Domoic Acid Epileptic Disease

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Case Definition
2. Concept of Domoic Acid Epileptic Disease
3. Acute Poisoning
3.1. Amnesic Shellfish Poisoning
3.2. Acute Domoic Acid Toxicosis
4. Domoic Acid Epileptic Disease
4.1. Human Case Study
4.1.1. Clinical Evaluation
4.1.2. Neuropathological Findings
4.1.3. Kainic Acid Model for Temporal Lobe Epilepsy
4.2. California Sea Lion Chronic Epileptic Syndrome
4.2.1. Clinical Evaluation
4.2.2. Neuropathological Findings
4.3. Rat Model for Domoic Acid Epileptic Disease
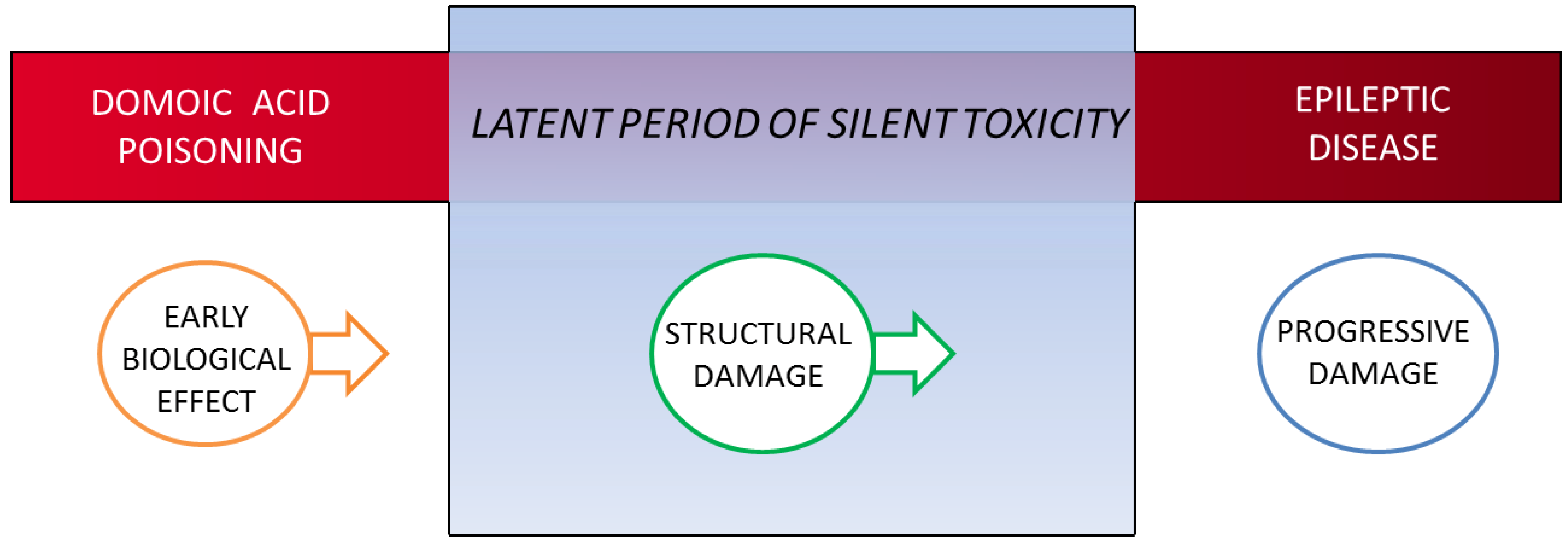
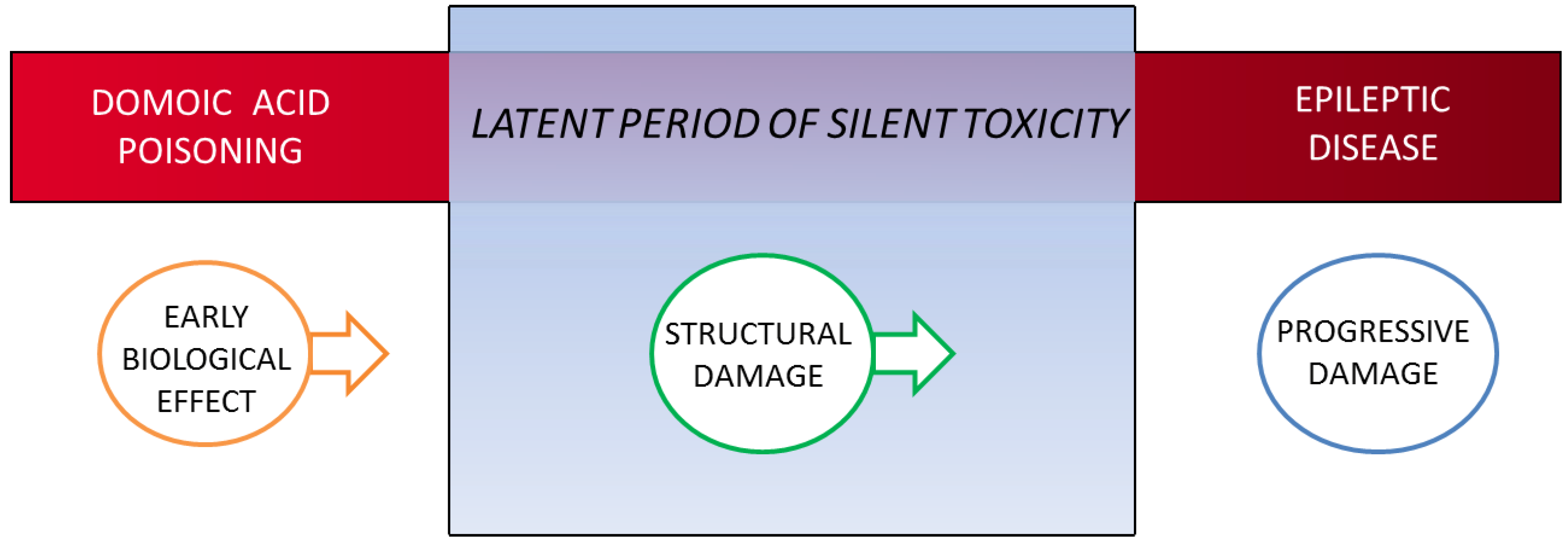
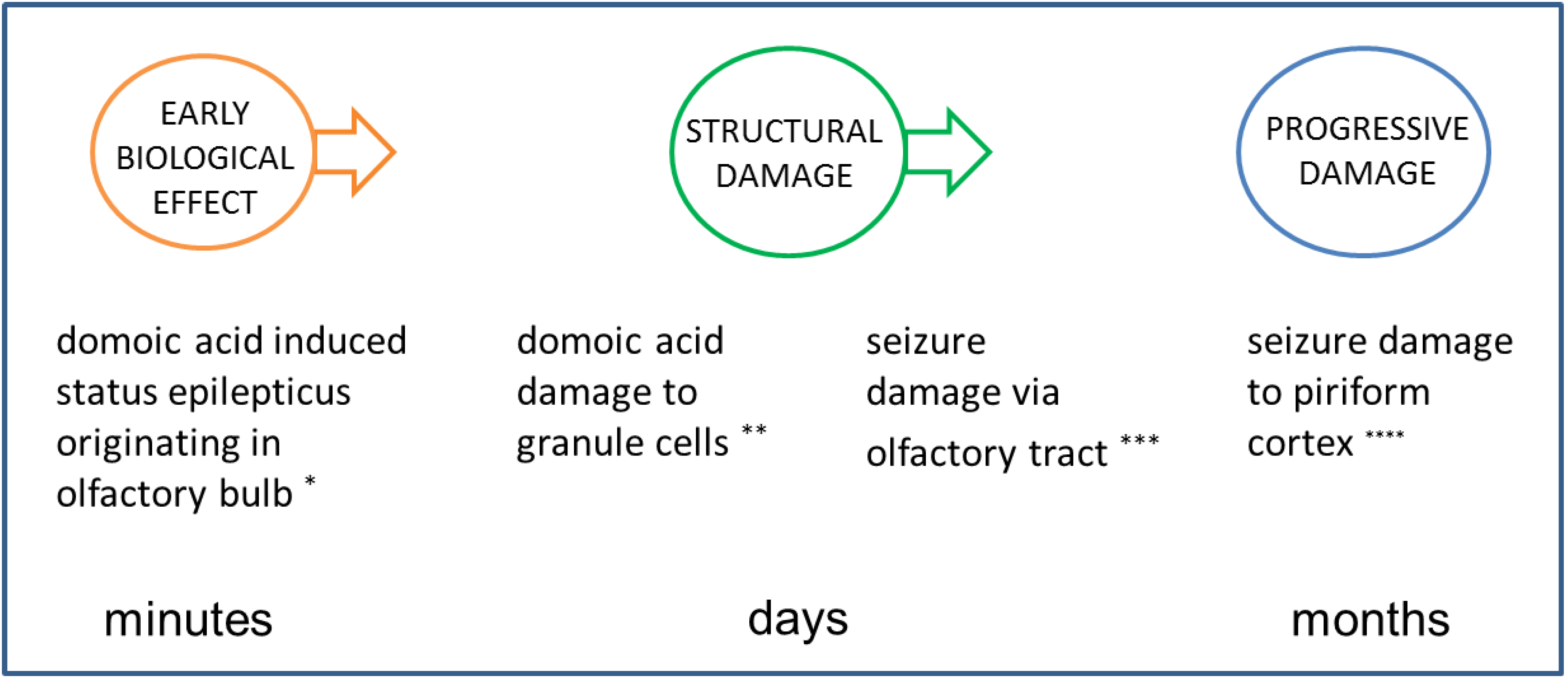
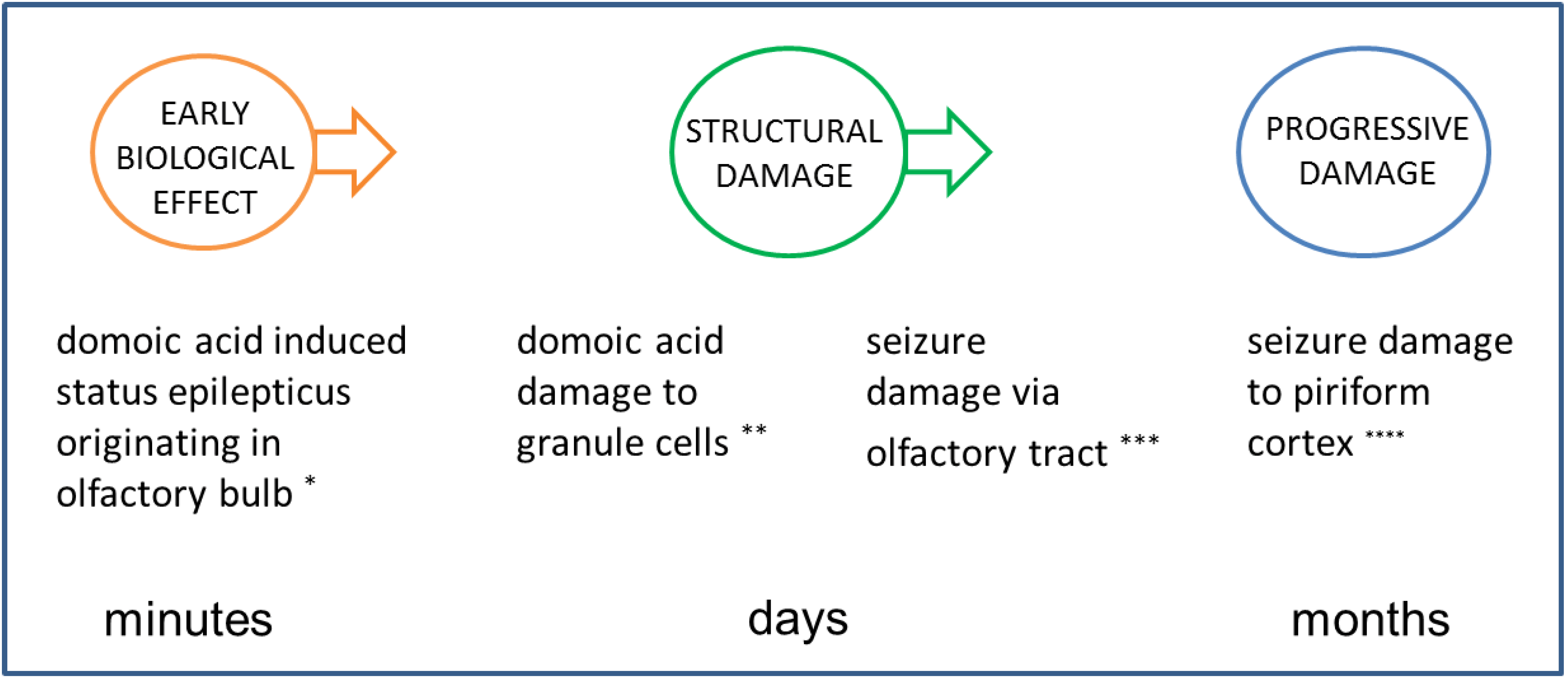
4.3.1. Early Biological Effect
4.3.2. Structural/Functional Damage Evident in the Latent Period
4.3.3. Behavioral Changes during Expression of Disease State
4.3.4. Structural Damage during Expression of Disease State
4.4. Concept for Olfactory Origin of Epileptic Disease
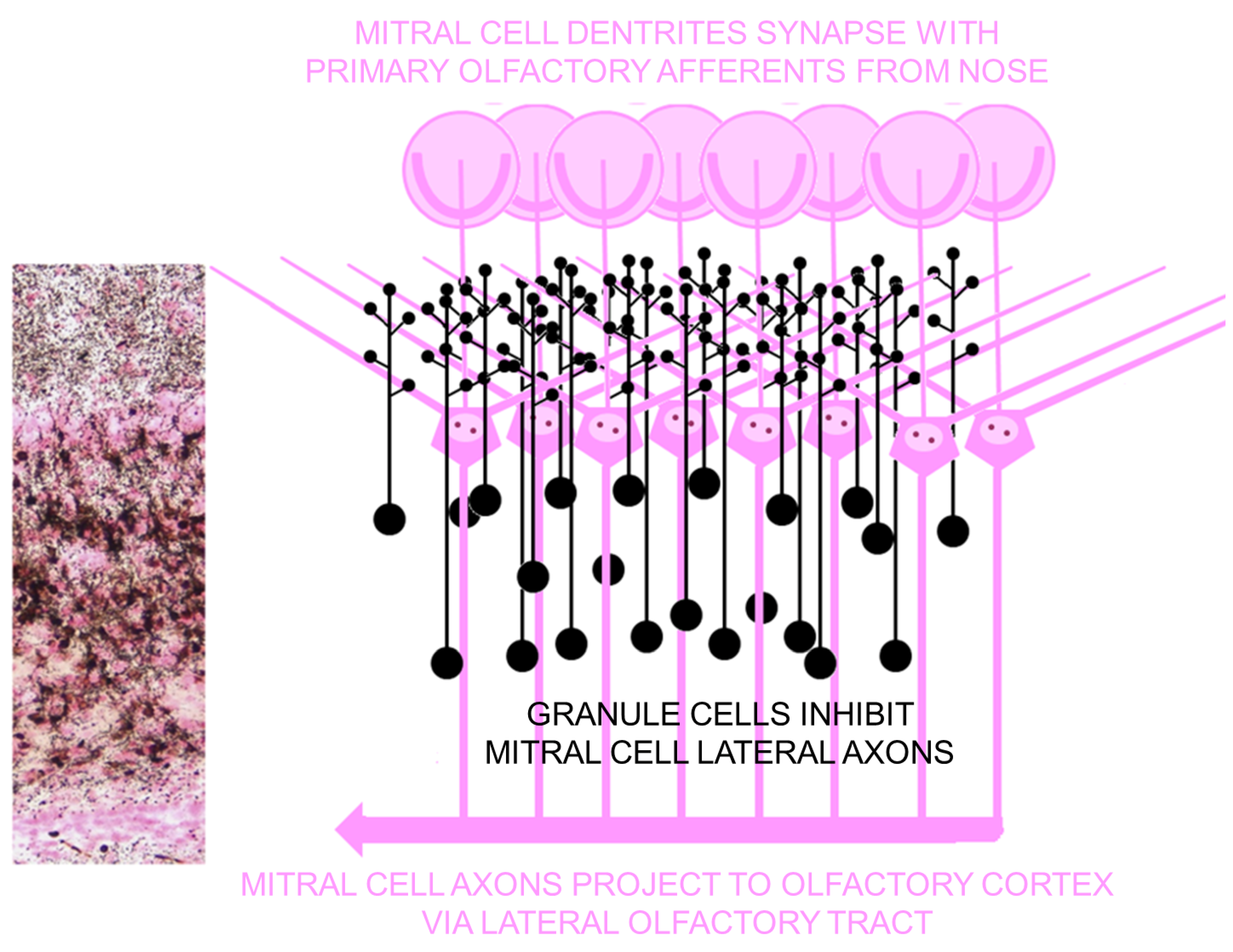
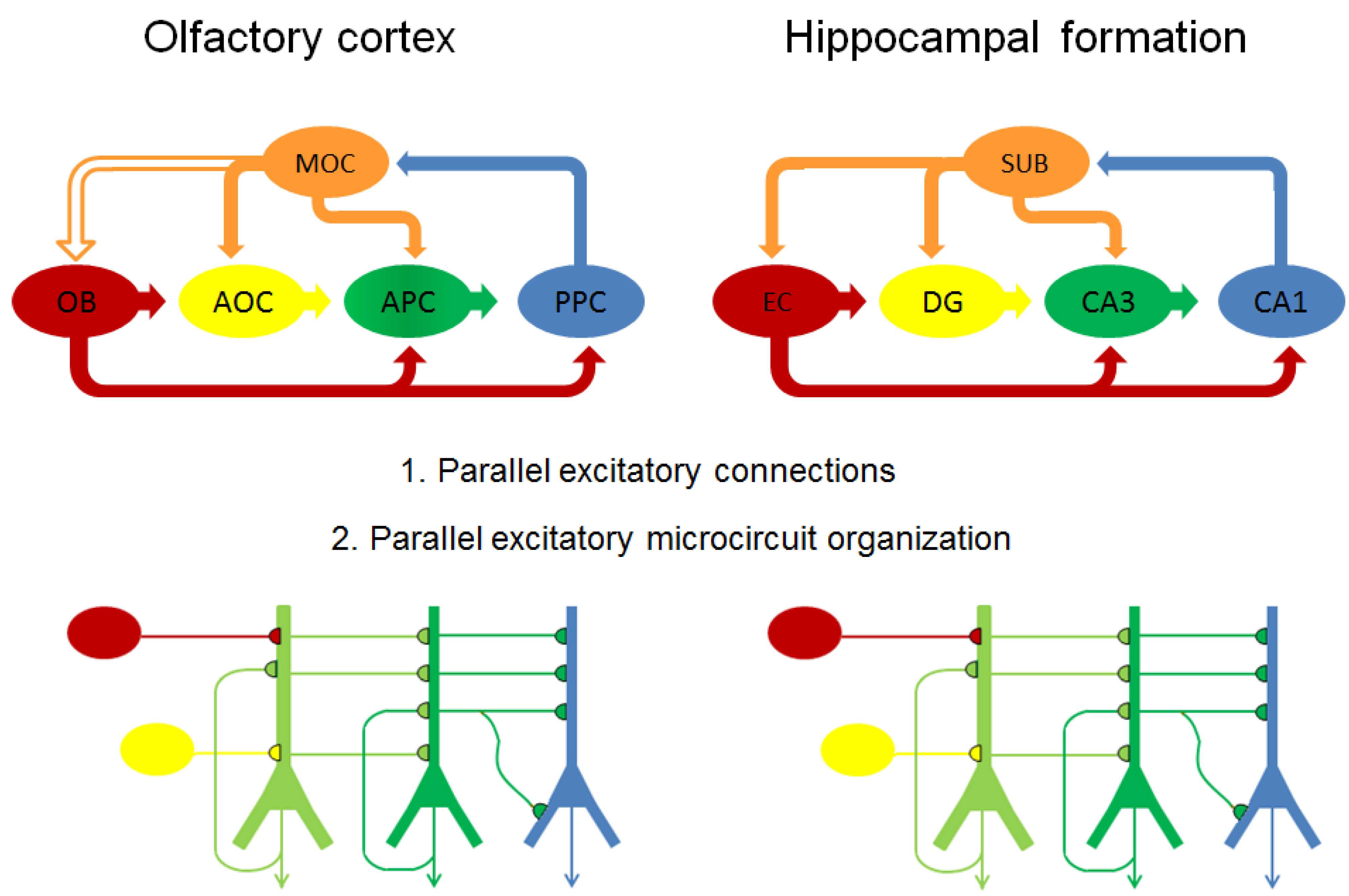
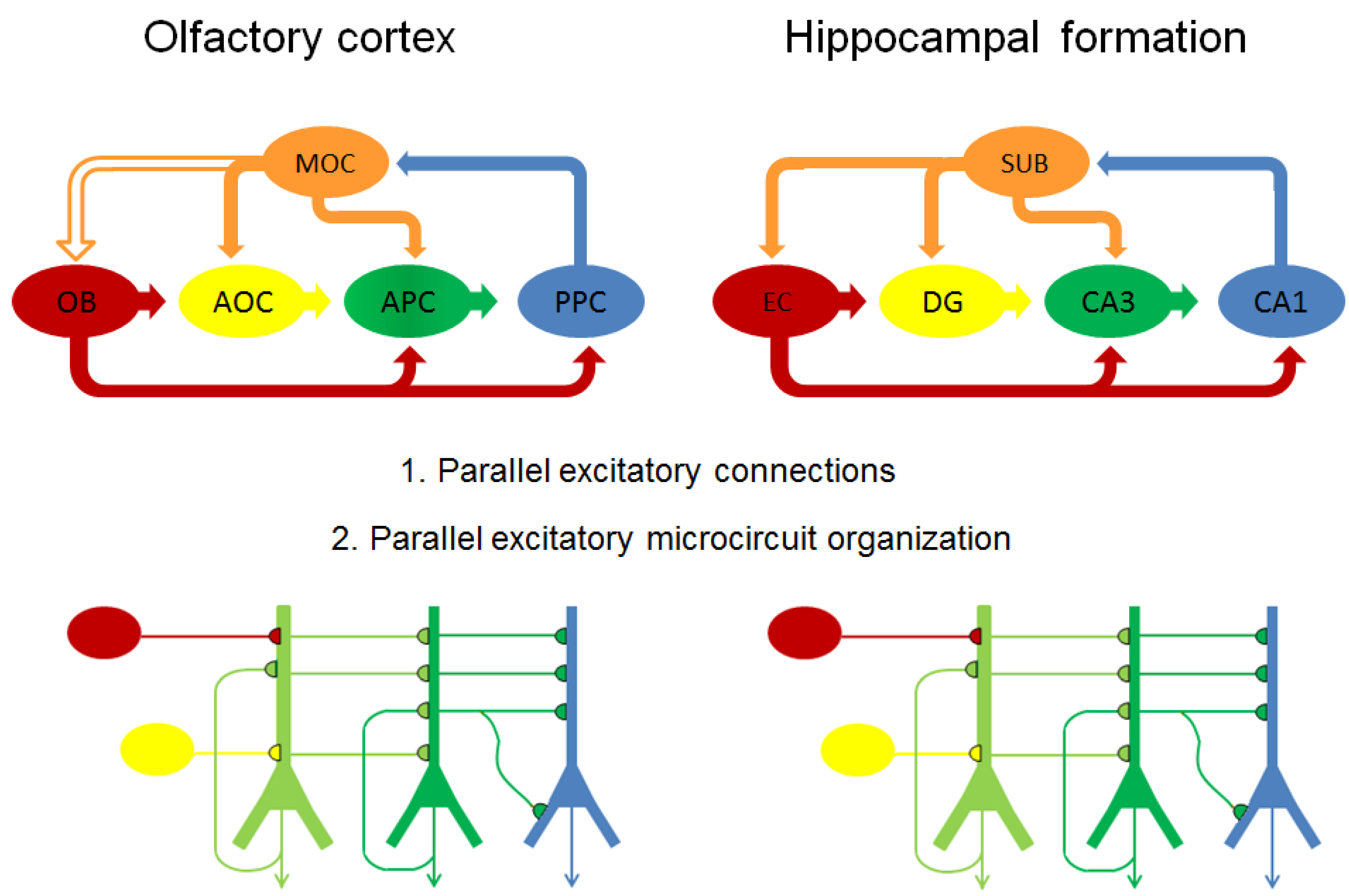
4.4.1. Parallels between Olfactory Cortex and Hippocampal Formation
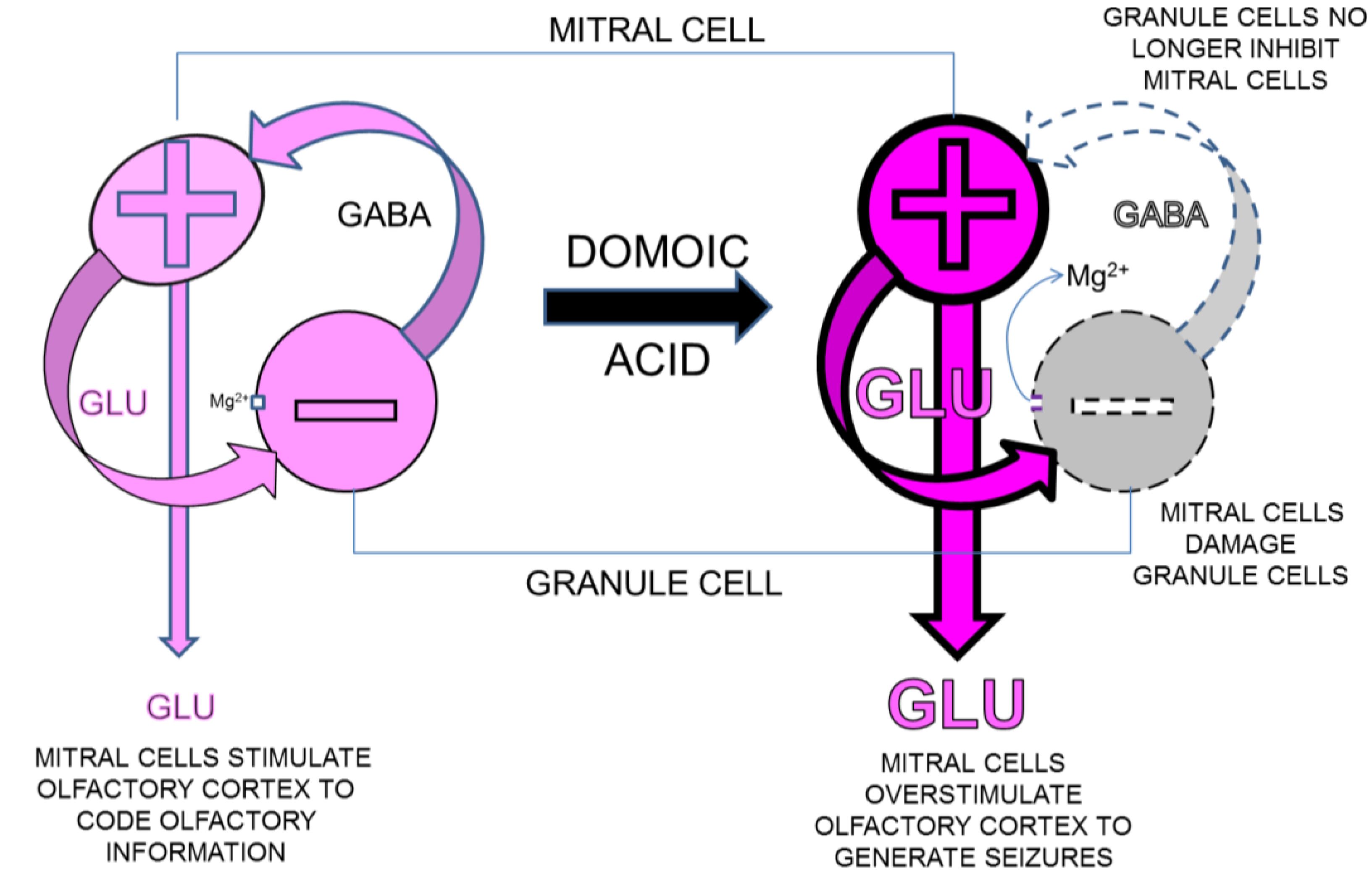
4.4.2. Seizures Originating in Olfactory Bulb
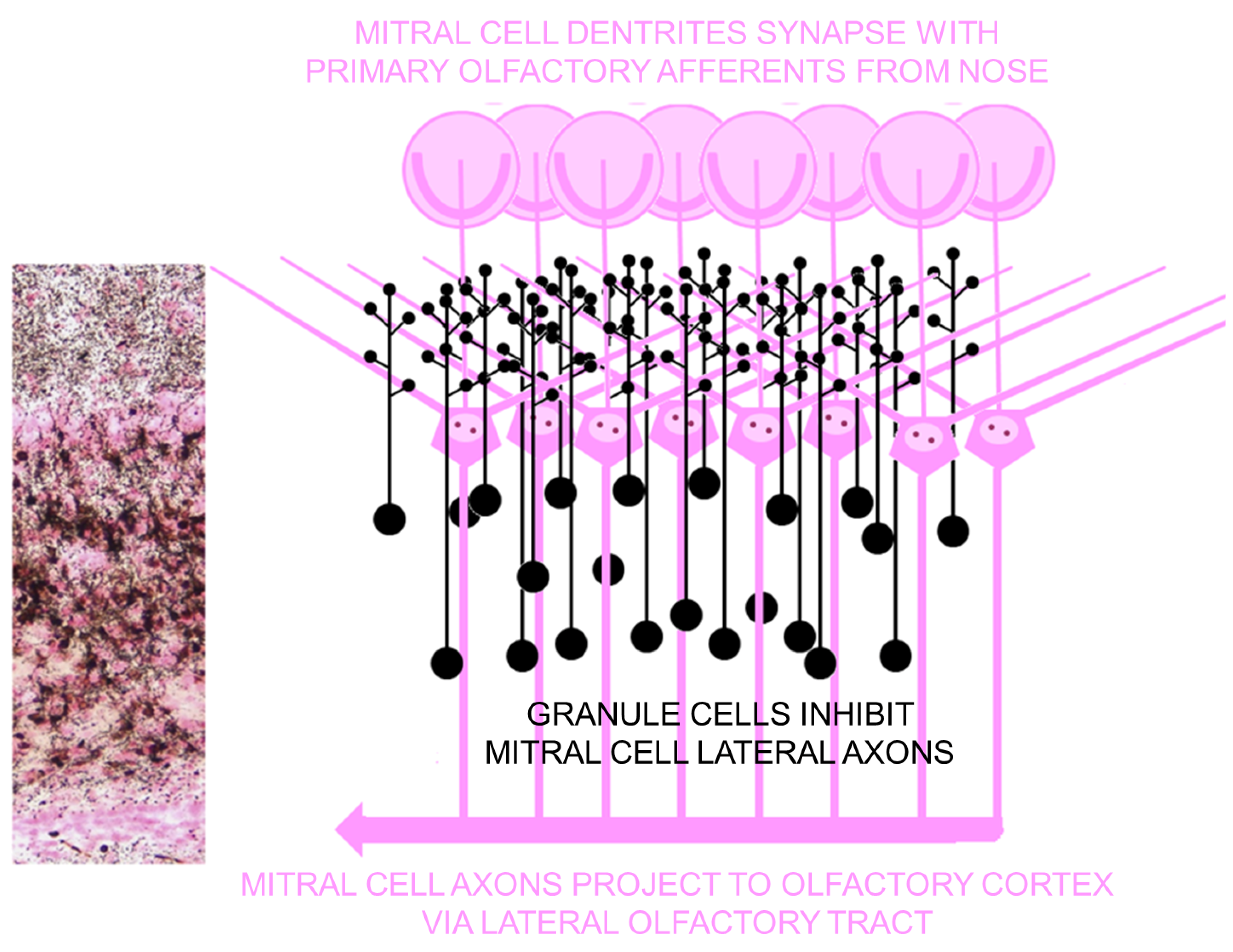
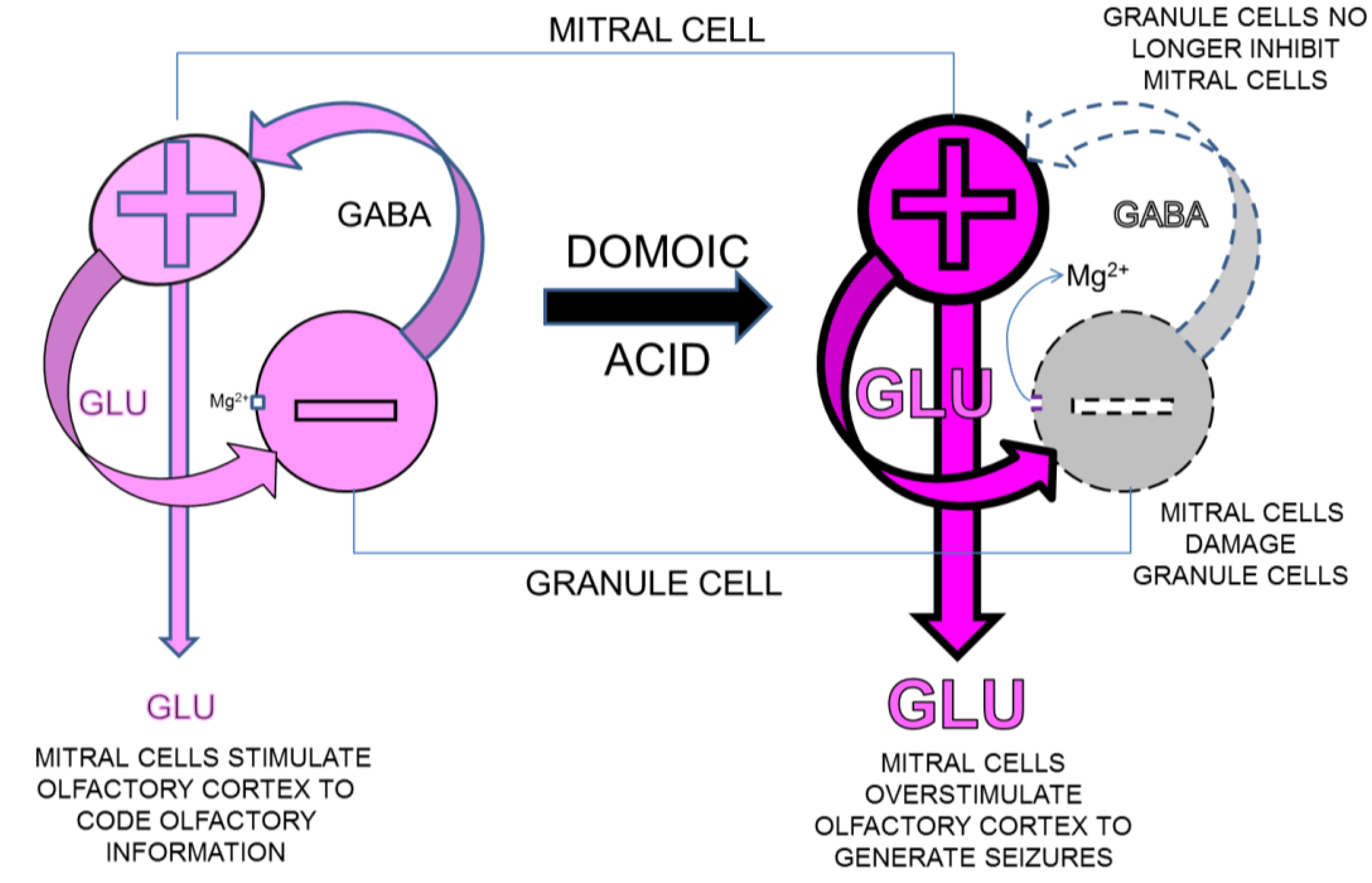
4.4.3. Olfactory Outcome Pathway for Rat Model for Domoic Acid Epileptic Disease
4.4.4. Role for Hippocampus and Olfactory Bulb in Domoic Acid Epileptic Disease
5. Populations at Risk
5.1. Observational Studies
5.1.1. Human Studies
5.1.2. Sea Lion Studies
5.2. Toxicokinetic Susceptibility Factors
5.2.1. Renal Clearance
5.2.2. Maternal Toxicokinetics
5.2.3. Prenatal Toxicokinetics
5.2.4. Postnatal Toxicokinetics
5.3. Intrinsic Susceptibility Factors
5.3.1. Age Susceptibility
5.3.2. Developmental Susceptibility
6. Medical Diagnosis and Treatment
6.1. Diagnostic Criteria
6.2. Diagnostic Tests
6.3. Comorbidity
Cardiomyopathy
6.4. Management and Treatment
6.4.1. Human Poisoning and Disease
6.4.2. Sea Lion Poisoning and Disease
6.4.3. Prognosis
7. Conclusions
Supplementary Files
Conflicts of Interest
References
- Perl, T.M.; Bedard, L.; Kosatsky, T.; Hockin, J.C.; Todd, E.C.; McNutt, L.A.; Remis, R.S. Amnesic shellfish poisoning: A new clinical syndrome due to domoic acid. Can. Dis. Wkly. Rep. 1990, 16, 7–8. [Google Scholar]
