Trabectedin and Plitidepsin: Drugs from the Sea that Strike the Tumor Microenvironment


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Tumor Microenvironment

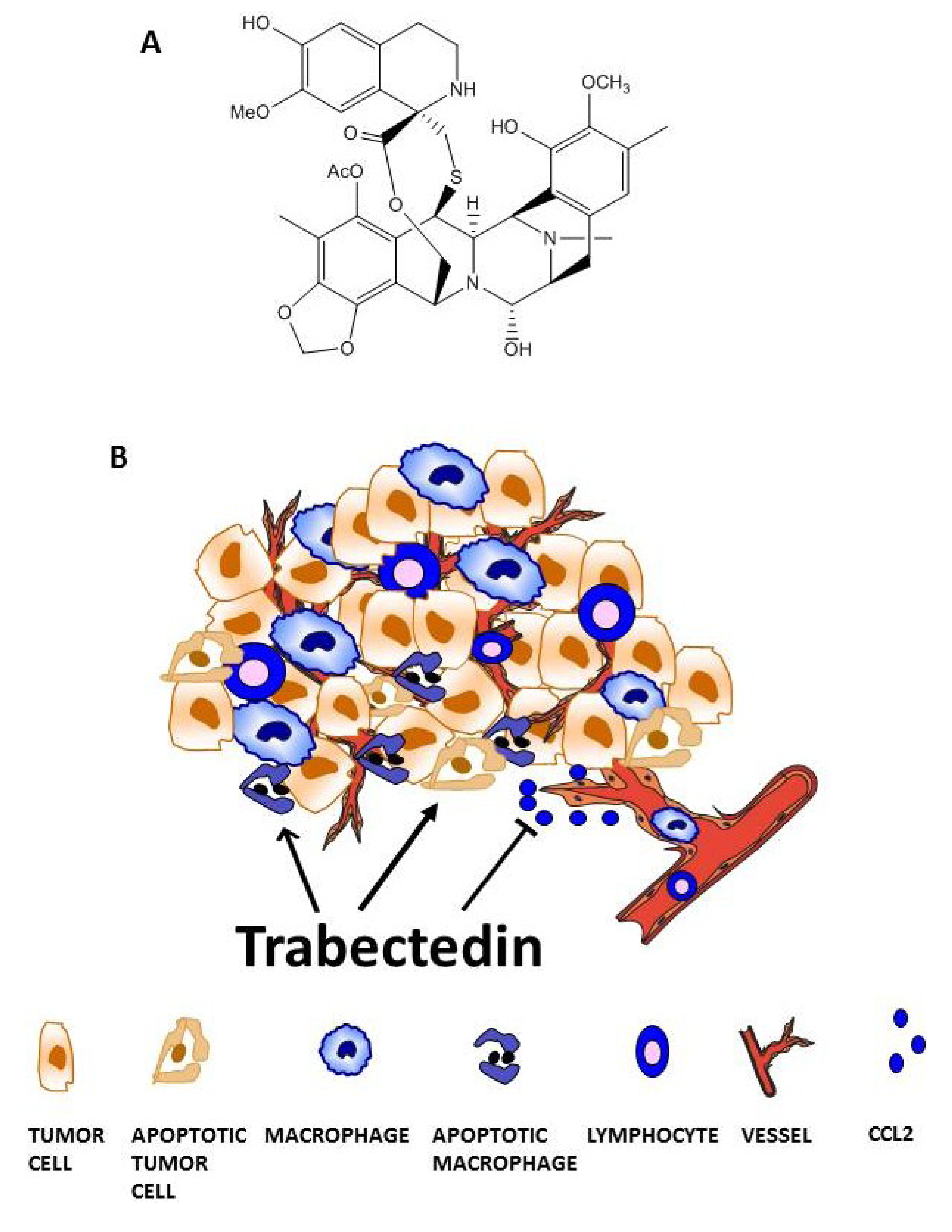
3. Trabectedin Targets Tumor-Associated Macrophages (TAMs)
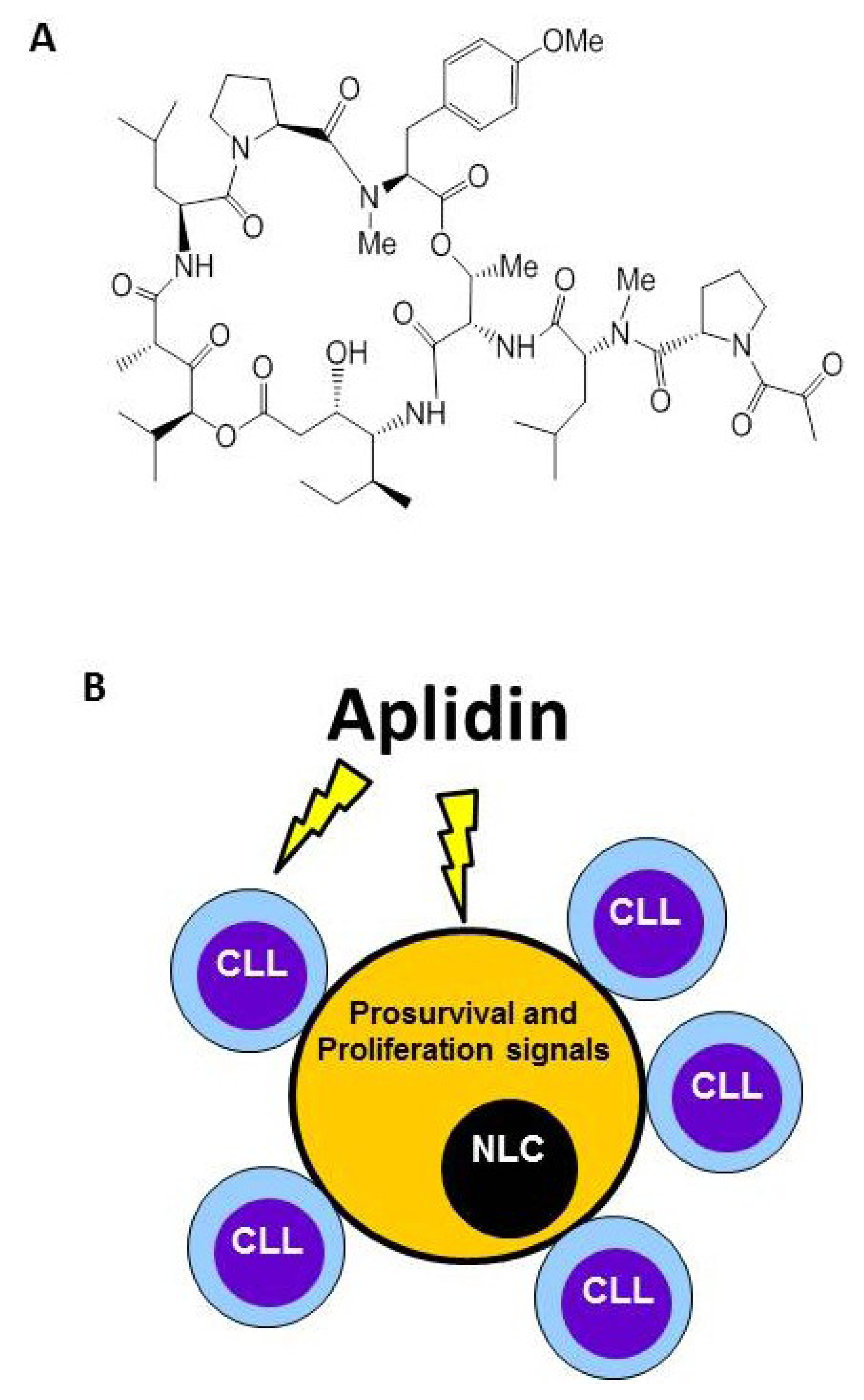
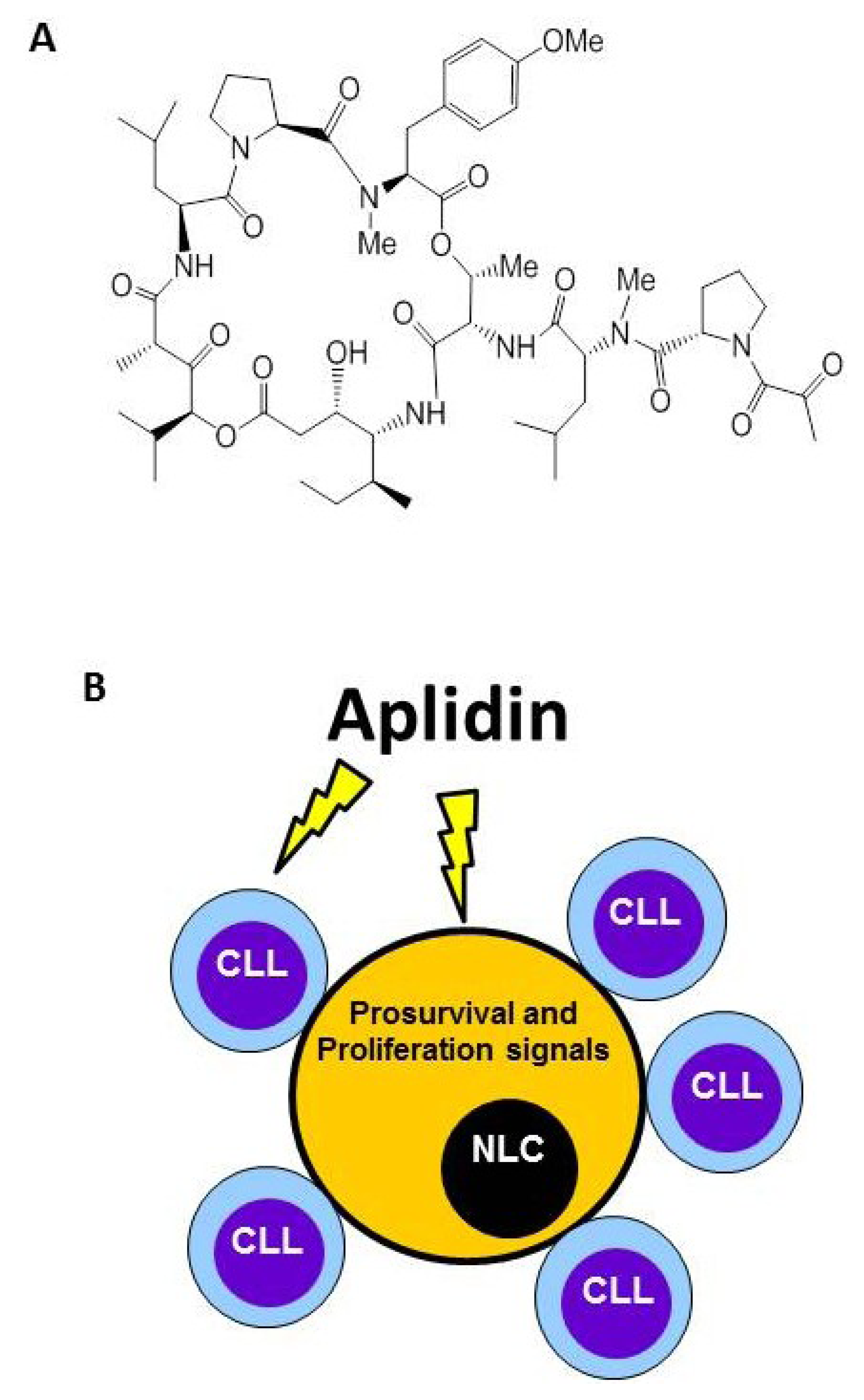
4. Plitidepsin Targets Monocytes and Nurse-Like Cells (NLCs)
5. Conclusions
6. Future Directions
Conflicts of Interest
References
- Farber, E. The multistep nature of cancer development. Cancer Res. 1984, 44, 4217–4223. [Google Scholar]
- Weinberg, R.A. Oncogenes, antioncogenes, and the molecular bases of multistep carcinogenesis. Cancer Res. 1989, 49, 3713–3721. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Albini, A.; Sporn, M.B. The tumour microenvironment as a target for chemoprevention. Nat. Rev. Cancer 2007, 7, 139–147. [Google Scholar]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as organs: Complex tissues that interface with the entire organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef]
- Mueller, M.M.; Fusenig, N.E. Friends or foes—bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef]
- Joyce, J.A. Therapeutic targeting of the tumor microenvironment. Cancer Cell 2005, 7, 513–520. [Google Scholar] [CrossRef]
- O’Brien, L.E.; Zegers, M.M.; Mostov, K.E. Opinion: Building epithelial architecture: Insights from three-dimensional culture models. Nat. Rev. Mol. Cell Biol. 2002, 3, 531–537. [Google Scholar] [CrossRef]
- Bissell, M.J.; Weaver, V.M.; Lelievre, S.A.; Wang, F.; Petersen, O.W.; Schmeichel, K.L. Tissue structure, nuclear organization, and gene expression in normal and malignant breast. Cancer Res. 1999, 59, 1757s–1763s. [Google Scholar]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Allavena, P.; Mantovani, A. Immunology in the clinic review series; focus on cancer: Tumour-associated macrophages: Undisputed stars of the inflammatory tumour microenvironment. Clin. Exp. Immunol. 2012, 167, 195–205. [Google Scholar] [CrossRef]
