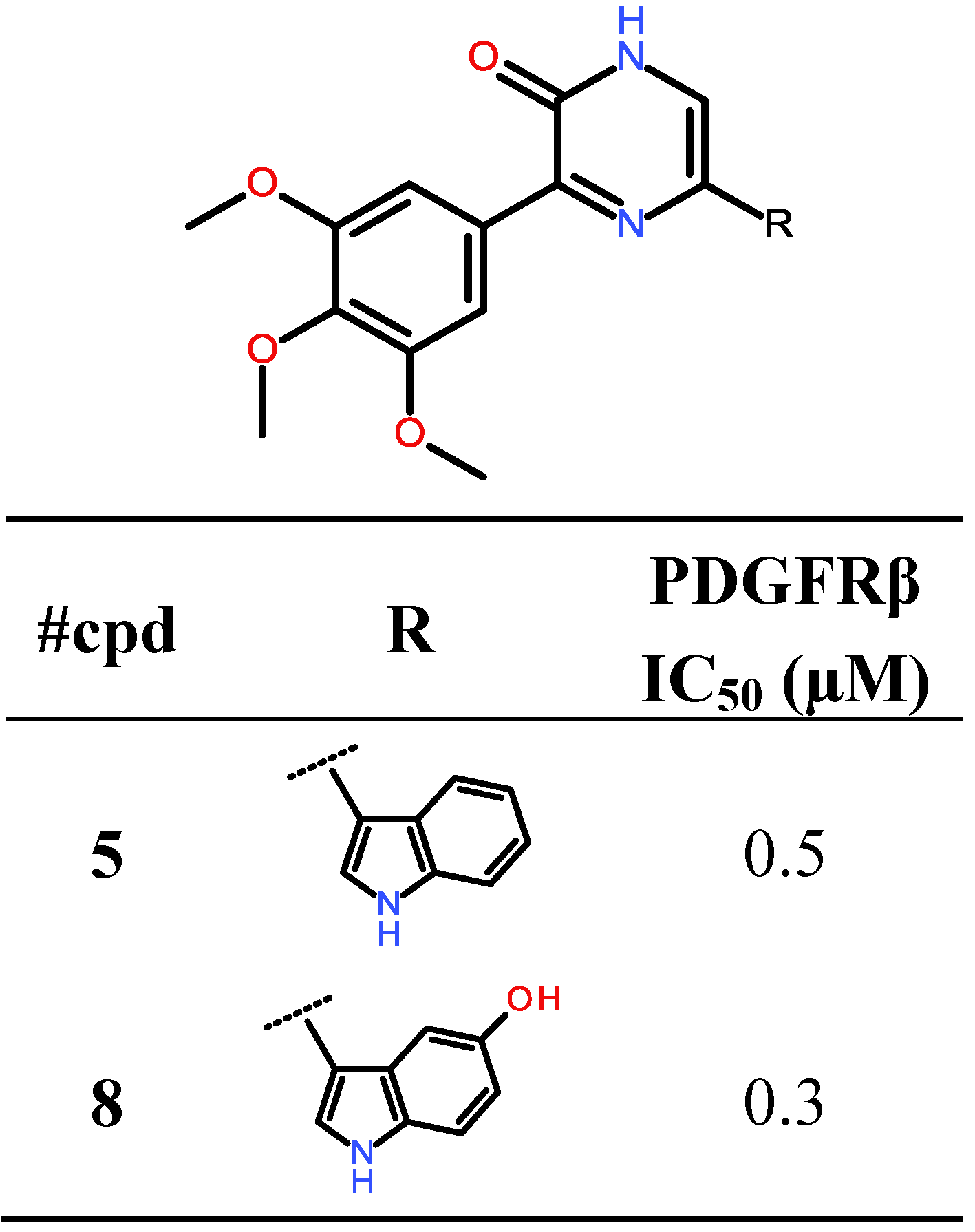
Marine Derived Hamacanthins as Lead for the Development of Novel PDGFRβ Protein Kinase Inhibitors

Abstract
:1. Introduction
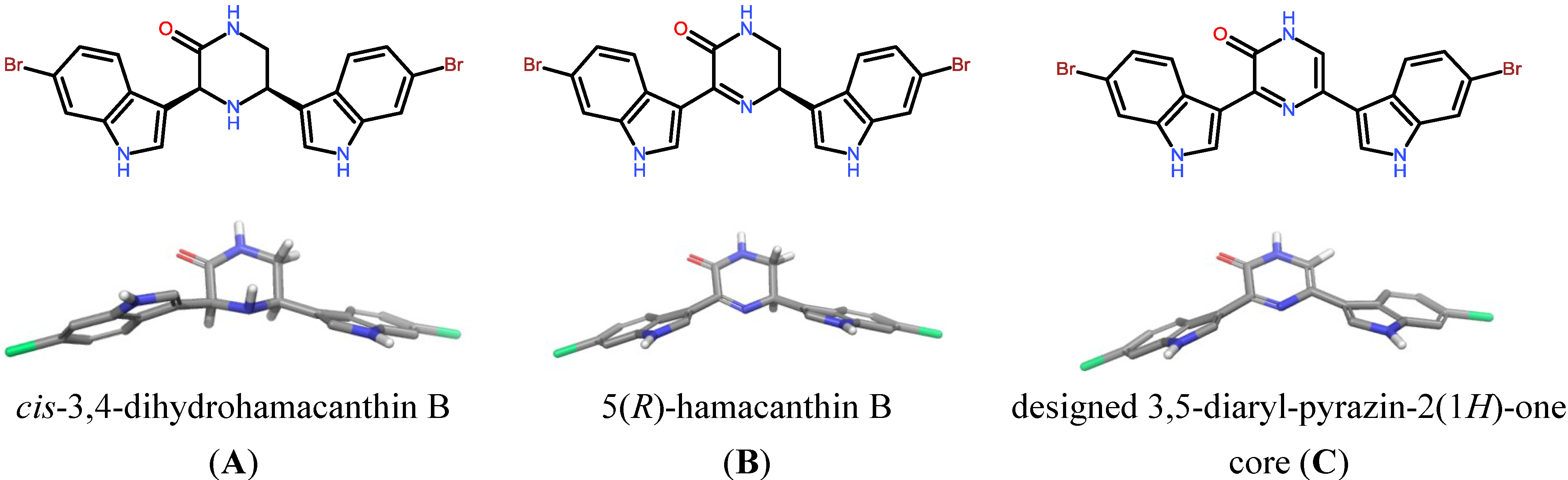

2. Results and Discussion
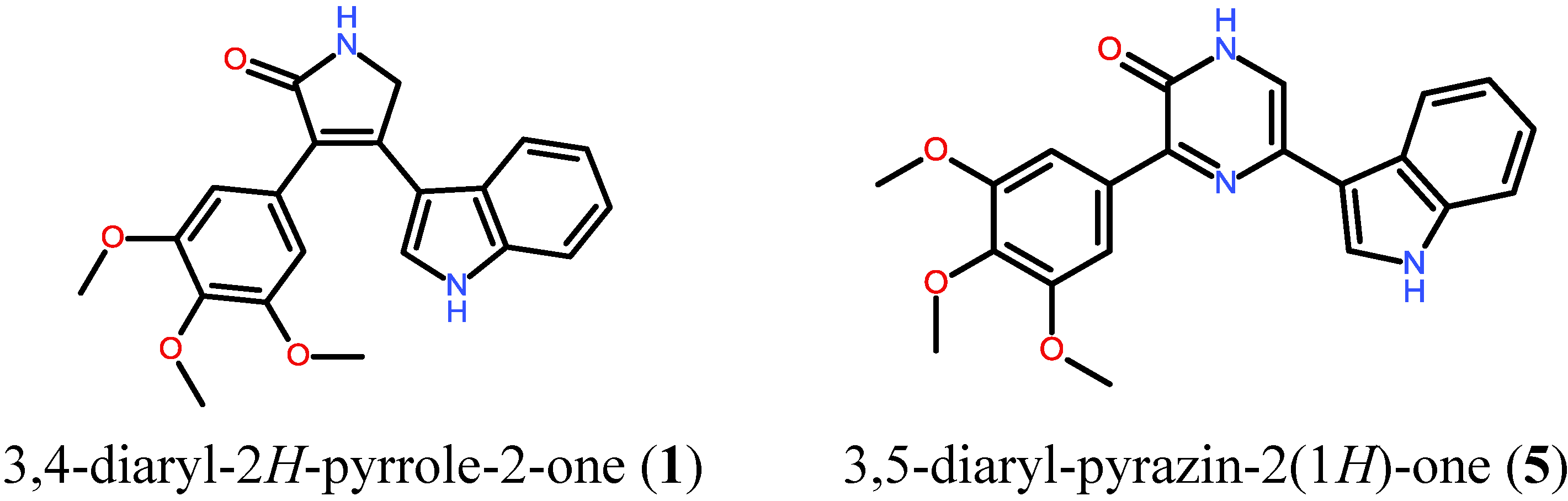
2.1. Synthesis of Designed Compound 5
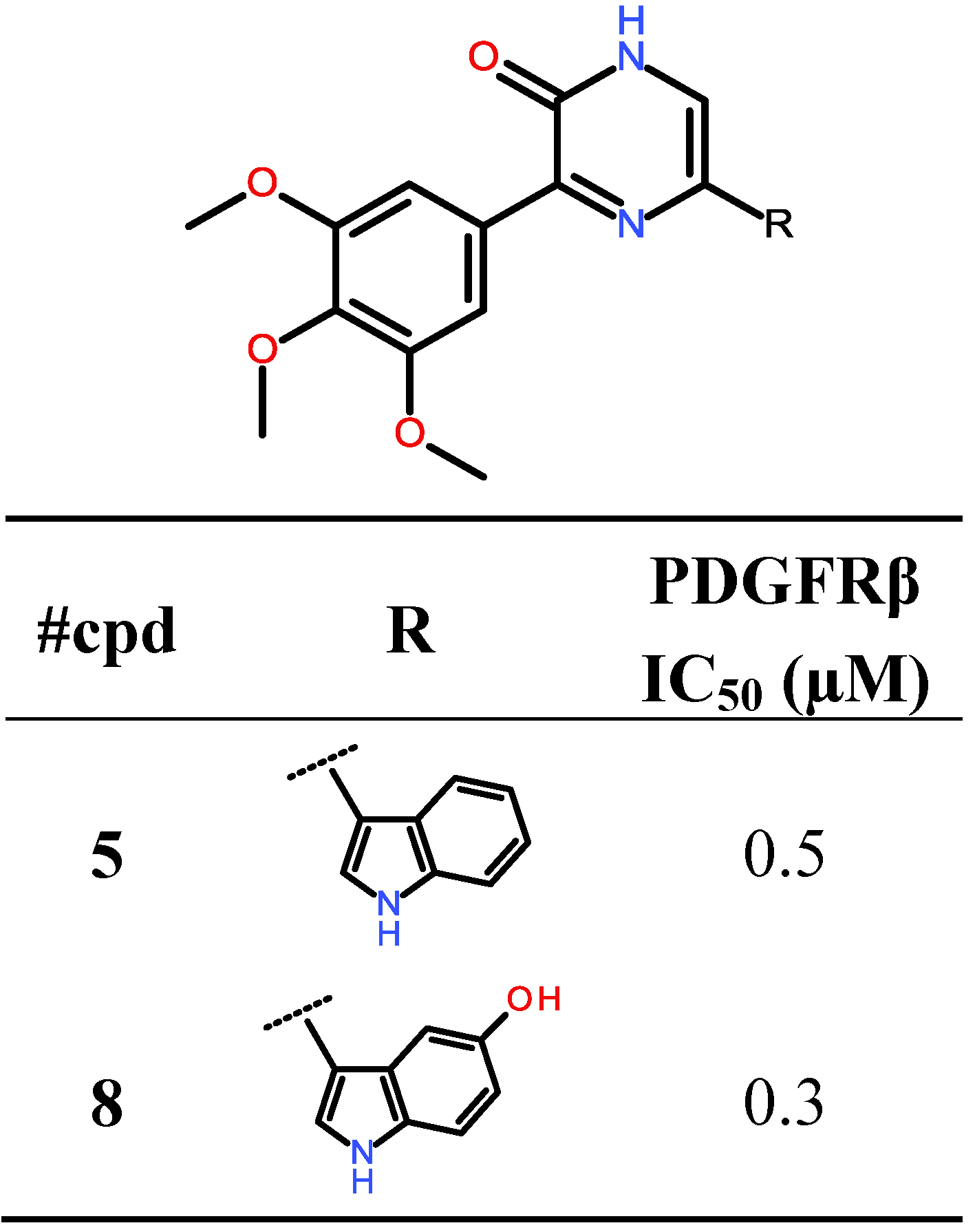
2.2. Biological Evaluation: Activity against PKs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein kinase | IC50 (µM) for Compound 1 | IC50 (µM) for Compound 5 |
|---|---|---|
| AKT1 | - | - |
| ARK5 | 16 | - |
| Aurora-A | 46 | - |
| Aurora-B | 66 | - |
| B-RAF-VE | - | - |
| CDK2/CycA | 66 | - |
| CDK4/CycD1 | 56 | - |
| COT | 32 | - |
| EGFR | 23 | - |
| EPHB4 | 53 | - |
| ERBB2 | 51 | - |
| FAK | 9 | - |
| IGF1R | 22 | - |
| SRC | 14 | - |
| VEGF-R2 | 0.031 | 4 |
| VEGF-R3 | 0.037 | 5 |
| FLT3 | 61 | 36 |
| INSR | 43 | - |
| MET | 60 | - |
| PDGFRβ | 11 | 0.5 |
| PLK1 | - | - |
| SAK | 10 | - |
| TIE2 | 5 | - |
| CK2a1 | - | - |
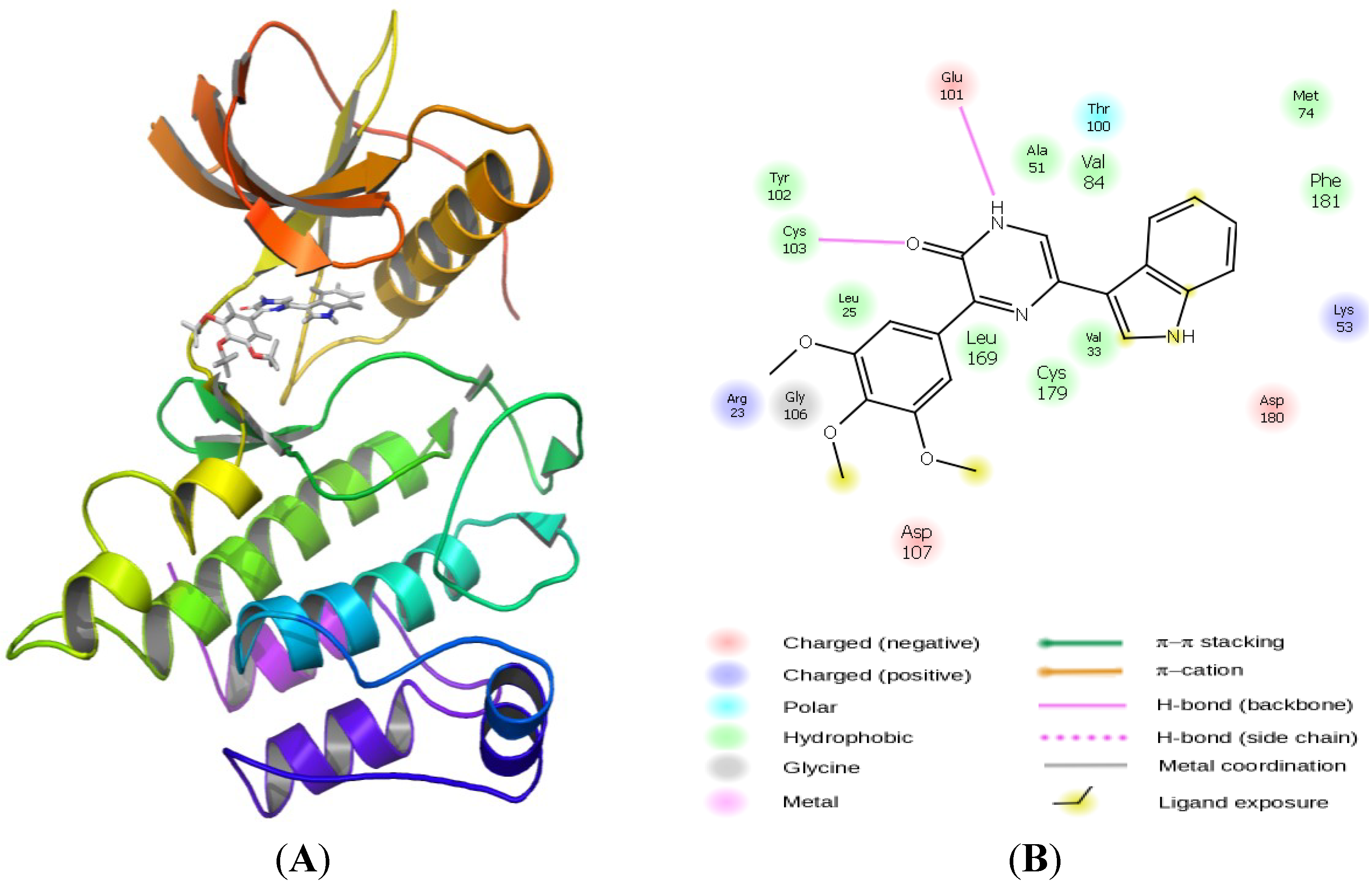
2.3. Molecular Modeling


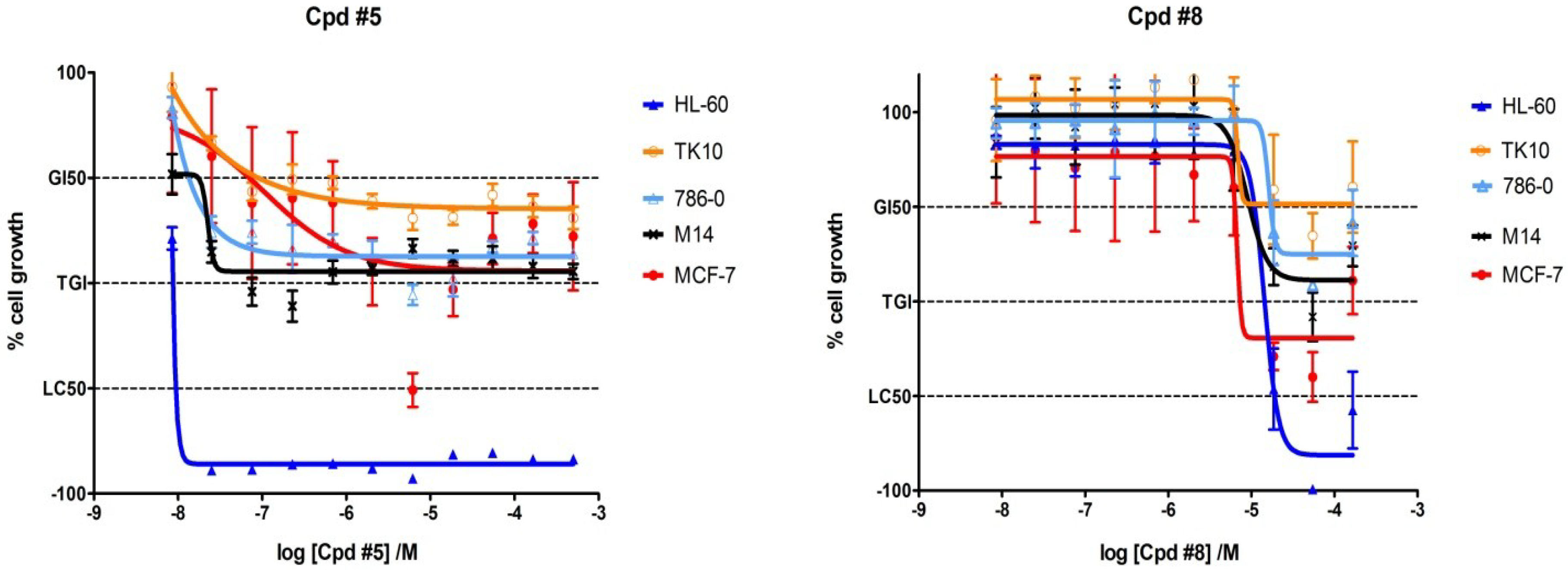
2.4. Biological Activity in Cancer Cell Lines of Compounds 5 and 8
3. Experimental Section
3.1. Chemistry and Synthesis of Test Compounds