- Gulland, F.M.; Haulena, M.; Fauquier, D.; Langlois, G.; Lander, M.E.; Zabka, T.; Duerr, R. Domoic acid toxicity in Californian sea lions (Zalophus californianus): Clinical signs, treatment and survival. Vet. Rec. 2002, 150, 475–480. [Google Scholar] [CrossRef]
- Pulido, O.M. Domoic acid toxicologic pathology: A review. Mar. Drugs 2008, 6, 180–219. [Google Scholar] [CrossRef]
- Lefebvre, K.A.; Robertson, A. Domoic acid and human exposure risks: A review. Toxicon 2010, 56, 218–230. [Google Scholar] [CrossRef]
- Costa, L.G.; Giordano, G.; Faustman, E.M. Domoic acid as a developmental neurotoxin. Neurotoxicology 2010, 31, 409–423. [Google Scholar] [CrossRef]
- Grant, K.S.; Burbacher, T.M.; Faustman, E.M.; Gratttan, L. Domoic acid: Neurobehavioral consequences of exposure to a prevalent marine biotoxin. Neurotoxicol. Teratol. 2010, 32, 132–141. [Google Scholar] [CrossRef]
- Ramsdell, J.S. The molecular and integrative basis to domoic acid toxicity. In Phycotoxins: Chemisty and Biochemistry; Botana, L., Ed.; Blackwell Publishing: Ames, IA, USA, 2007; pp. 223–250. [Google Scholar]
- Cendes, F.; Andermann, F.; Carpenter, S. Temporal lobe epilepsy caused by domoic acid intoxication: Evidence for glutamate receptor-mediated excitotoxicity in humans. Ann. Neurol. 1995, 37, 123–126. [Google Scholar] [CrossRef]
- Goldstein, T.; Mazet, J.A.K.; Zabka, T.S.; Langlois, G.; Colegrove, K.M.; Silver, M.; Bargu, S.; Van Dolah, F.; Leighfield, T.; Conrad, P.A.; et al. Novel symptomatology and changing epidemiology of domoic acid toxicosis in California sea lions (Zalophus californianus): An increasing risk to marine mammal health. Proc. R. Soc. B 2008, 275, 267–276. [Google Scholar] [CrossRef]
- Muha, N.; Ramsdell, J.S. Domoic acid induced seizures progress to a chronic state of epilepsy in rats. Toxicon 2011, 57, 168–171. [Google Scholar] [CrossRef]
- Fuquay, J.M.; Muha, N.; Pennington, P.L.; Ramsdell, J.S. Domoic acid induced status epilepticus promotes aggressive behavior in rats. Physiol. Behav. 2012, 105, 315–320. [Google Scholar] [CrossRef]
- Pitkänen, A.; Sutula, T.P. Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurol. 2002, 1, 173–181. [Google Scholar] [CrossRef]
- Sloviter, R.S.; Bumanglag, A.V. Defining “epileptogenesis” and identifying “antiepileptogenic targets” in animal models of acquired temporal lobe epilepsy is not as simple as it might seem. Neuropharmacology 2013, 69, 3–15. [Google Scholar] [CrossRef]
