3.2. Synthetic Procedures
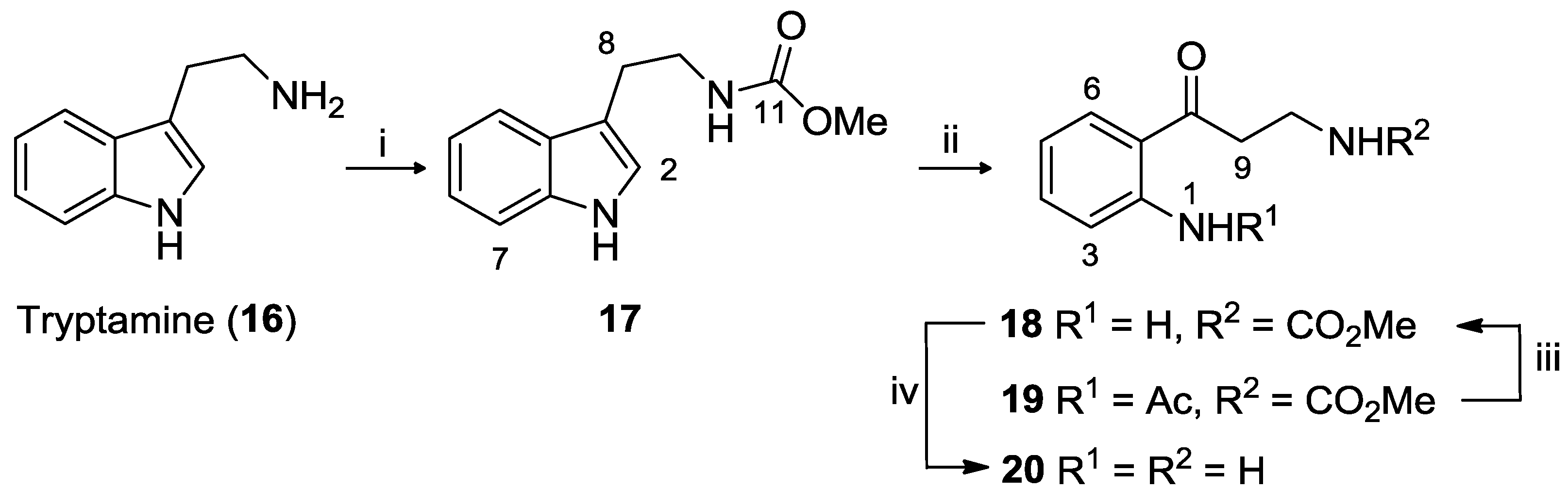
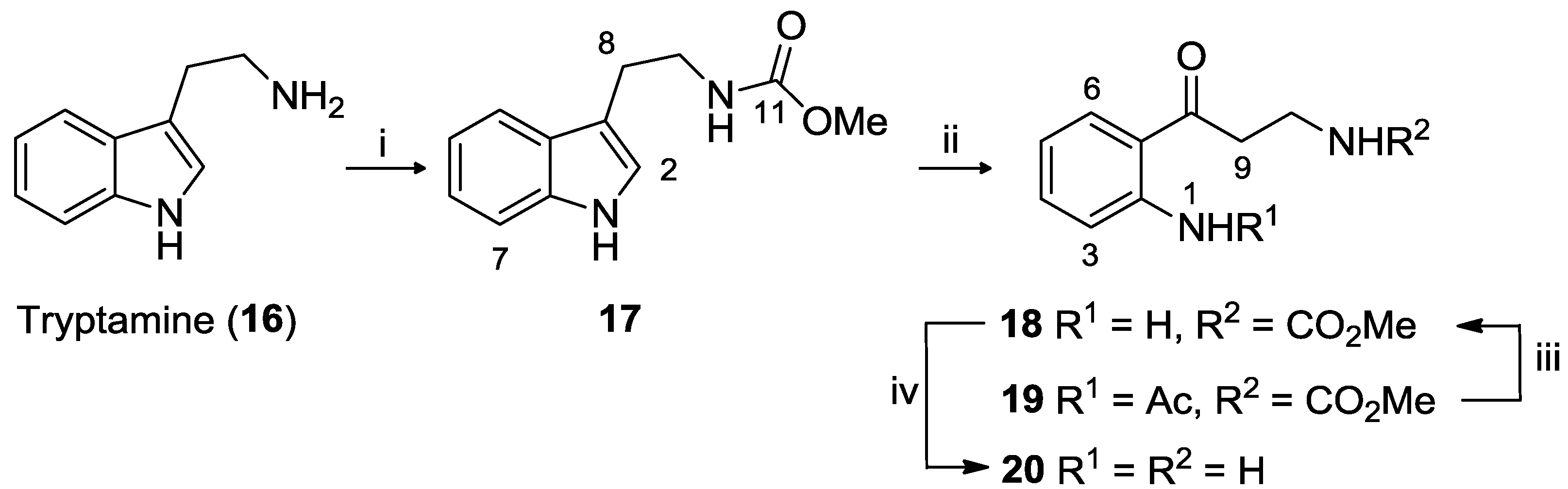
3.2.1. Tryptamine Methyl Carbamate (17)
To a stirred solution of tryptamine (2.0 g, 0.01 mol) in a degassed biphasic mixture of NaOH (1 N, 12.5 mL) and EtOAc (20 mL) was added methyl chloroformate (1.01 mL, 13.13 mmol) dropwise, under N2. The brown solution was stirred for 30 min at room temperature, after which it was washed with H2O (2 × 40 mL) and the organic phase dried in vacuo. The residue was dissolved in EtOAc (10 mL) and then added to hexane (50 mL) to yield 17 as brown crystals (2.47 g, 73% yield).
Mp 82.9–83.9 °C (lit. [
19] 79.0–81.0 °C);
Rf (1 Hex:2 EtOAc) 0.64; IR ν
max (ATR) 3400, 1686, 1544, 1264 cm
−1;
1H NMR (CDCl
3, 400 MHz) δ
H 8.02 (1H, br s, NH-1), 7.61 (1H, d,
J = 7.4 Hz, H-4), 7.38 (1H, d,
J = 7.6 Hz, H-7), 7.21 (1H, td,
J = 7.6, 1.2 Hz, H-6), 7.14 (1H, td,
J = 7.4, 0.9 Hz, H-5), 7.03 (1H, d,
J = 2.1 Hz, H-2), 4.73 (1H, br s, NH-10), 3.65 (3H, s, OMe), 3.53 (2H, dt,
J = 6.8, 6.8 Hz, H
2-9), 2.98 (2H, t,
J = 6.8 Hz, H
2-8);
13C NMR (CDCl
3, 100 MHz) δ