- Coussens, L.M.; Raymond, W.W.; Bergers, G.; Laig-Webster, M.; Behrendtsen, O.; Werb, Z.; Caughey, G.H.; Hanahan, D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999, 13, 1382–1397. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Allavena, P.; Germano, G.; Marchesi, F.; Mantovani, A. Chemokines in cancer related inflammation. Exp. Cell Res. 2011, 317, 664–673. [Google Scholar] [CrossRef]
- Mantovani, A. Molecular pathways linking inflammation and cancer. Curr. Mol. Med. 2010, 10, 369–373. [Google Scholar] [CrossRef]
- Germano, G.; Mantovani, A.; Allavena, P. Targeting of the innate immunity/inflammation as complementary anti-tumor therapies. Ann. Med. 2011, 43, 581–593. [Google Scholar] [CrossRef]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Eliceiri, K.W.; Campbell, J.M.; Inman, D.R.; White, J.G.; Keely, P.J. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006, 4, 38:1–38:16. [Google Scholar]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef]
- Allen, M.; Louise Jones, J. Jekyll and Hyde: the role of the microenvironment on the progression of cancer. J. Pathol. 2011, 223, 162–176. [Google Scholar]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef]
- Roycik, M.D.; Fang, X.; Sang, Q.X. A fresh prospect of extracellular matrix hydrolytic enzymes and their substrates. Curr. Pharm. Des. 2009, 15, 1295–1308. [Google Scholar] [CrossRef]
- Giraudo, E.; Inoue, M.; Hanahan, D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J. Clin. Invest. 2004, 114, 623–633. [Google Scholar]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef]
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef]
- Albini, A.; Tosetti, F.; Benelli, R.; Noonan, D.M. Tumor inflammatory angiogenesis and its chemoprevention. Cancer Res. 2005, 65, 10637–10641. [Google Scholar] [CrossRef]
- Bissell, M.J.; Labarge, M.A. Context, tissue plasticity, and cancer: Are tumor stem cells also regulated by the microenvironment? Cancer Cell 2005, 7, 17–23. [Google Scholar]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef]
- D’Incalci, M.; Galmarini, C.M. A review of trabectedin (ET-743): a unique mechanism of action. Mol. Cancer Ther. 2010, 9, 2157–2163. [Google Scholar] [CrossRef]
- Demetri, G.D.; Chawla, S.P.; von Mehren, M.; Ritch, P.; Baker, L.H.; Blay, J.Y.; Hande, K.R.; Keohan, M.L.; Samuels, B.L.; Schuetze, S.; et al. Efficacy and safety of trabectedin in patients with advanced or metastatic liposarcoma or leiomyosarcoma after failure of prior anthracyclines and ifosfamide: results of a randomized phase II study of two different schedules. J. Clin. Oncol. 2009, 27, 4188–4196. [Google Scholar] [CrossRef]
