13-Acetoxysarcocrassolide Induces Apoptosis on Human Gastric Carcinoma Cells Through Mitochondria-Related Apoptotic Pathways: p38/JNK Activation and PI3K/AKT Suppression

 and
and 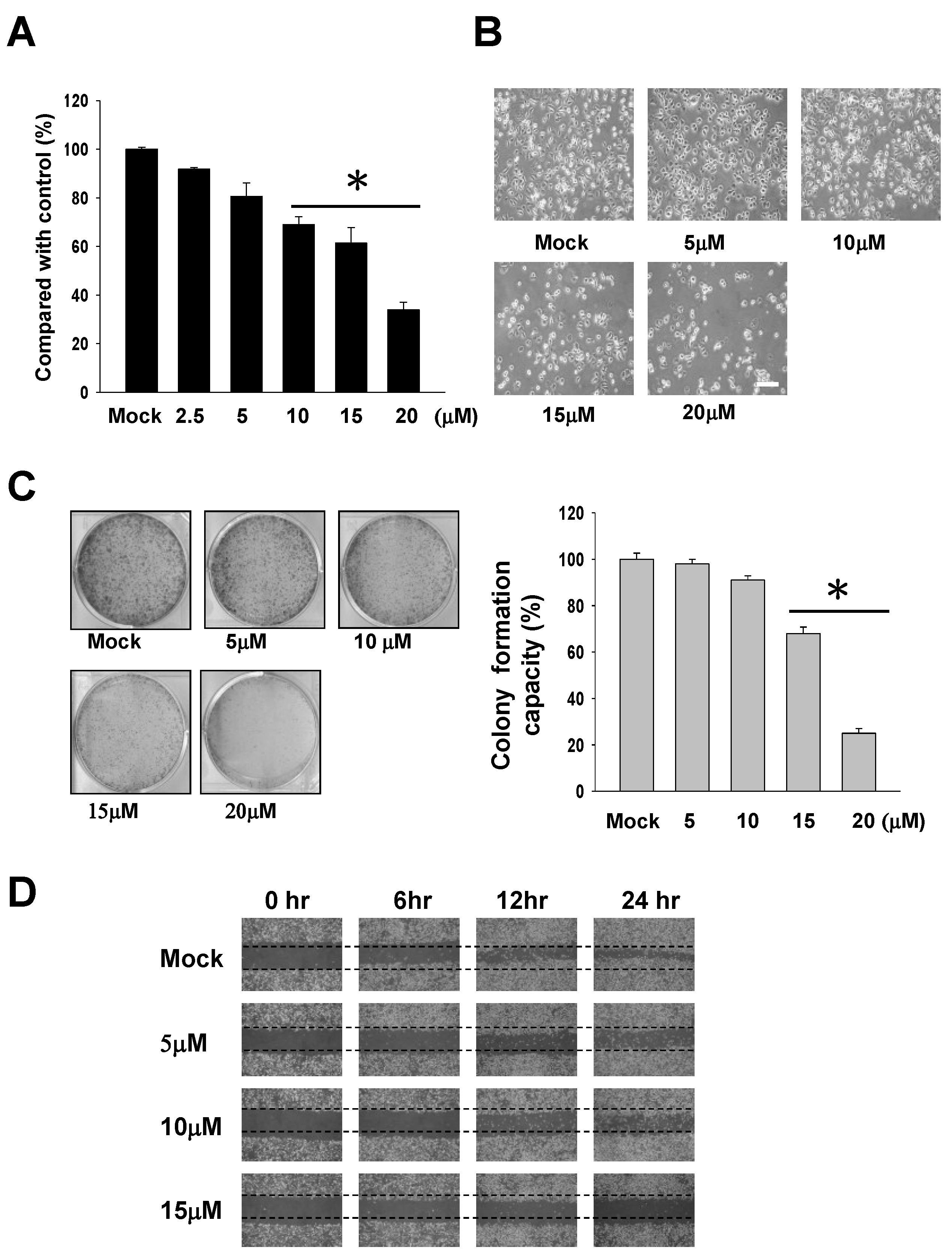{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
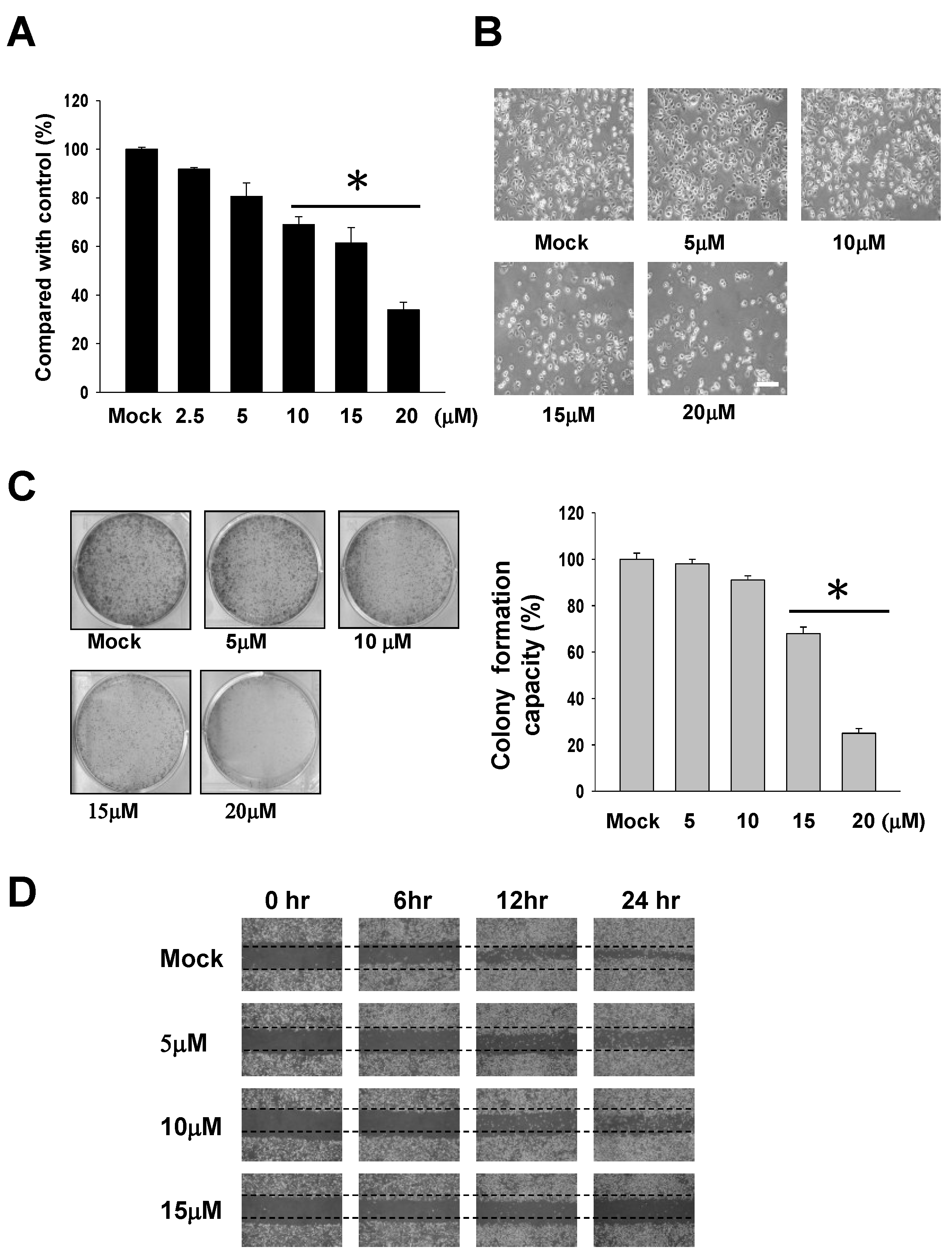
2.1. The Cytotoxic Effects of 13-AC on AGS Gastric Carcinoma Cells
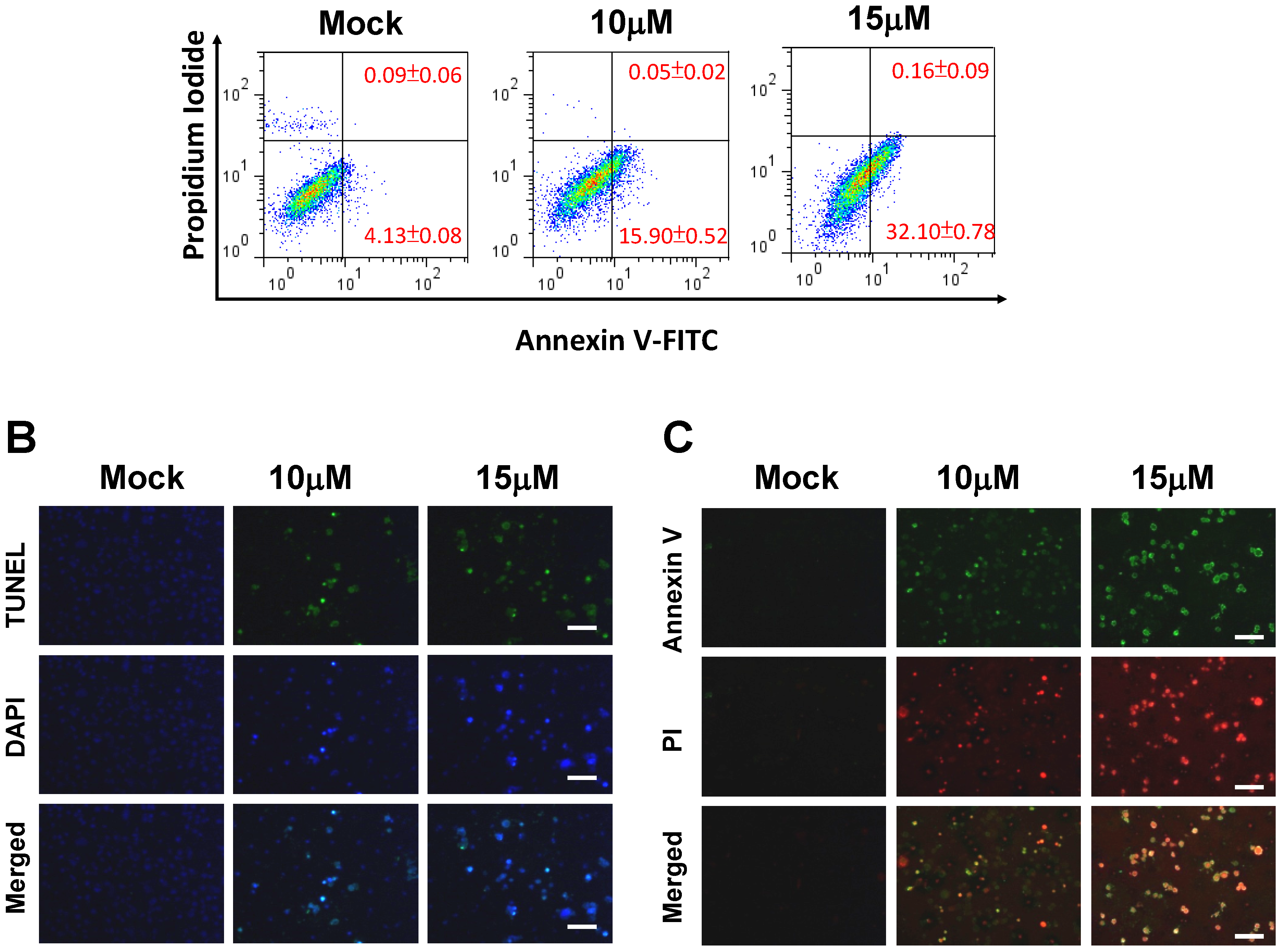
2.2. 13-AC Induces Apoptosis of AGS Cells

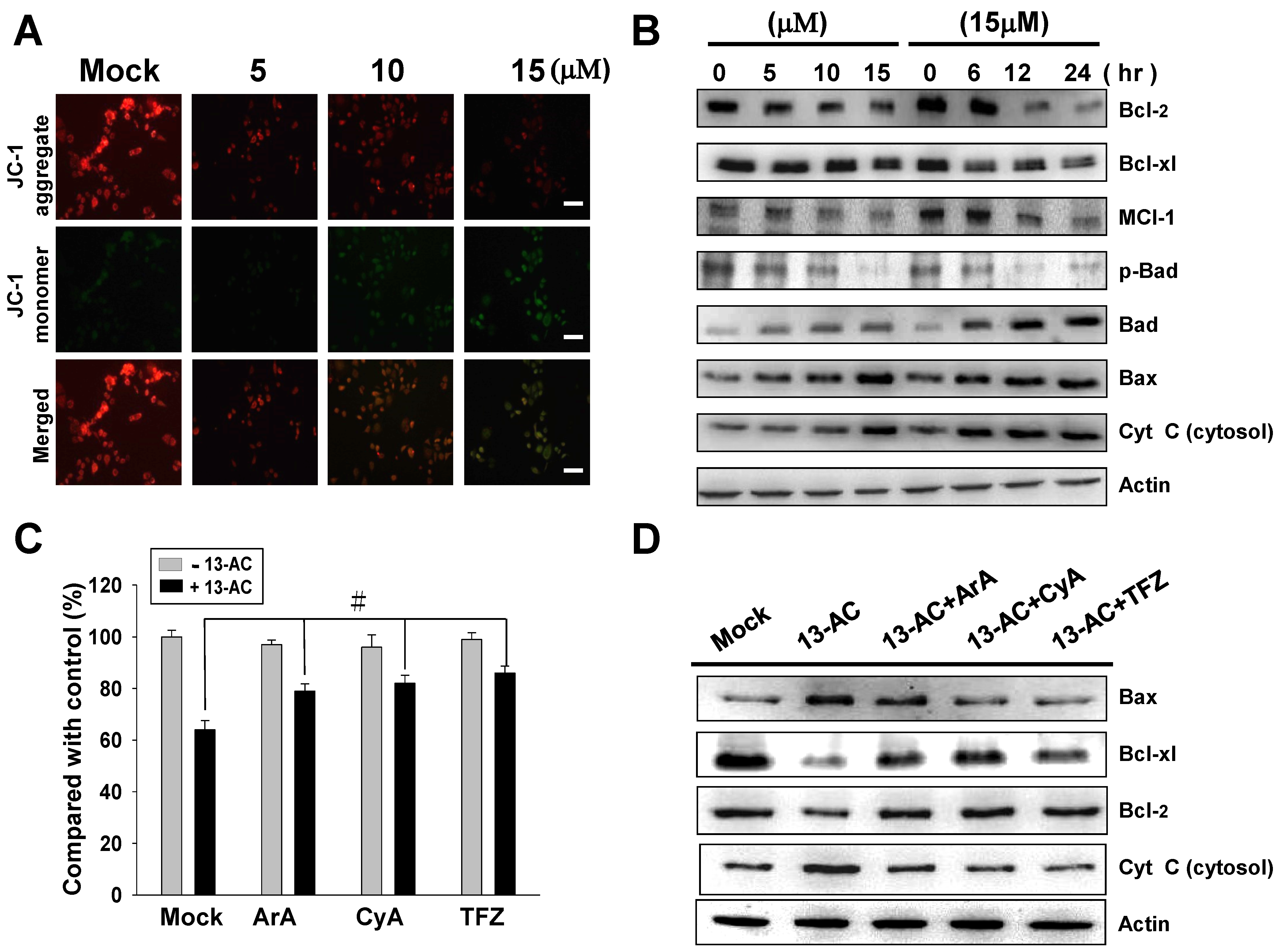
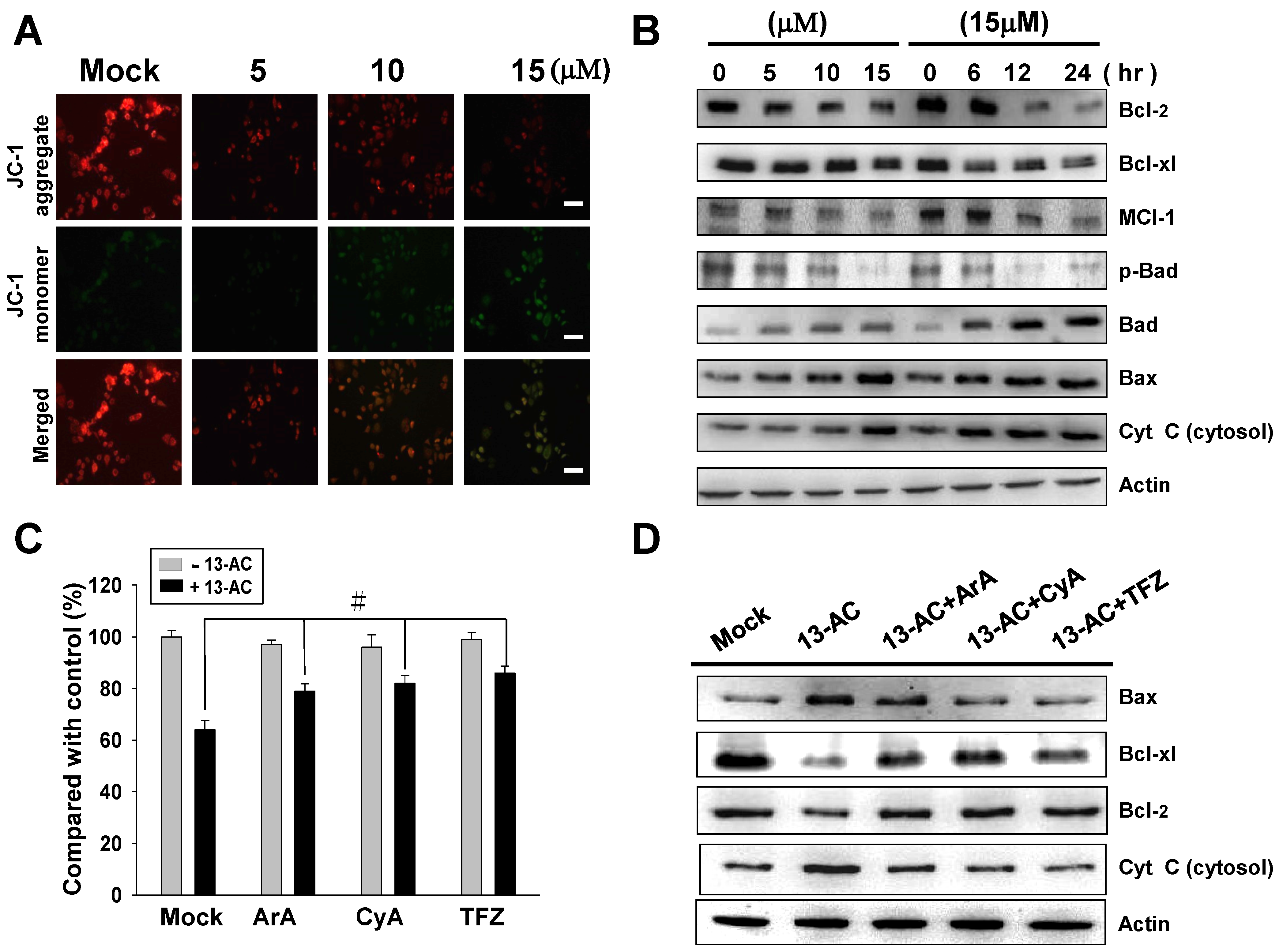
2.3. 13-AC Induces Apoptosis and Causes a Mitochondria Membrane Potential Change in AGS Cells
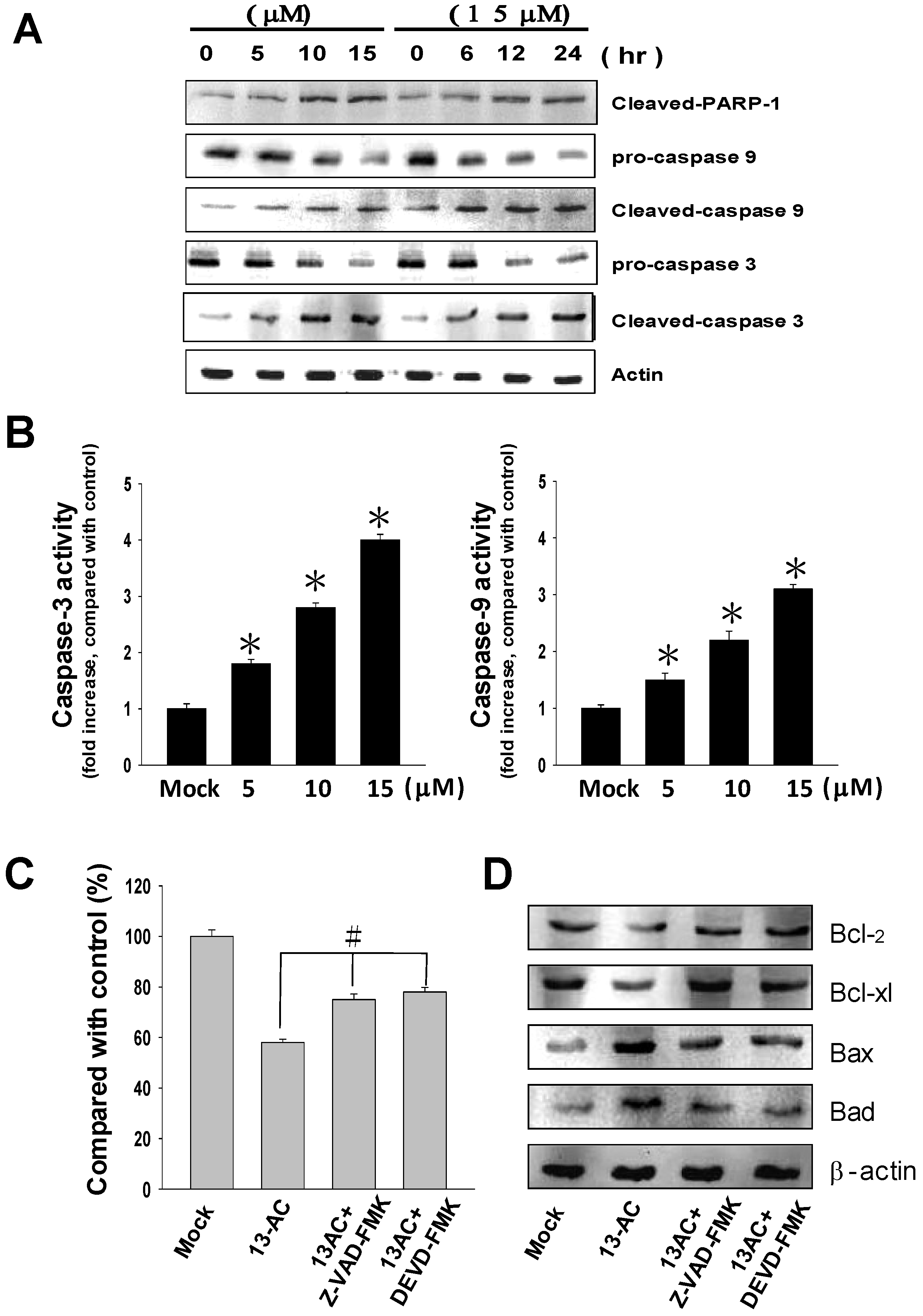
2.4. 13-AC Activates the Caspase-Dependent Pathway Causing Cell Apoptosis in AGS Cells
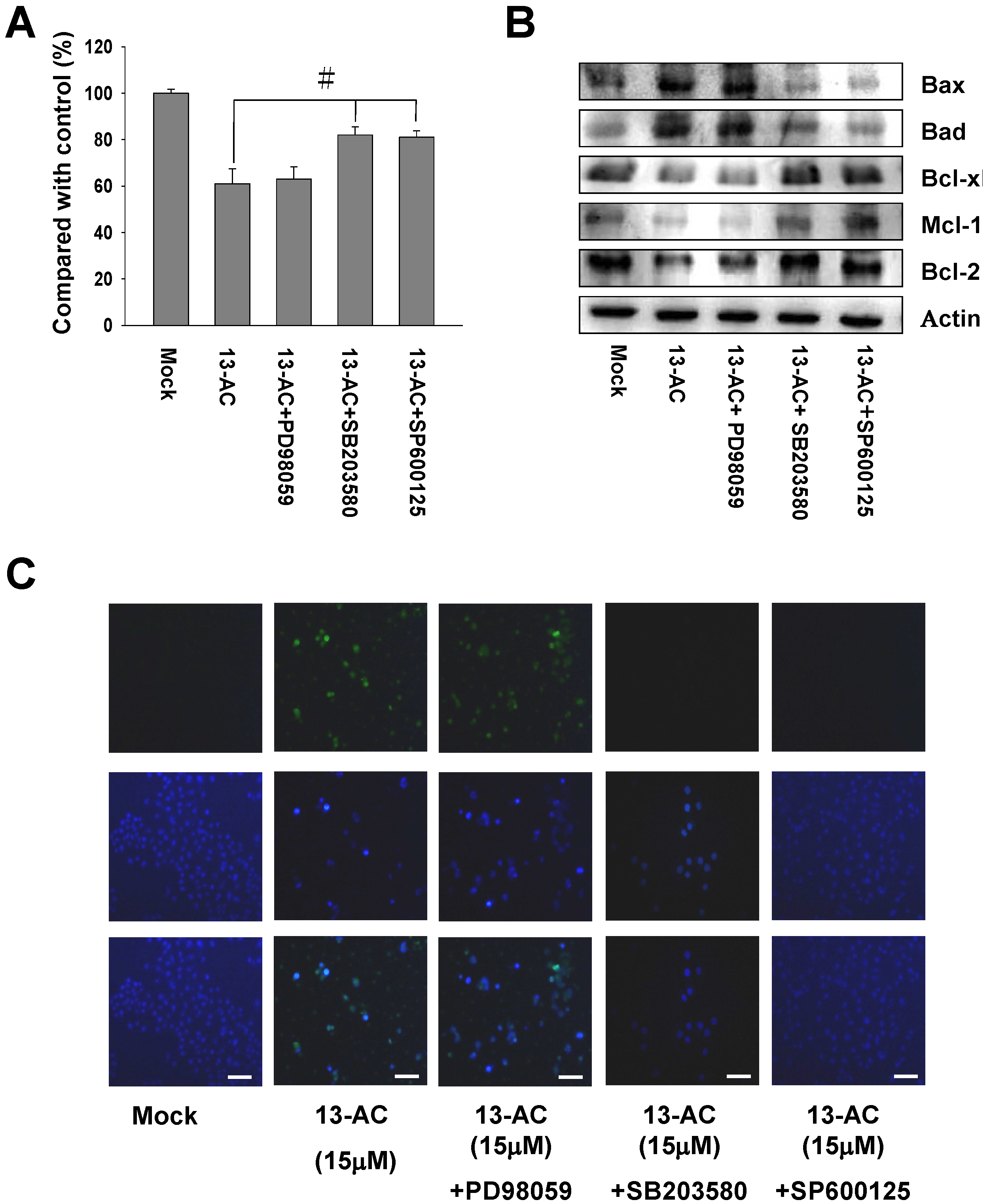
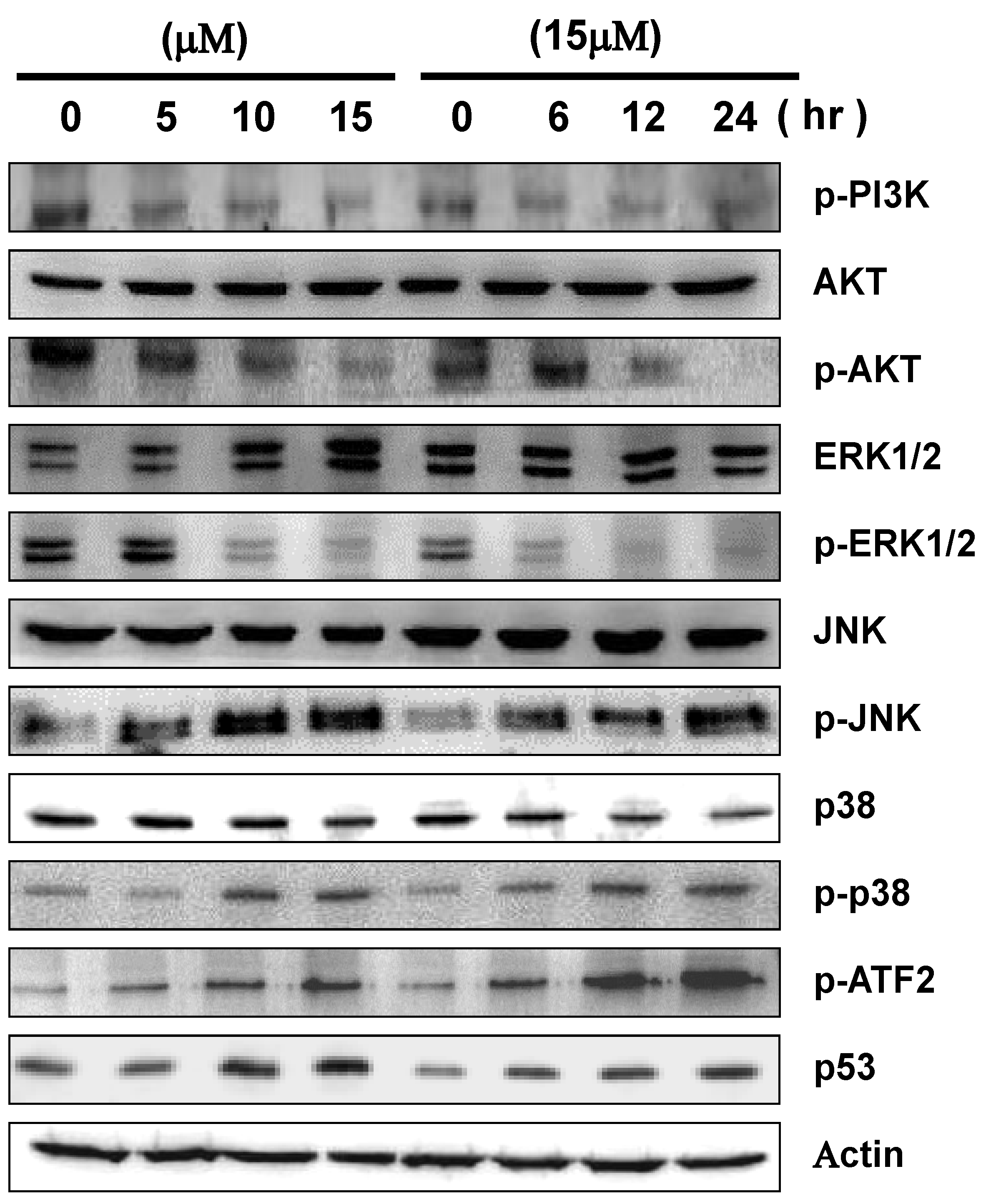
2.5. 13-AC Induces Activation of the p38 and JNK Pathways and Suppression of PI3K/AKT
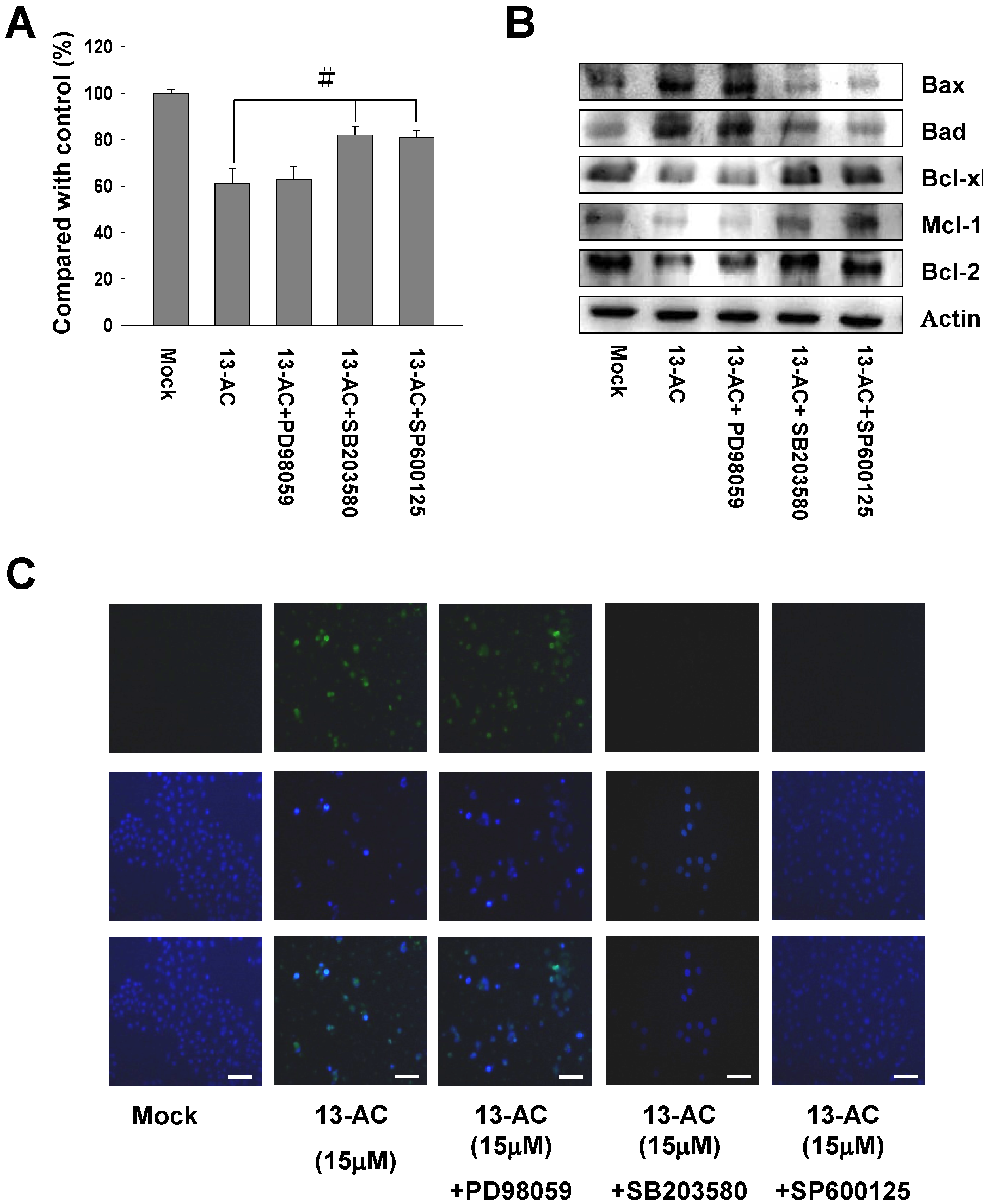
2.6. Inhibition of p38 and JNK Activities Rescued the Cell Cytotoxicity of AGS Cells by 13-AC
3. Discussion
3.1. 13-AC Induces Apoptosis and Causes Mitochondria Dysfunction in AGS Cells
3.2. The MAPK Signaling Pathways are Involved in 13-AC-Induced Apoptosis
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and 13-AC Treatment
4.3. MTT Assay for Cellular Cytotoxicity
4.4. Wound-Healing Assay
4.5. Colony Formation Assay
4.6. Antibody and Western Blot Analyses
4.7. Annexin V-FITC Apoptosis Assay
4.8. Immunofluorescence Microscopy
4.9. Mitochondrial Membrane Potential (ΔΨm) Assay Using Fluorescence Microscopy
4.10. Inhibitors Assessment
4.11. Statistical Analysis
5. Conclusion
Supplementary Files
Supplementary File 1Acknowledgements
Conflict of Interests
References
- Hu, Y.; Fang, J.Y.; Xiao, S.D. Can the incidence of gastric cancer be reduced in the new century? J. Dig. Dis. 2013, 14, 11–15. [Google Scholar] [CrossRef]
- Thiel, A.; Ristimaki, A. Gastric cancer: Basic aspects. Helicobacter 2012, 17 (Suppl. 1), 26–29. [Google Scholar] [CrossRef]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: Globocan 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Truong Minh, P.; Fujino, Y.; Yoshimura, T.; Tokui, N.; Mizoue, T.; Yatsuya, H.; Toyoshima, H.; Sakata, K.; Kikuchi, S.; Hoshiyama, Y.; et al. Mortality and incidence rates of stomach cancer in the jacc study. J. Epidemiol. 2005, 15 (Suppl. 2), S89–S97. [Google Scholar]
