Self-Association of Okadaic Acid: Structural and Pharmacological Significance


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Dimerization of Okadaic Acid Overview
2.1. Complexation of Okadaic Acid

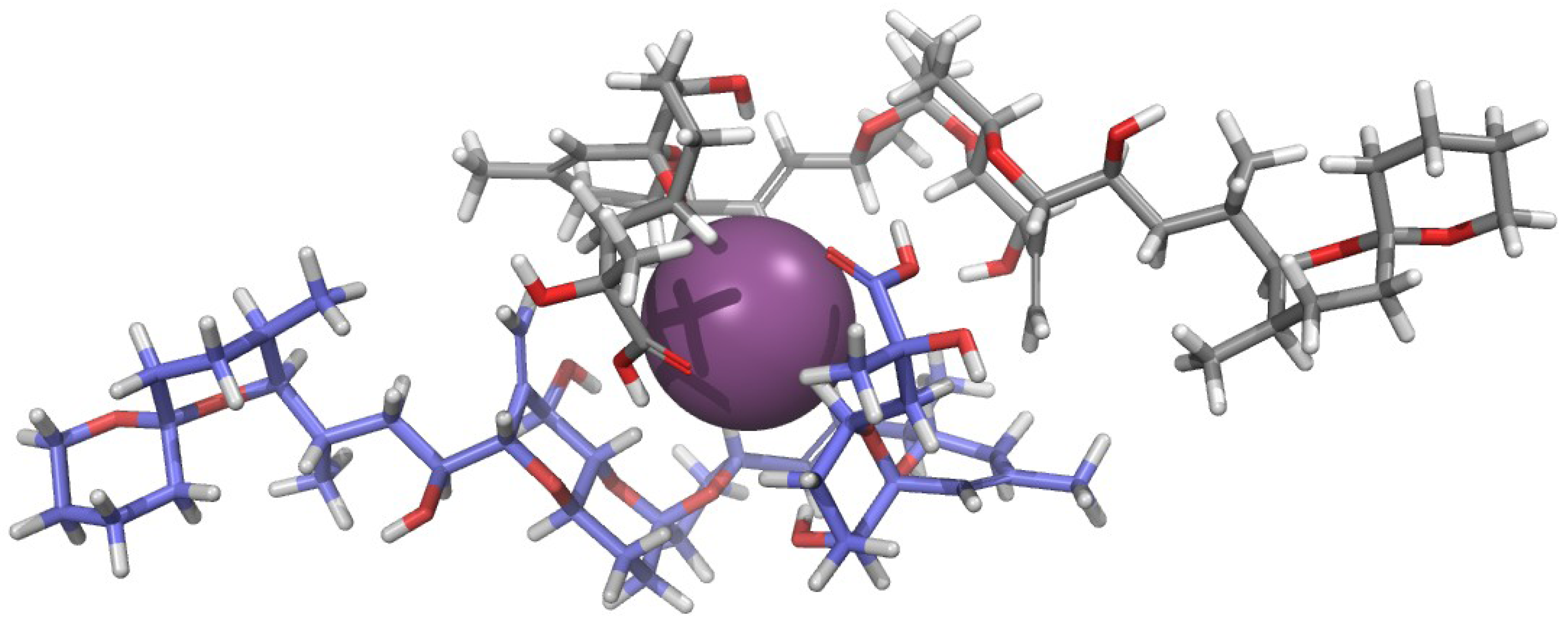
2.2. Physiological Importance of the Potassium Ion
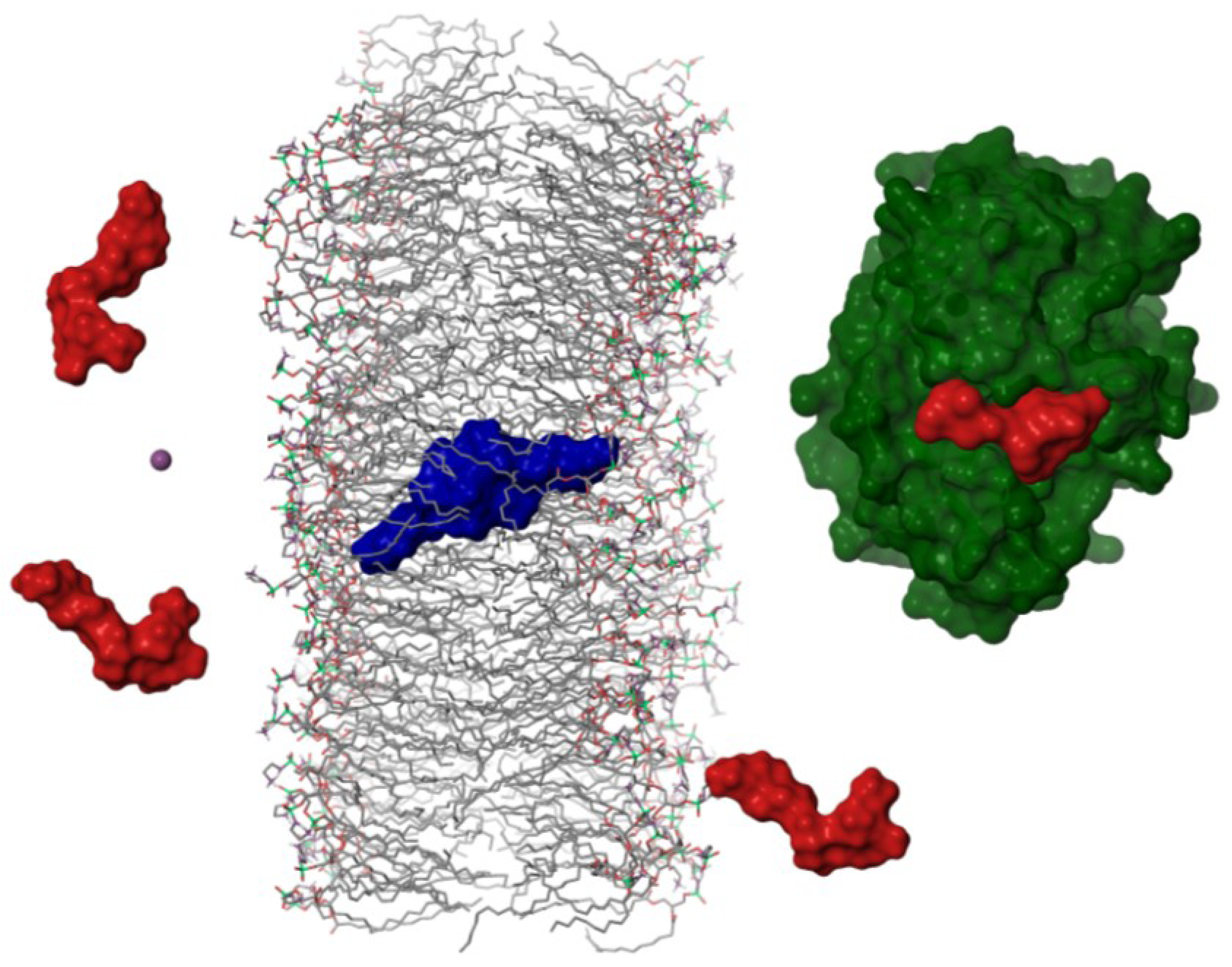
2.3. Structure of the Okadaic Acid—Potassium Complex

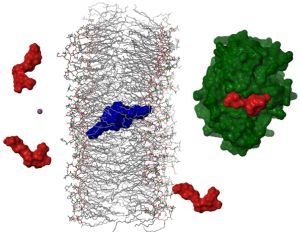
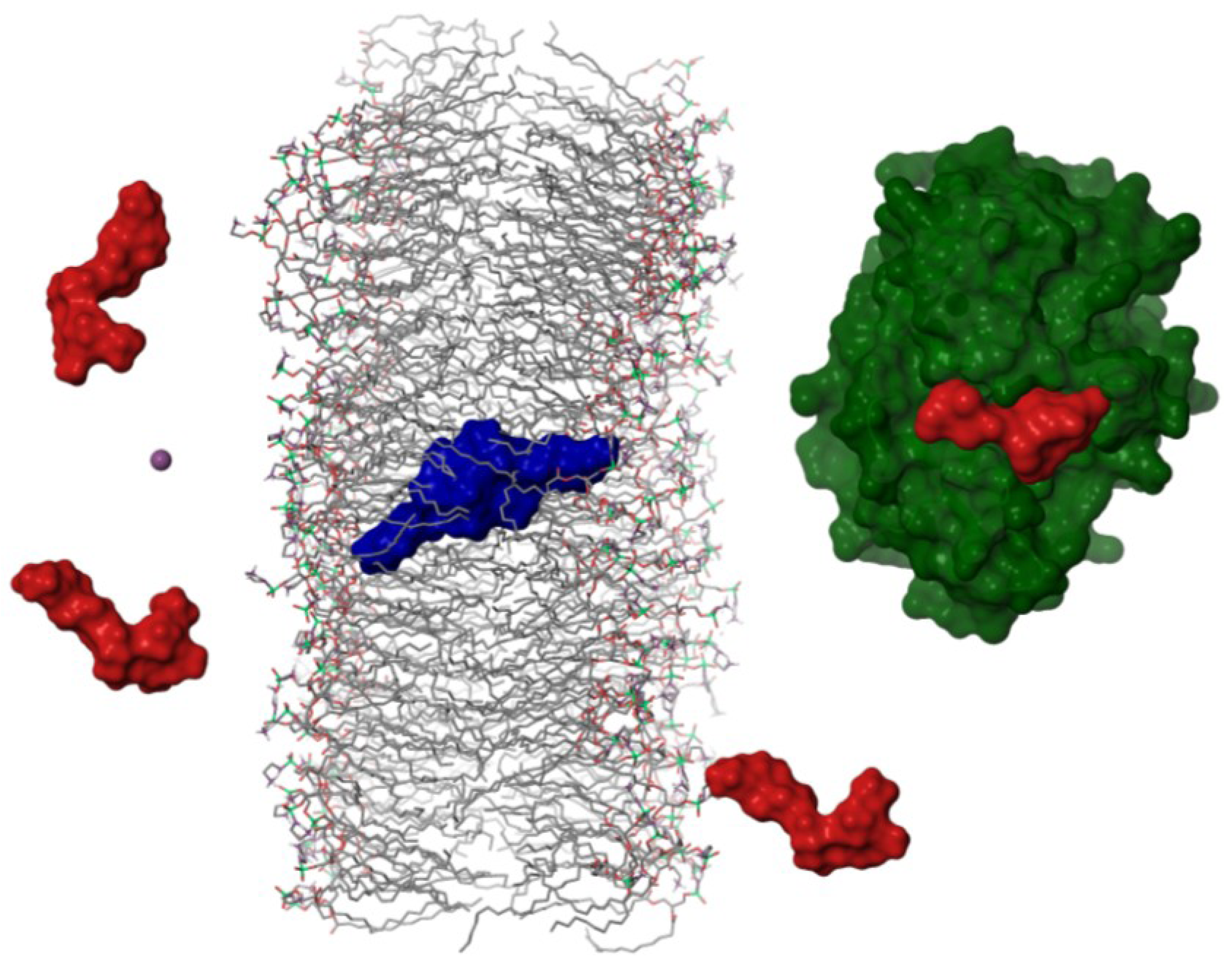
2.4. Dimerization as a Pathway to the Cell
3. Results and Discussion
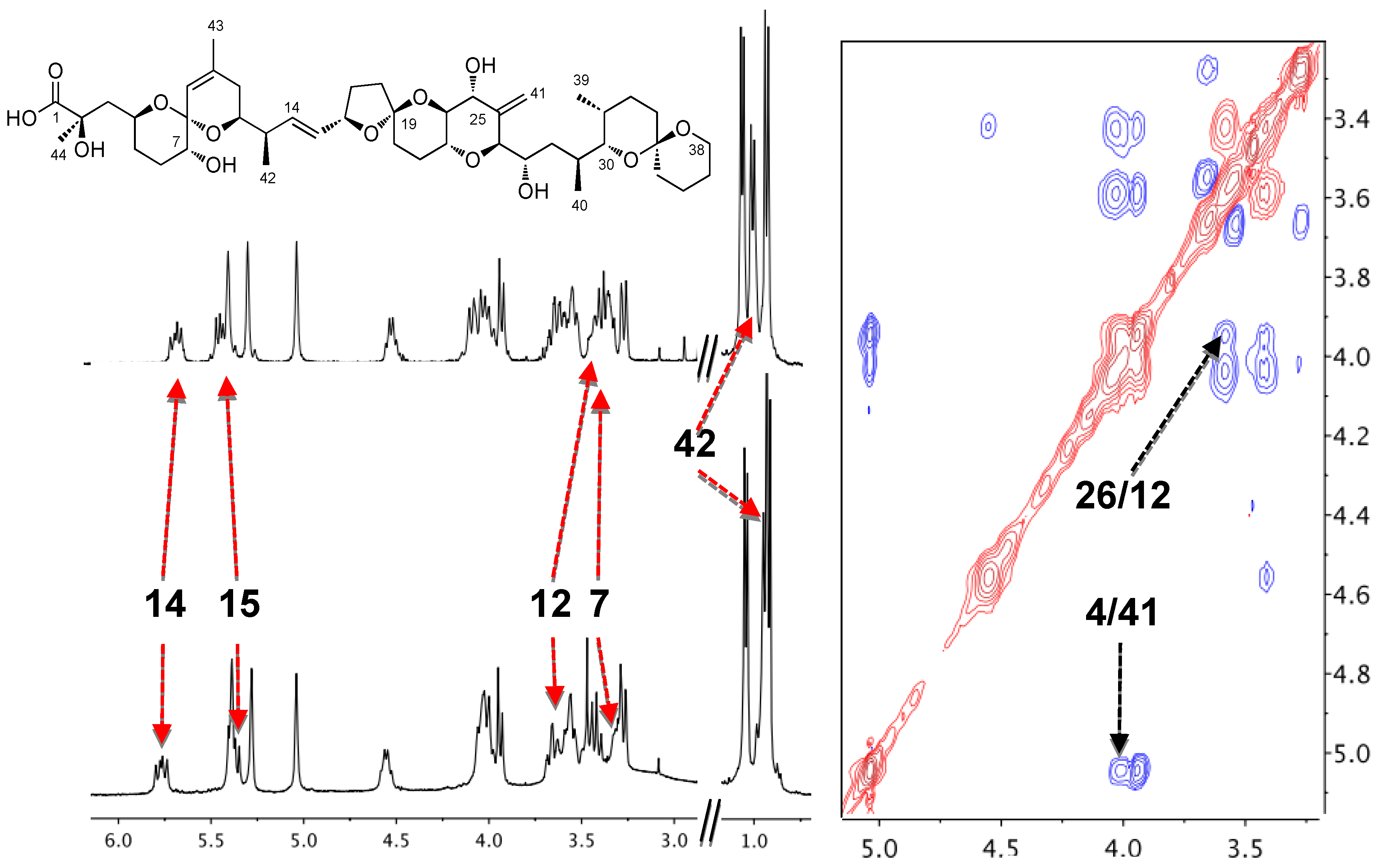
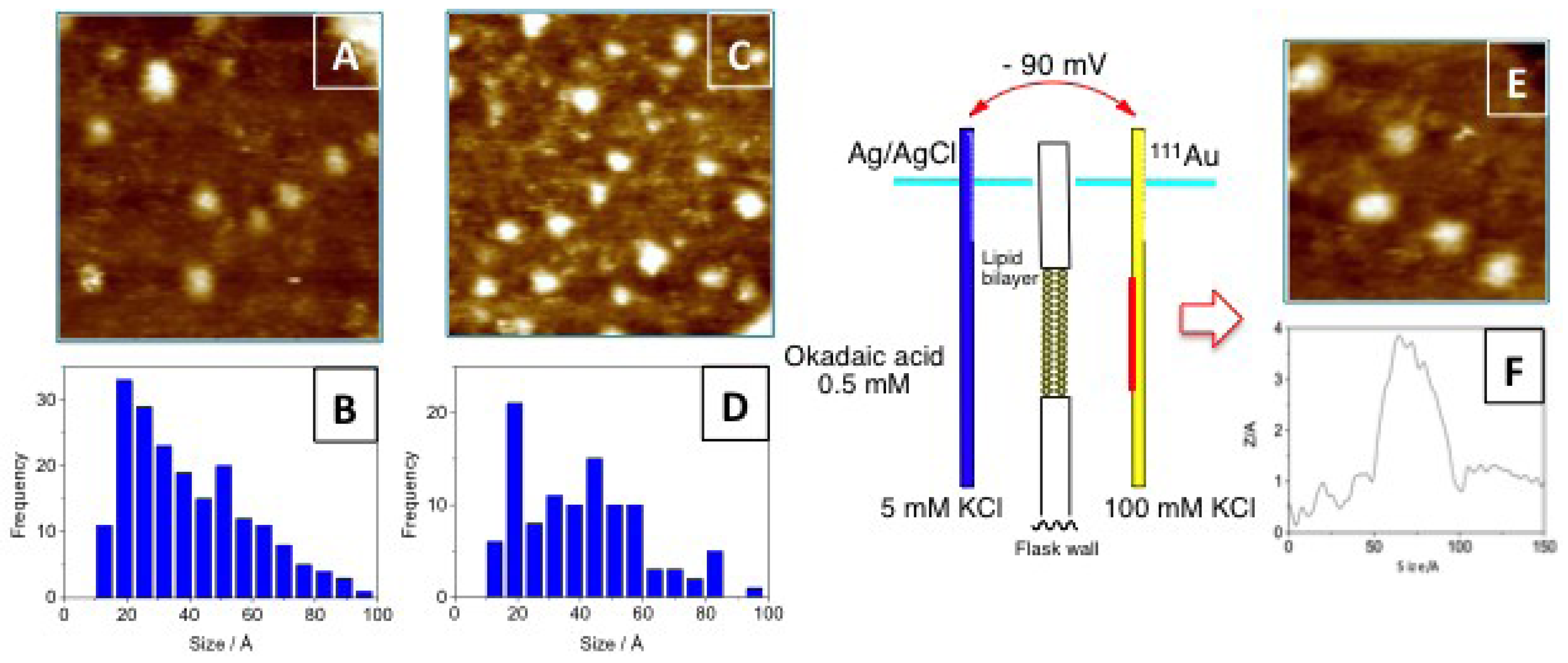
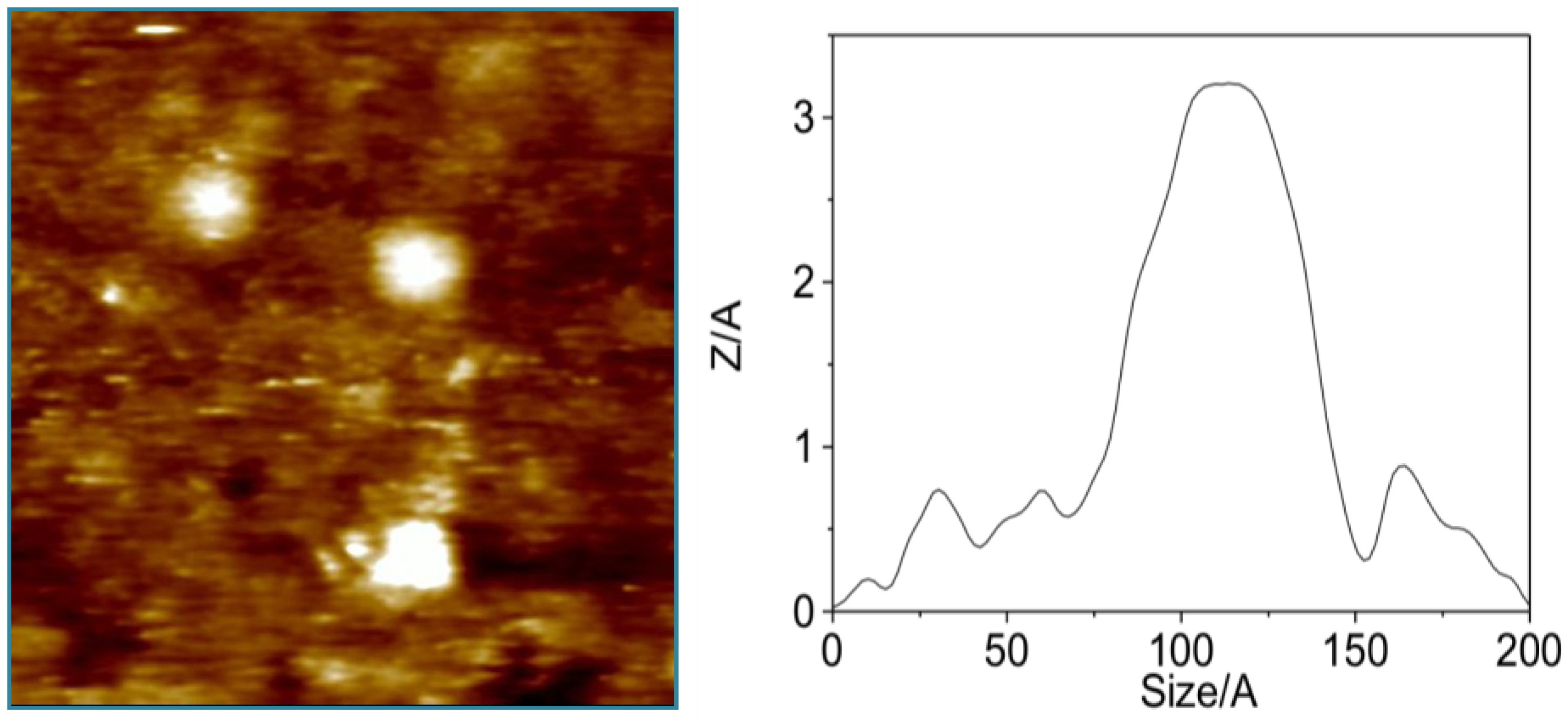
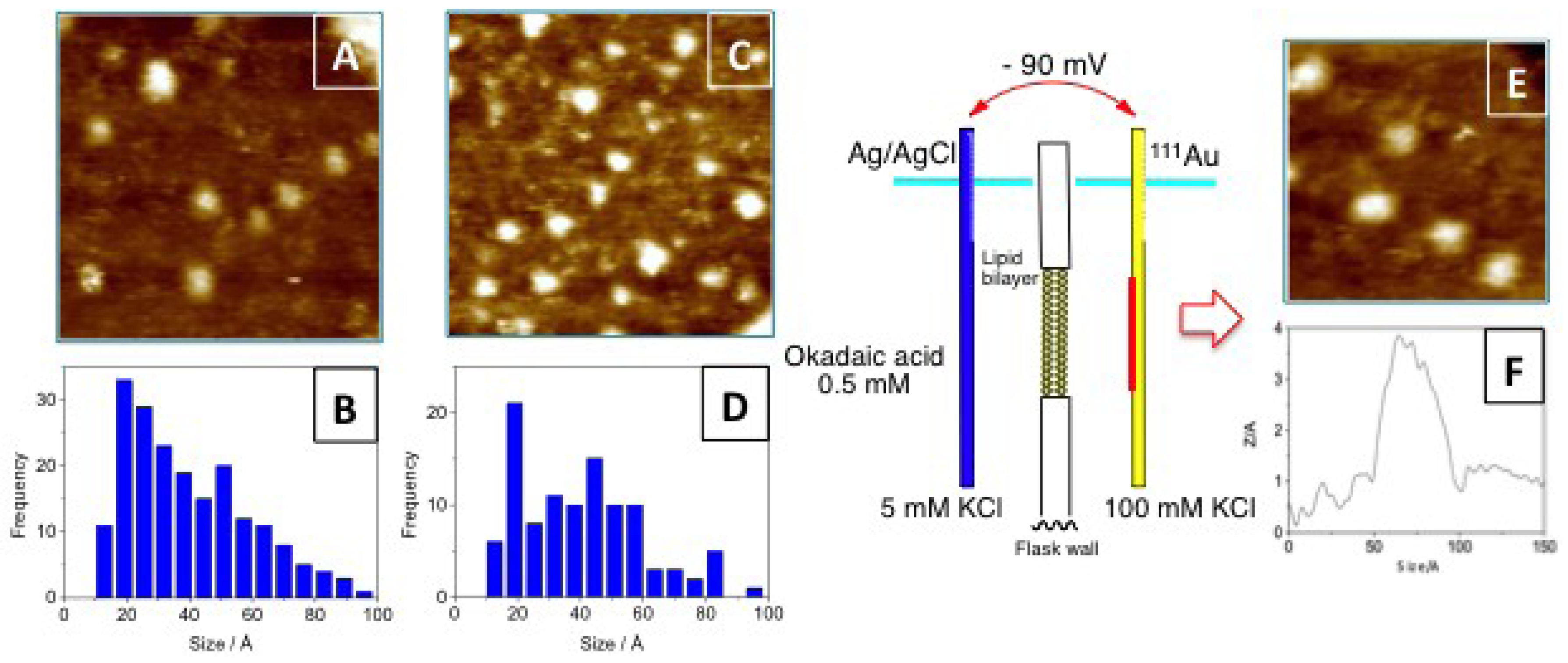
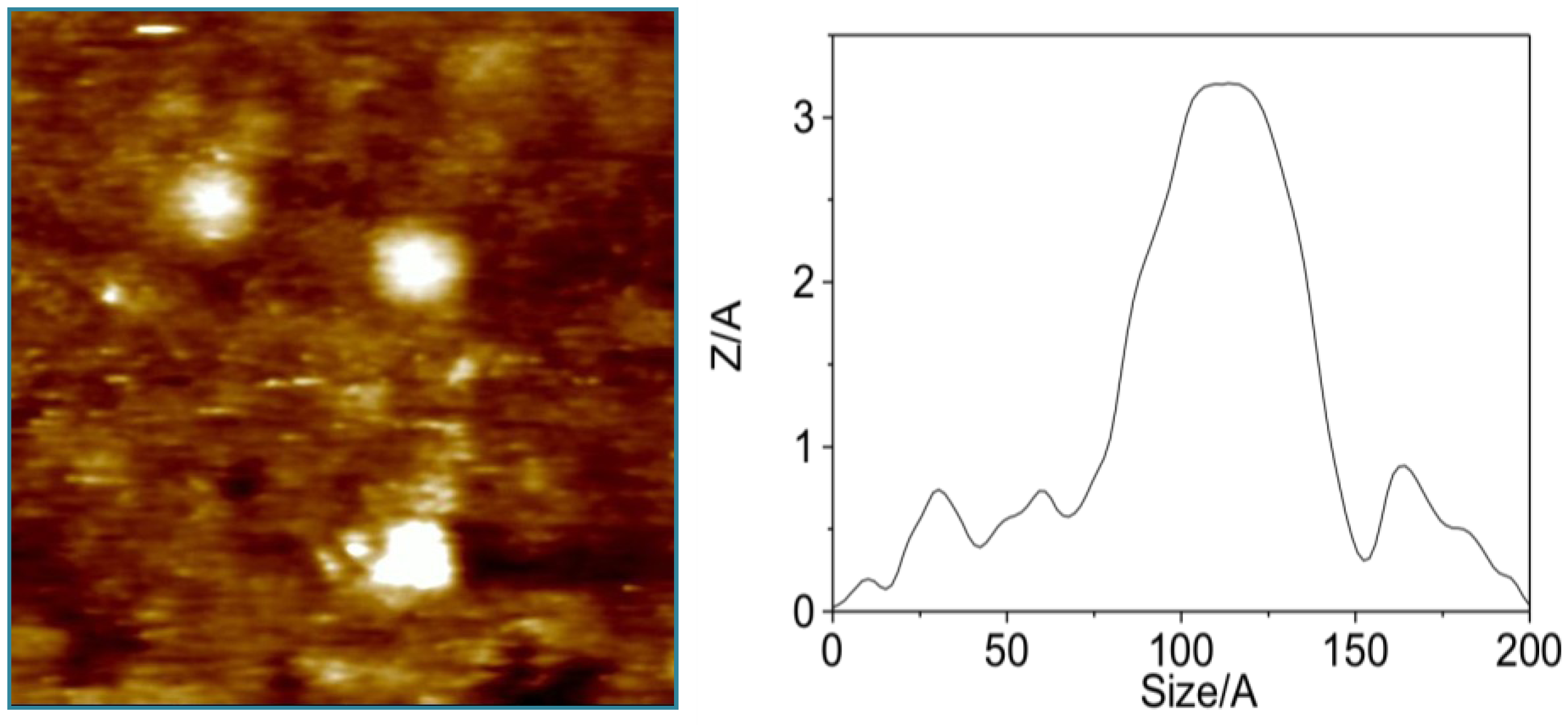
3.1. Higher Oligomerization States of Okadaic Acid
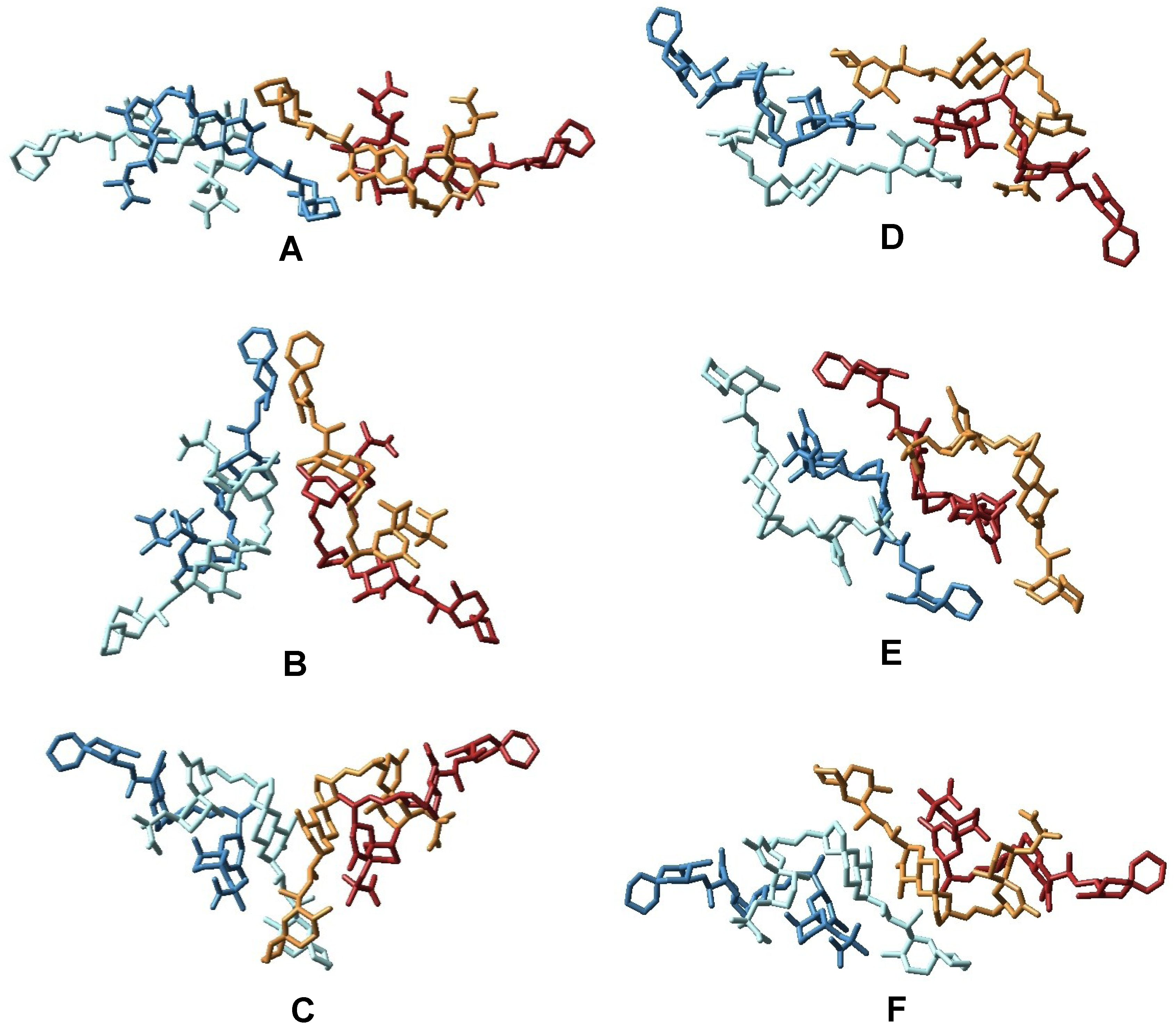
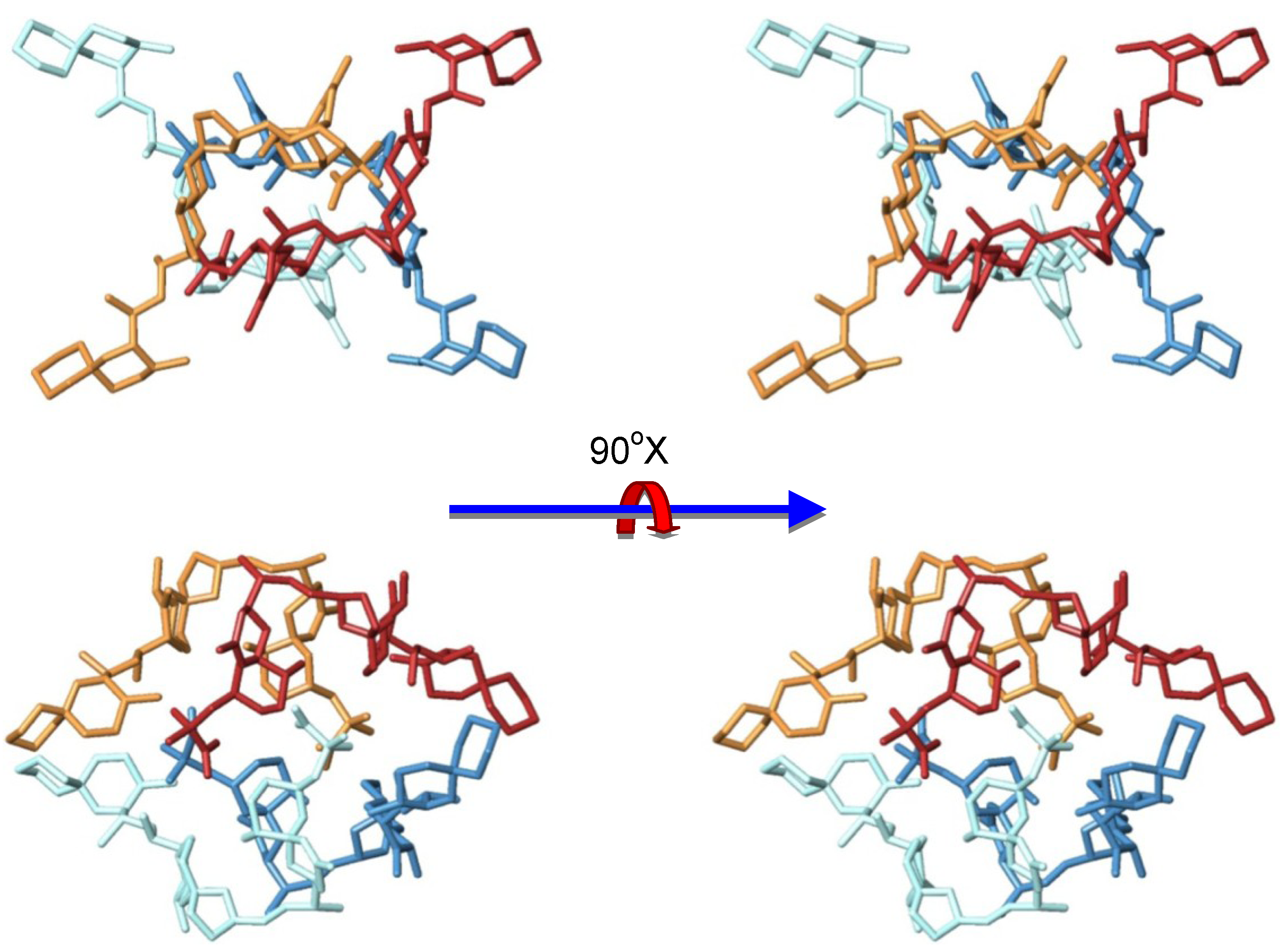
3.2. Structure of Okadaic Acid Tetramers
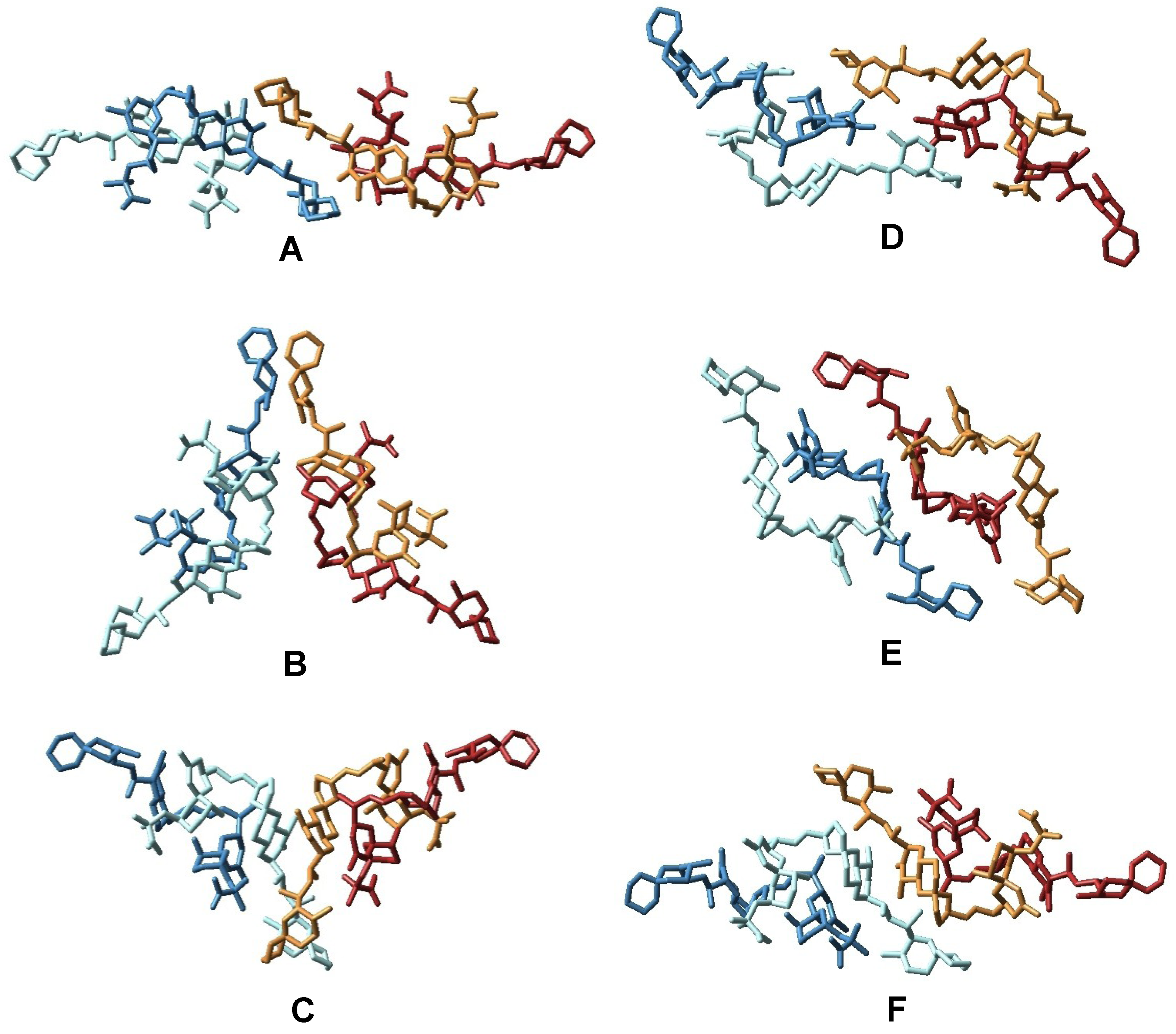