- Monk, B.J.; Herzog, T.J.; Kaye, S.B.; Krasner, C.N.; Vermorken, J.B.; Muggia, F.M.; Pujade-Lauraine, E.; Lisyanskaya, A.S.; Makhson, A.N.; Rolski, J.; et al. Trabectedin plus pegylated liposomal Doxorubicin in recurrent ovarian cancer. J. Clin. Oncol. 2010, 28, 3107–3114. [Google Scholar] [CrossRef]
- Aune, G.J.; Takagi, K.; Sordet, O.; Guirouilh-Barbat, J.; Antony, S.; Bohr, V.A.; Pommier, Y. Von Hippel-Lindau-coupled and transcription-coupled nucleotide excision repair-dependent degradation of RNA polymerase II in response to trabectedin. Clin. Cancer Res. 2008, 14, 6449–6455. [Google Scholar] [CrossRef]
- Bueren-Calabuig, J.A.; Giraudon, C.; Galmarini, C.M.; Egly, J.M.; Gago, F. Temperature-induced melting of double-stranded DNA in the absence and presence of covalently bonded antitumour drugs: insight from molecular dynamics simulations. Nucleic Acids Res. 2011, 39, 8248–8257. [Google Scholar] [CrossRef]
- Feuerhahn, S.; Giraudon, C.; Martinez-Diez, M.; Bueren-Calabuig, J.A.; Galmarini, C.M.; Gago, F.; Egly, J.M. XPF-dependent DNA breaks and RNA polymerase II arrest induced by antitumor DNA interstrand crosslinking-mimetic alkaloids. Chem. Biol. 2011, 18, 988–999. [Google Scholar] [CrossRef]
- Friedman, D.; Hu, Z.; Kolb, E.A.; Gorfajn, B.; Scotto, K.W. Ecteinascidin-743 inhibits activated but not constitutive transcription. Cancer Res. 2002, 62, 3377–3381. [Google Scholar]
- Charytonowicz, E.; Terry, M.; Coakley, K.; Telis, L.; Remotti, F.; Cordon-Cardo, C.; Taub, R.N.; Matushansky, I. PPARgamma agonists enhance ET-743-induced adipogenic differentiation in a transgenic mouse model of myxoid round cell liposarcoma. J. Clin. Invest. 2012, 122, 886–898. [Google Scholar] [CrossRef]
- Grohar, P.J.; Griffin, L.B.; Yeung, C.; Chen, Q.R.; Pommier, Y.; Khanna, C.; Khan, J.; Helman, L.J. Ecteinascidin 743 interferes with the activity of EWS-FLI1 in Ewing sarcoma cells. Neoplasia 2011, 13, 145–153. [Google Scholar]