3.1.1. Synthesis of Compound 3
3.1.2. Synthesis of Compound 4
3.1.3. Synthesis of Compound 5
3.1.4. Synthesis of Compound 6
3.1.5. Synthesis of Compound 7
3.1.6. Synthesis of Compound 8a
3.1.7. Synthesis of Compound 8
3.2. Molecular Modeling
3.3. Biological Evaluation
3.3.1. Selectivity Profiling of Compounds by IC50 Values Using 24 Protein Kinases
3.3.2. Cell Culture and Proliferative Assays using HL-60, TK 10, 786-0, M 14, and MCF-7 Cells
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Haefner, B. Drugs from the deep: Marine natural products as drug candidates. Drug Discov. Today 2003, 8, 536–544. [Google Scholar] [CrossRef]
- Gupta, L.; Talwar, A.; Chauhan, P.M. Bis and tris indole alkaloids from marine organisms: New leads for drug discovery. Curr. Med. Chem. 2007, 14, 1789–1803. [Google Scholar] [CrossRef]
- Skropeta, D.; Pastro, N.; Zivanovic, A. Kinase inhibitors from marine sponges. Mar. Drugs 2011, 9, 2131–2154. [Google Scholar] [CrossRef]
- Kouko, T.; Matsumura, K.; Kawasaki, T. Total synthesis of marine bisindole alkaloids, (+)-hamacanthins A, B and (−)-antipode of cis-dihydrohamacanthin B. Tetrahedron 2005, 61, 2309–2318. [Google Scholar] [CrossRef]