- Yamashita, K.; Sakuramoto, S.; Nemoto, M.; Shibata, T.; Mieno, H.; Katada, N.; Kikuchi, S.; Watanabe, M. Trend in gastric cancer: 35 years of surgical experience in Japan. World J. Gastroenterol. 2011, 17, 3390–3397. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.J.; Wilke, H. Treatment strategies in gastric cancer. Deutsches Arzteblatt Int. 2011, 108, 698–705. [Google Scholar]
- Proserpio, I.; Rausei, S.; Barzaghi, S.; Frattini, F.; Galli, F.; Iovino, D.; Rovera, F.; Boni, L.; Dionigi, G.; Pinotti, G. Multimodal treatment of gastric cancer. World J. Gastrointest. Surg. 2014, 6, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.S.; Chen, M.J.; Shih, S.C.; Bair, M.J.; Fang, C.J.; Wang, H.Y. Management of infection after gastric surgery. World J. Gastroenterol. 2014, 20, 5274–5282. [Google Scholar] [CrossRef] [PubMed]
- Montori, G.; Coccolini, F.; Ceresoli, M.; Catena, F.; Colaianni, N.; Poletti, E.; Ansaloni, L. The treatment of peritoneal carcinomatosis in advanced gastric cancer: State of the art. Int. J. Surg. Oncol. 2014, 2014, 912418. [Google Scholar] [PubMed]
- Zhang, X.; Yashiro, M.; Ren, J.; Hirakawa, K. Histone deacetylase inhibitor, trichostatin a, increases the chemosensitivity of anticancer drugs in gastric cancer cell lines. Oncol. Rep. 2006, 16, 563–568. [Google Scholar] [PubMed]
- Faulkner, D.J. Marine natural products. Nat. Prod. Rep. 2001, 18, 1–49. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, D.J. Marine natural products. Nat. Prod. Rep. 2002, 19, 1–48. [Google Scholar] [PubMed]
- Ojika, M.; Islam, M.K.; Shintani, T.; Zhang, Y.; Okamoto, T.; Sakagami, Y. Three new cytotoxic acylspermidines from the soft coral, Sinularia sp. Biosci. Biotechnol. Biochem. 2003, 67, 1410–1412. [Google Scholar] [CrossRef] [PubMed]
- Poza, J.J.; Fernandez, R.; Reyes, F.; Rodriguez, J.; Jimenez, C. Isolation, biological significance, synthesis, and cytotoxic evaluation of new natural parathiosteroids a–c and analogues from the soft coral Paragorgia sp. J. Org. Chem. 2008, 73, 7978–7984. [Google Scholar] [CrossRef] [PubMed]