- Perl, T.M.; Bédard, L.; Kosatsky, T.; Hockin, J.C.; Todd, E.; Remis, R.S. An outbreak of toxic encephalopathy caused by eating mussels contaminated with domoic acid. N. Engl. J. Med. 1990, 322, 1775–1780. [Google Scholar] [CrossRef]
- Teitelbaum, J.S.; Zatorre, R.J.; Carpenter, S.; Gendron, D.; Evans, A.C.; Gjedde, A.; Cashman, N.R. Neurologic sequelae of domoic acid intoxication due to the ingestion of contaminated mussels. N. Engl. J. Med. 1990, 322, 1781–1787. [Google Scholar] [CrossRef]
- Teitelbaum, J. Acute manifestations of domoic acid poisoning: case presentations. Can. Dis. Wkly. Rep. 1990, 16, 5–6. [Google Scholar]
- Scholin, C.A.; Gulland, F.; Doucette, G.J.; Benson, S.; Busman, M.; Chavez, F.P.; Cordaro, J.; DeLong, R.; De Vogelaere, A.; Harvey, J.; et al. Mortality of sea lions along the central California coast linked to a toxic diatom bloom. Nature 2000, 403, 80–84. [Google Scholar] [CrossRef]
- Torres de la Riva, G.T.; Johnson, C.K.; Gulland, F.M.; Langlois, G.W.; Heyning, J.E.; Rowles, T.K.; Mazet, J.A. Association of an unusual marine mammal mortality event with Pseudo-nitzschia spp. Blooms along the southern California coastline. J. Wildl. Dis. 2009, 45, 109–121. [Google Scholar] [CrossRef]
- Carpenter, S. The human neuropathology of encephalopathic mussel toxin poisoning. Can. Dis. Wkly. Rep. 1990, 16, 73–74. [Google Scholar]
- Hauser, W.A.; Hesdorffer, D.C. Epilepsy: Frequency, Causes, and Consequences; Epilepsy Foundation of America: Landover, MD, USA, 1990. [Google Scholar]
- Nadler, J.V. Minireview. Kainic acid as a tool for the study of temporal lobe epilepsy. Life Sci. 1981, 29, 2031–2042. [Google Scholar] [CrossRef]
- Dudek, F.E.; Clark, S.; Williams, P.A.; Grabenstatter, H.L. Kainate-Induced Status Epilepticus: A Chronic Model of Acquired Epilepsy. In Models Seizures and Epilepsy; Sutula, T., Pitkanen, A., Eds.; Elsevier Academic Press: London, UK, 2005; pp. 415–432. [Google Scholar]
- Vincent, P.; Mulle, C. Kainate receptors in epilepsy and excitotoxicity. Neuroscience 2009, 158, 309–323. [Google Scholar] [CrossRef]
- Silvagni, P.A.; Lowenstine, L.J.; Spraker, T.; Lipscomb, T.P.; Gulland, F.M. Pathology of domoic acid toxicity in California sea lions (Zalophus californianus). Vet. Pathol. 2005, 42, 184–191. [Google Scholar] [CrossRef]