- Di Giandomenico, S.; Frapolli, R.; Bello, E.; Uboldi, S.; Licandro, S.A.; Marchini, S.; Beltrame, L.; Brich, S.; Mauro, V.; Tamborini, E.; et al. Mode of action of trabectedin in myxoid liposarcomas. Oncogene 2013, in press. [Google Scholar]
- Allavena, P.; Signorelli, M.; Chieppa, M.; Erba, E.; Bianchi, G.; Marchesi, F.; Olimpio, C.O.; Bonardi, C.; Garbi, A.; Lissoni, A.; et al. Anti-inflammatory properties of the novel antitumor agent yondelis (trabectedin): Inhibition of macrophage differentiation and cytokine production. Cancer Res. 2005, 65, 2964–2971. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar]
- Louneva, N.; Saitta, B.; Herrick, D.J.; Jimenez, S.A. Transcriptional inhibition of type I collagen gene expression in scleroderma fibroblasts by the antineoplastic drug ecteinascidin 743. J. Biol. Chem. 2003, 278, 40400–40407. [Google Scholar]
- Liguori, M.; Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages as incessant builders and destroyers of the cancer stroma. Cancers 2011, 3, 3740–3761. [Google Scholar] [CrossRef]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Johansson, M.; Coussens, L.M. Immune cells as mediators of solid tumor metastasis. Cancer Metastas. Rev. 2008, 27, 11–18. [Google Scholar] [CrossRef]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stange, G.; Van den Bossche, J.; Mack, M.; Pipeleers, D.; In’t Veld, P.; De Baetselier, P.; et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef]
- Pollard, J.W. Macrophages define the invasive microenvironment in breast cancer. J. Leukoc. Biol. 2008, 84, 623–630. [Google Scholar] [CrossRef]
- Mantovani, A.; Romero, P.; Palucka, A.K.; Marincola, F.M. Tumour immunity: effector response to tumour and role of the microenvironment. Lancet 2008, 371, 771–783. [Google Scholar] [CrossRef]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef]
- Clear, A.J.; Lee, A.M.; Calaminici, M.; Ramsay, A.G.; Morris, K.J.; Hallam, S.; Kelly, G.; Macdougall, F.; Lister, T.A.; Gribben, J.G. Increased angiogenic sprouting in poor prognosis FL is associated with elevated numbers of CD163+ macrophages within the immediate sprouting microenvironment. Blood 2010, 115, 5053–5056. [Google Scholar] [CrossRef]
- Heusinkveld, M.; van der Burg, S.H. Identification and manipulation of tumor associated macrophages in human cancers. J. Transl. Med. 2011, 9, 216:1–216:13. [Google Scholar]
