Hyaluromycin, a New Hyaluronidase Inhibitor of Polyketide Origin from Marine Streptomyces sp.

Abstract
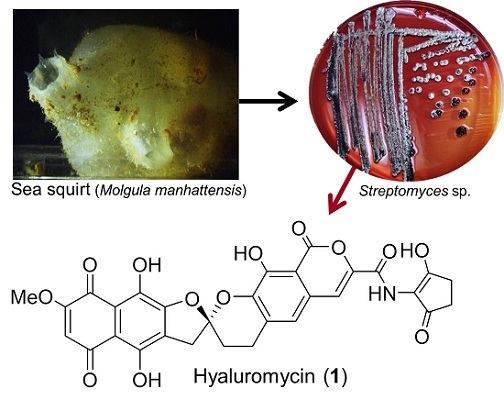
:
1. Introduction
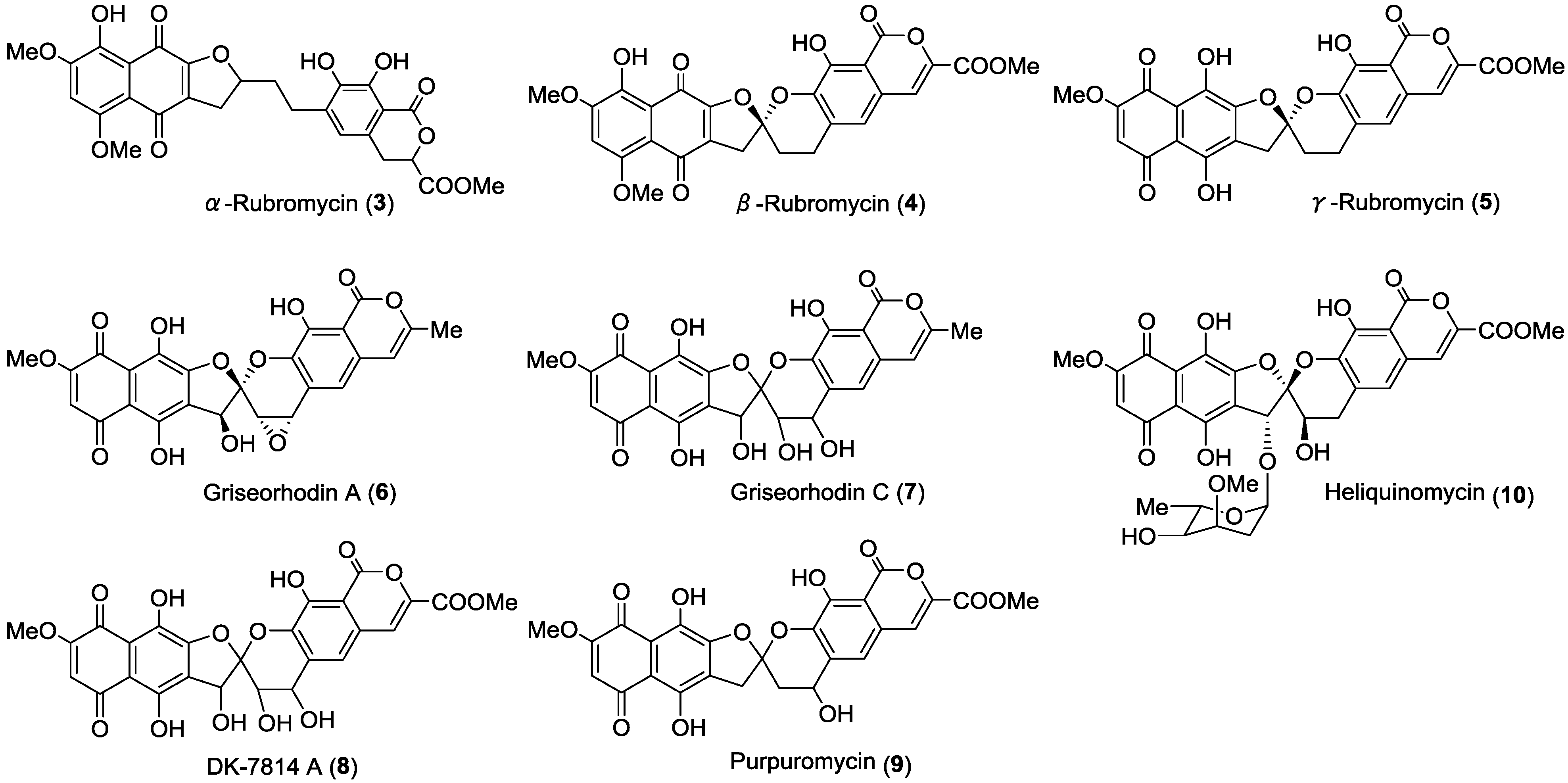
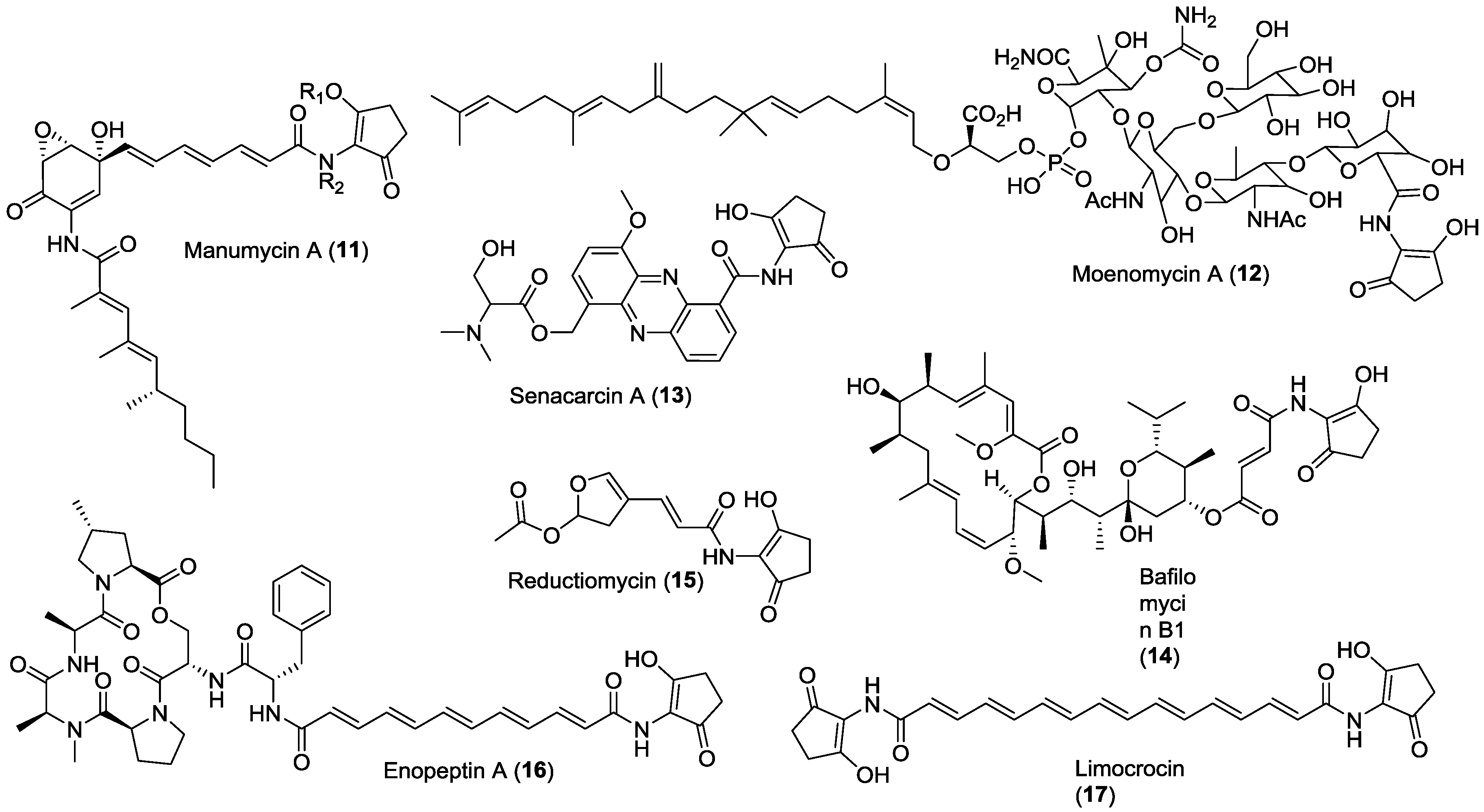
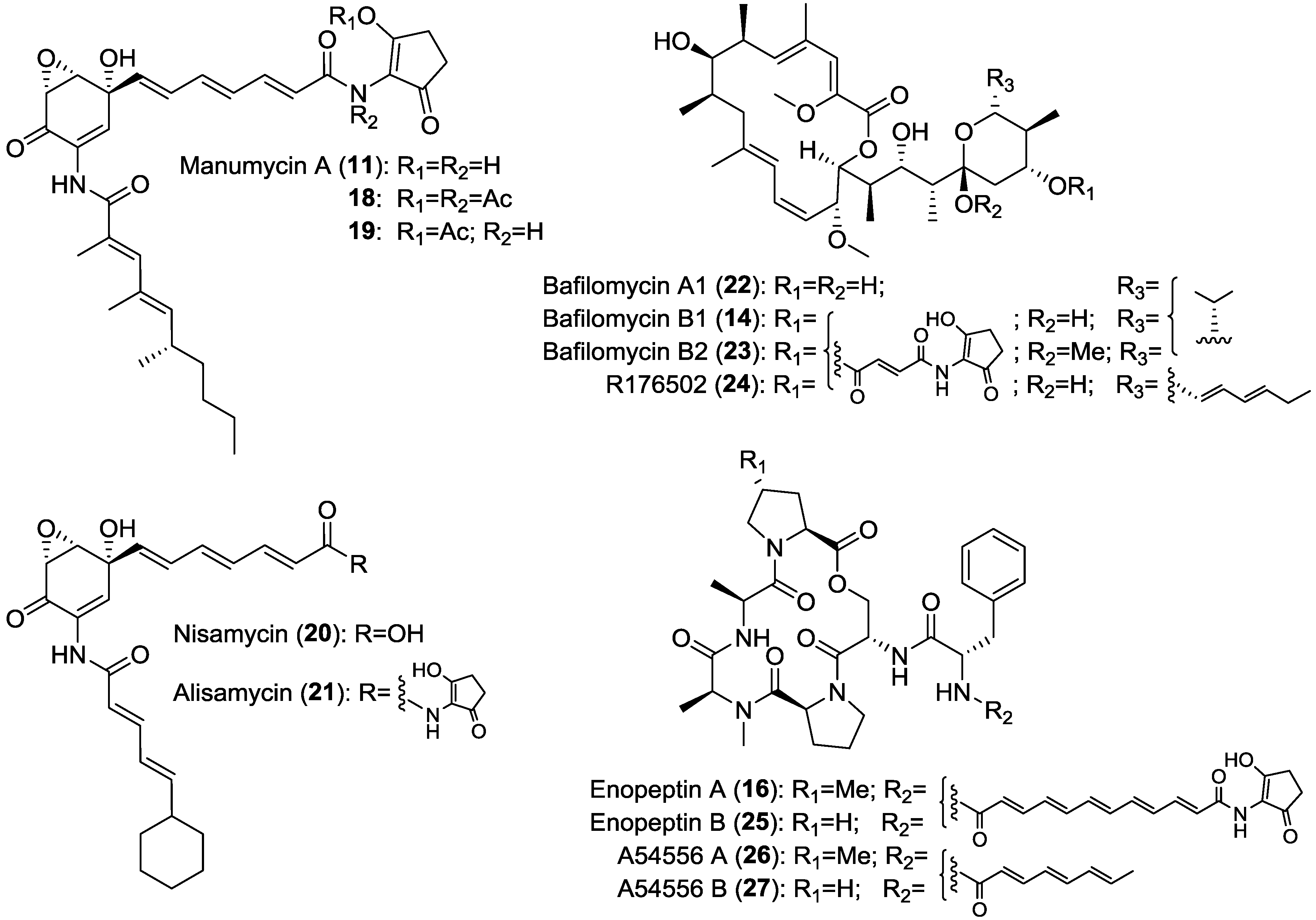
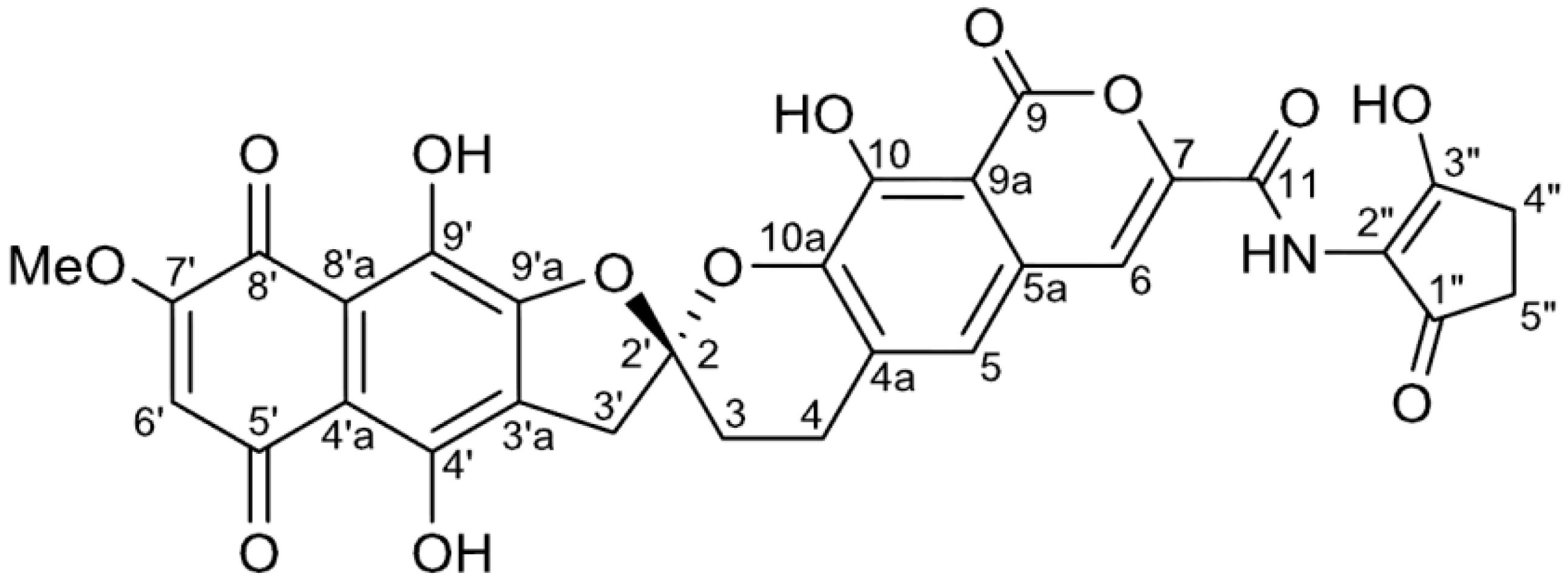
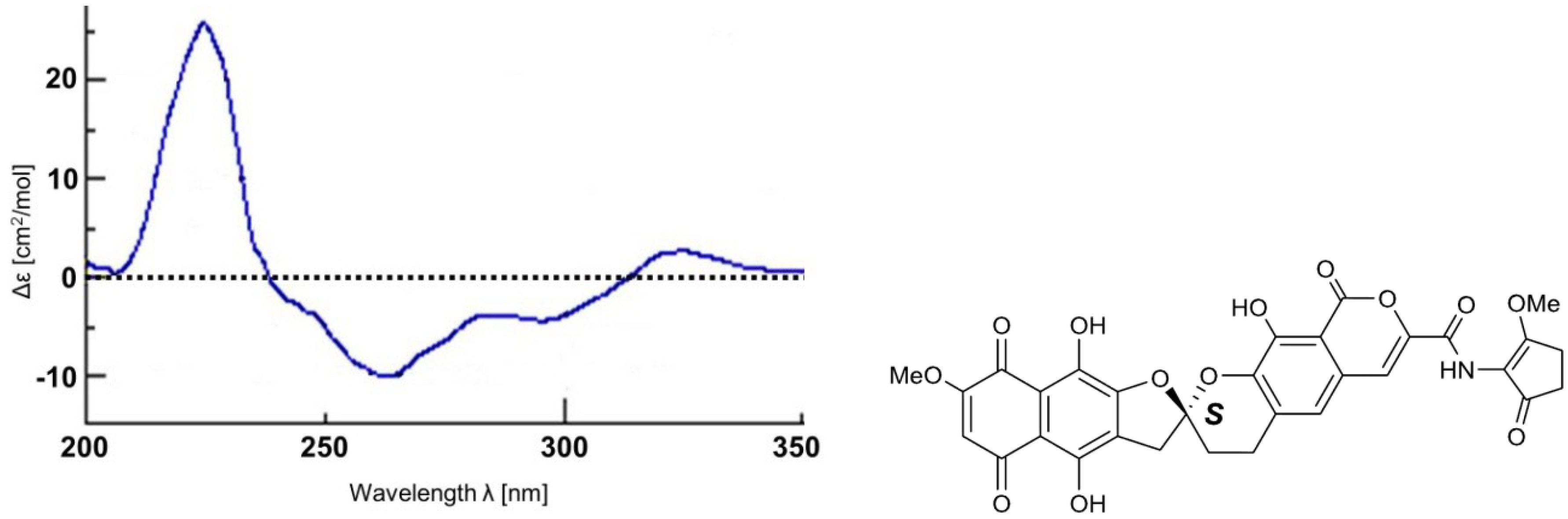
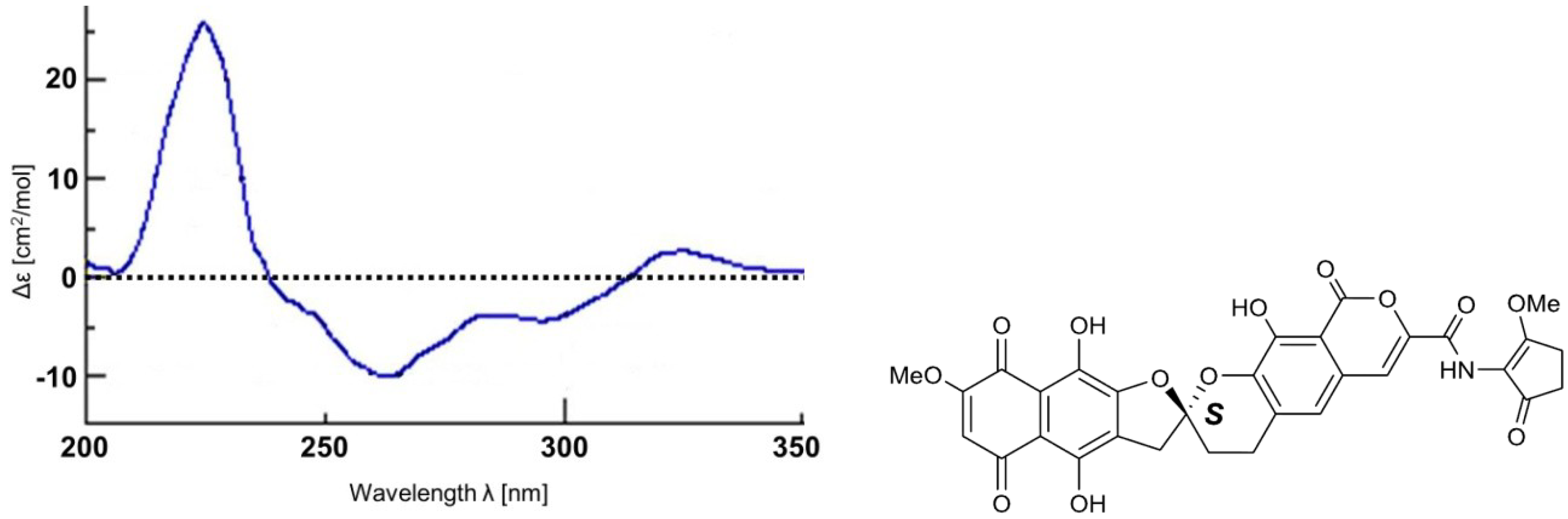
2. Results and Discussion
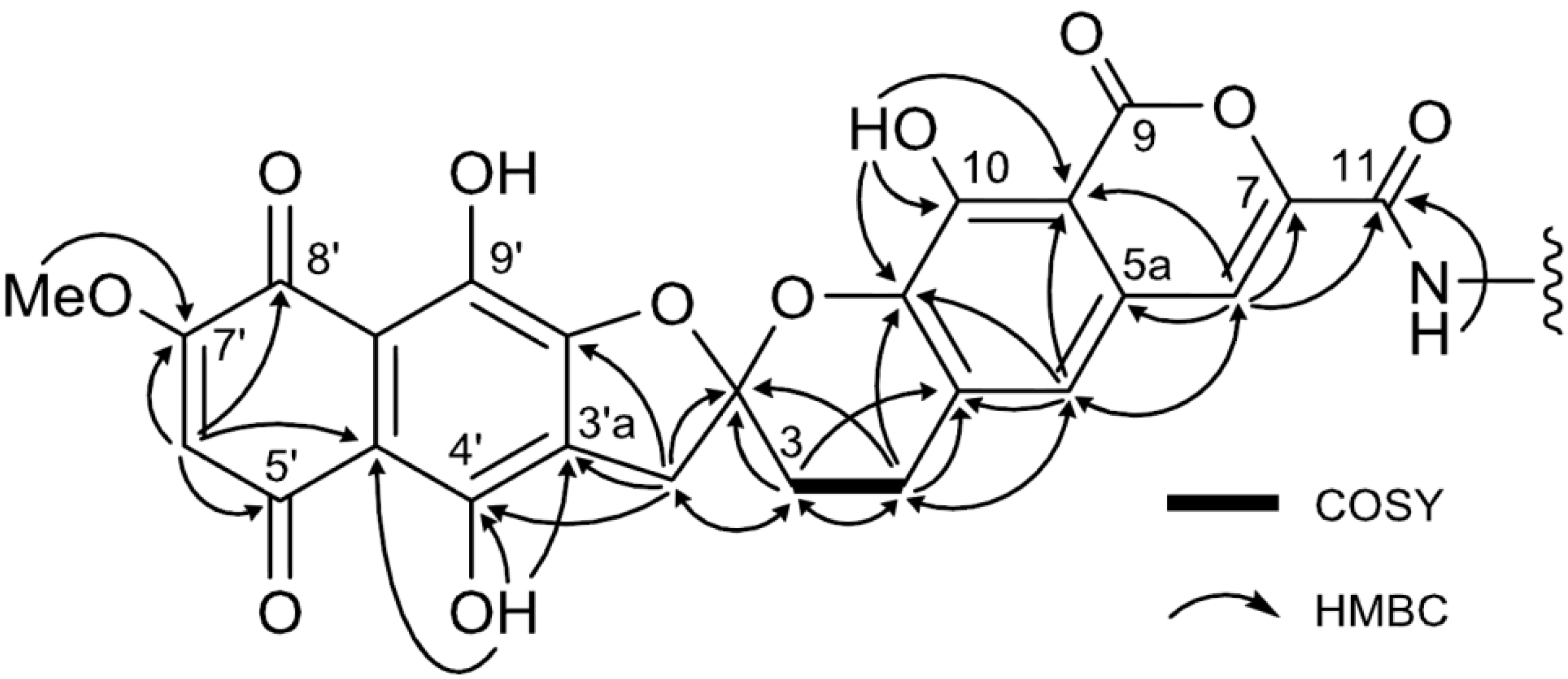
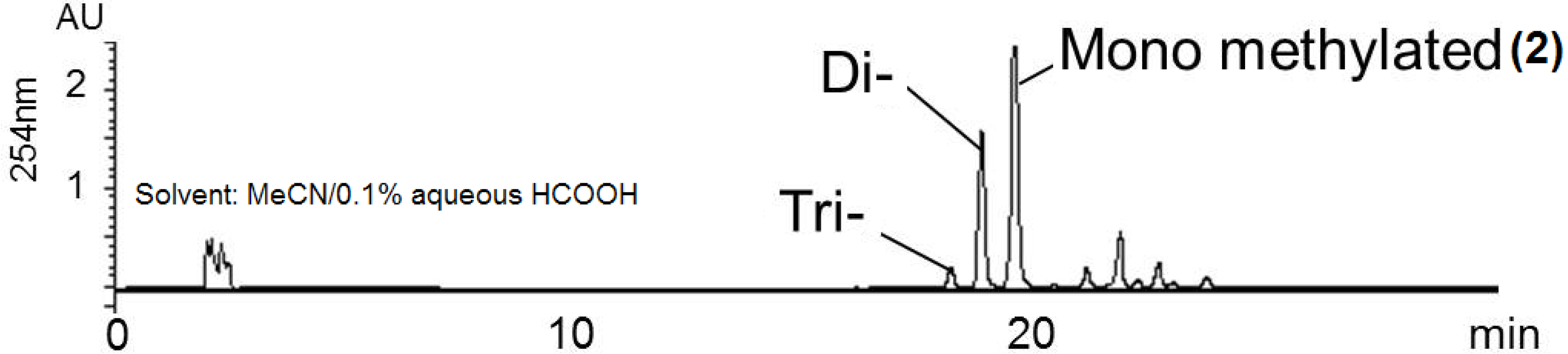
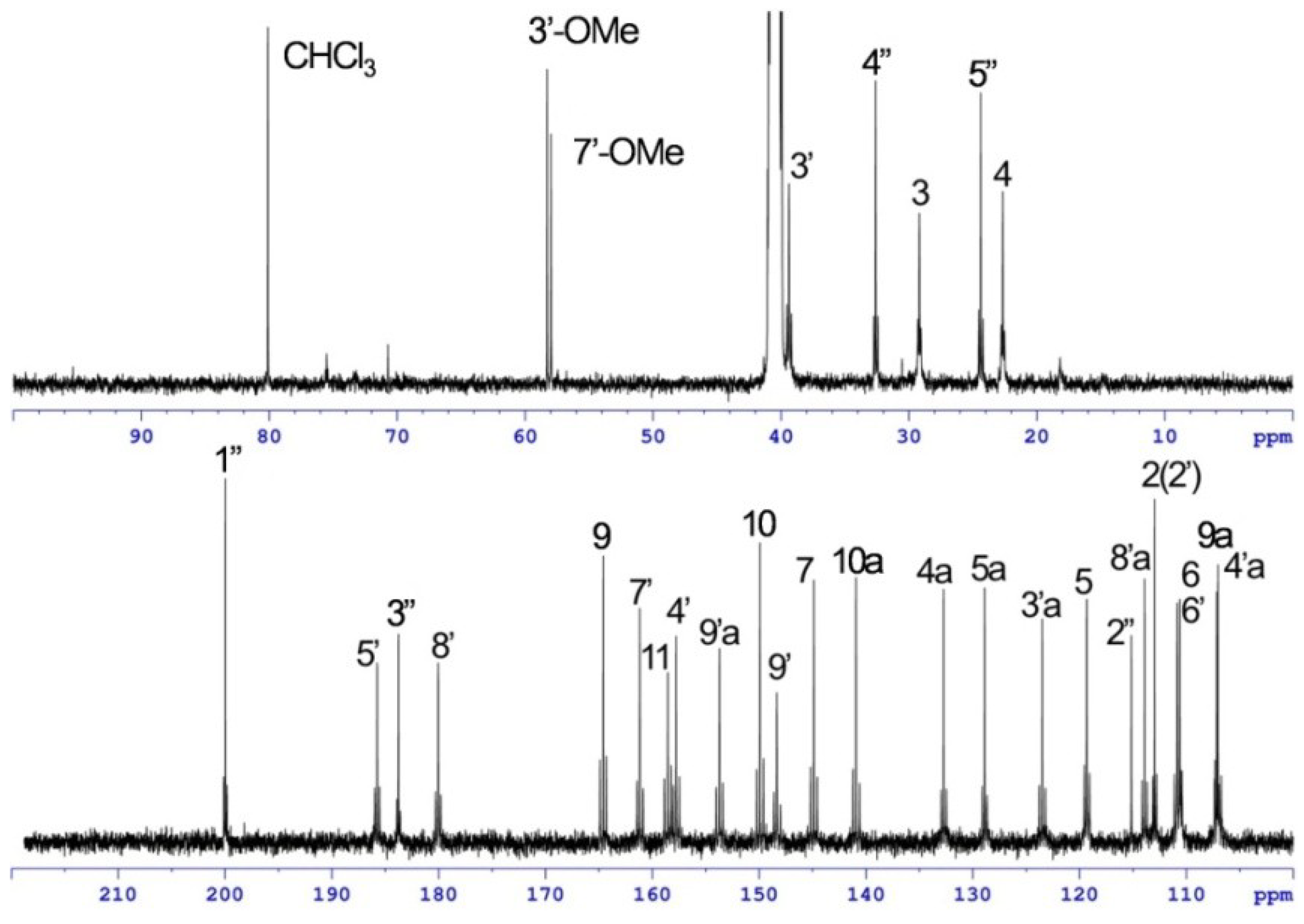

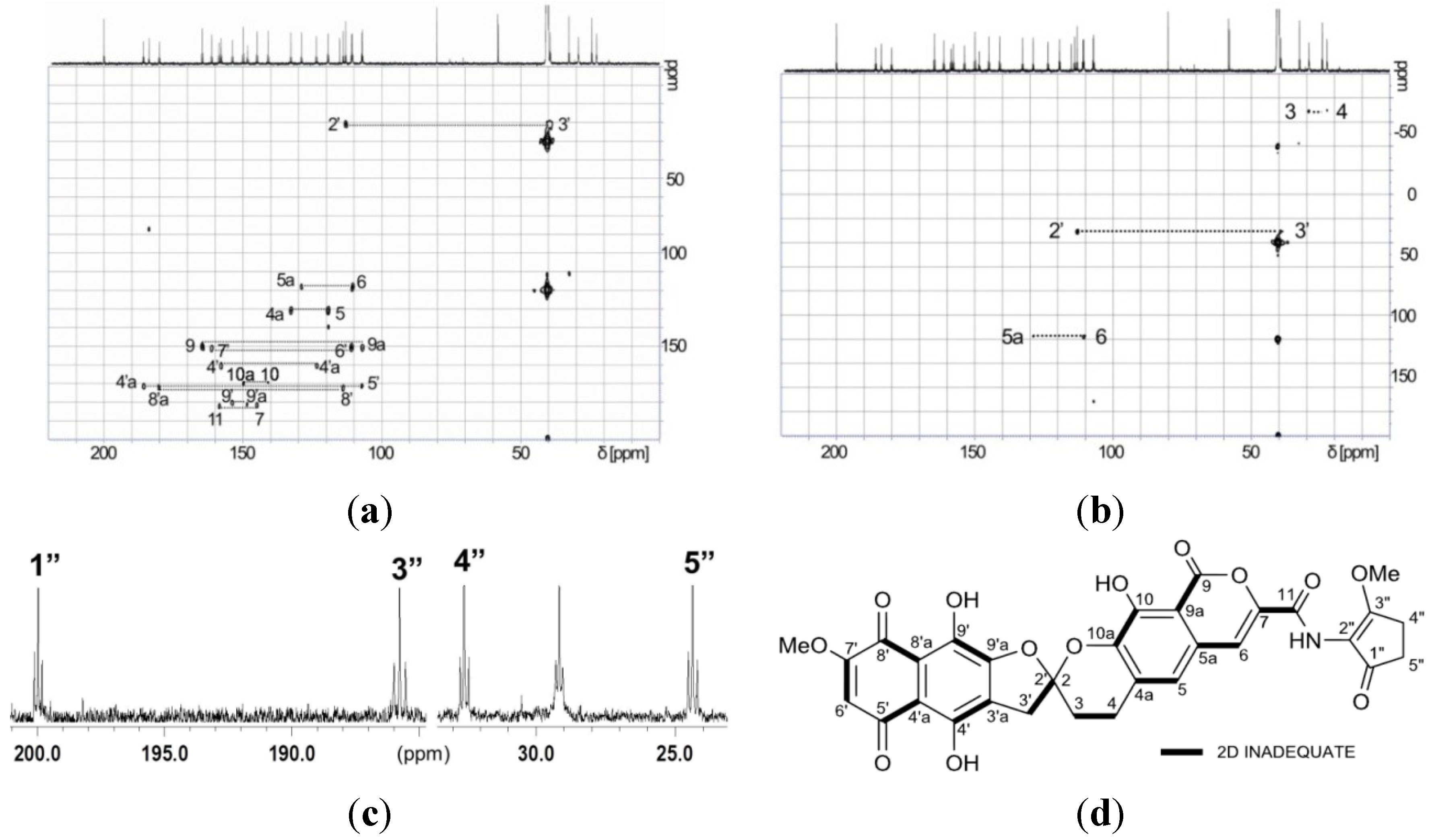

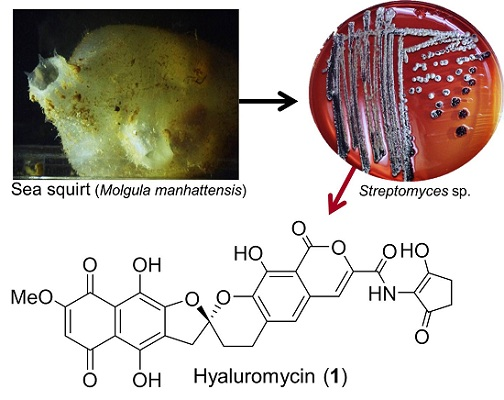{kind=link}
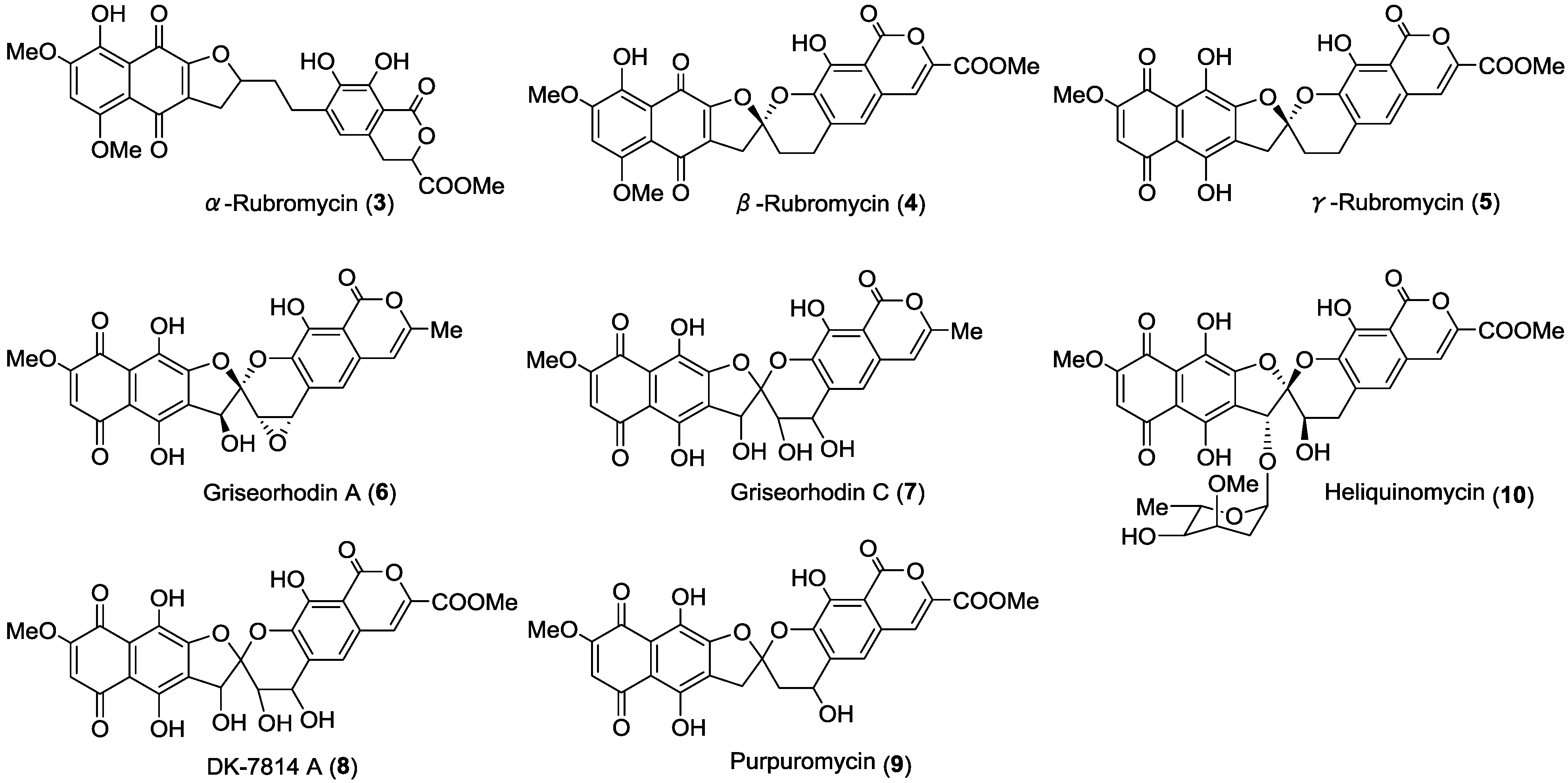{kind=link}
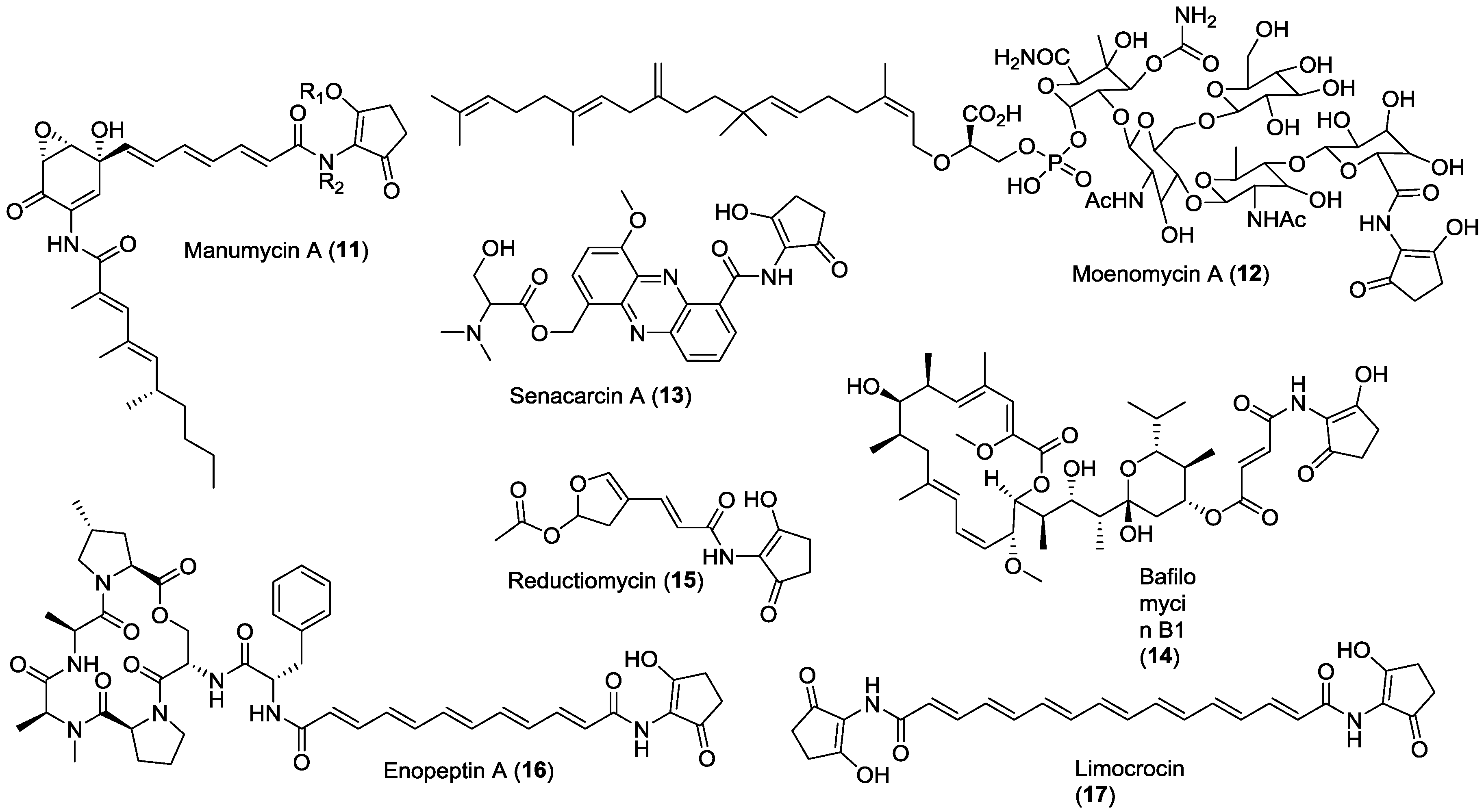{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
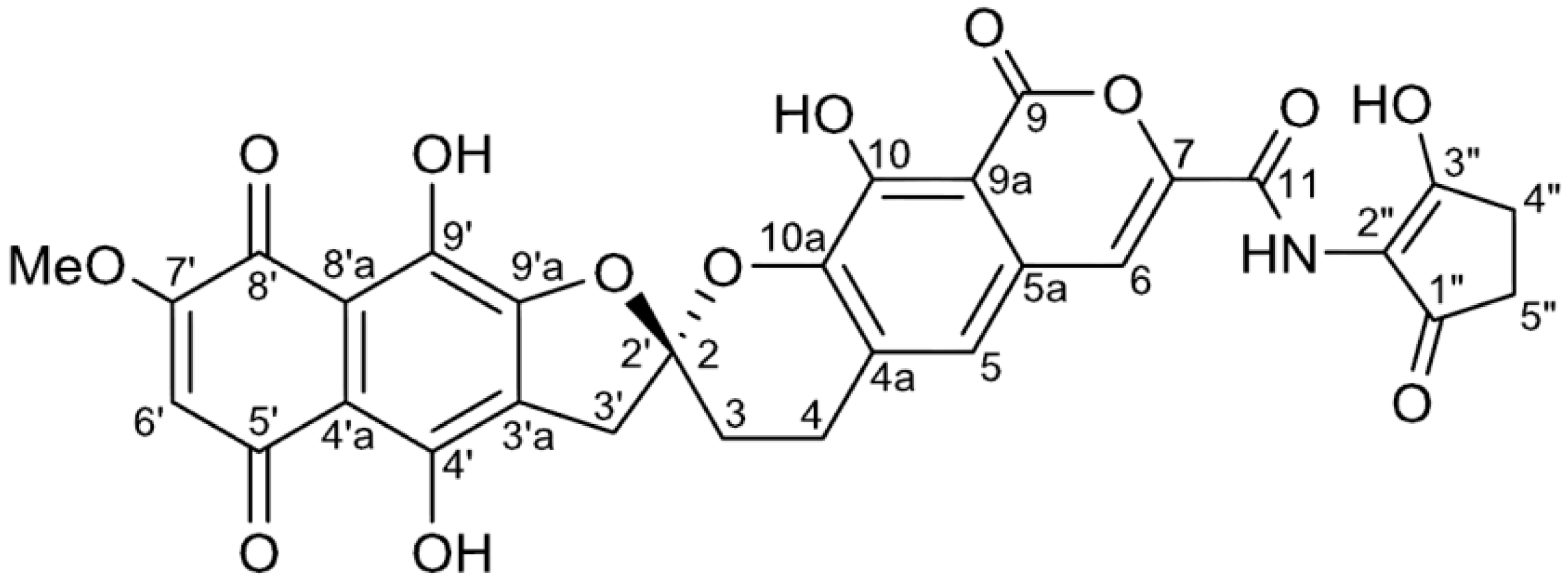
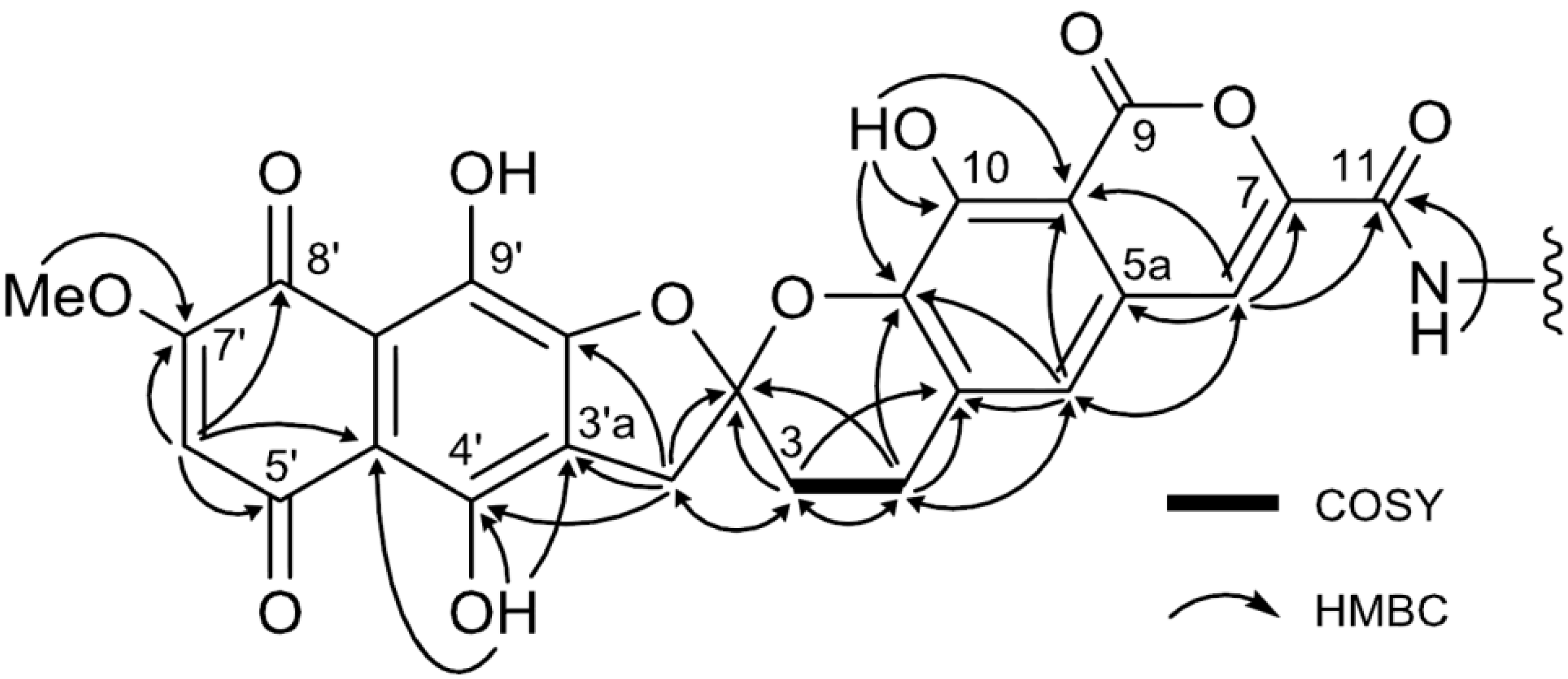
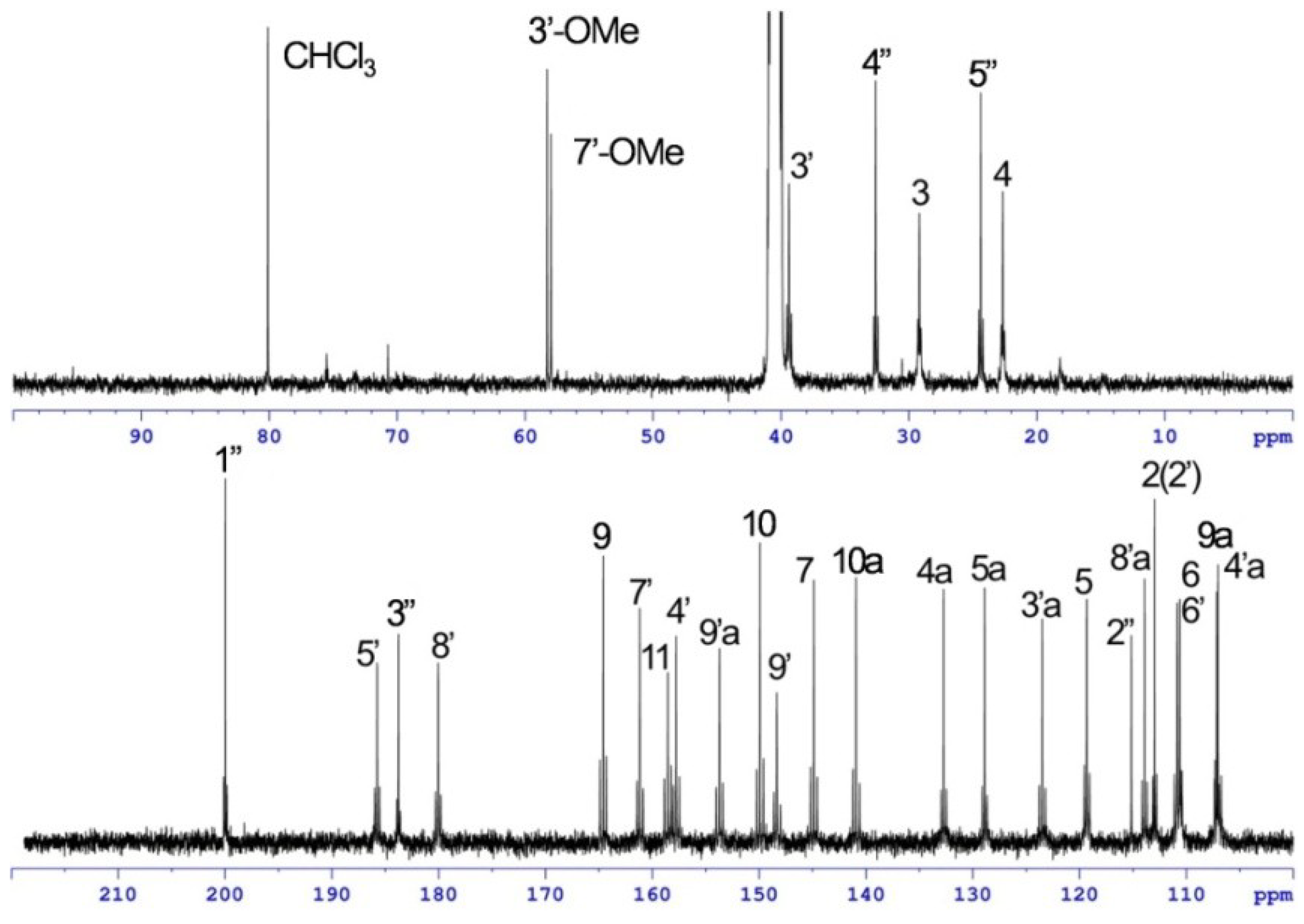
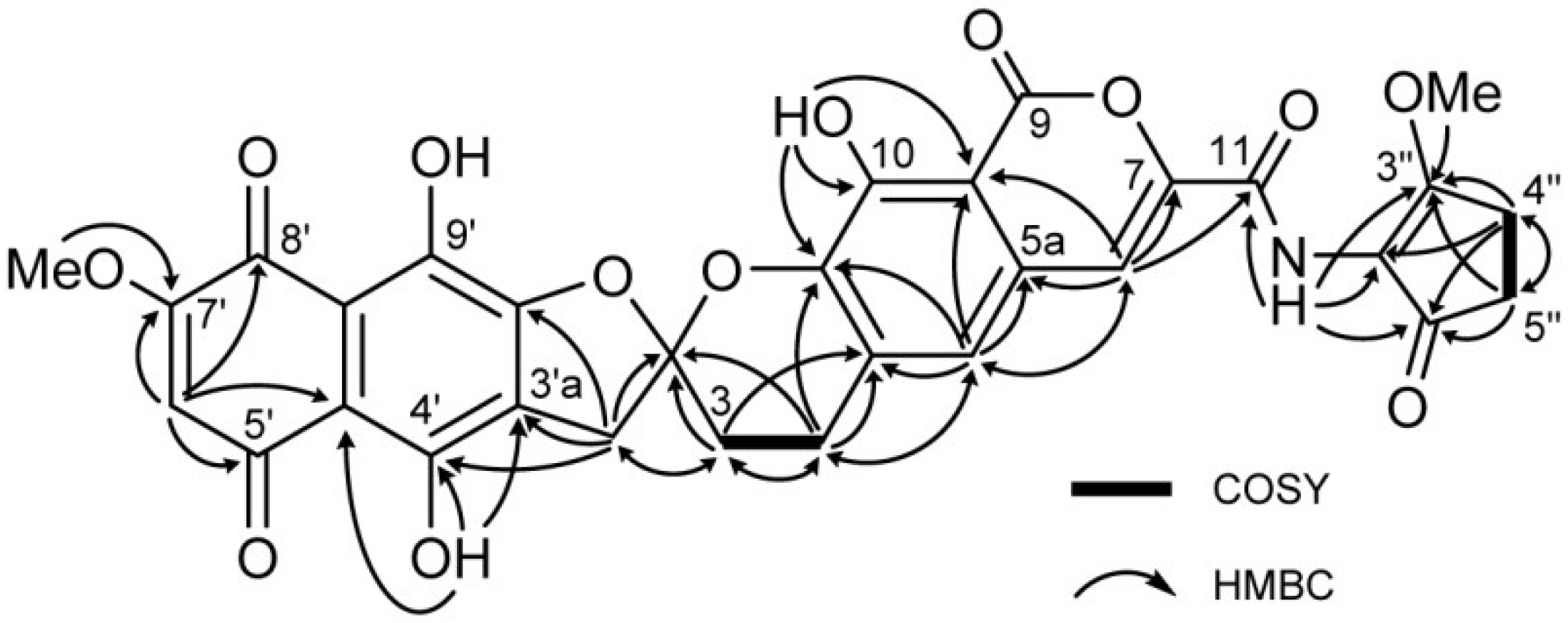
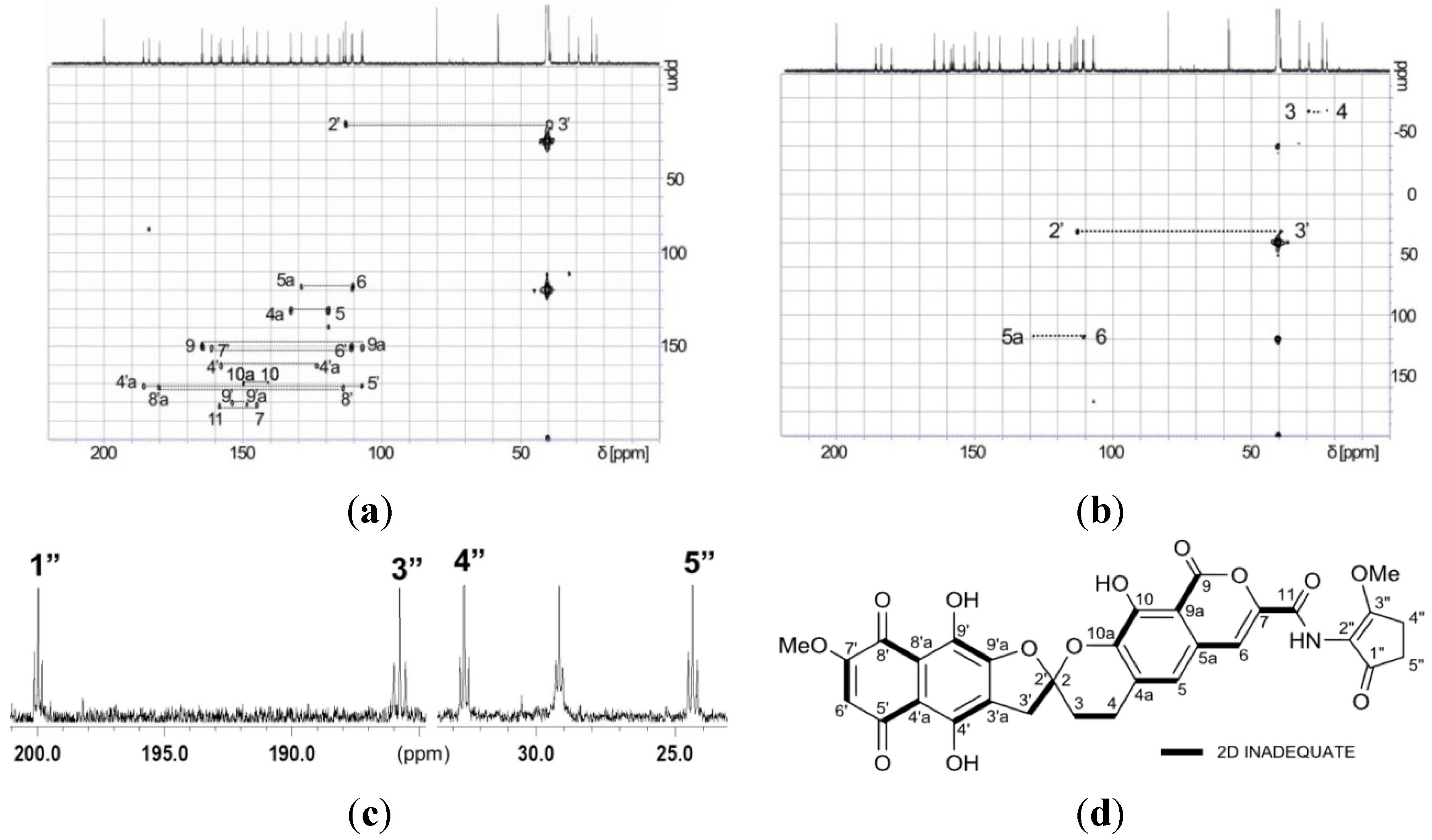
| Position | 1 | [1,2-13C2]Acetate-labeled 2 | |||||
|---|---|---|---|---|---|---|---|
| δC a, Type | δH, Mult. (J in Hz) b | HMBC c | δC a, Type | 1JCC (Hz), Mult. | δH, Mult. (J in Hz) b | HMBC c | |
| 2 (2′) | 113.0, qC | 113.0, qC | 42.4, dd | ||||
| 3 | 29.1, CH2 | 2.37, m; 2.56, m | 2, 4a | 29.2, CH2 | 30.8, dd | 2.38–2.60, m | 2, 4a |
| 4 | 22.7, CH2 | 3.06, m; 3.17, m | 2, 3, 4a, 5, 10a, | 22.7, CH2 | 31.4, dd | 3.07, m; 3.19, m | 2, 3, 4a, 5, 10a |
| 4a | 132.8, qC | 132.7, qC | 59.8, dd | ||||
| 5 | 119.5, CH | 7.25, s | 4a, 5a, 6, 9a, 10a | 119.3, CH | 59.9, dd | 7.24, s | 4a, 5a, 6, 9a, 10a |
| 5a | 128.9, qC | 128.9, qC | 54.6, dd | ||||
| 6 | 110.9, CH | 7.52, s | 5, 5a, 7, 9a, 11 | 110.6, CH | 56.0, dd | 7.50, s | 5, 5a, 7, 9a, 11 |
| 7 | 144.7, qC | 144.9, qC | 78.6, dd | ||||
| 9 | 164.4, qC | 164.6, qC | 72.5, dd | ||||
| 9a | 107.0, qC | 106.9, qC | 76.3, dd | ||||
| 10 | 150.0, qC | 149.9, qC | 79.1, dd | ||||
| 10a | 141.1, qC | 140.9, qC | 78.9, dd | ||||
| 11 | 158.8, qC | 158.5, qC | 78.4, dd | ||||
| 2′ (2) | 113.0, qC | 113.0, qC | 42.4, dd | ||||
| 3′ | 39.4, CH2 | 3.55, d (18.0) | 3, 2′, 3′a, 4′, 9′a | 39.4, CH2 | 42.8, dd | 3.49, d (17.9) | 3, 2′, 3′a, 4′, 9′a |
| 3.48, d (18.0) | 3.63, d (17.9) | ||||||
| 3′a | 123.6, qC | 123.5, qC | 71.6, dd | ||||
| 4′ | 157.8, qC | 157.8, qC | 71.4, dd | ||||