- Buckmaster, P.S.; Wen, X.; Toyoda, I.; Gulland, F.M.D.; Van Bonn, W. Hippocampal neuropathology of domoic acid-induced epilepsy in California sea lions (Zalophus californianus). J. Comp. Neurol. 2013, in press. [Google Scholar]
- Madl, J.E.; Duncan, C.G.; Stanhill, J.E.; Tai, P.Y.; Spraker, T.R.; Gulland, F.M. Oxidative stress and redistribution of glutamine synthetase in California sea lions (Zalophus californianus) with domoic acid toxicosis. J. Comp. Pathol. 2013, in press. [Google Scholar]
- Montie, E.W.; Wheeler, E.; Pussini, N.; Battey, T.W.K.; Van Bonn, W.; Gulland, F. Magnetic resonance imaging reveals that brain atrophy is more severe in older California sea lions with domoic acid toxicosis. Harmful Algae 2012, 20, 19–29. [Google Scholar] [CrossRef]
- Ramsdell, J.S.; Stafstrom, C.E. Rat kainic acid model provides unexpected insight into an emerging epilepsy syndrome in sea lions. Epilepsy Curr. 2009, 9, 142–143. [Google Scholar] [CrossRef]
- Perez-Mendes, P.; Cinini, S.M.; Medeiros, M.A.; Tufik, S.; Mello, L.E. Behavioral and histopathological analysis of domoic acid administration in marmosets. Epilepsia 2005, 46, 148–151. [Google Scholar] [CrossRef]
- Hellier, J.L.; Dudek, F.E. Chemoconvulsant Model of Chronic Spontaneous Seizures. In Current Protocols in Neuroscience; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Tiedeken, J.A.; Muha, N.; Ramsdell, J.S. A cupric silver histochemical analysis of domoic acid damage to olfactory pathways following status epilepticus in a rat model for chronic recurrent spontaneous seizures and aggressive behavior. Toxicol. Pathol. 2012, 41, 454–469. [Google Scholar] [CrossRef]
- Egger, V.; Urban, N.N. Dynamic connectivity in the mitral cell–granule cell microcircuit. Semin. Cell Dev. Biol. 2006, 17, 424–432. [Google Scholar]
- Barnett, S.A. An analysis of social behavior in wild rats. Proce. Zool. Soc. Lond. 1958, 130, 107–152. [Google Scholar] [CrossRef]
- Blanchard, R.J.; Fukunaga, K.; Blanchard, D.C.; Kelley, M.J. Conspecific aggression in the laboratory rat. J. Comp. Physiol. Psychol. 1975, 89, 1204–1209. [Google Scholar] [CrossRef]
- Tiedeken, J.A.; Ramsdell, J.S. Persistent neurological damage associated with spontaneous recurrent seizures and atypical aggressive behavior of domoic acid epileptic disease. Toxicol. Sci. 2013, 133, 133–143. [Google Scholar] [CrossRef]