- Chiang, P.C.; Chien, C.L.; Pan, S.L.; Chen, W.P.; Teng, C.M.; Shen, Y.C.; Guh, J.H. Induction of endoplasmic reticulum stress and apoptosis by a marine prostanoid in human hepatocellular carcinoma. J. Hepatol. 2005, 43, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Kamel, H.N.; Ferreira, D.; Garcia-Fernandez, L.F.; Slattery, M. Cytotoxic diterpenoids from the hybrid soft coral Sinularia maxima x Sinularia polydactyla. J. Nat. Prod. 2007, 70, 1223–1227. [Google Scholar] [CrossRef] [PubMed]
- Hassan, H.M.; Khanfar, M.A.; Elnagar, A.Y.; Mohammed, R.; Shaala, L.A.; Youssef, D.T.A.; Hifnawy, M.S.; El Sayed, K.A. Pachycladins A–E, prostate cancer invasion and migration inhibitory eunicellin-based diterpenoids from the red sea soft coral Cladiella pachyclados. J. Nat. Prod. 2010, 73, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.I.; Chen, C.C.; Chen, J.C.; Su, J.H.; Huang, H.H.; Chen, J.Y.; Wu, Y.J. Proteomic analysis of anti-tumor effects of 11-dehydrosinulariolide on cal-27 cells. Mar. Drugs 2011, 9, 1254–1272. [Google Scholar] [CrossRef] [PubMed]
- Su, C.C.; Su, J.H.; Lin, J.J.; Chen, C.C.; Hwang, W.I.; Huang, H.H.; Wu, Y.J. An investigation into the cytotoxic effects of 13-acetoxysarcocrassolide from the soft coral Sarcophyton crassocaule on bladder cancer cells. Mar. Drugs 2011, 9, 2622–2642. [Google Scholar] [CrossRef] [PubMed]
- Neoh, C.A.; Wang, R.Y.; Din, Z.H.; Su, J.H.; Chen, Y.K.; Tsai, F.J.; Weng, S.H.; Wu, Y.J. Induction of apoptosis by sinulariolide from soft coral through mitochondrial-related and p38mapk pathways on human bladder carcinoma cells. Mar. Drugs 2012, 10, 2893–2911. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Lin, A.W. Apoptosis in cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Denicourt, C.; Dowdy, S.F. Targeting apoptotic pathways in cancer cells. Science 2004, 305, 1411–1413. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.C.; Tan, S.K.; Lieu, C.H.; Jung, H.K. Involvement of endoplasmic reticulum in paclitaxel‐induced apoptosis. J. Cell. Biochem. 2008, 104, 1509–1523. [Google Scholar] [CrossRef] [PubMed]
