyces seoulensis. J. Appl. Microbiol. 2013, 114, 1046–1053. [Google Scholar] [CrossRef]
- Engelhardt, K.; Degnes, K.F.; Kemmler, M. Production of a new thiopeptide antibiotic, TP-1161, by a marine-derived Nocardiopsis species. Appl. Environ. Microbiol. 2010. [Google Scholar] [CrossRef] [Green Version]
- Pimentel-Elardo, S.M.; Kozytska, S.; Bugni, T.S.; Ireland, C.M.; Moll, H.; Hentschel, U. Anti-parasitic compounds from Streptomyces sp. strains isolated from Mediterranean sponges. Mar. Drugs 2010, 8, 373–380. [Google Scholar] [CrossRef]
- Olano, C.; Mendez, C.; Salas, J.A. Antitumor compounds from marine actinomycetes. Mar. Drugs 2009, 7, 210–248. [Google Scholar] [CrossRef]
- Abdelmohsen, U.R.; Szesny, M.; Othman, E.M.; Schirmeister, T.; Grond, S.; Stopper, H.; Hentschel, U. Antioxidant and anti-protease activities of diazepinomicin from the sponge-associated Micromonospora strain RV115. Mar. Drugs 2012, 10, 2208–2221. [Google Scholar] [CrossRef]
- Tabares, P.; Pimentel-Elardo, S.M.; Schirmeister, T.; Hunig, T.; Hentschel, U. Anti-protease and immunomodulatory activities of bacteria associated with Caribbean sponges. Mar. Biotechnol. 2011, 13, 883–892. [Google Scholar] [CrossRef]
- Schneemann, I.; Kajahn, I.; Ohlendorf, B.; Zinecker, H.; Erhard, A.; Nagel, K.; Wiese, J.; Imhoff, J.F. Mayamycin, a cytotoxic polyketide from a Streptomyces strain isolated from the marine sponge Halichondria panicea. J. Nat. Prod. 2010, 73, 1309–1312. [Google Scholar] [CrossRef] [Green Version]
- Asolkar, R.N.; Kirkland, T.N.; Jensen, P.R.; Fenical, W. Arenimycin, an antibiotic effective against rifampin- and methicillin-resistant Staphylococcus aureus from the marine actinomycete Salinispora arenicola. J. Antibiot. 2010, 63, 37–39. [Google Scholar] [CrossRef]
- Abdelmohsen, U.R.; Zhang, G.L.; Philippe, A.; Schmitz, W.; Pimentel-Elardo, S.M.; Hertlein-Amslinger, B.; Hentschel, U.; Bringmann, G. Cyclodysidins A–D, cyclic lipopeptides from the marine sponge-derived Streptomyces strain RV15. Tetrahedron Lett. 2012, 53, 23–29. [Google Scholar] [CrossRef]
- Tang, X.; Zhou, Y.; Zhang, J.; Ming, H.; Nie, G.X.; Yang, L.L.; Tang, S.K.; Li, W.J. Actinokineospora soli sp. nov., a thermotolerant actinomycete isolated from soil, and emended description of the genus Actinokineospora. Int. J. Syst. Evol. Microbiol. 2012, 62, 1845–1849. [Google Scholar] [CrossRef]
- Lisdiyanti, P.; Otoguro, M.; Ratnakomala, S.; Lestari, Y.; Hastuti, R.D.; Triana, E.; Katsuhiko, A.; Widyastuti, Y. Actinokineospora baliensis sp. nov., Actinokineospora cibodasensis sp. nov. and Actinokineospora cianjurensis sp. nov., isolated from soil and plant litter. Int. J. Syst. Evol. Microbiol. 2010, 60, 2331–2335. [Google Scholar] [CrossRef]
- Abdelmohsen, U.R.; Pimentel-Elardo, S.M.; Hanora, A.; Radwan, M.; Abou-El-Ela, S.H.; Ahmed, S.; Hentschel, U. Isolation, phylogenetic analysis and anti-infective activity screening of marine sponge-associated actinomycetes. Mar. Drugs 2010, 8, 399–412. [Google Scholar] [CrossRef]
- Kharel, M.K.; Pahari, P.; Shepherd, M.D.; Tibrewal, N.; Nybo, S.E.; Shaaban, K.A.; Rohr, J. Angucyclines: Biosynthesis, mode-of-action, new natural products, and synthesis. Nat. Prod. Rep. 2012, 29, 264–325. [Google Scholar] [CrossRef]
- Rohr, J.; Thiericke, R. Angucycline group antibiotics. Nat. Prod. Rep. 1992, 9, 103–137. [Google Scholar] [CrossRef]
- Kharel, M.K.; Pahari, P.; Lian, H.; Rohr, J. Enzymatic total synthesis of rabelomycin, an angucycline group antibiotic. Org. Lett. 2010, 12, 2814–2817. [Google Scholar] [CrossRef]
- Shaaban, K.A.; Srinivasan, S.; Kumar, R.; Damodaran, C.; Rohr, J.; Landomycins, P.-W. Cytotoxic angucyclines from Streptomyces cyanogenus S-136. J. Nat. Prod. 2011, 74, 2–11. [Google Scholar] [CrossRef]
- Korynevska, A.; Heffeter, P.; Matselyukh, B.; Elbling, L.; Micksche, M.; Stoika, R.; Berger, W. Mechanisms underlying the anticancer activities of the angucycline landomycin E. Biochem. Pharmacol. 2007, 74, 1713–1726. [Google Scholar] [CrossRef]
- Shaaban, K.A.; Stamatkin, C.; Damodaran, C.; Rohr, J. 11-Deoxylandomycinone and landomycins X-Z, new cytotoxic angucyclin(on)es from a Streptomyces cyanogenus K62 mutant strain. J. Antibiot. 2011, 64, 141–150. [Google Scholar] [CrossRef]