- Bao, B.; Sun, Q.; Yao, X.; Hong, J.; Lee, C.-O.; Cho, H.Y.; Jung, J.H. Bisindole alkaloids of the topsentin and hamacanthin classes from a marine sponge Spongosorites sp. J. Nat. Prod. 2007, 70, 2–8. [Google Scholar] [CrossRef]
- Casapullo, A.; Bifulco, G.; Bruno, I.; Riccio, R. New bisindole alkaloids of the topsentin and hamacanthin classes from the Mediterranean marine sponge Rhaphisia lacazei. J. Nat. Prod. 2000, 63, 447–451. [Google Scholar] [CrossRef]
- Peifer, C.; Bühler, S.; Hauser, D.; Kinkel, K.; Totzke, F.; Schaechtele, C.; Laufer, S. Design, synthesis and characterization of N9/N7-substituted 6-aminopurines as VEGF-R and EGF-R inhibitors. Eur. J. Med. Chem. 2009, 44, 1788–1793. [Google Scholar] [CrossRef]
- Peifer, C.; Kinkel, K.; Abadleh, M.; Schollmeyer, D.; Laufer, S. From five- to six-membered rings: 3,4-diarylquinolinone as lead for novel p38MAP kinase inhibitors. J. Med. Chem. 2007, 50, 1213–1221. [Google Scholar] [CrossRef]
- Peifer, C.; Krasowski, A.; Hämmerle, N.; Kohlbacher, O.; Dannhardt, G.; Totzke, F.; Schaechtele, C.; Laufer, S. Profile and molecular modeling of 3-(indole-3-yl)-4-(3,4,5-trimethoxyphenyl)-1H-pyrrole-2,5-dione(1) as a highly selective VEGF-R2/3 inhibitor. J. Med. Chem. 2006, 49, 7549–7553. [Google Scholar] [CrossRef]