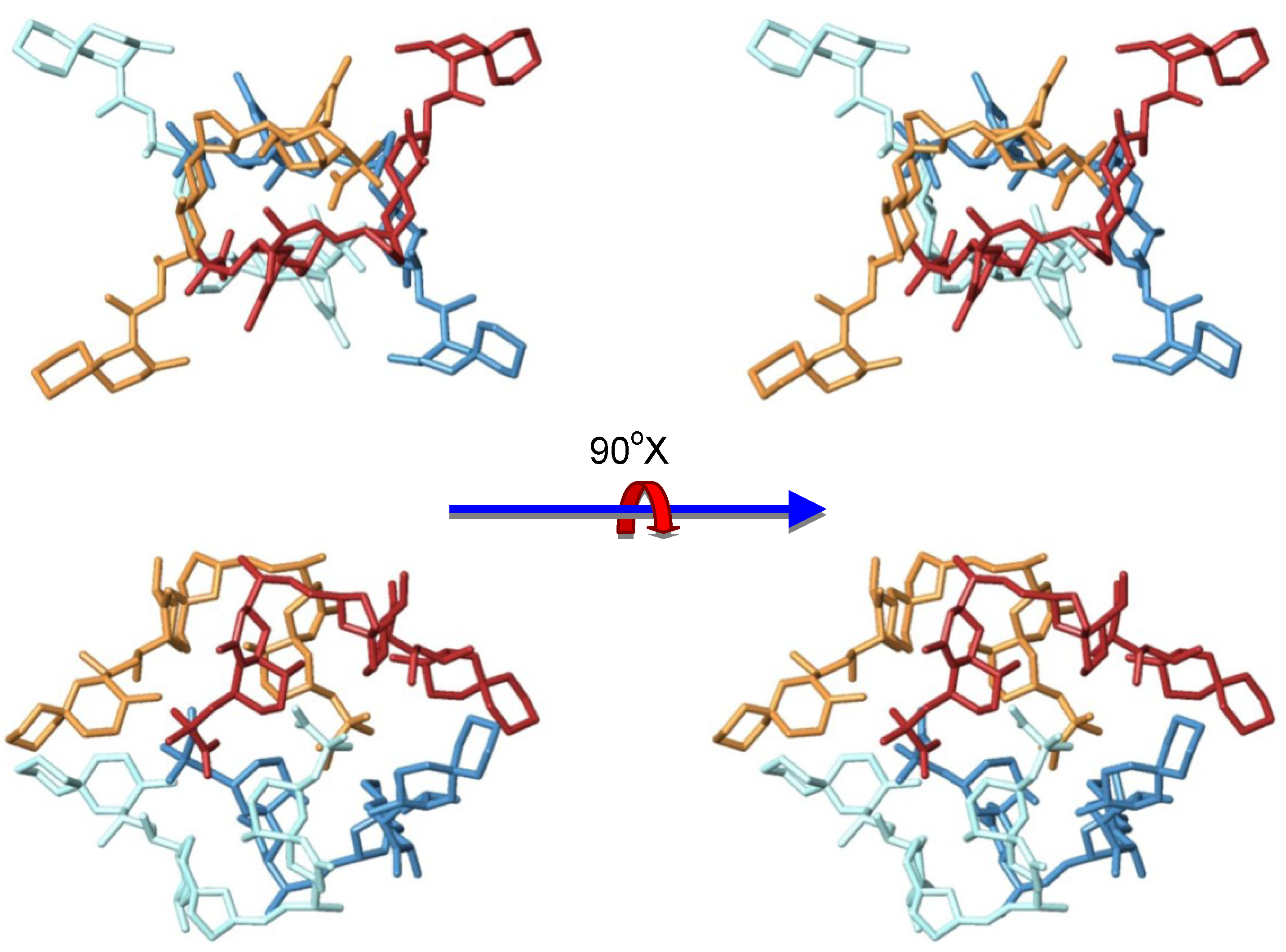
4. Experimental Section
4.1. Spectroscopic and Spectrometric Techniques
4.2. Prorocentrum Belizeanum Cultures
4.3. Extraction, Isolation and Purification
4.4. Preparation of STM Gold Plate
5. Conclusions
Acknowledgments
References
- Franchini, A.; Malagoli, D.; Ottaviani, E. Targets and effects of yessotoxin, okadaic acid and palytoxin: A differential review. Mar. Drugs 2010, 8, 658–677. [Google Scholar] [CrossRef]
- Domínguez, H.J.; Paz, B.; Daranas, A.H.; Norte, M.; Franco, J.M.; Fernández, J.J. Dinoflagellate polyether within the yessotoxin, pectenotoxin and okadaic acid toxin group: Characterization, analysis and human health implications. Toxicon 2010, 56, 191–217. [Google Scholar] [CrossRef]
- Daranas, A.H.; Norte, M.; Fernandez, J.J. Toxic marine microalgae. Toxicon 2001, 39, 1101–1132. [Google Scholar] [CrossRef]
