Cytotoxicity, Fractionation and Dereplication of Extracts of the Dinoflagellate Vulcanodinium rugosum, a Producer of Pinnatoxin G

 ,
,  ,
, 
Abstract
:1. Introduction
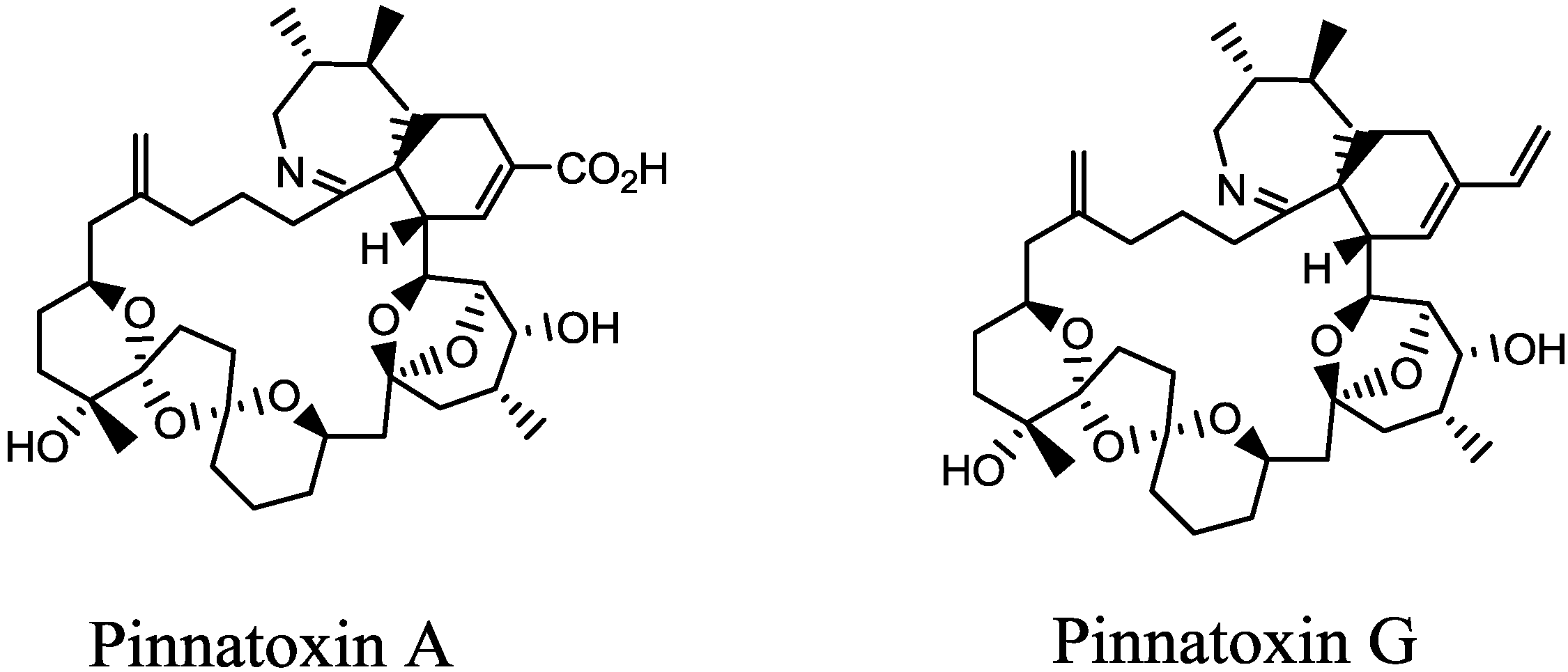
2. Results
2.1. Toxin Content of V. rugosum and Purification of PnTX-G

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
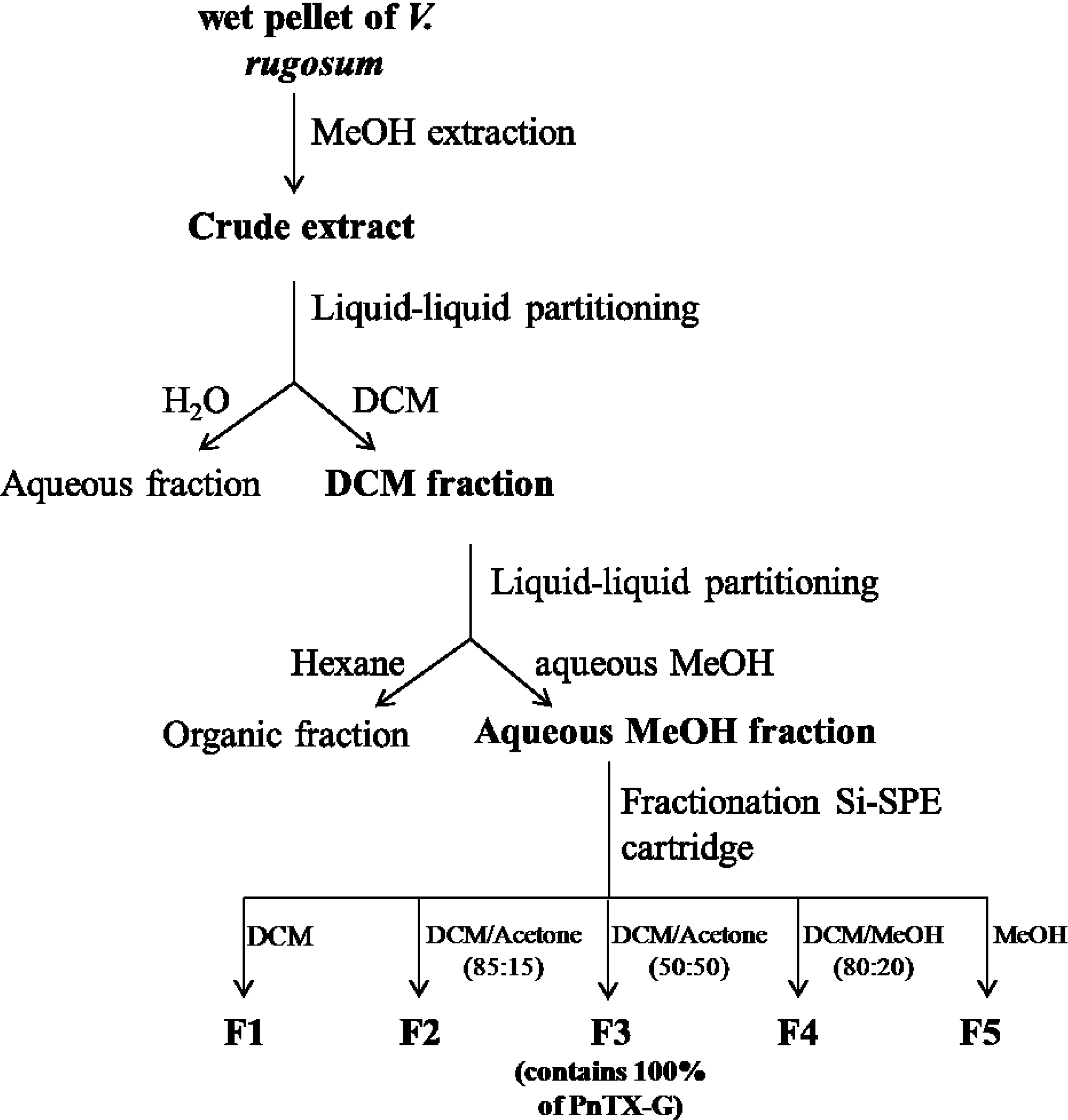{kind=link}
| Fraction | Sample mass | PnTX-G | |
|---|---|---|---|
| (mg) | (µg) | ||
| Crude extract | n/a | 2050 | 844 |
| Partitioning 1 | DCM phase | 442 | 620 |
| Aqueous phase | 1618 | 0.80 | |
| Interface | 168 | 5.80 | |
| Partitioning 2 | Aq. MeOH phase | 168 | 571 |
| Hexane phase | 274 | 21.2 | |
| Fractions | F1 | 7.00 | 0.00 |
| F2 | 73.1 | 0.05 | |
| F3 | 38.8 | 649 | |
| F4 | 6.10 | 0.27 | |
| F5 | 21.7 | 0.16 |
2.2. In Vitro Toxic Effects of Crude and Partitioned Extracts of Vulcanodinium rugosum
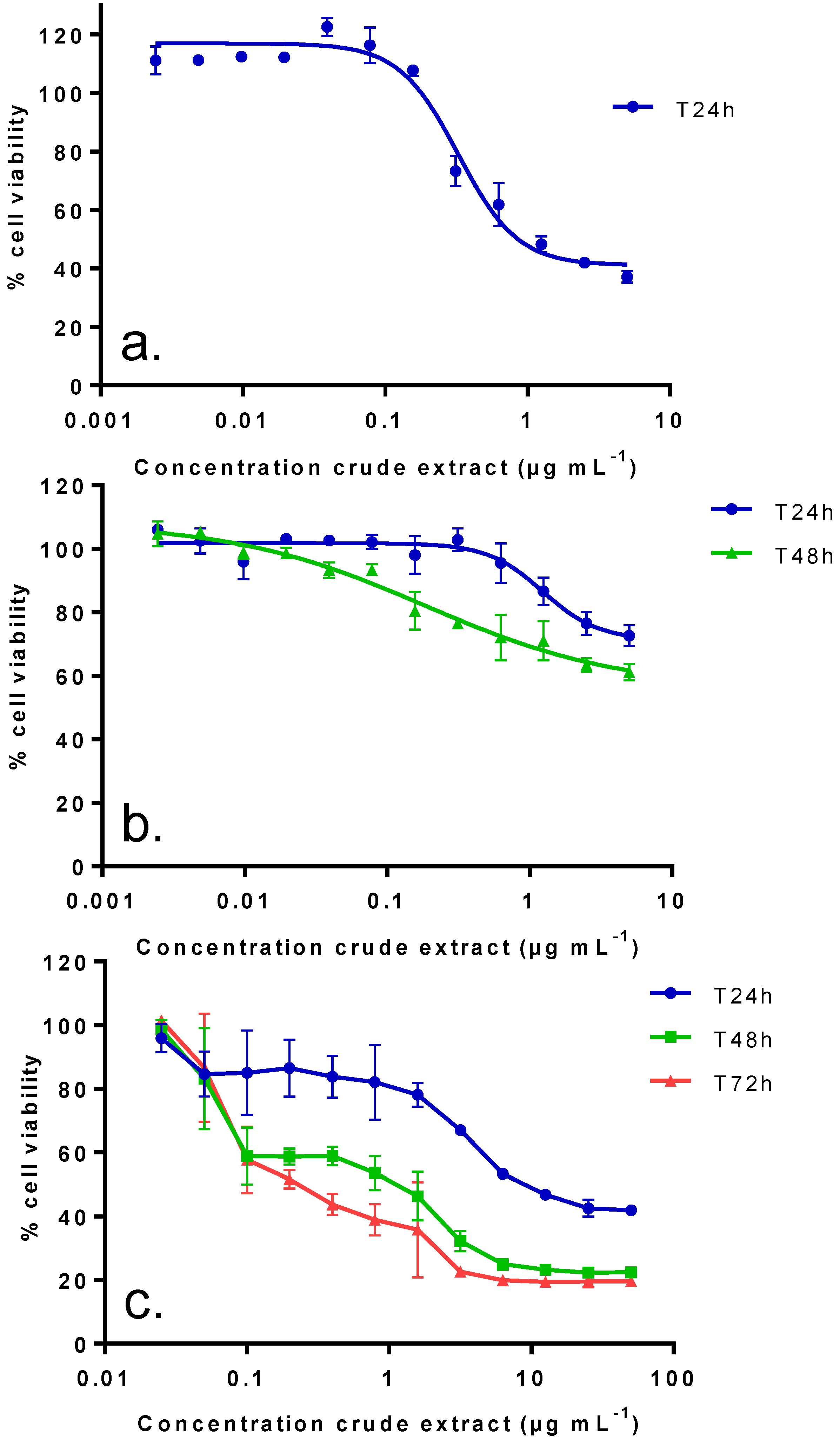
| IC50 (µg mL−1): median (values obtained with two independent experiments) | |||
|---|---|---|---|
| Neuro2A cells (24 h of incubation) | KB cells (72 h of incubation) | Undifferentiated Caco-2 cells (48 h of incubation) | |
| Crude extract | 0.38 (0.24–0.51) | 0.19 (0.26–0.12) | N.R. (40%) |
| DCM fraction | 0.15 (0.14–0.15) | 0.007 (0.006–0.008) | N.R. (40%) |
| aq.MeOH fraction | 0.03 (0.01–0.04) | 0.004 (0.003–0.004) | N.R. (45%) |
2.3. In Vitro Effects of Various Fractions of V. rugosum Extract