C 157.1 (C-11), 136.4 (C-7a), 127.2 (C-3a), 122.2 (C-6 or C-2), 122.0 (C-6 or C-2), 119.4 (C-5), 118.7 (C-4), 112.9 (C-3), 111.2 (C-7), 52.0 (OMe), 41.2 (C-9), 25.8 (C-8); (+)-ESIMS
m/z 219 [M + H]
+; (+)-HRESIMS [M + H]
+ 219.1131 (calcd. for C
12H
15N
2O
2, 219.1128).
1H and
13C NMR data agreed with literature [
19].
3.2.2. Kynuramine Methyl Carbamate (18) and N-Acetyl-kynuramine Methyl Carbamate (19)
Ozone was bubbled into a solution of tryptamine methyl carbamate (17) (1.00 g, 4.59 mmol) in AcOH (20 mL) that was stirred in an ice bath. The reaction was stopped once the solution became frozen. The frozen solution was degassed with N2 for 5 min and then conc. HCl (1 mL) was added to the solution and warmed to 40 °C for 1.5 h. After this time the solution was dried in vacuo, the residue dissolved in CH2Cl2 (20 mL), and washed with phosphate buffer until neutral (3 × 20 mL). The organic phase was dried (MgSO4), solvent removed in vacuo and the mixture purified using silica gel flash chromatography (hexane/EtOAc) to afford kynuramine methyl carbamate 18 as a yellow solid (0.42 g, 42% yield) and 19 also as a yellow solid (0.13 g, 10% yield).