- Schmitz, F.J.; Prasad, R.S.; Gopichand, Y.; Hossain, M.B.; van der Helm, D.; Schmidt, P. Acanthifolicin, a new episulfide-containing polyether carboxylic acid from extracts of the marine sponge Pandaros acanthifolium. J. Am. Chem. Soc. 1981, 103, 2467–2469. [Google Scholar] [CrossRef]
- Tachibana, K.; Scheuer, P.J.; Tsukitani, Y.; Kikuchi, H.; Egen, D.V.; Clardy, J.; Gopichand, Y.; Schmitz, F.J. Okadaic acid, a cytotoxic polyether from two marine sponges of the genus Halichondria. J. Am. Chem. Soc. 1981, 103, 2469–2471. [Google Scholar] [CrossRef]
- Norte, M.; González, R.; Fernández, J.J.; Rico, M. Okadaic acid: A proton and carbon NMR study. Tetrahedron 1991, 47, 7437–7446. [Google Scholar] [CrossRef]
- Matsumori, N.; Murata, M.; Tachibana, K. Conformational analysis of natural products using long range carbon-proton coupling constants: Three-dimensional structure of okadaic acid in solution. Tetrahedron 1995, 51, 12229–12238. [Google Scholar] [CrossRef]
- Takai, A.; Bialojan, C.; Troschka, M.; Rüegg, J.C. Smooth muscle myosin phosphatase inhibition and force enhancement by black sponge toxin. FEBS Lett. 1987, 217, 81–84. [Google Scholar] [CrossRef]
- Bialojan, C.; Rüegg, J.C.; Takai, A. Effects of okadaic acid on isometric tension and myosin phosphorylation of chemically skinned guinea-pig taenia coli. J. Physiol. 1988, 398, 81–95. [Google Scholar]
- Bialojan, C.; Takai, A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem. J. 1988, 256, 283–290. [Google Scholar]
- Fernandez, J.J.; Candenas, M.L.; Souto, M.L.; Trujillo, M.M.; Norte, M. Okadaic acid, a useful tool for studying cellular processes. Curr. Med. Chem. 2002, 9, 229–262. [Google Scholar] [CrossRef]
- Quinn, R.J.; Taylor, C.; Suganuma, M.; Fujiki, H. The conserved acid binding domain model of inhibitors of protein phosphatases 1 and 2A: Molecular modelling aspects. Bioorg. Med. Chem. Lett. 1993, 3, 1029–1034. [Google Scholar] [CrossRef]
- Dounay, A.B.; Forsyth, C.J. Okadaic acid: The archetypal serine/threonine protein phosphatase inhibitor. Curr. Med. Chem. 2002, 9, 1939–1980. [Google Scholar] [CrossRef]