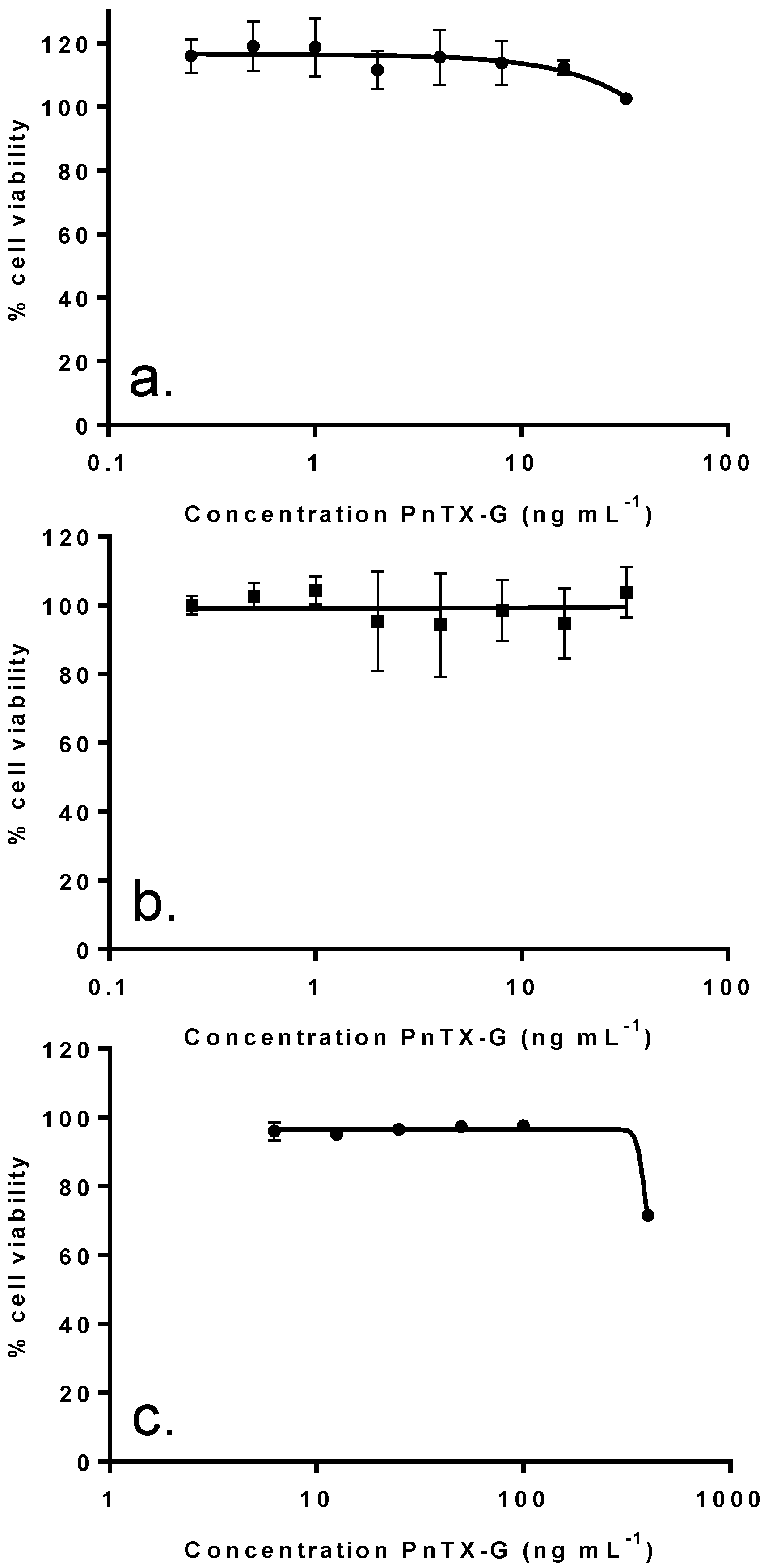
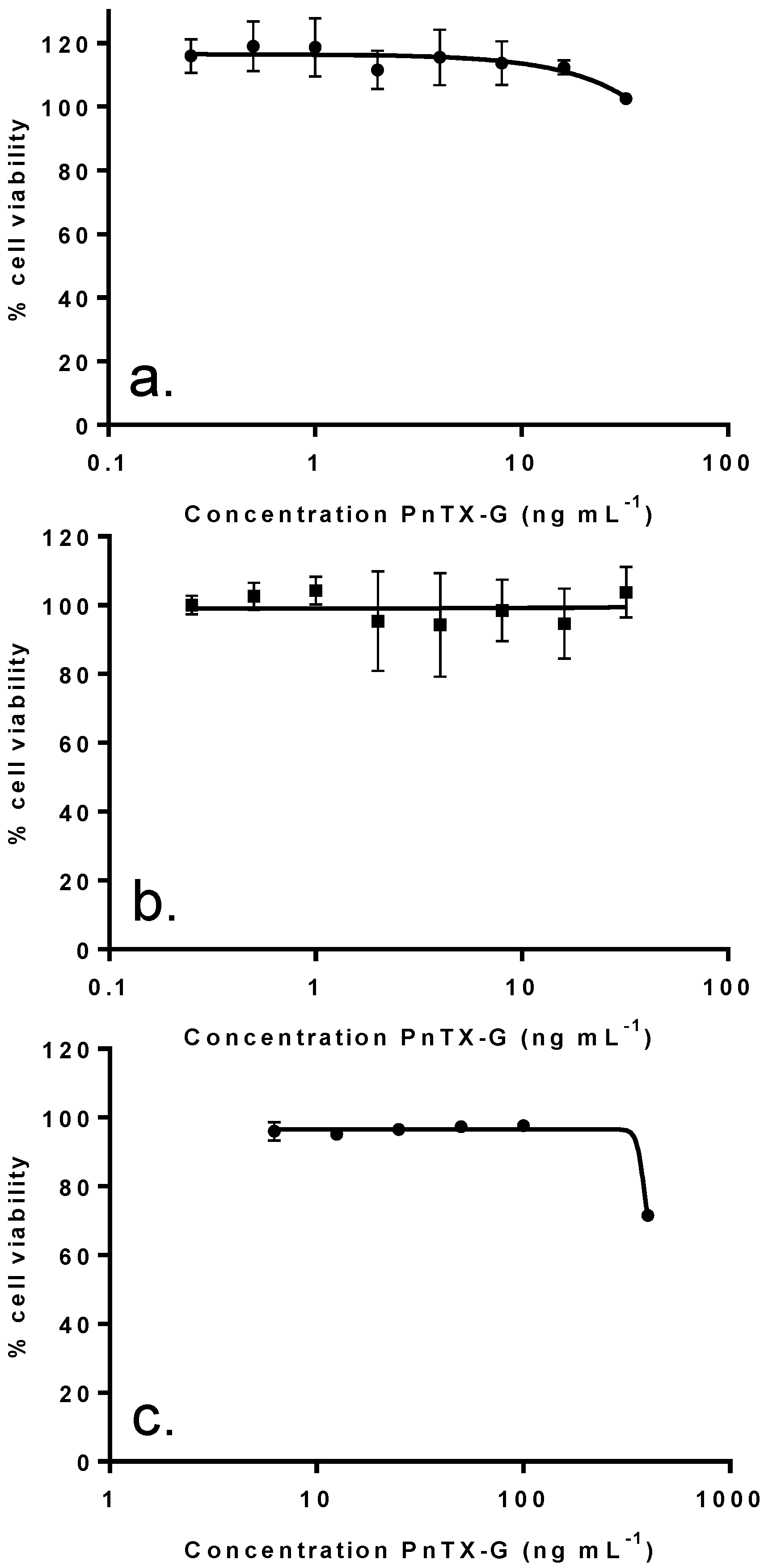
2.4. In Vitro Effects of Pinnatoxin G
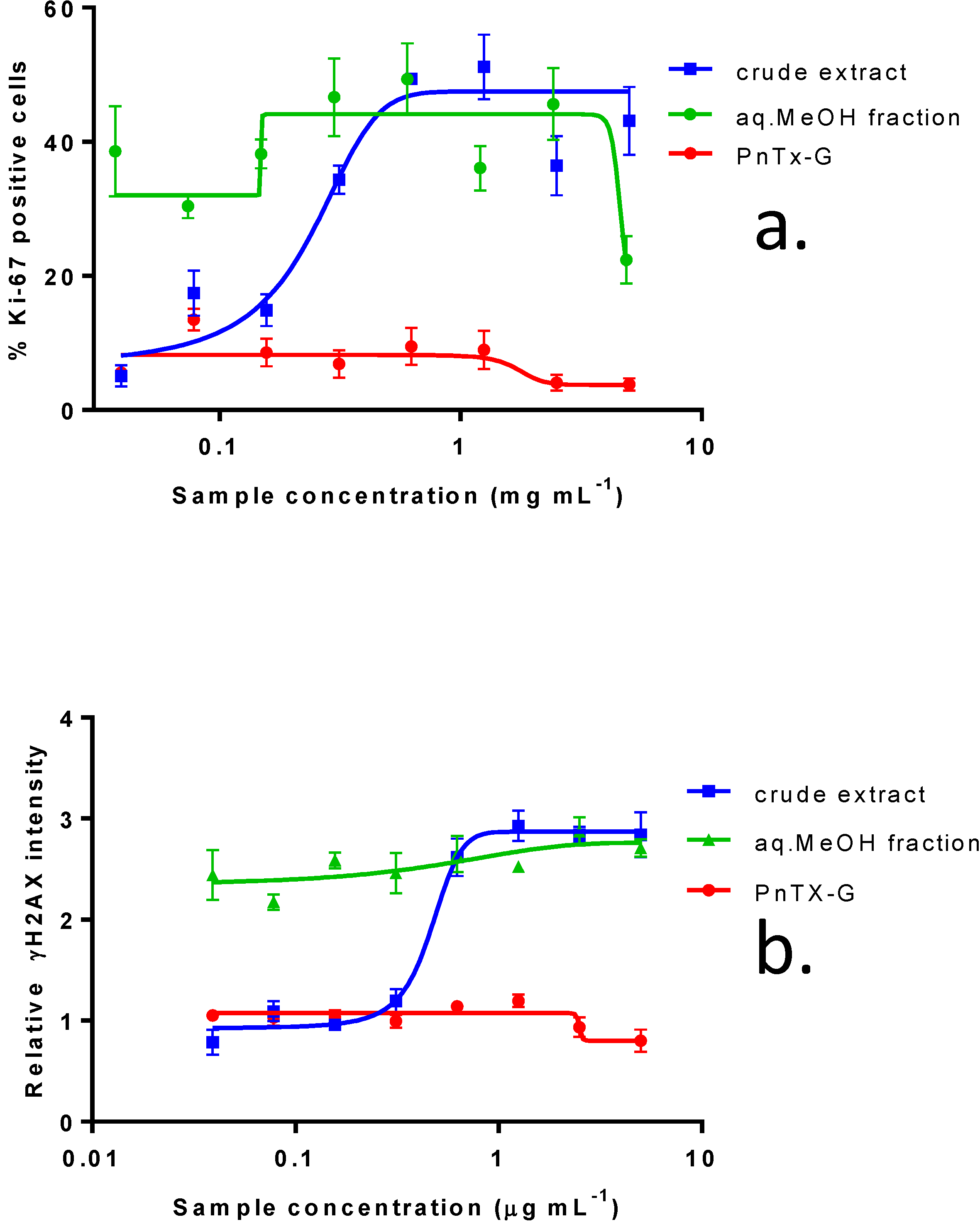
| IC50 (ng mL−1): median (values obtained with two independent experiments) | |||||
|---|---|---|---|---|---|
| Fraction Weight (%) | Distribution of PnTX-G (%) | KB cells (72 h of incubation) | Neuro2A cells (24 h of incubation) | Undifferentiated Caco-2 cells (48 h of incubation) | |
| F1 | 4.9 | 0 | 1023 (958, 1088) | N.R. (45%) | N.R. (44.5%) |
| F2 | 50.3 | 0 | 0.21 (0.18, 0.24) | 11 (5–17) | N.R. (41.6%) |
| F3 | 26.5 | 100 | 2.59 (2.55, 2.63) | 25 (44–6) | N.R. (42.6%) |
| F4 | 4.6 | 0 | 53.6 (45.4, 61.8) | 575 (760–390) | N.R. (25.2%) |
| F5 | 13.8 | 0 | 62.7 (55.9, 69.5) | 1140 (800–1480) | N.R. (41.1%) |
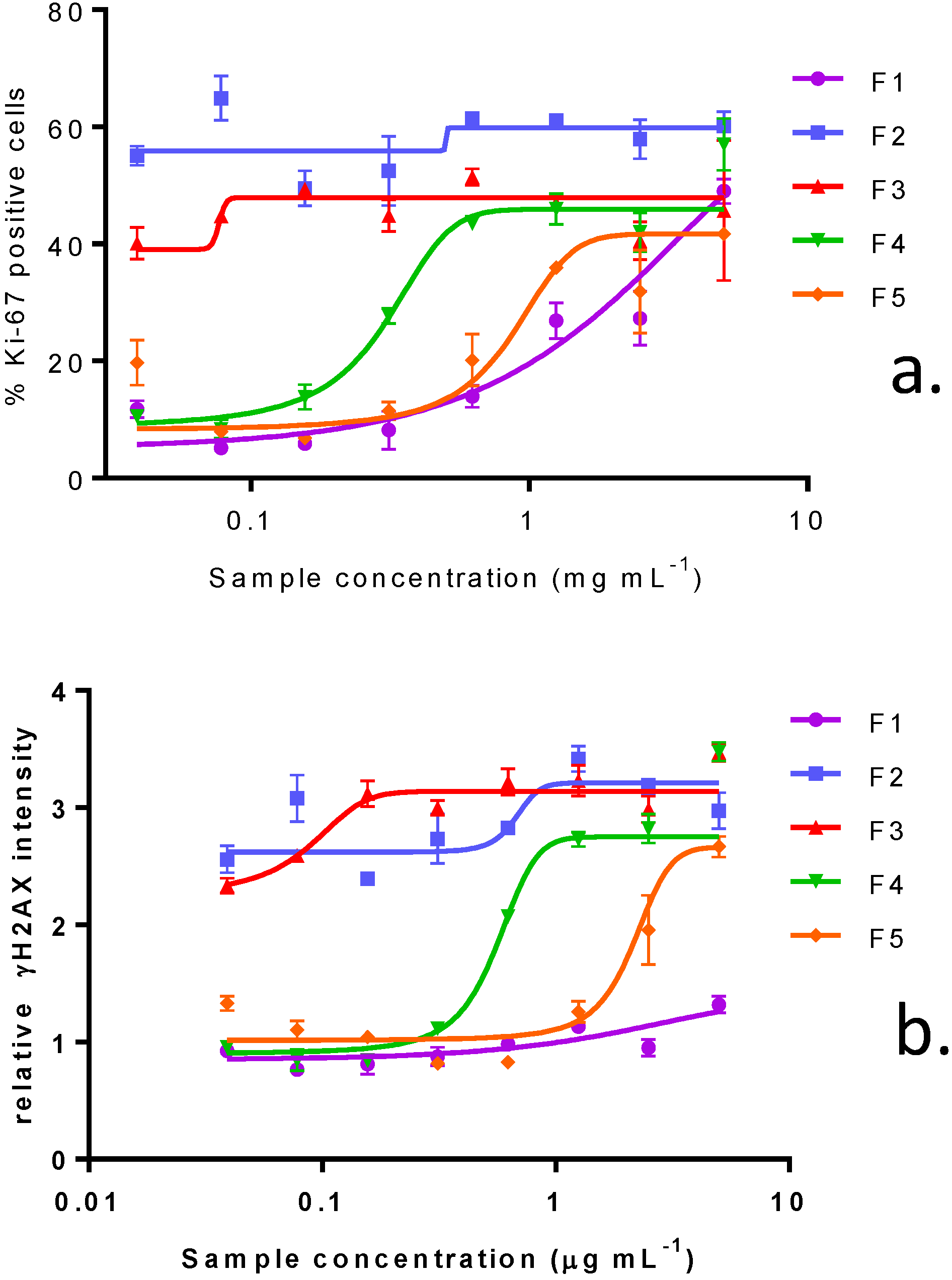
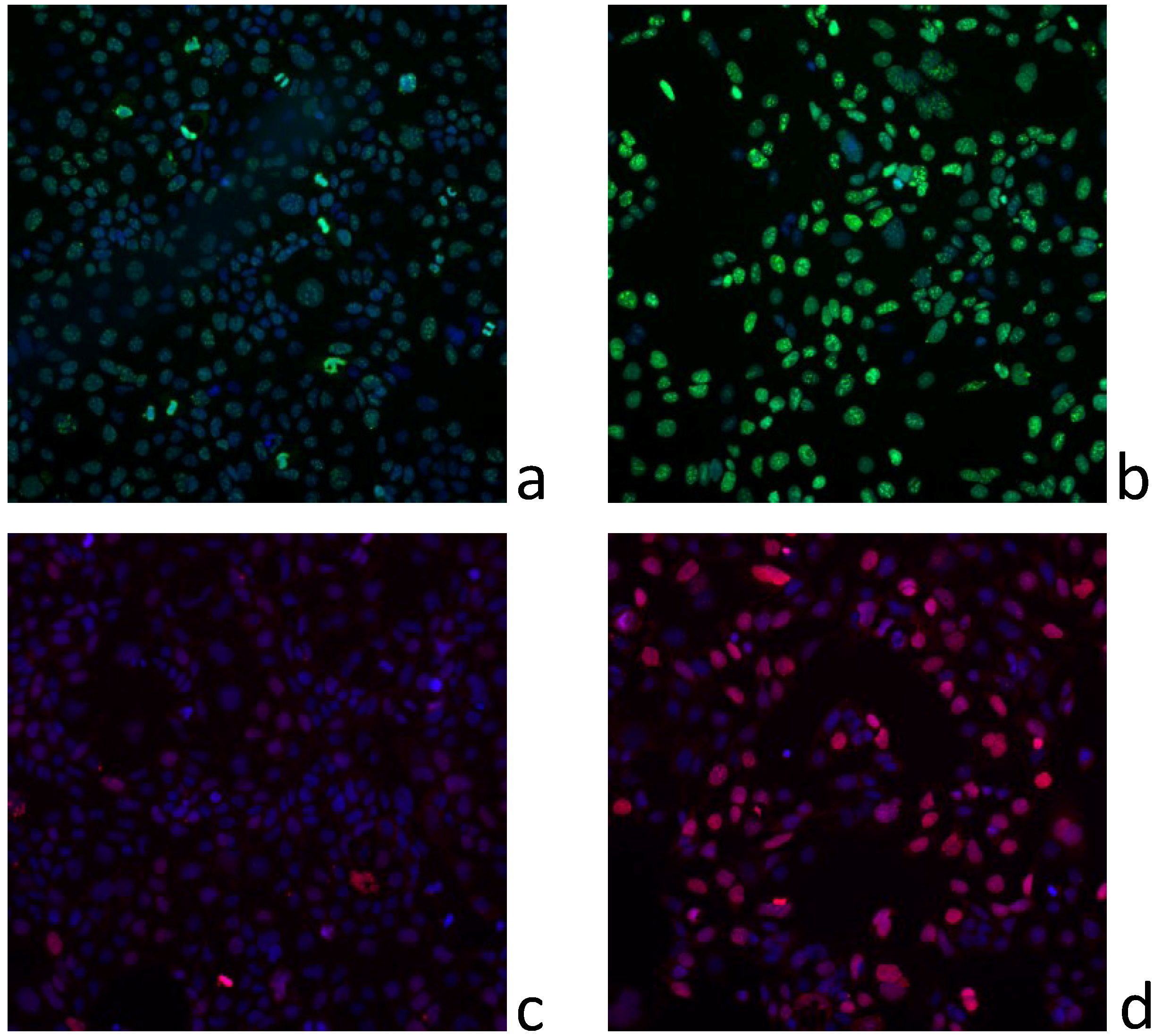
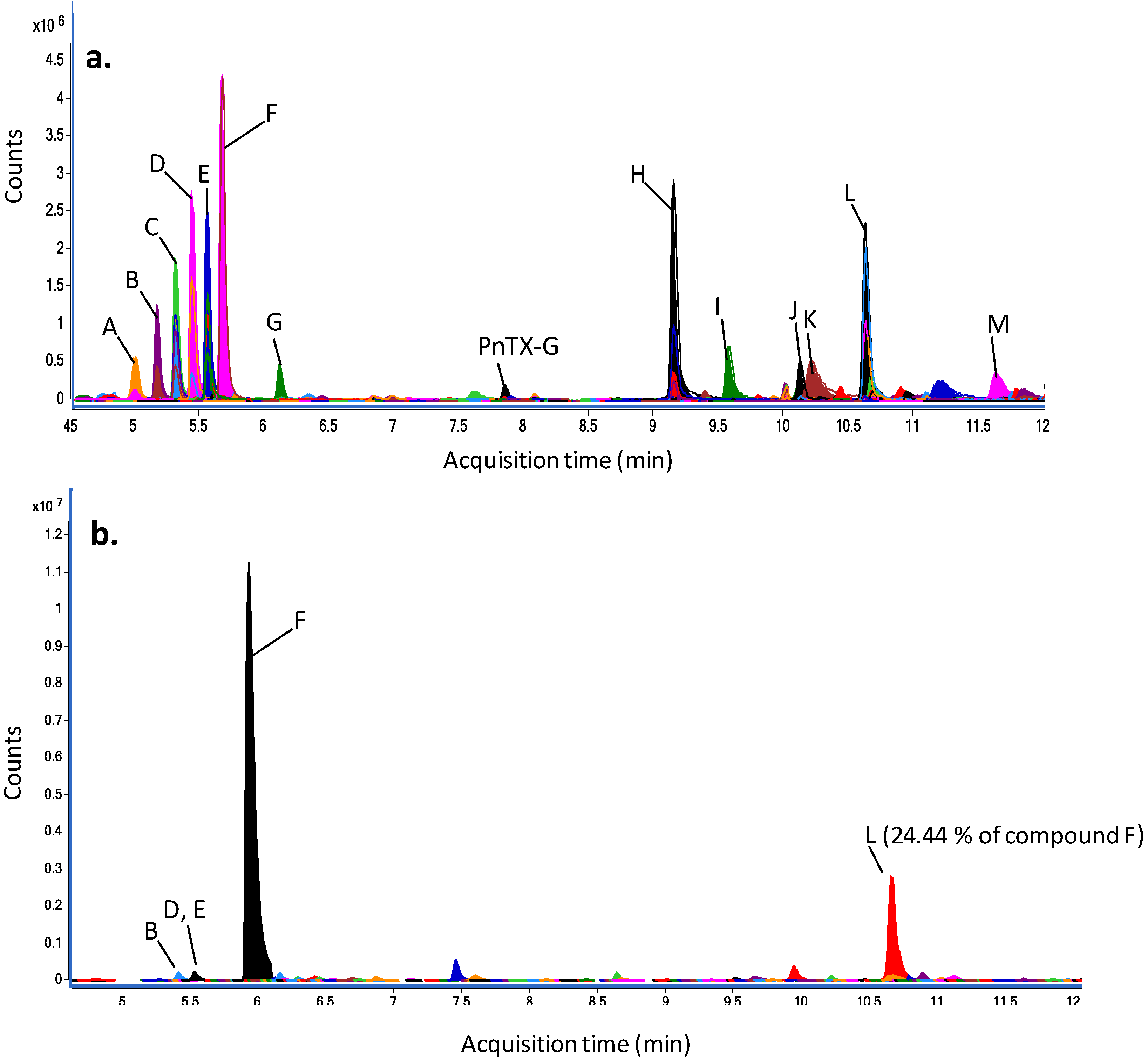
2.5. Dereplication Study
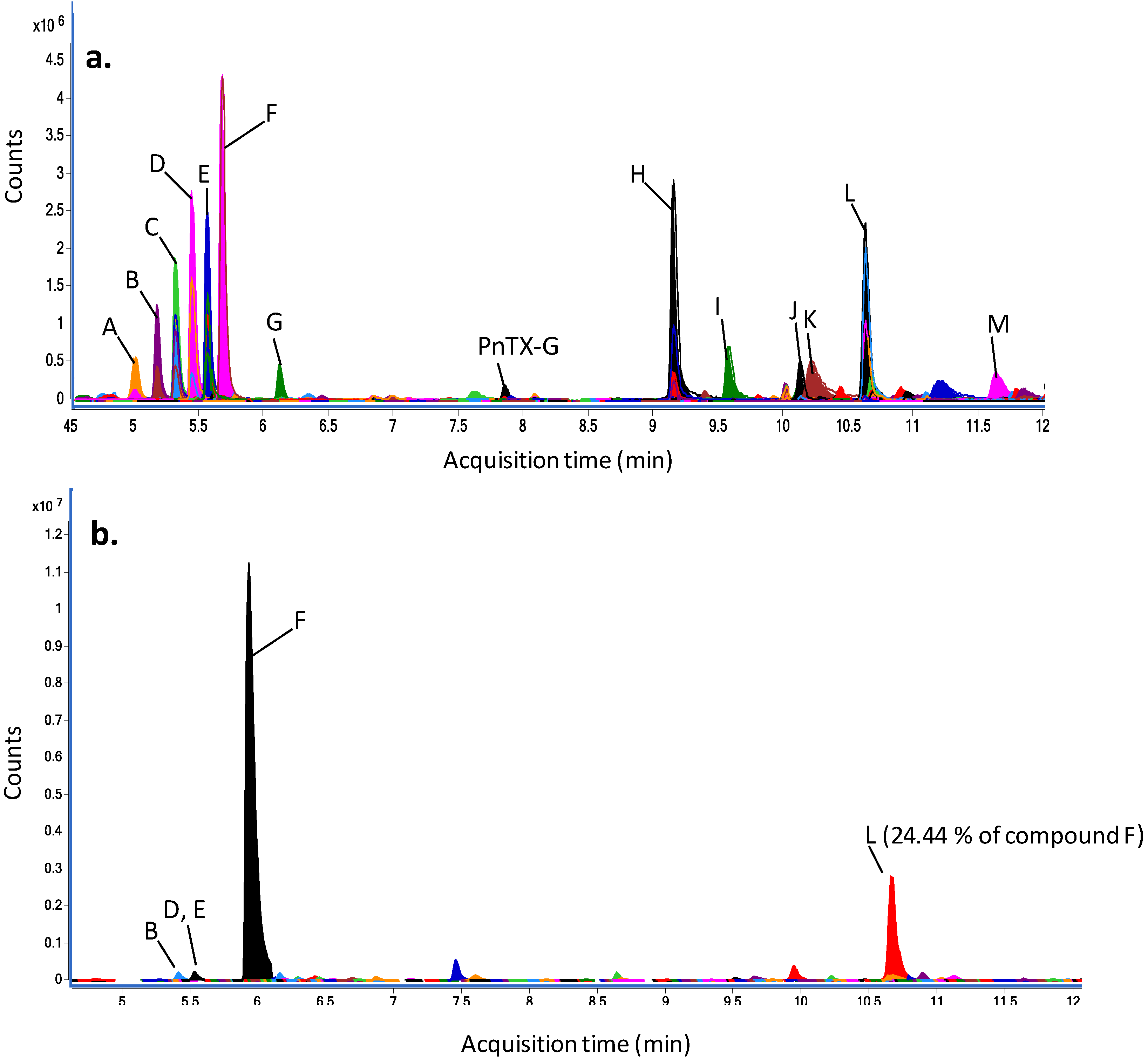
| Peak | Experimental m/z [M + H]+ | Relative abundance of compound F (%) | Putative annotation using MarinLit database | Score (DB) | Diff (DB, ppm) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Mean ( N = 3) | SD | Mean ( N = 3) | SD | Mean ( N = 3) | SD | Mean ( N = 3) | SD | ||
| A | 393.2102 | 0.0001 | 16.74 | 0.72 | unknown | - | - | - | - |
| B | 437.2362 | 0.0002 | 26.65 | 0.62 | unknown | - | - | - | - |
| 432.2810 | 0.0002 | 9.97 | 0.47 | unknown | - | - | - | - | |