- Yokouchi, M.; Hiramatsu, N.; Hayakawa, K.; Kasai, A.; Takano, Y.; Yao, J.; Kitamura, M. Atypical, bidirectional regulation of cadmium-induced apoptosis via distinct signaling of unfolded protein response. Cell Death Differ. 2007, 14, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, D.W.; Thornberry, N.A. Life and death decisions. Science 2003, 299, 214–215. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Ellerby, H.; Bredesen, D. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004, 11, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Yuan, J. Cellular response to endoplasmic reticulum stress: A matter of life or death. Cell Death Differ. 2006, 13, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.I.; Wang, R.Y.; Lin, J.J.; Su, J.H.; Chiu, C.C.; Chen, J.C.; Chen, J.Y.; Wu, Y.J. Proteomic profiling of the 11-dehydrosinulariolide-treated oral carcinoma cells ca9-22: Effects on the cell apoptosis through mitochondrial-related and er stress pathway. J. Proteomics 2012, 75, 5578–5589. [Google Scholar] [CrossRef] [PubMed]
- Moenner, M.; Pluquet, O.; Bouchecareilh, M.; Chevet, E. Integrated endoplasmic reticulum stress responses in cancer. Cancer Res. 2007, 67, 10631–10634. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Invest. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, K.M.; Quinn, D.M.; Kellett, G.L.; Warr, J.R. Ly294002, an inhibitor of phosphatidylinositol-3-kinase, causes preferential induction of apoptosis in human multidrug resistant cells. Cancer Lett. 2003, 190, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Affar, E.B.; Germain, M.; Winstall, E.; Vodenicharov, M.; Shah, R.G.; Salvesen, G.S.; Poirier, G.G. Caspase-3-mediated processing of poly(adp-ribose) glycohydrolase during apoptosis. J. Biol. Chem. 2001, 276, 2935–2942. [Google Scholar] [CrossRef] [PubMed]
- Miloso, M.; Scuteri, A.; Foudah, D.; Tredici, G. Mapks as mediators of cell fate determination: An approach to neurodegenerative diseases. Curr. Med. Chem. 2008, 15, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Suganuma, T.; Workman, J.L. Map kinases and histone modification. J. Mol. Cell Biol. 2012, 4, 348–350. [Google Scholar] [CrossRef] [PubMed]
- Leanza, L.; Henry, B.; Sassi, N.; Zoratti, M.; Chandy, K.G.; Gulbins, E.; Szabo, I. Inhibitors of mitochondrial kv1.3 channels induce bax/bak-independent death of cancer cells. EMBO Mol. Med. 2012, 4, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Ribe, E.M.; Serrano-Saiz, E.; Akpan, N.; Troy, C.M. Mechanisms of neuronal death in disease: Defining