- Maynes, J.M.; Bateman, K.S.; Cherney, M.M.; Das, A.K.; Luu, H.A.; Holmes, C.F.B.; James, M.N.G. Crystal structure of the tumor-promoter okadaic acid bound to protein phosphatase-1. J. Biol. Chem. 2001, 276, 44078–44082. [Google Scholar]
- Xing, Y.; Xu, Y.; Chen, Y.; Jeffrey, P.D.; Chao, Y.; Lin, Z.; Li, Z.; Strack, S.; Stock, J.B.; Shi, Y. Structure of protein phosphatase 2A core enzyme bound to tumor-inducing toxins. Cell 2006, 127, 341–353. [Google Scholar] [CrossRef]
- Larsen, K.; Petersen, D.; Wilkins, A.L.; Samdal, I.A.; Sandvik, M.; Rundberget, T.; Goldstone, D.; Arcus, V.; Hovgaard, P.; Rise, F.; et al. Clarification of the C-35 stereochemistries of dinophysistoxin-1 and dinophysistoxin-2 and its consequences for binding to protein phosphatase. Chem. Res. Toxicol. 2007, 20, 868–875. [Google Scholar] [CrossRef]
- Cruz, P.; Daranas, A.H.; Fernandez, J.J.; Norte, M. 19-epi-okadaic acid, a novel protein phosphatase inhibitor with enhanced selectivity. Org. Lett. 2007, 9, 3045–3048. [Google Scholar] [CrossRef]
- Cruz, P.; Fernandez, J.J.; Norte, M.; Daranas, A.H. Belizeanic acid: A potent protein phosphatase 1 inhibitor belonging to the okadaic acid class, with an unusual skeleton. Chem. Eur. J. 2008, 14, 6948–6956. [Google Scholar] [CrossRef]
- McConnell, J.L.; Wadzinski, B.E. Targeting protein serine/threonine phosphatases for drug development. Mol. Pharmacol. 2009, 75, 1249–1261. [Google Scholar] [CrossRef]
- Norte, M.; Fernández, J.J.; Souto, M.L.; Gavín, J.A.; Candenas, M.L.; Ausina, P. Complexation of okadaic acid: A preliminary study. Bioorg. Med. Chem. Lett. 1998, 8, 1007–1012. [Google Scholar] [CrossRef]
- Daranas, H.D.; Fernandez, J.J.; Morales, E.Q.; Norte, M.; Gavín, J.A. Self-association of okadaic acid upon complexation with potassium ion. J. Med. Chem. 2004, 47, 10–13. [Google Scholar] [CrossRef]