| 4′a | 107.2, qC | 107.2, qC | 56.5, dd | ||||
| 5′ | 185.9, qC | 185.8, qC | 38.4, dd | ||||
| 6′ | 110.9, CH | 6.41, s | 4′a, 5′, 7′, 8′ | 110.9, CH | 65.5, dd | 6.42, s | 4′a, 5′, 7′, 8′ |
| 7′ | 161.2, qC | 161.2, qC | 71.3, dd | ||||
| 8′ | 180.2, qC | 180.1, qC | 59.1, dd | ||||
| 8′a | 114.0, qC | 113.9, qC | 59.1, dd | ||||
| 9′ | 148.3, qC | 148.3, qC | 73.9, dd | ||||
| 9′a | 153.7, qC | 153.7, qC | 74.8, dd | ||||
| 7′-OMe | 58.0, CH3 | 3.89, s | 7′ | 58.0, CH3 | 3.91, s | 7′ | |
| 1″ | n.d. d | 200.0, qC | 38.4, dd | ||||
| 2″ | 113.9, qC | 115.1, qC | |||||
| 3″ | n.d. d | 183.8, qC | 39.8, dd | ||||
| 4″ | n.d. d | 32.6, CH2 | 40.5, dd | 2.41, m | 1″, 3″, 4″ | ||
| 5″ | n.d. d | 24.4, CH2 | 38.3, dd | 2.83, dd (3.7, 3.7) | 1″, 2″, 3″, 5″ | ||
| 3″-OMe | 58.3, CH3 | 4.01, s | 3″ | ||||
| 10-OH | 10.67, s | 9a, 10, 10a | 10.71, s | 9a, 10, 10a | |||
| 4′-OH | 13.14, s | 3′a, 4′, 4′a | 11.88, s | 3′a, 4′, 4′a | |||
| 9′-OH | 11.90, s | 13.15, s | |||||
| 11-NH | 9.44, s | 11 | 9.44, s | 11, 1″, 2″, 3″ | |||
| Compound | 0.0031 | 0.0063 | 0.013 | 0.025 | 0.050 | 0.10 | 0.25 | 0.50 | 1.0 | 2.0 | (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 7.1 | 33.5 | 70.5 | 94.2 | 94.9 | 96.7 | |||||
| 2 | 0 | 0 | 0 | 0 | 0 | 0 | |||||
| β-Rubromycin (4) | 0 | 0 | 0 | 0 | 0 | 0 | |||||
| γ-Rubromycin (5) | 0 | 0 | 0 | 0 | 0 | 0 | |||||
| Glycyrrhizin | 10.6 | 20.0 | 37.9 | 87.6 | 99.1 | 99.2 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Microorganism
3.3. Fermentation
3.4. Extraction and Isolation
3.5. Hyaluromycin (1)
3.6. Feeding Experiment
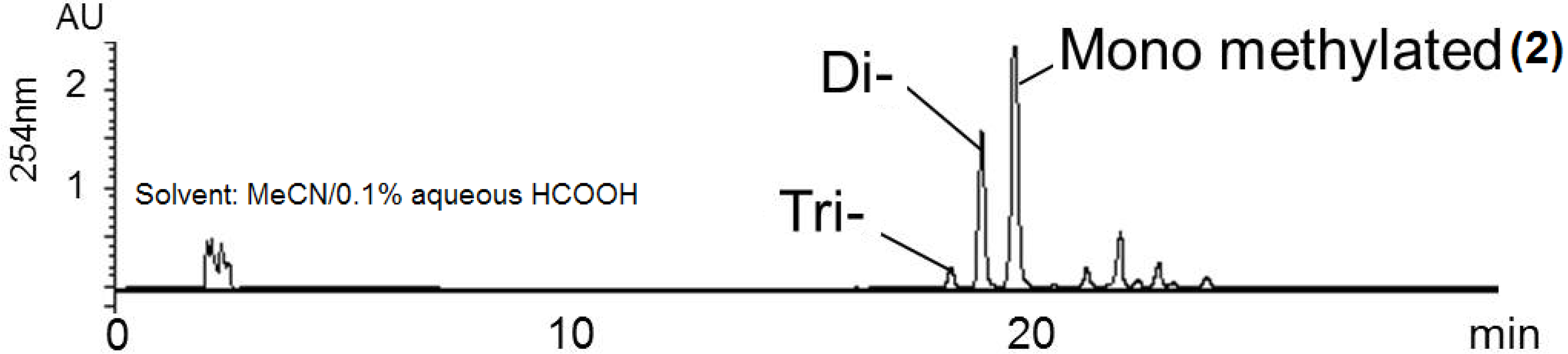
3.7. Methylation of [1,2-13C2]Acetate-Labeled Hyaluromycin (1)
3.8. Methylation of Hyaluromycin (1)
3.9. Hyaluronidase Inhibitory Activity
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ohya, T.; Kaneko, Y. Novel hyaluronidase from streptomyces. Biochim. Biophys. Acta 1970, 198, 607–609. [Google Scholar] [CrossRef]
- Hamai, A.; Morikawa, K.; Horei, K.; Tokuyasu, K. Purification and characterization of hyaluronidase from Streptococcus dysgalactiae. Agric. Biol. Chem. 1989, 53, 2163–2168. [Google Scholar] [CrossRef]
- Baker, J.R.; Dong, S.; Pritchard, D.G. The hyaluronan lyase of Streptococcus pyogenes bacteriophage H4489A. Biochem. J. 2002, 365, 317–322. [Google Scholar] [CrossRef]