Kynuramine methyl carbamate 18: Mp 90.0–91.0 °C (lit. [
20] 98.0–99.0 °C);
Rf (10% MeOH/CH
2Cl
2) 0.89; IR ν
max (ATR) 3360, 1685, 1619, 1531, 1264 cm
−1;
1H NMR (CDCl
3, 400 MHz) δ
H 7.68 (1H, d,
J = 7.6 Hz, H-6), 7.26 (1H, dt,
J = 7.6, 1.6 Hz, H-4), 6.65–6.61 (2H, m, H-3 and H-5), 3.64 (3H, s, OMe), 3.57 (2H, dt,
J = 5.6, 5.6 Hz, H
2-10), 3.17 (2H, t,
J = 5.6 Hz, H
2-9);
13C NMR (CDCl
3, 100 MHz) δ
C 201.1 (C-8), 157.1 (C-12), 150.4 (C-2), 134.6 (C-4), 131.0 (C-6), 117.7 (C-5), 117.4 (C-7), 115.9 (C-3), 52.0 (OMe), 38.9 (C-9), 36.2 (C-10); (+)-ESIMS
m/z 223 [M + H]
+; (+)-HRESIMS [M + H]
+ 223.1076 (calcd. for C
11H
15N
2O
3, 223.1077).
N-Acetyl kynuramine methyl carbamate 19: Mp 120.0–121.0 °C; Rf (1 Hex:2 EtOAc) 0.30; IR νmax (ATR) 3332, 3220, 3112, 2947, 1700, 1686, 1544, 1292, 1195, 760 cm−1; 1H NMR (CDCl3, 400 MHz) δH 11.62 (1H, br s, NH-1), 8.70 (1H, d, J = 7.3 Hz, H-3), 7.90 (1H, d, J = 6.8 Hz, H-6), 7.55 (1H, td, J = 7.3, 1.5 Hz, H-4), 7.11 (1H, td, J = 6.8, 1.3 Hz, H-5), 5.29 (1H, br s, NH-11), 3.66 (3H, s, OMe), 3.58 (2H, dt, J = 5.6, 5.6 Hz, H2-10), 3.29 (2H, t, J = 5.6 Hz, H2-9), 2.23 (3H, s, H3-14); 13C NMR (CDCl3, 100 MHz) δC 203.4 (C-8), 169.4 (C-13), 157.0 (C-12), 141.1 (C-2), 135.4 (C-4), 130.8 (C-6), 122.4 (C-5), 121.1 (C-7), 120.8 (C-3), 52.1 (OMe), 39.8 (C-9), 36.0 (C-10), 25.6 (C-14); (+)-ESIMS m/z 265 [M + H]+; (+)-HRESIMS [M + H]+ 265.1191 (calcd. for C13H17N2O4, 265.1183).
An alternative method to bypass the formation of acetamide 19 was to take the crude reaction product containing both 18 and 19, dissolve it in aq. HCl (10%, 40 mL), and heat at reflux for 4 h. Removal of solvents in vacuo afforded 18 as a yellow solid (1.35 g, 66% yield over two steps).
3.2.3. Kynuramine Dihydrobromide (20)
A solution of kynuramine methyl carbamate 18 (1.346 g, 6.06 mmol) in HBr saturated AcOH (20 mL) was heated to 80 °C and stirred for 18 h under N2. The brown solution was cooled to room temperature and THF (80 mL) was added which resulted in the formation of a precipitate. The mixture was stirred in an ice bath for 20 min, then filtered. The brown solid was dried under N2 to afford 20 (1.89 g, 96% yield).