- Sasaki, E.; Ogasawara, Y.; Liu, H.W. A biosynthetic pathway for BE-7585A, a 2-thiosugar-containing angucycline-type natural product. J. Am. Chem. Soc. 2010, 132, 7405–7417. [Google Scholar] [CrossRef]
- Fotso, S.; Mahmud, T.; Zabriskie, T.M.; Santosa, D.A.; Sulastri, D.A.; Proteau, P.J. Angucyclinones from an Indonesian Streptomyces sp. J. Nat. Prod. 2008, 71, 61–65. [Google Scholar] [CrossRef]
- Guo, Z.K.; Liu, S.B.; Jiao, R.H.; Wang, T.; Tan, R.X.; Ge, H.M. Angucyclines from an insect-derived actinobacterium Amycolatopsis sp. HCa1 and their cytotoxic activity. Bioorg. Med. Chem. Lett. 2012, 22, 7490–7493. [Google Scholar] [CrossRef]
- Huang, H.; Yang, T.; Ren, X.; Liu, J.; Song, Y.; Sun, A.; Ma, J.; Wang, B.; Zhang, Y.; Huang, C.; et al. Cytotoxic angucycline class glycosides from the deep sea actinomycete Streptomyces lusitanus SCSIO LR32. J. Nat. Prod. 2012, 75, 202–208. [Google Scholar] [CrossRef]
- Bode, H.B.; Bethe, B.; Höfs, R.; Zeeck, A. Big effects from small changes: Possible ways to explore nature′s chemical diversity. Chem. Biol. Chem. 2002, 3, 619–627. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinf. 2010, 11, 395–397. [Google Scholar] [CrossRef]
- Pluskal, T.; Uehara, T.; Yanagida, M. Highly accurate chemical formula prediction tool utilizing high-resolution mass spectra, MS/MS fragmentation, heuristic rules, and isotope pattern matching. Anal. Chem. 2012, 84, 4396–4403. [Google Scholar] [CrossRef]
- Hoet, S.; Opperdoes, F.; Brun, R.; Quetin-Leclercq, J. Natural products active against African trypanosomes: A step towards new drugs. Nat. Prod. Rep. 2004, 21, 353–364. [Google Scholar] [CrossRef]
- Tang, H.; Mechref, Y.; Novotny, M.V. Automated interpretation of MS/MS spectra of oligosaccharides. Bioinformatics 2005, 21, 431–439. [Google Scholar] [CrossRef]
- Vanhamme, L.; Paturiaux-Hanocq, F.; Poelvoorde, P.; Nolan, D.P.; Lins, L.; van Den Abbeele, J.; Pays, A.; Tebabi, P.; van Xong, H.; Jacquet, A.; et al. Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature 2003, 422, 83–87. [Google Scholar] [CrossRef]
- Jacobs, R.T.; Nare, B.; Phillips, M.A. State of the art in African trypanosome drug discovery. Curr. Top. Med. Chem. 2011, 11, 1255–1274. [Google Scholar] [CrossRef]
- ProteoWizard Software Foundation. ProteoWizard Homepage. Available online: http://proteowizard.sourceforge.net/ (accessed on 22 April 2012).
- Sourceforge Project Website. Available online: http://sourceforge.net/projects/mzmine/ (accessed on 22 April 2012).
- Craig, A.; Cloarec, O.; Holmes, E.; Nicholson, J.K.; Lindon, J.C. Scaling and normalization effects in NMR spectroscopic metabonomic data sets. Anal. Chem. 2006, 78, 2262–2267. [Google Scholar] [CrossRef]
- Huber, W.; Koella, J.C. A comparison of three methods of estimating EC50 in studies of drug resistance of malaria parasites. Acta Trop. 1993, 55, 257–261. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Abdelmohsen, U.R.; Cheng, C.; Viegelmann, C.; Zhang, T.; Grkovic, T.; Ahmed, S.; Quinn, R.J.; Hentschel, U.; Edrada-Ebel, R. Dereplication Strategies for Targeted Isolation of New Antitrypanosomal Actinosporins A and B from a Marine Sponge Associated-Actinokineospora sp. EG49. Mar. Drugs 2014, 12, 1220-1244. https://doi.org/10.3390/md12031220
Abdelmohsen UR, Cheng C, Viegelmann C, Zhang T, Grkovic T, Ahmed S, Quinn RJ, Hentschel U, Edrada-Ebel R. Dereplication Strategies for Targeted Isolation of New Antitrypanosomal Actinosporins A and B from a Marine Sponge Associated-Actinokineospora sp. EG49. Marine Drugs. 2014; 12(3):1220-1244. https://doi.org/10.3390/md12031220
Chicago/Turabian StyleAbdelmohsen, Usama Ramadan, Cheng Cheng, Christina Viegelmann, Tong Zhang, Tanja Grkovic, Safwat Ahmed, Ronald J. Quinn, Ute Hentschel, and RuAngelie Edrada-Ebel. 2014. "Dereplication Strategies for Targeted Isolation of New Antitrypanosomal Actinosporins A and B from a Marine Sponge Associated-Actinokineospora sp. EG49" Marine Drugs 12, no. 3: 1220-1244. https://doi.org/10.3390/md12031220