- Peifer, C.; Selig, R.; Kinkel, K.; Ott, D.; Totzke, F.; Schaechtele, C.; Heidenreich, R.; Röcken, M.; Schollmeyer, D.; Laufer, S. Design, synthesis, and biological evaluation of novel 3-aryl-4-(1H-indole-3yl)-1,5-dihydro-2H-pyrrole-2-ones as vascular endothelial growth factor receptor (VEGF-R) inhibitors. J. Med. Chem. 2008, 51, 3814–3824. [Google Scholar] [CrossRef]
- Jiang, B.; Yang, C.G.; Wang, J. Enantioselective synthesis for the (−)-antipode of the pyrazinone marine alkaloid, hamacanthin A. J. Org. Chem. 2001, 66, 4865–4869. [Google Scholar] [CrossRef]
- Miyake, F.Y.; Yakushijin, K.; Horne, D.A. Synthesis of marine sponge bisindole alkaloids dihydrohamacanthins. Org. Lett. 2002, 4, 941–943. [Google Scholar] [CrossRef]
- Zoraghi, R.; Worrall, L.; See, R.H.; Strangman, W.; Popplewell, W.L.; Gong, H.; Samaai, T.; Swayze, R.D.; Kaur, S.; Vuckovic, M.; et al. Methicillin-resistant Staphylococcus aureus (MRSA) pyruvate kinase as a target for bis-indole alkaloids with antibacterial activities. J. Biol. Chem. 2011, 286, 44716–44725. [Google Scholar] [CrossRef]
- Caldwell, J.J.; Veillard, N.; Collins, I. Design and synthesis of 2(1H)-pyrazinones as inhibitors of protein kinases. Tetrahedron 2012, 68, 9713–9728. [Google Scholar] [CrossRef]