- Kemeny, D.M.; Dalton, N.; Lawrence, A.J.; Pearce, F.L.; Vernon, C.A. The purification and characterisation of hyaluronidase from the venom of the honey bee, Apis mellifera. Eur. J. Biochem. 1984, 139, 217–223. [Google Scholar] [CrossRef]
- Kolarich, D.; Léonard, R.; Hemmer, W.; Altmann, F. The N-glycans of yellow jacket venom hyaluronidases and the protein sequence of its major isoform in Vespula vulgaris. FEBS J. 2005, 272, 5182–5190. [Google Scholar] [CrossRef]
- Pessini, A.C.; Takao, T.T.; Cavalheiro, E.C.; Vichnewski, W.; Sampaio, S.V.; Giglio, J.R.; Arantes, E.C. A hyaluronidase from Tityus serrulatus scorpion venom: Isolation, characterization and inhibition by flavonoids. Toxicon 2001, 39, 1495–1504. [Google Scholar] [CrossRef]
- Girish, K.S.; Kemparaju, K. Inhibition of Naja naja venom hyaluronidase: Role in the management of poisonous bite. Life Sci. 2006, 78, 1433–1440. [Google Scholar] [CrossRef]
- Tu, A.T.; Hendon, R.R. Characterization of lizard venom hyaluronidase and evidence for its action as a spreading factor. Comp. Biochem. Physiol. B 1983, 76, 377–383. [Google Scholar]
- Karlstam, B.; Ljungloef, A. Purification and partial characterization of a novel hyaluronic acid-degrading enzyme from Antarctic krill (Euphausia superba). Polar Biol. 1991, 11, 501–507. [Google Scholar]
- Krishnapillai, A.M.; Taylor, K.D.A.; Morris, A.E.J.; Quantick, P.C. Characterisation of Norway lobster (Nephrops norvegicus) hyaluronidase and comparison with sheep and bovine testicular hyaluronidase. Food Chem. 1999, 65, 515–521. [Google Scholar] [CrossRef]
- Hopkins, B.J.; Hodgson, W.C. Enzyme and biochemical studies of stonefish (Synacneja trachynis) and solidfish (Gymnapistes marmoratus). Toxicon 1998, 36, 791–793. [Google Scholar] [CrossRef]
- Noble, P.W.; McKee, C.M.; Cowman, M.; Shin, H.S. Hyaluronan fragments activate an NF-kappa B/I-kappa B alpha autoregulatory loop in murine macrophages. J. Exp. Med. 1996, 183, 2373–2378. [Google Scholar] [CrossRef]
- Termei, R.; Laschinger, C.; Lee, W.; McCulloch, C.A. Intercellular interactions between mast cells and fibroblasts promote pro-inflammatory signaling. Exp. Cell Res. 2013, 319, 1839–1851. [Google Scholar] [CrossRef]
- Feinberg, R.N.; Beebe, D.C. Hyaluronate in vasculogenesis. Science 1983, 220, 1177–1179. [Google Scholar]