- Loscher, W.; Ebert, U. The role of the piriform cortex in kindling. Prog. Neurobiol. 1996, 50, 427–481. [Google Scholar] [CrossRef]
- Racine, R.J.; Mosher, M.; Kairiss, E.W. The role of the pyriform cortex in the generation of interictal spikes in the kindled preparation. Brain Res. 1988, 454, 251–263. [Google Scholar] [CrossRef]
- Pereira, P.M.G.; Insausti, R.; Artacho-Pérula, E.; Salmenperä, T.; Kälviäinen, R.; Pitkänen, A. MR volumetric analysis of the piriform cortex and cortical amygdala in drug-refractory temporal lobe epilepsy. Am. J. Neuroradiol. 2005, 26, 319–332. [Google Scholar]
- Wacker, D.W.; Tobin, V.A.; Noack, J.; Bishop, V.R.; Duszkiewicz, A.J.; Engelmann, M.; Meddle, S.L.; Ludwig, M. Expression of early growth response protein 1 in vasopressin neurones of the rat anterior olfactory nucleus following social odour exposure. J. Physiol. 2010, 588, 4705–4717. [Google Scholar] [CrossRef]
- Colman, J.R.; Nowocin, K.J.; Switzer, R.C.; Trusk, T.C.; Ramsdell, J.S. Mapping and reconstruction of domoic acid-induced neurodegeneration in the mouse brain. Neurotoxicol. Teratol. 2005, 27, 753–767. [Google Scholar] [CrossRef]
- Tryphonas, L.; Truelove, J.; Nera, E.; Iverson, F. Acute neurotoxicity of domoic acid in the rat. Toxicol. Pathol. 1989, 18, 1–9. [Google Scholar]
- Rowe, T.B.; Macrini, T.E.; Luo, Z.-X. Fossil evidence on origin of the mammalian brain. Science 2011, 332, 955–957. [Google Scholar] [CrossRef]
- Haberly, L.B. Parallel-distributed processing in olfactory cortex: New insights from morphological and physiological analysis of neuronal circuitry. Chem. Senses 2001, 26, 551–576. [Google Scholar] [CrossRef]
- Shepherd, G.M. The microcircuit concept applied to cortical evolution: From three-layer to six-layer cortex. Front. Neuroanat. 2011, 5. [Google Scholar] [CrossRef]
- Goddard, G.V.; McIntyre, D.C.; Leech, C.K. A permanent change in brain function resulting from daily electrical stimulation. Exp. Neurol. 1969, 25, 295–330. [Google Scholar] [CrossRef]
- Restrepo, D.; Hellier, J.L.; Salcedo, E. Complex metabolically demanding sensory processing in the olfactory system: Implications for epilepsy. Epilepsy Behav. 2013. [Google Scholar] [CrossRef]
- Araki, T.; Kato, M.; Kobayashi, T. Limbic seizures originating in the olfactory bulb: An electro-behavioral annd glucose metabolism study. Brain Res. 1995, 693, 207–216. [Google Scholar] [CrossRef]