- Leek, R.D.; Lewis, C.E.; Whitehouse, R.; Greenall, M.; Clarke, J.; Harris, A.L. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996, 56, 4625–4629. [Google Scholar]
- Hagemann, T.; Wilson, J.; Burke, F.; Kulbe, H.; Li, N.F.; Pluddemann, A.; Charles, K.; Gordon, S.; Balkwill, F.R. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. J. Immunol. 2006, 176, 5023–5032. [Google Scholar]
- Lee, C.H.; Espinosa, I.; Vrijaldenhoven, S.; Subramanian, S.; Montgomery, K.D.; Zhu, S.; Marinelli, R.J.; Peterse, J.L.; Poulin, N.; Nielsen, T.O.; et al. Prognostic significance of macrophage infiltration in leiomyosarcomas. Clin. Cancer Res. 2008, 14, 1423–1430. [Google Scholar] [CrossRef]
- Lissbrant, I.F.; Stattin, P.; Wikstrom, P.; Damber, J.E.; Egevad, L.; Bergh, A. Tumor associated macrophages in human prostate cancer: relation to clinicopathological variables and survival. Int. J. Oncol. 2000, 17, 445–451. [Google Scholar]
- Jensen, T.O.; Schmidt, H.; Moller, H.J.; Hoyer, M.; Maniecki, M.B.; Sjoegren, P.; Christensen, I.J.; Steiniche, T. Macrophage markers in serum and tumor have prognostic impact in American Joint Committee on Cancer stage I/II melanoma. J. Clin. Oncol. 2009, 27, 3330–3337. [Google Scholar] [CrossRef]
- van Dongen, M.; Savage, N.D.; Jordanova, E.S.; Briaire-de Bruijn, I.H.; Walburg, K.V.; Ottenhoff, T.H.; Hogendoorn, P.C.; van der Burg, S.H.; Gelderblom, H.; van Hall, T. Anti-inflammatory M2 type macrophages characterize metastasized and tyrosine kinase inhibitor-treated gastrointestinal stromal tumors. Int. J. Cancer 2010, 127, 899–909. [Google Scholar]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Nagai, T.; Tanaka, M.; Tsuneyoshi, Y.; Xu, B.; Michie, S.A.; Hasui, K.; Hirano, H.; Arita, K.; Matsuyama, T. Targeting tumor-associated macrophages in an experimental glioma model with a recombinant immunotoxin to folate receptor beta. Cancer Immunol. Immunother. 2009, 58, 1577–1586. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Simone, M.; Tavecchio, M.; Erba, E.; Pesce, S.; Pasqualini, F.; Grosso, F.; Sanfilippo, R.; Casali, P.G.; et al. Antitumor and anti-inflammatory effects of trabectedin on human myxoid liposarcoma cells. Cancer Res. 2010, 70, 2235–2244. [Google Scholar] [CrossRef]