- McKee, C.M.; Penno, M.B.; Cowman, M.; Burdick, M.D.; Strieter, R.M.; Bao, C.; Noble, P.W. Hyaluronan (HA) fragments induce chemokine gene expression in alveolar macrophages. The role of HA size and CD44. J. Clin. Investig. 1996, 98, 2403–2413. [Google Scholar] [CrossRef]
- Kobayashi, H.; Terao, T. Hyaluronic acid-specific regulation of cytokines by human uterine fibroblasts. Am. J. Physiol. 1997, 273, 1151–1159. [Google Scholar]
- Nakamura, K.; Yokohama, S.; Yoneda, M.; Okamoto, S.; Tamaki, Y.; Ito, T.; Okada, M.; Aso, K.; Makino, I. High, but not low, molecular weight hyaluronan prevents T-cell-mediated liver injury by reducing proinflammatory cytokines in mice. J. Gastroenterol. 2004, 39, 346–354. [Google Scholar] [CrossRef]
- Asari, A.; Kanemitsu, T.; Kurihara, H. Oral administration of high molecular weight hyaluronan (900 kDa) controls immune system via toll-like receptor 4 in the intestinal epithelium. J. Biol. Chem. 2010, 285, 24751–24758. [Google Scholar]
- Delmage, J.M.; Powars, D.R.; Jaynes, P.K.; Allerton, S.E. The selective suppression of immunogenicity by hyaluronic acid. Ann. Clin. Lab. Sci. 1986, 16, 303–310. [Google Scholar]
- Tian, X.; Azpurua, J.; Hine, C.; Vaidya, A.; Myakishev-Rempel, M.; Ablaeva, J.; Mao., Z.; Nevo, E.; Gorbunova, V.; Seluanov, A. High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature 2013, 499, 346–349. [Google Scholar] [CrossRef]
- Holmes, M.W.; Bayliss, M.T.; Muir, H. Hyaluronic acid in human articular cartilage. Age-related changes in content and size. Biochem. J. 1988, 250, 435–441. [Google Scholar]
- Sherman, P.W.; Javis, J.U.M. Extraordinary life spans of naked mole-rats (Heterocephalus glaber). J. Zool. 2002, 258, 307–311. [Google Scholar] [CrossRef]
- Buffenstein, R. Negligible senescence in the longest living rodent, the naked mole-rat: Insights from a successfully aging species. J. Comp. Physiol. B 2008, 178, 439–445. [Google Scholar] [CrossRef]
- Sakamoto, K.; Nagai, H.; Koda, A. Role of hyaluronidase in immediate hypersensitivity reaction. Immunopharmacology 1980, 2, 139–146. [Google Scholar] [CrossRef]
- Kakegawa, H.; Mitsuo, N.; Matsumoto, H.; Satoh, H.; Akagi, M.; Tasaka, K. Hyaluronidase-inhibitory and anti-allergic activities of the photo-irradiated products of tranilast. Chem. Pharm. Bull. 1983, 33, 3738–3744. [Google Scholar]
- Furuya, T.; Yamagata, S.; Shimoyama, Y.; Fujihara, M.; Morishita, N.; Ohtsuki, K. Biochemical characterization of glycyrrhizin as an effective inhibitor for hyaluronidase from bovine testis. Biol. Pharm. Bull. 1997, 20, 973–977. [Google Scholar] [CrossRef]