the models and the players. Biochem. J. 2008, 415, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Srivastava, R.K. Bax and bak genes are essential for maximum apoptotic response by curcumin, a polyphenolic compound and cancer chemopreventive agent derived from turmeric, curcuma longa. Carcinogenesis 2007, 28, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, S.; Ong, C.N.; Shen, H.M. Critical role of pro-apoptotic bcl-2 family members in andrographolide-induced apoptosis in human cancer cells. Biochem. Pharmacol. 2006, 72, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L.; Yeh, T.H.; Chou, A.H.; Kuo, Y.L.; Luo, L.J.; He, C.Y.; Huang, P.C.; Li, A.H. Polyglutamine-expanded ataxin-7 activates mitochondrial apoptotic pathway of cerebellar neurons by upregulating bax and downregulating bcl-x(l). Cell. Signal. 2006, 18, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Ola, M.S.; Nawaz, M.; Ahsan, H. Role of bcl-2 family proteins and caspases in the regulation of apoptosis. Mol. Cell. Biochem. 2011, 351, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Su, T.R.; Lin, J.J.; Chiu, C.C.; Chen, J.Y.; Su, J.H.; Cheng, Z.J.; Hwang, W.I.; Huang, H.H.; Wu, Y.J. Proteomic investigation of anti-tumor activities exerted by sinularin against a2058 melanoma cells. Electrophoresis 2012, 33, 1139–1152. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Karin, M. Mammalian map kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the mapk-ras-raf signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Boronkai, A.; Brubel, R.; Racz, B.; Tamas, A.; Kiss, P.; Horvath, G.; Lubics, A.; Szigeti, A.; Bellyei, S.; Toth, G.; et al. Effects of pituitary adenylate cyclase activating polypeptide on the survival and signal transduction pathways in human choriocarcinoma cells. Ann. N. Y. Acad. Sci. 2009, 1163, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. P38(mapk): Stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Yanase, S.; Yasuda, K.; Ishii, N. Adaptive responses to oxidative damage in three mutants of caenorhabditis elegans (age-1, mev-1 and daf-16) that affect life span. Mech. Ageing Dev. 2002, 123, 1579–1587. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lu, J.; Liu, Y.; Meng, Q.; Xie, J.; Wang, Z.; Teng, L. Liquiritigenin induces tumor cell death through mitogen-activated protein kinase- (mpaks-) mediated pathway in hepatocellular carcinoma cells. BioMed. Res. Int. 2014, 2014, 965316. [Google Scholar] [PubMed]