| C | 481.2622 | 0.0001 | 38.61 | 1.04 | unknown | - | - | - | - |
| 476.3067 | 0.0001 | 18.21 | 1.49 | unknown | - | - | - | - | |
| D | 520.3331 | 0.0002 | 33.91 | 0.50 | unknown | - | - | - | - |
| 525.2886 | 0.0002 | 59.92 | 0.90 | unknown | - | - | - | - | |
| E | 564.3593 | 0.0001 | 36.13 | 3.03 | unknown | - | - | - | - |
| 569.3147 | 0.0001 | 59.10 | 5.14 | unknown | - | - | - | - | |
| F | 613.3408 | 0.0003 | 13.70 | 5.11 | unknown | - | - | - | - |
| 402.2281 | 0.0003 | 100.00 | 100.00 | Nakijiquinone A | 98.14 | 1.34 | −1.38 | 0.56 | |
| N-carboxy-methyl-smenospongine | |||||||||
| Stachybotrin | |||||||||
| G | 370.2748 | 0.0000 | 10.83 | 0.23 | N-methyl-xestamine D | 99.44 | 0.03 | −0.84 | 0.02 |
| PnTX-G | 694.4664 | 0.0003 | 1.06 | 0.06 | - | - | - | - | - |
| H | 343.2958 | 0.0001 | 66.03 | 1.95 | unknown | - | - | - | - |
| 240.2327 | 0.0001 | 8.74 | 0.50 | 2-hydroxypentadecanoic acid | 98.13 | 0.44 | −1.60 | 0.34 | |
| I | 272.2589 | 0.0001 | 20.50 | 0.70 | 2-amino-1,3,4-hexadecanetriol; (2 S,3R,4R)-form | 99.11 | 0.21 | −1.18 | 0.14 |
| J | 692.3843 | 0.0003 | 4.04 | 0.38 | unknown | - | - | - | - |
| K | 286.1444 | 0.0002 | 11.69 | 0.80 | Solanapyrone B; 7ct-Hydroxy, 4′-demethoxy, 4′-amino, 1-aldehyde | 99.03 | 0.71 | −1.12 | 0.53 |
| L | 453.1680 | 0.0001 | 61.23 | 0.14 | 7,11-dihydroxy-16-oxo-12-spongien-17-al; (713,1113)-form, Di-Ac | 97.96 | 0.94 | −1.44 | 0.36 |