Mp 192.0–193.0 °C (lit. [
21] 214.0–216.0 °C); IR ν
max (ATR) 3400, 1705, 1619, 1543, 1261 cm
−1;
1H NMR (CD
3OD, 400 MHz) δ
H 8.14 (1H, dd,
J = 7.8, 1.4 Hz, H-6), 7.69 (1H, td,
J = 7.9, 1.4 Hz, H-4), 7.51 (1H, td,
J = 7.8, 1.1 Hz, H-5), 7.42 (1H, dd,
J = 7.9, 1.1 Hz, H-3), 3.55 (2H, t,
J = 6.4 Hz, H
2-9), 3.34 (2H, t,
J = 6.4 Hz, H
2-10);
13C NMR (CD
3OD, 100 MHz) δ
C 200.8 (C-8), 162.8 (C-2), 136.3 (C-4), 132.8 (C-6), 128.9 (C-5), 128.5 (C-7), 125.4 (C-3), 37.8 (C-9), 35.9 (C-10); (+)-ESIMS
m/z 165 [M + H]
+; (+)-HRESIMS [M + H]
+ 165.1016 (calcd. for C
9H
13N
2O, 165.1022).
3.2.4. N-(3,4-Dimethoxyphenethyl)acetamide (22)
Et3N (1.54 mL, 0.01 mol) and acetic anhydride (1.56 mL, 0.02 mol) was added to 2-(3,4-dimethoxyphenyl)ethylamine (21) (0.93 mL, 5.52 mmol). The reaction mixture was yellow, and was stirred at room temperature for 1 h under N2. CH2Cl2 (100 mL) was added then washed with H2O (50 mL) and the organic phase dried in vacuo, to afford 22 as a yellow solid (1.17 g, 95% yield).
Mp 97.8–98.6 °C (lit. [
22] 100.0–101.0 °C);
Rf (5% MeOH/CH
2Cl
2) 0.39; IR ν
max (ATR) 3250, 3080, 2928, 2840, 1631, 1590, 1516, 1261, 1232, 1155, 1019 cm
−1;
1H NMR (CDCl
3, 400 MHz) δ
H 6.80 (1H, d,
J = 8.0 Hz, H-8), 6.72 (1H, d,
J = 1.8 Hz, H-5), 6.70 (1H, dd,
J = 8.0, 1.8 Hz, H-9), 5.55 (1H, br s, NH-1), 3.87 (6H, s, OMe), 3.30 (2H, dt,
J = 7.0, 7.0 Hz, H
2-2), 2.60 (2H, t,
J = 7.0 Hz, H
2-3), 1.92 (3H, s, H
3-11);
13C NMR (CDCl
3, 100 MHz) δ
C 170.0 (C-10), 149.1 (C-6), 147.7 (C-7), 131.3 (C-4), 120.6 (C-9), 111.9 (C-8), 111.4 (C-5), 55.9 (OMe × 2), 40.7 (C-2), 35.2 (C-3), 23.3 (C-11); (+)-ESIMS
m/z 224 [M + H]
+; (+)-HRESIMS [M + H]
+ 224.1279 (calcd. for C
12H
18NO
3, 224.1281).
1H and
13C NMR data agreed with literature [
22].
3.2.5. N-(3,4-Dimethoxyphenethyl)-3-methylbut-2-enamide (23)
To a solution of 3,3-dimethylacrylic acid (100 mg, 1.00 mmol) in dry DMF (3 mL) was added 2-(3,4-dimethoxyphenyl)ethylamine (21) (0.17 mL, 1.00 mmol), PyBOP (520 mg, 1.00 mmol) and Et3N (0.42 mL, 3.00 mmol). The mixture was stirred under N2 at room temperature for 20 h. CH2Cl2 (20 mL) was added, washed with H2O (30 mL), and the organic phase dried in vacuo. EtOAc (75 mL) was added and washed with 5% aq. K2CO3 (50 mL), 10% HCl (10 mL) and brine (20 mL). The organic phase was then dried (MgSO4) and solvent removed in vacuo to give 23 as an orange-brown solid (0.24 g, 92% yield).