- Peng, Y.G.; Taylor, T.B.; Finch, R.E.; Switzer, R.C.; Ramsdell, J.S. Neuroexcitatory and neurotoxic actions of the amnesic shellfish poison, domoic acid. Neuroreport 1994, 5, 981–985. [Google Scholar] [CrossRef]
- Wacker, D.W.; Engelmann, M.; Tobin, V.A.; Meddle, S.L.; Ludwig, M. Vasopressin and social odor processing in the olfactory bulb and anterior olfactory nucleus. Ann. N. Y. Acad. Sci. 2011, 1220, 106–116. [Google Scholar]
- Brodie, E.C.; Gulland, F.M.D.; Greig, D.J.; Hunter, M.; Jaakola, J.; Leger, J.S.; Leighfield, T.A.; Van Dolah, F.M. Domoic acid causes reproductive failure in California sea lions (Zalophus californianus). Mar. Mamm. Sci. 2006, 22, 700–707. [Google Scholar] [CrossRef]
- Gulland, F. Domoic Acid Toxicity in California sea lions (Zalophus californianus) Stranded along the Central California Coast, May October 1998. Report to the National Marine Fisheries Service Working Group on Unusual Marine Mammal Mortality Events; U.S. Department of Commerce, National Oceanic and Atmospheric Administration, National Marine Fisheries Service: Seattle, WA, USA, 2000; p. 45. [Google Scholar]
- Suzuki, C.A.; Hierlihy, S.L. Renal clearance of domoic acid in the rat. Food Chem. Toxicol. 1993, 31, 701–706. [Google Scholar] [CrossRef]
- Truelove, J.; Iverson, F. Serum domoic acid clearance and clinical observations in the Cynomolgus monkey and Sprague-Dawley rat following a single IV-dose. Bull. Environ. Contam. Toxicol. 1994, 52, 479–486. [Google Scholar]
- Maucher Fuquay, J.; Muha, N.; Wang, Z.; Ramsdell, J.S. Toxicokinetics of domoic acid in the fetal rat. Toxicology 2012, 294, 36–41. [Google Scholar] [CrossRef]
- Maucher Fuquay, J.; Muha, N.; Wang, Z.; Ramsdell, J.S. Elimination kinetics of domoic acid from brain and CSF of the pregnant rat. Chem. Res. Toxicol. 2012. [Google Scholar] [CrossRef]
- Maucher, J.M.; Ramsdell, J.S. Domoic acid transfer to milk: Evaluation of a potential route of neonatal exposure. Environ. Health Perspect 2005, 113, 461–464. [Google Scholar] [CrossRef]
- Ramsdell, J.S.; Zabka, T.S. In utero domoic acid toxicity: A fetal basis to adult disease in the California sea lion (Zalophus californianus). Mar. Drugs 2008, 6, 262–290. [Google Scholar] [CrossRef]
- Ramsdell, J.S. Neurological disease rises from ocean to bring model for human epilepsy to life. Toxins 2010, 2, 1646–1675. [Google Scholar] [CrossRef]