| Furcellataepoxylactone | |||||||||
| Branacenal | |||||||||
| 288.2538 | 0.0001 | 9.02 | 0.52 | unknown | - | - | - | - | |
| M | 402.3575 | 0.0001 | 6.97 | 0.28 | unknown | - | - | - | - |
3. Discussion
4. Experimental Section
4.1. Reagents
4.2. Vulcanodinium rugosum Growth Conditions
4.3. Extraction, Purification and Fractionation of a Crude Extract of V. rugosum
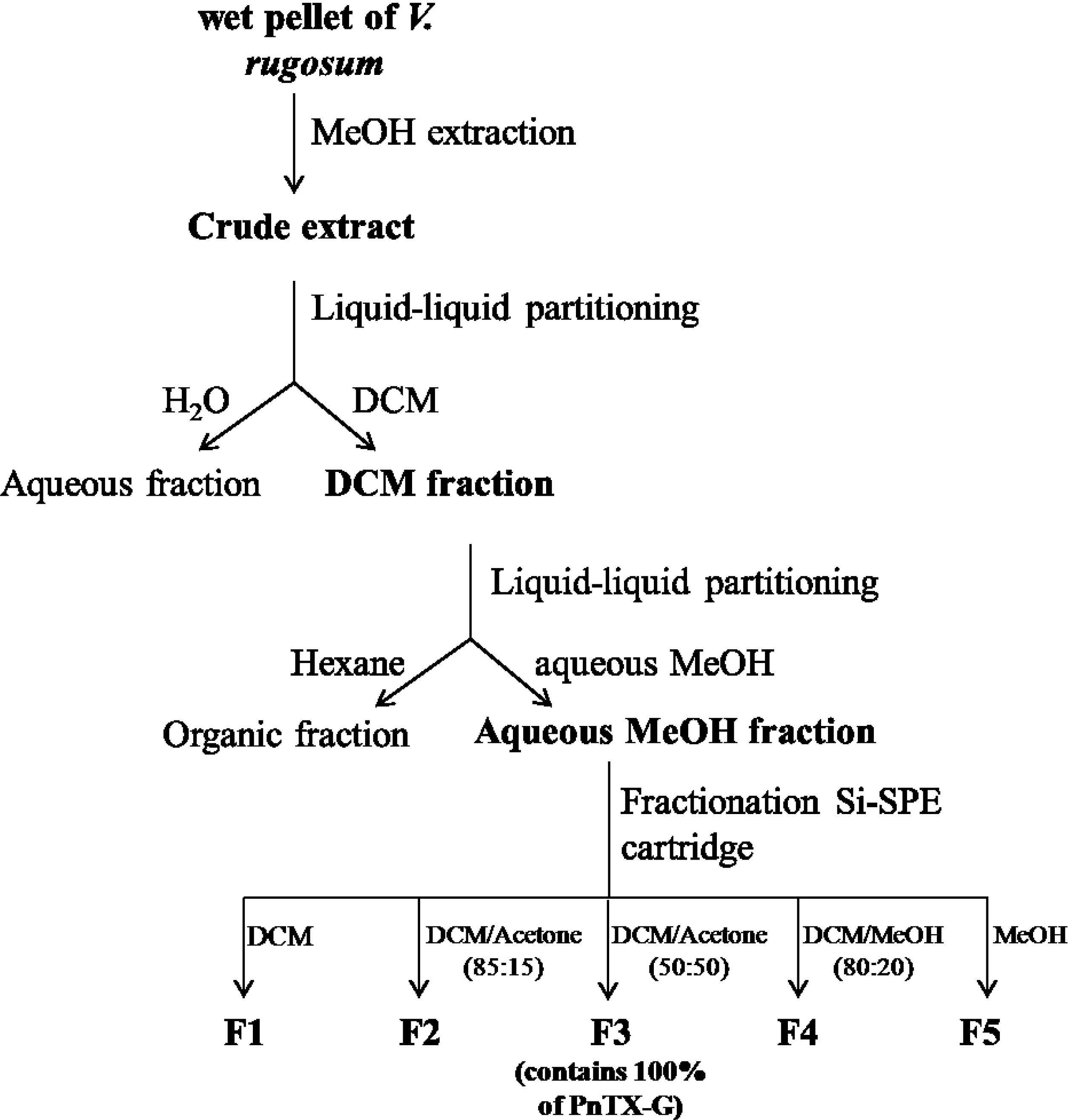
4.4. Quantification of PnTX-G Using a Triple Stage Quadrupole Mass Spectrometer
4.5. Growth Conditions of the Different Cell Lines
4.6. Cytotoxicity Assays
4.7. Assessment of the Cell Viability
4.8. IC50 Determination
4.9. DNA Damage and Cell Cycle Progression Measurements
4.10. Dereplication Study Using a Quadrupole-Time-of-Flight Hybrid Mass Spectrometer
5. Conclusions
Acknowledgements
Conflicts of Interest
References
- Zheng, S.; Huang, F.; Chen, S.; Tan, X.; Zuo, J.; Peng, J.; Xie, R. The isolation and bioactivities of pinnatoxin. Chin. J. Mar. Drugs 1990, 9, 33–35. [Google Scholar]
- Uemura, D.; Chou, T.; Haino, T.; Nagatsu, A.; Fukuzawa, S.; Zheng, S.Z.; Chen, H.S. Pinnatoxin-A a toxic amphoteric macrocycle from the okinawan bivalve Pinna muricata. J. Am. Chem. Soc. 1995, 117, 1155–1156. [Google Scholar] [CrossRef]
- Chou, T.; Haino, T.; Kuramoto, M.; Uemura, D. Isolation and structure of pinnatoxin D, a new shellfish poison from the okinawan bivalve Pinna muricata. Tetrahedron Lett. 1996, 37, 4027–4030. [Google Scholar] [CrossRef]