- Dar, A.C.; Shokat, K.M. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Ann. Rev. Biochem. 2011, 80, 769–795. [Google Scholar] [CrossRef]
- Appelmann, I.; Liersch, R.; Kessler, T.; Mesters, R.; Berdel, W. Angiogenesis inhibition in cancer therapy: Platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF) and their receptors: Biological functions and role in malignancy. Recent Results Cancer Res. 2010, 180, 51–81. [Google Scholar] [CrossRef]
- Koch, S.; Tugues, S.; Li, X.; Gualandi, L.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 2011, 437, 169–183. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, Y.; Tong, L.; Xie, H.; Wen, W.; Zhang, J.; Xi, Y.; Shen, Y.; Geng, M.; Wang, Y.; et al. AL3810, a multi-tyrosine kinase inhibitor, exhibits potent anti-angiogenic and anti-tumour activity via targeting VEGFR, FGFR and PDGFR. J. Cell. Mol. Med. 2012, 16, 2321–2330. [Google Scholar] [CrossRef]
- Noble, M.E.M.; Endicott, J.A.; Johnson, L.N. Protein kinase inhibitors: Insights into drug design from structure. Science 2004, 303, 1800–1805. [Google Scholar] [CrossRef]
- Norman, R.A.; Toader, D.; Ferguson, A.D. Structural approaches to obtain kinase selectivity. Trends Pharmacol. Sci. 2012, 33, 273–278. [Google Scholar] [CrossRef]