- Lv, C.; Hong, Y.; Miao, L.; Li, C.; Xu, G.; Wei, S.; Wang, B.; Huang, C.; Jiao, B. Wentilactone a as a novel potential antitumor agent induces apoptosis and g2/m arrest of human lung carcinoma cells, and is mediated by hras-gtp accumulation to excessively activate the ras/raf/erk/p53-p21 pathway. Cell Death Dis. 2013, 4, e952. [Google Scholar] [CrossRef] [PubMed]
- Bu, H.Q.; Liu, D.L.; Wei, W.T.; Chen, L.; Huang, H.; Li, Y.; Cui, J.H. Oridonin induces apoptosis in sw1990 pancreatic cancer cells via p53- and caspase-dependent induction of p38 mapk. Oncol. Rep. 2014, 31, 975–982. [Google Scholar] [PubMed]
- Yang, F.; Chen, H.; Liu, Y.; Yin, K.; Wang, Y.; Li, X.; Wang, G.; Wang, S.; Tan, X.; Xu, C.; et al. Doxorubicin caused apoptosis of mesenchymal stem cells via p38, jnk and p53 pathway. Cell. Physiol. Biochem. 2013, 32, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Deng, Y.; Rong, Y.; Lou, W.; Mao, Z.; Feng, Y.; Xie, D.; Jin, D. Irf-2 is over-expressed in pancreatic cancer and promotes the growth of pancreatic cancer cells. Tumour Biol. 2012, 33, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Masmoudi-Kouki, O.; Douiri, S.; Hamdi, Y.; Kaddour, H.; Bahdoudi, S.; Vaudry, D.; Basille, M.; Leprince, J.; Fournier, A.; Vaudry, H.; et al. Pituitary adenylate cyclase-activating polypeptide protects astroglial cells against oxidative stress-induced apoptosis. J. Neurochem. 2011, 117, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Makkinje, A.; Quinn, D.A.; Chen, A.; Cadilla, C.L.; Force, T.; Bonventre, J.V.; Kyriakis, J.M. Gene 33/mig-6, a transcriptionally inducible adapter protein that binds gtp-cdc42 and activates sapk/jnk. A potential marker transcript for chronic pathologic conditions, such as diabetic nephropathy. Possible role in the response to persistent stress. J. Biol. Chem. 2000, 275, 17838–17847. [Google Scholar] [CrossRef] [PubMed]