- Takada, N.; Umemura, N.; Suenaga, K.; Chou, T.; Nagatsu, A.; Haino, T.; Yamada, K.; Uemura, D. Pinnatoxins B and C, the most toxic components in the pinnatoxin series from the okinawan bivalve Pinna muricata. Tetrahedron Lett. 2001, 42, 3491–3494. [Google Scholar] [CrossRef]
- Selwood, A.I.; Miles, C.O.; Wilkins, A.L.; van Ginkel, R.; Munday, R.; Rise, F.; McNabb, P. Isolation, structural determination and acute toxicity of pinnatoxins E, F and G. J. Agric. Food Chem. 2010, 58, 6532–6542. [Google Scholar]
- Rundberget, T.; Aasen, J.A.B.; Selwood, A.I.; Miles, C.O. Pinnatoxins and spirolides in norwegian blue mussels and seawater. Toxicon 2011, 58, 700–711. [Google Scholar] [CrossRef]
- Rhodes, L.; Smith, K.; Selwood, A.; McNabb, P.; van Ginkel, R.; Holland, P.; Munday, R. Production of pinnatoxins by a peridinoid dinoflagellate isolated from Northland, New Zealand. Harmful Algae 2010, 9, 384–389. [Google Scholar] [CrossRef]
- Smith, K.F.; Rhodes, L.L.; Suda, S.; Selwood, A.I. A dinoflagellate producer of pinnatoxin G, isolated from sub-tropical Japanese waters. Harmful Algae 2011, 10, 702–705. [Google Scholar] [CrossRef]
- Rhodes, L.; Smith, K.; Selwood, A.; McNabb, P.; Molenaar, S.; Munday, R.; Wilkinson, C.; Hallegraeff, G. Production of pinnatoxins E, F and G by scrippsielloid dinoflagellates isolated from Franklin Harbour, South Australia. N. Z. J. Mar. Freshw. Res. 2011, 45, 703–709. [Google Scholar] [CrossRef]