- Sachsenmaier, C.; Schachtele, C. Integrated technology platform protein kinases for drug development in oncology. Biotechniques 2002, 33, S101–S106. [Google Scholar]
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar]
- Hodous, B.L.; Geuns-Meyer, S.; Hughes, P.; Albrecht, B.; Bellon, S.; Bready, J.; Caenepeel, S.; Cee, V.; Chaffee, S.; Coxon, A.; et al. Evolution of a highly selective and potent 2-(pyridin-2-yl)-1,3,5-triazine tie-2 kinase inhibitor. J. Med. Chem. 2007, 50, 611–626. [Google Scholar] [CrossRef]
- Garuti, L.; Roberti, M.; Bottegoni, G. Non-ATP competitive protein kinase inhibitors. Curr. Med. Chem. 2010, 17, 2804–2821. [Google Scholar] [CrossRef]
- Peifer, C.; Stoiber, T.; Unger, E.; Totzke, F.; Schaechtele, C.; Marmé, D.; Brenk, R.; Klebe, G.; Schollmeyer, D.; Dannhardt, G. Design, synthesis, and biological evaluation of 3,4-diarylmaleimides as angiogenesis inhibitors. J. Med. Chem. 2006, 49, 1271–1281. [Google Scholar] [CrossRef]
- Reiterer, G.; Yen, A. Platelet-derived growth factor receptor regulates myeloid and monocytic differentiation of HL-60 cells. Cancer Res. 2007, 67, 7765–7772. [Google Scholar] [CrossRef]