- Munoz-Alonso, M.J.; Gonzalez-Santiago, L.; Martinez, T.; Losada, A.; Galmarini, C.M.; Munoz, A. The mechanism of action of plitidepsin. Curr. Opin. Investig. Drugs 2009, 10, 536–542. [Google Scholar]
- Suarez, Y.; Gonzalez-Santiago, L.; Zarich, N.; Davalos, A.; Aranda, J.F.; Alonso, M.A.; Lasuncion, M.A.; Rojas, J.M.; Munoz, A. Plitidepsin cellular binding and Rac1/JNK pathway activation depend on membrane cholesterol content. Mol. Pharmacol. 2006, 70, 1654–1663. [Google Scholar] [CrossRef]
- Garcia-Fernandez, L.F.; Losada, A.; Alcaide, V.; Alvarez, A.M.; Cuadrado, A.; Gonzalez, L.; Nakayama, K.; Nakayama, K.I.; Fernandez-Sousa, J.M.; Munoz, A.; et al. Aplidin induces the mitochondrial apoptotic pathway via oxidative stress-mediated JNK and p38 activation and protein kinase C delta. Oncogene 2002, 21, 7533–7544. [Google Scholar] [CrossRef]
- Gonzalez-Santiago, L.; Suarez, Y.; Zarich, N.; Munoz-Alonso, M.J.; Cuadrado, A.; Martinez, T.; Goya, L.; Iradi, A.; Saez-Tormo, G.; Maier, J.V.; et al. Aplidin induces JNK-dependent apoptosis in human breast cancer cells via alteration of glutathione homeostasis, Rac1 GTPase activation, and MKP-1 phosphatase downregulation. Cell Death Differ. 2006, 13, 1968–1981. [Google Scholar] [CrossRef]
- Cuadrado, A.; Garcia-Fernandez, L.F.; Gonzalez, L.; Suarez, Y.; Losada, A.; Alcaide, V.; Martinez, T.; Fernandez-Sousa, J.M.; Sanchez-Puelles, J.M.; Munoz, A. Aplidin induces apoptosis in human cancer cells via glutathione depletion and sustained activation of the epidermal growth factor receptor, Src, JNK, and p38 MAPK. J. Biol. Chem. 2003, 278, 241–250. [Google Scholar]
- Losada, A.; Martínez-Leal, J.F.; Bejarano, A.; Cuevas, C.; García-Fernández, L.F.; Galmarini, C.M. Aplidin triggers the activation of molecular components of the UPR as part of its pro-apoptotic program in tumor cells. In Proceedings of the AACR 104th Annual Meeting of the American Association for Cancer Research Washington, Washington, DC, USA, 6–10 April 2013. [CrossRef]