- Nézan, E.; Chomerat, N. Vulcanodinium rugosum gen. Nov., sp. Nov. (dinophyceae): A new marine dinoflagellate from the French mediterranean coast. Cryptogam. Algol. 2011, 32, 3–18. [Google Scholar]
- Hess, P.; Abadie, E.; Hervé, F.; Berteaux, T.; Séchet, V.; Aráoz, R.; Molgó, J.; Zakarian, A.; Sibat, M.; Rundberget, T.; et al. Pinnatoxin G is responsible for atypical toxicity in mussels (Mytilus galloprovincialis) and clams (Venerupis decussata) from Ingril, a French mediterranean lagoon. Toxicon 2013. [Google Scholar] [CrossRef]
- Rhodes, L.; Smith, K.; Selwood, A.; McNabb, P.; Munday, R.; Suda, S.; Molenaar, S.; Hallegraeff, G. Dinoflagellate Vulcanodinium rugosum identified as the causative organism of pinnatoxins in Australia, New Zealand and Japan. Phycologia 2011, 50, 624–628. [Google Scholar] [CrossRef]
- McCarron, P.; Rourke, W.A.; Hardstaff, W.; Pooley, B.; Quilliam, M.A. Identification of pinnatoxins and discovery of their fatty acid ester metabolites in mussels (Mytilus edulis) from eastern canada. J. Agric. Food Chem. 2012, 60, 1437–1446. [Google Scholar] [CrossRef]
- European Food Safety Authority. Opinion of the scientific panel on contaminants in the food chain on a request from the european commission on marine biotoxins in shellfish–cyclic imines (spirolides, gymnodimines, pinnatoxins and pteriatoxins). Eur. Food Saf. Auth. J. 2010, 8, 1628.