Mp 67.0–68.2 °C; Rf (5% MeOH/CH2Cl2) 0.22; IR νmax (ATR) 3302, 3002, 2939, 1666, 1141, 1027 cm−1; 1H NMR (CDCl3, 400 MHz) δH 6.79 (1H, d, J = 8.4 Hz, H-8), 6.73–6.71 (2H, m, H-5 and H-9), 5.48 (1H, s, H-11), 3.84 (6H, s, OMe), 3.50 (2H, dt, J = 7.2, 7.2 Hz, H2-2), 2.76 (2H, t, J = 7.2 Hz, H2-3), 2.12 (3H, s, H3-14), 1.79 (3H, s, H3-13); 13C NMR (CDCl3, 100 MHz) δC 166.9 (C-10), 150.8 (C-12), 148.9 (C-6), 147.5 (C-7), 131.5 (C-4), 120.6 (C-9), 118.4 (C-11), 111.9 (C-5), 111.3 (C-8), 55.8 (OMe × 2), 40.3 (C-2), 35.3 (C-3), 27.0 (C-13), 19.7 (C-14); (+)-ESIMS m/z 264 [M + H]+; (+)-HRESIMS [M + H]+ 264.1595 (calcd. for C15H22NO3, 264.1594).
3.2.6. N-(3,4-Dimethoxyphenethyl)benzamide (24)
To a cold (0 °C) solution of 2-(3,4-dimethoxyphenyl)ethylamine (21) (0.093 mL, 0.55 mmol) and Et3N (0.35 mL, 2.48 mmol) in THF (7.0 mL) was added benzoyl chloride (0.223 mL, 1.93 mmol). The milky white solution was warmed to room temperature and solvents were removed in vacuo. CHCl3 (20 mL) was added, the solution washed with 10% aq. NaCO3 (50 mL), H2O (20 mL) and brine (20 mL) and then the organic phase was dried in vacuo. The residue was triturated with hexane (6 mL) and EtOAc (2 mL) to give 24 as a greenish-white solid (90 mg, 57% yield).
Mp 85.0–85.8 °C (lit. [
23] 85.0–86.0 °C);
Rf (5% MeOH/CH
2Cl
2) 0.67; IR ν
max (ATR) 3236, 2981, 1634, 1590, 1231 cm
−1;
1H NMR (CDCl
3, 400 MHz) δ
H 7.71–7.69 (2H, m, H-12 and H-16), 7.44 (1H, tt,
J = 6.4, 1.2 Hz, H-14), 7.38–7.34 (2H, m, H-13 and H-15), 6.78 (1H, d,
J = 8.0 Hz, H-8), 6.74–6.72 (2H, m, H-5 and H-9), 3.82 (3H, s, OMe), 3.79 (3H, s, OMe), 3.66 (2H, t,
J = 7.2 Hz, H
2-2), 2.85 (2H, t,
J = 7.2 Hz, H
2-3);
13C NMR (CDCl
3, 100 MHz) δ
C 167.6 (C-10), 148.9 (C-6), 147.5 (C-7), 134.2 (C-11), 131.4 (C-14), 131.3 (C-4), 128.4 (C-13 and C-15), 126.8 (C-12 and C-16), 120.6 (C-9), 111.9 (C-5), 111.3 (C-8), 55.7 (OMe × 2), 41.3 (C-2), 35.1 (C-3); (+)-ESIMS
m/z 286 [M + H]
+; (+)-HRESIMS [M + H]
+ 286.1437 (calcd. for C
17H
20NO
3, 286.1438).
1H NMR data agreed with literature [
23].