- Morande, P.E.; Zanetti, S.R.; Borge, M.; Nannini, P.; Jancic, C.; Bezares, R.F.; Bitsmans, A.; Gonzalez, M.; Rodriguez, A.L.; Galmarini, C.M.; et al. The cytotoxic activity of Aplidin in chronic lymphocytic leukemia (CLL) is mediated by a direct effect on leukemic cells and an indirect effect on monocyte-derived cells. Invest. New Drugs 2012, 30, 1830–1840. [Google Scholar] [CrossRef]
- Burger, J.A.; Tsukada, N. leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood 2000, 96, 2655–2663. [Google Scholar]
- Nishio, M.; Endo, T.; Tsukada, N.; Ohata, J.; Kitada, S.; Reed, J.C.; Zvaifler, N.J.; Kipps, T.J. Nurselike cells express BAFF and APRIL, which can promote survival of chronic lymphocytic leukemia cells via a paracrine pathway distinct from that of SDF-1alpha. Blood 2005, 106, 1012–1020. [Google Scholar] [CrossRef]
- Tsukada, N.; Burger, J.A.; Zvaifler, N.J.; Kipps, T.J. Distinctive features of “nurselike” cells that differentiate in the context of chronic lymphocytic leukemia. Blood 2002, 99, 1030–1037. [Google Scholar] [CrossRef]
- Baran, C.P.; Zeigler, M.M.; Tridandapani, S.; Marsh, C.B. The role of ROS and RNS in regulating life and death of blood monocytes. Curr. Pharm. Des. 2004, 10, 855–866. [Google Scholar] [CrossRef]
- Zurgil, N.; Solodeev, I.; Gilburd, B.; Shafran, Y.; Afrimzon, E.; Avtalion, R.; Shoenfeld, Y.; Deutsch, M. Monitoring the apoptotic process induced by oxidized low-density lipoprotein in Jurkat T-lymphoblast and U937 monocytic human cell lines. Cell Biochem. Biophys. 2004, 40, 97–113. [Google Scholar] [CrossRef]
- Sanfilippo, R.; Casali, P.G. The intriguing patterns of tumor response to trabectedin. Expert Rev. Anticancer Ther. 2013, 13, 21–24. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Galmarini, C.M.; D'Incalci, M.; Allavena, P. Trabectedin and Plitidepsin: Drugs from the Sea that Strike the Tumor Microenvironment. Mar. Drugs 2014, 12, 719-733. https://doi.org/10.3390/md12020719
Galmarini CM, D'Incalci M, Allavena P. Trabectedin and Plitidepsin: Drugs from the Sea that Strike the Tumor Microenvironment. Marine Drugs. 2014; 12(2):719-733. https://doi.org/10.3390/md12020719
Chicago/Turabian StyleGalmarini, Carlos M., Maurizio D'Incalci, and Paola Allavena. 2014. "Trabectedin and Plitidepsin: Drugs from the Sea that Strike the Tumor Microenvironment" Marine Drugs 12, no. 2: 719-733. https://doi.org/10.3390/md12020719