- Cembella, A.; Krock, B. Cyclic Imine Toxins: Chemistry, Biogeography, Biosynthesis, and Pharmacology. In Seafood and Freshwater Toxins—Pharmacology, Physiology and Detection, 2nd ed.; Botana, L.M., Ed.; CRC Press Taylor & Francis Group: Boca Raton, FL, USA, 2008; p. 941. [Google Scholar]
- Munday, R.; Selwood, A.I.; Rhodes, L. Acute toxicity of pinnatoxins E, F and G to mice. Toxicon 2012, 60, 995–999. [Google Scholar] [CrossRef]
- Jackson, J.J.; Stivala, C.E.; Iorga, B.I.; Molgo, J.; Zakarian, A. Stability of cyclic imine toxins: Interconversion of pinnatoxin amino ketone and pinnatoxin a in aqueous media. J. Org. Chem. 2012, 77, 10435–10440. [Google Scholar] [CrossRef]
- Kuramoto, M.; Arimoto, H.; Uemura, D. Bioactive alkaloids from the sea: A review. Mar. Drugs 2004, 2, 39–54. [Google Scholar] [CrossRef]
- Araoz, R.; Servent, D.; Molgo, J.; Iorga, B.I.; Fruchart-Gaillard, C.; Benoit, E.; Gu, Z.; Stivala, C.; Zakarian, A. Total synthesis of pinnatoxins A and G and revision of the mode of action of pinnatoxin A. J. Am. Chem. Soc. 2011, 133, 10499–10511. [Google Scholar] [CrossRef]
- Hellyer, S.D.; Selwood, A.I.; Rhodes, L.; Kerr, D.S. Marine algal pinnatoxins E and F cause neuromuscular block in an in vitro hemidiaphragm preparation. Toxicon 2011, 58, 693–699. [Google Scholar] [CrossRef]
- Marine Literature Database; Version 13.5; Marine Chemistry Group, Department of Chemistry, University of Canterbury: Christchurch, New Zealand, 2007.