- Johannes, E.; Horbert, R.; Schlosser, J.; Schmidt, D.; Peifer, C. Effective synthesis of 3,5-diaryl-(1H)-pyrazin-2-ones via microwave mediated ring closure. Tetrahedron Lett. 2013, 54, 4067–4072. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- ProQinase Targeting Cancer. Available online: http://www.proqinase.com (accessed on 19 August 2013).
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pinchuk, B.; Johannes, E.; Gul, S.; Schlosser, J.; Schaechtele, C.; Totzke, F.; Peifer, C. Marine Derived Hamacanthins as Lead for the Development of Novel PDGFRβ Protein Kinase Inhibitors. Mar. Drugs 2013, 11, 3209-3223. https://doi.org/10.3390/md11093209
Pinchuk B, Johannes E, Gul S, Schlosser J, Schaechtele C, Totzke F, Peifer C. Marine Derived Hamacanthins as Lead for the Development of Novel PDGFRβ Protein Kinase Inhibitors. Marine Drugs. 2013; 11(9):3209-3223. https://doi.org/10.3390/md11093209
Chicago/Turabian StylePinchuk, Boris, Eugen Johannes, Sheraz Gul, Joachim Schlosser, Christoph Schaechtele, Frank Totzke, and Christian Peifer. 2013. "Marine Derived Hamacanthins as Lead for the Development of Novel PDGFRβ Protein Kinase Inhibitors" Marine Drugs 11, no. 9: 3209-3223. https://doi.org/10.3390/md11093209