- Kakegawa, H.; Matsumoto, H.; Satoh, T. Activation of hyaluronidase by metallic salts and compound 48/80, and inhibitory effect of anti-allergic agents on hyaluronidase. Chem. Pharm. Bull. 1985, 33, 642–646. [Google Scholar] [CrossRef]
- Girish, K.S.; Kemparaju, K.; Nagaraju, S.; Vishwanath, B.S. Hyaluronidase inhibitors: A biological and therapeutic perspective. Curr. Med. Chem. 2009, 16, 2261–2288. [Google Scholar] [CrossRef]
- Brockmann, H.; Lenk, W.; Schwantje, G.; Zeeck, A. Rubromycin II. Chem. Ber. 1969, 102, 126–151. [Google Scholar] [CrossRef]
- Brockmann, H.; Zeeck, A. Rubromycins. 3. The constitution of alpha-rubromycin, beta-rubromycin, gamma-rubromycin, and gamma-iso-rubromycin. Chem. Ber. 1970, 103, 1709–1726. [Google Scholar] [CrossRef]
- Yan, J.X.; Fan, S.X.; Pei, H.S.; Zhu, B.Q.; Xu, W.S.; Naganawa, H.; Hamada, M.; Takeuchi, T. 8-Methoxygriseorhodin C, a new member of griseorhodin antibiotic. J. Antibiot. 1991, 44, 1277–1279. [Google Scholar] [CrossRef]
- Eckardt, K.; Tresselt, D.; Ihn, W. The structure of the antibiotic griseorhodin C. J. Antibiot. 1978, 31, 970–973. [Google Scholar] [CrossRef]
- Ohshima, M.; Ishizaki, N.; Horiuchi, T.; Marumoto, Y.; Sugiyama, N. Novel antibiotics substance DK-7814. Jpn. Patent 57,032,286, 20 February 1982. [Google Scholar]
- Coronelli, C.; Pagani, H.; Bardone, M.R.; Lancini, G.C. Purpuromycin, a new antibiotic isolated from Actinoplanes ianthinogenes N. sp. J. Antibiot. 1974, 27, 161–168. [Google Scholar] [CrossRef]
- Chino, M.; Nishikawa, K.; Umekita, M.; Hayashi, C.; Yamazaki, T.; Tsuchida, T.; Sawa, T.; Hamada, M.; Takeuchi, T. Heliquinomycin, a new inhibitor of DNA helicase, produced by Streptomyces sp. MJ929-SF2 I. Taxonomy, production, isolation, physico-chemical properties and biological activities. J. Antibiot. 1996, 49, 752–757. [Google Scholar] [CrossRef]
- Ueno, T.; Takahashi, H.; Oda, M.; Yokoyama, A.; Goto, Y.; Mizushima, Y.; Sakaguchi, K.; Hayashi, H. Inhibition of human telomerase by rubromycins: Implication of spiroketal system of the compounds as an active moiety. Biochemisrty 2000, 39, 5995–6002. [Google Scholar] [CrossRef]