- Munday, R. Toxicology of the Pectenotoxins. In Seafood and Freshwater Toxins–Pharmacology, Physiology and Detection, 2nd ed.; Botana, L.M., Ed.; CRC Press Taylor&Francis Group: Boca Raton, FL, USA, 2008; pp. 371–380. [Google Scholar]
- Kondracki, M.L.; Guyot, M. Smenospongine: A cytotoxic and antimicrobial aminoquinone isolated from Smenospongia sp. Tetrahedron Lett. 1987, 28, 5815–5818. [Google Scholar] [CrossRef]
- Shigemori, H.; Madono, T.; Sasaki, T.; Mikami, Y.; Kobayashi, J. Nakijiquinones A and B, new antifungal sesquiterpenoid quinones with an amino acid residue from an Okinawan marine sponge. Tetrahedron 1994, 50, 8347–8354. [Google Scholar] [CrossRef]
- Xu, X.; de Guzman, F.S.; Gloer, J.B. Stachybotrins A and B: Novel bioactive metabolites from a brackish water isolate of the fungus Stachybotrys sp. J. Org. Chem. 1992, 57, 6700–6703. [Google Scholar] [CrossRef]
- Stahl, P.; Kissau, L.; Mazitschek, R.; Huwe, A.; Furet, P.; Giannis, A.; Waldmann, H. Total synthesis and biological evaluation of the nakijiquinones. J. Am. Chem. Soc. 2001, 123, 11586–11593. [Google Scholar]
- Gordaliza, M. Cytotoxic terpene quinones from marine sponges. Mar. Drugs 2010, 8, 2849–2870. [Google Scholar] [CrossRef]
- Aoki, S.; Kong, D.; Matsui, K.; Rachmat, R.; Kobayashi, M. Sesquiterpene aminoquinones, from a marine sponge, induce erythroid differentiation in human chronic myelogenous leukemia, K562 cells. Chem. Pharm. Bull. 2004, 52, 935–937. [Google Scholar] [CrossRef]
- Kong, D.; Yamori, T.; Kobayashi, M.; Duan, H. Antiproliferative and antiangiogenic activities of smenospongine, a marine sponge sesquiterpene aminoquinone. Mar. Drugs 2011, 9, 154–161. [Google Scholar] [CrossRef]
- Kong, D.; Aoki, S.; Sowa, Y.; Sakai, T.; Kobayashi, M. Smenospongine, a sesquiterpene aminoquinone from a marine sponge, induces G1 arrest or apoptosis in different leukemia cells. Mar. Drugs 2008, 6, 480–488. [Google Scholar]
- Mayer, A.M.S.; Rodríguez, A.D.; Berlinck, R.G.S.; Hamann, M.T. Marine pharmacology in 2005–2006: Marine compounds with anthelmintic, antibacterial, anticoagulant, antifungal, anti-inflammatory, antimalarial, antiprotozoal, antituberculosis, and antiviral activities; affecting the cardiovascular, immune and nervous systems, and other miscellaneous mechanisms of action. Biochim. Biophys. Acta 2009, 1790, 283–308. [Google Scholar] [CrossRef]
- Guillard, R.R.L.; Hargraves, P.E. Stichochrysis immobilis is a diatom, not a chrysophyte. Phycologia 1993, 32, 234–236. [Google Scholar] [CrossRef]
- Ledreux, A.; Sérandour, A.-L.; Morin, B.; Derick, S.; Lanceleur, R.; Hamlaoui, S.; Furger, C.; Biré, R.; Krys, S.; Fessard, V. Collaborative study for the detection of toxic compounds in shellfish extracts using cell-based assays. Part II: Application to shellfish extracts spiked with lipophilic marine toxins. Anal. Bioanal. Chem. 2012, 403, 1995–2007. [Google Scholar] [CrossRef]
- Sérandour, A.L.; Ledreux, A.; Morin, B.; Derick, S.; Augier, E.; Lanceleur, R.; Hamlaoui, S.; Moukha, S.; Furger, C.; Bire, R.; et al. Collaborative study for the detection of toxic compounds in shellfish extracts using cell-based assays. Part I: Screening strategy and pre-validation study with lipophilic marine toxins. Anal. Bioanal. Chem. 2012, 403, 1983–1993. [Google Scholar] [CrossRef]
- Amzil, Z.; Pouchus, Y.F.; Le, B.J.; Roussakis, C.; Verbist, J.F.; Marcaillou-Lebaut, C.; Masselin, P. Short-time cytotoxicity of mussel extracts: A new bioassay for okadaic acid detection. Toxicon 1992, 30, 1419–1425. [Google Scholar] [CrossRef]
- Tubaro, A.; Florio, C.; Luxich, E.; Vertua, R.; Loggia, R.D.; Yasumoto, T. Suitability of the MTT-based cytotoxicity assay to detect okadaic acid contamination of mussels. Toxicon 1996, 34, 965–974. [Google Scholar] [CrossRef]
- Sana, T.R.; Roark, J.C.; Li, X.; Waddell, K.; Fischer, S.M. Molecular formula and metlin personal metabolite database matching applied to the identification of compounds generated by LC/TOF-MS. J. Biomol. Tech. 2008, 19, 258–266. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Geiger, M.; Desanglois, G.; Hogeveen, K.; Fessard, V.; Leprêtre, T.; Mondeguer, F.; Guitton, Y.; Hervé, F.; Séchet, V.; Grovel, O.; et al. Cytotoxicity, Fractionation and Dereplication of Extracts of the Dinoflagellate Vulcanodinium rugosum, a Producer of Pinnatoxin G. Mar. Drugs 2013, 11, 3350-3371. https://doi.org/10.3390/md11093350
Geiger M, Desanglois G, Hogeveen K, Fessard V, Leprêtre T, Mondeguer F, Guitton Y, Hervé F, Séchet V, Grovel O, et al. Cytotoxicity, Fractionation and Dereplication of Extracts of the Dinoflagellate Vulcanodinium rugosum, a Producer of Pinnatoxin G. Marine Drugs. 2013; 11(9):3350-3371. https://doi.org/10.3390/md11093350
Chicago/Turabian StyleGeiger, Marie, Gwenaëlle Desanglois, Kevin Hogeveen, Valérie Fessard, Thomas Leprêtre, Florence Mondeguer, Yann Guitton, Fabienne Hervé, Véronique Séchet, Olivier Grovel, and et al. 2013. "Cytotoxicity, Fractionation and Dereplication of Extracts of the Dinoflagellate Vulcanodinium rugosum, a Producer of Pinnatoxin G" Marine Drugs 11, no. 9: 3350-3371. https://doi.org/10.3390/md11093350
APA StyleGeiger, M., Desanglois, G., Hogeveen, K., Fessard, V., Leprêtre, T., Mondeguer, F., Guitton, Y., Hervé, F., Séchet, V., Grovel, O., Pouchus, Y.-F., & Hess, P. (2013). Cytotoxicity, Fractionation and Dereplication of Extracts of the Dinoflagellate Vulcanodinium rugosum, a Producer of Pinnatoxin G. Marine Drugs, 11(9), 3350-3371. https://doi.org/10.3390/md11093350