- Wozniak, D.F.; Stewart, G.R.; Miller, J.P.; Olney, J.W. Age-related sensitivity to kainate neurotoxicity. Exp. Neurol. 1991, 114, 250–253. [Google Scholar] [CrossRef]
- Hesp, B.R.; Clarkson, A.N.; Sawant, P.M.; Kerr, D.S. Domoic acid preconditioning and seizure induction in young and aged rats. Epilepsy Res. 2007, 76, 103–112. [Google Scholar]
- Stewart, I. Environmental risk factors for temporal lobe epilepsy—Is prenatal exposure to the marine algal neurotoxin domoic acid a potentially preventable cause? Med. Hypotheses 2010, 74, 466–481. [Google Scholar] [CrossRef]
- Doucette, T.; Bak-Jensen, H.H.; Perry, M.; Ryan, C.; Tasker, R.A. Developmental Animal Model of Temporal Lobe Epilepsy. Google Patents US7034201 B2, 25 April 2006. [Google Scholar]
- Dakshinamurti, K.; Sharma, S.K.; Sundaram, M.; Watanabe, T. Hippocampal changes in developing postnatal mice following intrauterine exposure to domoic acid. J. Neurosci. 1993, 13, 4486–4495. [Google Scholar]
- Levin, E.D.; Pizarro, K.; Pang, W.G.; Harrison, J.; Ramsdell, J.S. Persisting behavioral consequences of prenatal domoic acid exposure in rats. Neurotoxicol. Teratol. 2005, 27, 719–725. [Google Scholar] [CrossRef]
- Dobbing, J.; Sands, J. Comparative aspects of the brain growth spurt. Early Hum. Dev. 1979, 3, 79–83. [Google Scholar] [CrossRef]
- Doucette, T.A.; Bernard, P.B.; Husum, H.; Perry, M.A.; Ryan, C.L.; Tasker, R.A. Low doses of domoic acid during postnatal development produce permanent changes in rat behaviour and hippocampal morphology. Neurotox. Res. 2004, 6, 555–563. [Google Scholar] [CrossRef]
- Doucette, T.A.; Ryan, C.L.; Tasker, R.A. Gender-based changes in cognition and emotionality in a new rat model of epilepsy. Amino Acids 2007, 32, 317–322. [Google Scholar] [CrossRef]
- Gulland, F.M.D.; Hall, A.J.; Greig, D.J.; Frame, E.R.; Colegrove, K.M.; Booth, R.K.N.; Wasser, S.K.; Scott-Moncrieff, J.C.R. Evaluation of circulating eosinophil count and adrenal gland function in California sea lions naturally exposed to domoic acid. J. Am. Vet. Med. Assoc. 2012, 241, 943–949. [Google Scholar] [CrossRef]
- Zatorre, R.J. Memory loss following domoic acid intoxication from ingestion of toxic mussels. Can. Dis. Wkly. Rep. 1990, 16, 101–103; discussion 103–104. [Google Scholar]