- Yuen, T.Y.; Ng, Y.P.; Ip, F.C.F.; Chen, J.L.Y.; Atkinson, D.J.; Sperry, J.; Ip, N.Y.; Brimble, M.A. Telomerase inhibition studies of novel spiroketal-containing rubromycin derivatives. Aust. J. Chem. 2013, 66, 530–533. [Google Scholar]
- Ohta, E.; Ohta, S.; Kubota, N.K.; Suzuki, M.; Ogawa, T.; Yamasaki, A.; Ikegami, S. Micromonospolide A, a new macrolide from Micromonospora sp. Tetrahedron Lett. 2001, 42, 4179–4181. [Google Scholar] [CrossRef]
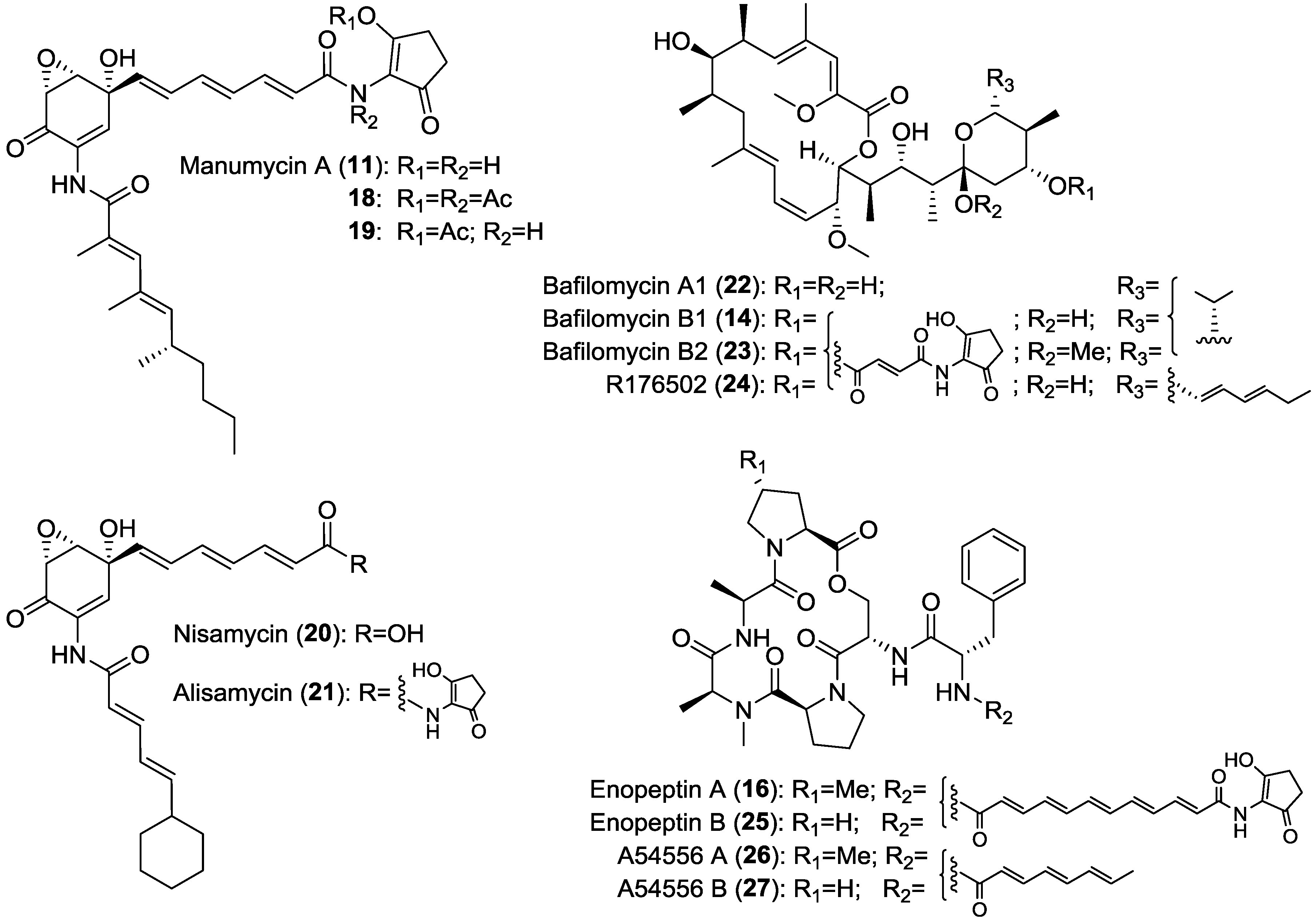
- Laakso, J.A.; Mocek, U.M.; Van, D.J.; Wouters, W.; Janicot, M. R176502, a new bafilolide metabolite with potent antiproliferative activity from a novel Micromonospora species. J. Antibiot. 2003, 56, 909–916. [Google Scholar] [CrossRef]
- Werner, G.; Hagenmaier, H.; Albert, K.; Kohlshorn, H. The structure of the bafilomycins, a new group of macrolide antibiotics. Tetrahedron Lett. 1983, 24, 5193–5196. [Google Scholar] [CrossRef]
- Yan, S.W.; Chan, T.M.; Terracciano, J.; Patel, R.; Loebenberg, D.; Chen, G.; Patel, M.; Gullo, V.; Pramanik, B.; Chu, M. New antibiotic Sch 725424 and its dehydration product Sch 725428 from Kitasatospora sp. J. Antibiot. 2005, 58, 192–195. [Google Scholar] [CrossRef]
- Banskota, A.H.; McAlpine, J.B.; Sørensen, D.; Ibrahim, A.; Aouidate, M.; Piraee, M.; Alarco, A.M.; Farnet, C.M.; Zazopoulos, E. Genomic analyses lead to novel secondary metabolites part 3. ECO-0501, a novel antibacterial of a new class. J. Antibiot. 2006, 59, 533–542. [Google Scholar] [CrossRef]
- Zeeck, A.; Schrӧder, K.; Frobel, K.; Grote, R.; Thiericke, R. The structure of manumycin. I. Characterization, structure elucidation and biological activity. J. Antibiot. 1987, 40, 1530–1540. [Google Scholar] [CrossRef]
- Huger, G. Moenomycin and related phosphorus-containing. Antibiotics 1979, 5, 135–153. [Google Scholar]
- Koshino, H.; Osada, H.; Yano, T.; Uzawa, J.; Isono, K. The structure of enopeptins A and B, novel depsipeptide antibiotics. Tetrahedron Lett. 1991, 32, 7707–7710. [Google Scholar] [CrossRef]
- Nakano, H.; Yoshida, M.; Shirahata, K.; Ishii, S.; Arai, Y.; Morimoto, M.; Tomita, F. Senacarcin A, a new antitumor antibiotic produced by Streptomyces endus subsp. aureus. J. Antibiot. 1982, 35, 760–762. [Google Scholar] [CrossRef]
- Brockmann, H.; Grothe, G. Über Actinomycetenfarbstoffe, II. mitteil.: Limocrocin, ein gelber actinomycetenfarbstoff. Chem. Ber. 1953, 36, 1110–1115. [Google Scholar] [CrossRef]