- Sweatt, J.D. The neuronal map kinase cascade: A biochemical signal integration system subserving synaptic plasticity and memory. J. Neurochem. 2001, 76, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Liu, J.; Wang, Y.; Lian, H.; Wang, J.; Xing, L.; Yan, X.; Wang, J.; Zhang, X. Aflatoxin g1-induced oxidative stress causes DNA damage and triggers apoptosis through mapk signaling pathway in a549 cells. Food Chem. Toxicol. 2013, 62, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Boucher, M.J.; Morisset, J.; Vachon, P.H.; Reed, J.C.; Laine, J.; Rivard, N. Mek/erk signaling pathway regulates the expression of bcl-2, bcl-x(l), and mcl-1 and promotes survival of human pancreatic cancer cells. J. Cell. Biochem. 2000, 79, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Balmanno, K.; Cook, S.J. Tumour cell survival signalling by the erk1/2 pathway. Cell Death Differ. 2009, 16, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Wheler, J.J.; Westin, S.N.; Moulder, S.L.; Naing, A.; Tsimberidou, A.M.; Fu, S.; Falchook, G.S.; Hong, D.S.; Garrido-Laguna, I.; et al. Pi3k/akt/mtor inhibitors in patients with breast and gynecologic malignancies harboring pik3ca mutations. J. Clin. Oncol. 2012, 30, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Cheung, M.; Testa, J.R. Diverse mechanisms of akt pathway activation in human malignancy. Curr. Cancer Drug Targets 2013, 13, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, C.M.; Chen, Z.F.; Ji, R.; Guo, Q.H.; Li, Q.; Zhang, H.L.; Zhou, Y.N. Celecoxib regulates apoptosis and autophagy via the pi3k/akt signaling pathway in sgc-7901 gastric cancer cells. Int. J. Mol. Med. 2014, 33, 1451–1458. [Google Scholar] [PubMed]
- Hong, S.W.; Shin, J.S.; Moon, J.H.; Kim, Y.S.; Lee, J.; Choi, E.K.; Ha, S.H.; Lee, D.H.; Chung, H.N.; Kim, J.E.; et al. Nvp-bez235, a dual pi3k/mtor inhibitor, induces cell death through alternate routes in prostate cancer cells depending on the pten genotype. Apoptosis 2014, 19, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Duh, C.Y.; Wang, S.K.; Chung, S.G.; Chou, G.C.; Dai, C.F. Cytotoxic cembrenolides and steroids from the formosan soft coral Sarcophyton crassocaule. J. Nat. Prod. 2000, 63, 1634–1637. [Google Scholar] [CrossRef] [PubMed]
- Su, T.R.; Tsai, F.J.; Lin, J.J.; Huang, H.H.; Chiu, C.C.; Su, J.H.; Yang, Y.T.; Chen, J.Y.; Wong, B.S.; Wu, Y.J. Induction of apoptosis by 11-dehydrosinulariolide via mitochondrial dysregulation and er stress pathways in human melanoma cells. Mar. Drugs 2012, 10, 1883–1898. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.Y.K.L. Host factors in the replication of positive-strand rna viruses. Chang Gung Med. J. 2012, 35, 14. [Google Scholar]