- Teitelbaum, J.S.; Zatorre, R.J.; Carpenter, S.; Gendron, D.; Evans, A.C.; Gjedde, A.; Cashman, N.R. Neurologic sequelae of domoic acid intoxication. Can. Dis. Wkly. Rep. 1990, 16, 9–11. [Google Scholar]
- Cook, P.; Reichmuth, C.; Gulland, F. Rapid behavioural diagnosis of domoic acid toxicosis in California sea lions. Biol. Lett. 2011, 7, 536–538. [Google Scholar] [CrossRef]
- Mancia, A.; Ryan, J.C.; Chapman, R.W.; Wu, Q.; Warr, G.W.; Gulland, F.M.D.; Van Dolah, F.M. Health status, infection and disease in California sea lions (Zalophus californianus) studied using a canine microarray platform and machine-learning approaches. Dev. Comp. Immunol. 2012, 36, 629–637. [Google Scholar] [CrossRef]
- Neely, B.; Soper, J.; Greig, D.; Carlin, K.; Favre, E.; Gulland, F.; Almeida, J.; Janech, M. Serum profiling by MALDI-TOF mass spectrometry as a diagnostic tool for domoic acid toxicosis in California sea lions. Prot. Sci. 2012, 10, 18. [Google Scholar] [CrossRef]
- Zabka, T.S.; Goldstein, T.; Cross, C.; Mueller, R.W.; Kreuder-Johnson, C.; Gill, S.; Gulland, F.M.D. Characterization of a degenerative cardiomyopathy associated with domoic acid toxicity in California sea lions (Zalophus californianus). Vet. Pathol. Online 2009, 46, 105–119. [Google Scholar] [CrossRef]
- Gill, S.S.; Pulido, O.M.; Mueller, R.W.; McGuire, P.F. Molecular and immunochemical characterization of the ionotropic glutamate receptors in the rat heart. Brain Res. Bull. 1998, 46, 429–434. [Google Scholar] [CrossRef]
- Thomas, K.; Harvey, J.T.; Goldstein, T.; Barakos, J.; Gulland, F. Movement, dive behavior, and survival of California sea lions (Zalophus californianus) posttreatment for domoic acid toxicosis. Mar. Mamm. Sci. 2010, 26, 36–52. [Google Scholar]
- Clayton, E.C.; Peng, Y.G.; Means, L.W.; Ramsdell, J.S. Working memory deficits induced by single but not repeated exposures to domoic acid. Toxicon 1999, 37, 1025–1039. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ramsdell, J.S.; Gulland, F.M. Domoic Acid Epileptic Disease. Mar. Drugs 2014, 12, 1185-1207. https://doi.org/10.3390/md12031185
Ramsdell JS, Gulland FM. Domoic Acid Epileptic Disease. Marine Drugs. 2014; 12(3):1185-1207. https://doi.org/10.3390/md12031185
Chicago/Turabian StyleRamsdell, John S., and Frances M. Gulland. 2014. "Domoic Acid Epileptic Disease" Marine Drugs 12, no. 3: 1185-1207. https://doi.org/10.3390/md12031185