- Ojika, M.; Shizuri, Y.; Niwa, H.; Yamada, K.; Iwadare, S. Structure and synthesis of reductiline, a novel metabolite from a variant of Streptomyces orientalis. Tetrahedron Lett. 1982, 23, 4977–4980. [Google Scholar] [CrossRef]
- Simizu, K.; Tamura, G. Reductiomycin, a new antibiotic. I. Taxonomy, fermentation, isolation, characterization and biological activities. J. Antibiot. 1981, 34, 649–653. [Google Scholar] [CrossRef]
- Omura, S.; Shimizu, H.; Iwai, Y.; Hinotozawa, K.; Otoguro, K.; Hashimoto, H.; Nakagawa, A. AM-2604 A, a new antiviral antibiotic produced by a strain of Streptomyces. J. Antibiot. 1982, 35, 1632–1637. [Google Scholar] [CrossRef]
- McAlpine, J.B.; Backmann, B.O.; Piraee, M.; Tremblay, S.; Alarco, A.M.; Zazopoudos, E.; Farnet, C.M. Microbial genomics as a guide to drug discovery and structural elucidation: ECO-02301, a novel antifungal agent, as an example. J. Nat. Prod. 2005, 68, 493–496. [Google Scholar] [CrossRef]
- Grote, R.; Zeek, A.; Drautz, H.; Zӓhner, H. Metabolic products of microorganisms. 246. 2880-II, a metabolite related to ferulic acid from Streptomyces griseoflavus. J. Antibiot. 1988, 41, 1275–1276. [Google Scholar] [CrossRef]
- Zeeck, A.; Frobel, K.; Heusel, C.; Schrӧder, K.; Thiericke, R. The structure of manumycin. II. Derivatives. J. Antibiot. 1987, 40, 1541–1548. [Google Scholar] [CrossRef]
- Hayashi, K.; Nakagawa, M.; Fujita, T.; Tanimori, S.; Nakayama, M. Nisamycin, a new manumycin group antibiotic from Streptomyces Sp K106 .2. Structure determination and structure-activity-relationships. J. Antibiot. 1994, 47, 1110–1115. [Google Scholar] [CrossRef]
- Hinzen, B.; Raddatz, S.; Paulsen, H.; Lampe, T.; Schumacher, A.; Häbich, D.; Hellwig, V.; Benet, B.J.; Endermann, R.; Labischinski, H.; et al. Medicinal chemistry optimization of acyldepsipeptides of the enopeptin class antibiotics. ChemMedChem 2006, 1, 689–693. [Google Scholar] [CrossRef]
- Puder, C.; Loya, S.; Hizi, A.; Zeeck, A. Structural and biosynthetic investigations of the rubromycins. Eur. J. Org. Chem. 2000, 5, 729–735. [Google Scholar] [CrossRef]
- Suetsuna, K.; Osajima, Y. Isolation of structure of dideoxygriseorhodin C produced by a Streptomyces sp. Agric. Biol. Chem. 1989, 53, 241–242. [Google Scholar] [CrossRef]
- Thiericke, R.; Zeeck, A.; Nakagawa, A.; Omura, S.; Herrold, R.E.; Wu, S.T.S.; Beale, J.M.; Floss, H.G. Biosynthesis of the manumycin group antibiotics. J. Am. Chem. Soc. 1990, 112, 3979–3987. [Google Scholar] [CrossRef]