- Daranas, H.D.; Cruz, P.; Creus, A.H.; Norte, M.; Fernández, J.J. Self-assembly of okadaic acid as pathway to the cell. Org. Lett. 2007, 9, 4191–4194. [Google Scholar] [CrossRef]
- Comeau, S.R.; Gatchell, D.W.; Vajda, S.; Camacho, C.J. ClusPro: A fully automated algorithm for protein-protein docking. Nucleic Acids Res. 2004, 32, W96–W99. [Google Scholar] [CrossRef]
- Macromodel, version 8.5; Schrödinger, LLC: New York, NY, USA, 2004.
- Horcas, I.; Fernández, R.; Gómez-Rodríguez, J.M.; Colchero, J.; Gómez-Herrero, J.; Baró, A.M. WSXM: A software for scanning probe microscopy and a tool for nanotechnology. Rev. Sci. Instrum. 2007, 78, 013705:1–013705:8. [Google Scholar]
- Matile, S.; Nakanishi, K. Selective cation movement across lipid bilayers containing brevetoxin-B. Angew. Chem. Int. Ed. 1996, 35, 757–759. [Google Scholar] [CrossRef]
- Inuzuka, T.; Fujisawa, T.; Arimoto, H.; Uemura, D. Molecular shape of palytoxin in aqueous solution. Org. Biom. Chem. 2007, 5, 897–899. [Google Scholar] [CrossRef]
- Inuzuka, T.; Uemura, D.; Arimoto, H. The conformational features of palytoxin in aqueous solutions. Tetrahedron 2008, 64, 7718–7723. [Google Scholar] [CrossRef]
- Samples Availability: Available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cruz, P.G.; Norte, M.; Creus, A.H.; Fernández, J.J.; Daranas, A.H. Self-Association of Okadaic Acid: Structural and Pharmacological Significance. Mar. Drugs 2013, 11, 1866-1877. https://doi.org/10.3390/md11061866
Cruz PG, Norte M, Creus AH, Fernández JJ, Daranas AH. Self-Association of Okadaic Acid: Structural and Pharmacological Significance. Marine Drugs. 2013; 11(6):1866-1877. https://doi.org/10.3390/md11061866
Chicago/Turabian StyleCruz, Patricia G., Manuel Norte, Alberto Hernández Creus, José J. Fernández, and Antonio Hernández Daranas. 2013. "Self-Association of Okadaic Acid: Structural and Pharmacological Significance" Marine Drugs 11, no. 6: 1866-1877. https://doi.org/10.3390/md11061866