- Chen, Y.J.; Su, J.H.; Tsao, C.Y.; Hung, C.T.; Chao, H.H.; Lin, J.J.; Liao, M.H.; Yang, Z.Y.; Huang, H.H.; Tsai, F.J.; et al. Sinulariolide induced hepatocellular carcinoma apoptosis through activation of mitochondrial-related apoptotic and perk/eif2alpha/atf4/chop pathway. Molecules 2013, 18, 10146–10161. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.C.; Haung, J.W.; Chang, F.R.; Huang, K.J.; Huang, H.M.; Huang, H.W.; Chou, C.K.; Wu, Y.C.; Chang, H.W. Golden berry-derived 4beta-hydroxywithanolide e for selectively killing oral cancer cells by generating ros, DNA damage, and apoptotic pathways. PLOS One 2013, 8, e64739. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, C.-C.; Chen, J.Y.-F.; Din, Z.-H.; Su, J.-H.; Yang, Z.-Y.; Chen, Y.-J.; Wang, R.Y.L.; Wu, Y.-J. 13-Acetoxysarcocrassolide Induces Apoptosis on Human Gastric Carcinoma Cells Through Mitochondria-Related Apoptotic Pathways: p38/JNK Activation and PI3K/AKT Suppression. Mar. Drugs 2014, 12, 5295-5315. https://doi.org/10.3390/md12105295
Su C-C, Chen JY-F, Din Z-H, Su J-H, Yang Z-Y, Chen Y-J, Wang RYL, Wu Y-J. 13-Acetoxysarcocrassolide Induces Apoptosis on Human Gastric Carcinoma Cells Through Mitochondria-Related Apoptotic Pathways: p38/JNK Activation and PI3K/AKT Suppression. Marine Drugs. 2014; 12(10):5295-5315. https://doi.org/10.3390/md12105295
Chicago/Turabian StyleSu, Ching-Chyuan, Jeff Yi-Fu Chen, Zhong-Hao Din, Jui-Hsin Su, Zih-Yan Yang, Yi-Jen Chen, Robert Y.L. Wang, and Yu-Jen Wu. 2014. "13-Acetoxysarcocrassolide Induces Apoptosis on Human Gastric Carcinoma Cells Through Mitochondria-Related Apoptotic Pathways: p38/JNK Activation and PI3K/AKT Suppression" Marine Drugs 12, no. 10: 5295-5315. https://doi.org/10.3390/md12105295
APA StyleSu, C.-C., Chen, J. Y.-F., Din, Z.-H., Su, J.-H., Yang, Z.-Y., Chen, Y.-J., Wang, R. Y. L., & Wu, Y.-J. (2014). 13-Acetoxysarcocrassolide Induces Apoptosis on Human Gastric Carcinoma Cells Through Mitochondria-Related Apoptotic Pathways: p38/JNK Activation and PI3K/AKT Suppression. Marine Drugs, 12(10), 5295-5315. https://doi.org/10.3390/md12105295