- He, H.; Shen, B.; Korshalla, J.; Siegel, M.M.; Carter, G.T. Isolation and structural elucidation of AC326-α, a new member of the moenomycin group. J. Antibiot. 2000, 53, 191–195. [Google Scholar] [CrossRef]
- Li, F.; Maskey, R.P.; Qin, S.; Sattler, I.; Fiebig, H.H.; Maier, A.; Zeeck, A.; Laatsch, H. Chinikomycins A and B: Isolation, structure elucidation, and biological activity of novel antibiotics from a marine Streptomyces sp. isolate M045. J. Nat. Prod. 2005, 68, 349–353. [Google Scholar] [CrossRef]
- Bringmann, G.; Kraus, J.; Schmitt, U.; Puder, C.; Zeeck, A. Determination of the absolute configurations of γ-rubromycin and related spiro compounds by quantum chemical CD calculations. Eur. J. Org. Chem. 2000, 15, 2729–2734. [Google Scholar]
- Yunt, Z.; Reinhardt, K.; Li, A.; Engeser, M.; Dahse, H.M.; Gϋtschow, M.; Bruhn, T.; Bringmann, G.; Piel, J. Cleavage of four carbon-carbon bonds during biosynthesis of the griseorhodin A spiroketal pharmacophore. J. Am. Chem. Soc. 2009, 131, 2297–2305. [Google Scholar] [CrossRef]
- Ferrante, N.D. Turbidimetric measurement of acid mucopolysaccharides and hyaluronidase activity. J. Biol. Chem. 1956, 220, 303–306. [Google Scholar]
Supplementary Files
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Harunari, E.; Imada, C.; Igarashi, Y.; Fukuda, T.; Terahara, T.; Kobayashi, T. Hyaluromycin, a New Hyaluronidase Inhibitor of Polyketide Origin from Marine Streptomyces sp. Mar. Drugs 2014, 12, 491-507. https://doi.org/10.3390/md12010491
Harunari E, Imada C, Igarashi Y, Fukuda T, Terahara T, Kobayashi T. Hyaluromycin, a New Hyaluronidase Inhibitor of Polyketide Origin from Marine Streptomyces sp. Marine Drugs. 2014; 12(1):491-507. https://doi.org/10.3390/md12010491
Chicago/Turabian StyleHarunari, Enjuro, Chiaki Imada, Yasuhiro Igarashi, Takao Fukuda, Takeshi Terahara, and Takeshi Kobayashi. 2014. "Hyaluromycin, a New Hyaluronidase Inhibitor of Polyketide Origin from Marine Streptomyces sp." Marine Drugs 12, no. 1: 491-507. https://doi.org/10.3390/md12010491
APA StyleHarunari, E., Imada, C., Igarashi, Y., Fukuda, T., Terahara, T., & Kobayashi, T. (2014). Hyaluromycin, a New Hyaluronidase Inhibitor of Polyketide Origin from Marine Streptomyces sp. Marine Drugs, 12(1), 491-507. https://doi.org/10.3390/md12010491