3.2.7. N-(3,4-Dimethoxyphenethyl)-2-phenylacetamide (25)
To a solution of phenylacetic acid (200 mg, 1.47 mmol) in dry DMF (3 mL) was added 2-(3,4-dimethoxyphenyl)ethylamine (21) (0.25 mL, 1.47 mmol), PyBOP (764 mg, 1.47 mmol) and Et3N (0.62 mL, 4.41 mmol). The mixture was stirred under N2 at room temperature for 26 h. CH2Cl2 (20 mL) was added, washed with H2O (30 mL), and the organic phase dried in vacuo. The crude product was triturated with hexane (15 mL) and EtOAc (7 mL) to yield a white solid that was recrystallized from ethanol (3 mL) to afford 25 as white crystals (0.438 g, 99% yield).
Mp 111.1–112.2 °C (lit. [
24] 110–111 °C);
Rf (3 EtOAc:1 Hex) 0.40; IR ν
max (ATR) 3245, 3008, 2994, 1660, 1605, 1232 cm
−1;
1H NMR (CDCl
3, 400 MHz) δ
H 7.32–7.25 (3H, m, H-14, H-15 and H-16), 7.16 (2H, dd,
J = 8.0, 2.0 Hz, H-13 and H-17), 6.71 (1H, d,
J = 8.2 Hz, H-8), 6.59 (1H, d,
J = 2.0 Hz, H-5), 6.54 (1H, dd,
J = 8.2, 2.0 Hz, H-9), 5.36 (1H, br s, NH-1), 3.85 (3H, s, OMe), 3.81 (3H, s, OMe), 3.53 (2H, s, H
2-11), 3.44 (2H, dt,
J = 6.9, 6.9 Hz, H
2-2), 2.67 (2H, t,
J = 6.9 Hz, H
2-3);
13C NMR (CDCl
3, 100 MHz) δ
C 170.9 (C-10), 149.0 (C-6 or C-7), 147.6 (C-6 or C-7), 134.8 (C-12), 131.1 (C-4), 129.4 (C-13 and C-17), 129.0 (C-14 and C-16), 127.3 (C-15), 120.5 (C-9), 111.7 (C-5), 111.3 (C-8), 55.9 (OMe), 55.8 (OMe), 43.9 (C-11), 40.7 (C-2), 35.0 (C-3); (+)-ESIMS
m/z 300 [M + H]
+; (+)-HRESIMS [M + H]
+ 300.1593 (calcd. for C
18H
22NO
3, 300.1594).
1H NMR data agreed with literature [
24].
3.2.8. N-(3,4-Dimethoxyphenethyl)-3-phenylpropanamide (26)
To a cold (0 °C) solution of 2-(3,4-dimethoxyphenyl)ethylamine (21) (90 μL, 0.55 mmol) and Et3N (0.35 mL, 2.48 mmol) in THF (4.5 mL) was added dihydrocinnamoyl chloride (0.29 mL, 1.93 mmol). The milky white solution was warmed to room temperature and then the solvents were removed in vacuo. CHCl3 (20 mL) was added, the solution washed with 10% aq. NaCO3 (50 mL), H2O (20 mL) and brine (20 mL) and then the organic phase was dried in vacuo. The residue was triturated with hexane (7 mL) and EtOAc (2 mL) to give 26 as a pale yellow solid (0.16 g, 93% yield).
Mp 123.1–124.0 °C; Rf (5% MeOH/CH2Cl2) 0.45; IR νmax (ATR) 3249, 2974, 1631, 1534, 1232 cm−1; 1H NMR (CDCl3, 400 MHz) δH 7.29–7.26 (2H, m, H-15, H-17), 7.24–7.16 (3H, m, H-14, H-16, H-18), 6.77 (1H, d, J = 8.0 Hz, H-8), 6.65 (1H, d, J = 2.0 Hz, H-5), 6.61 (1H, dd, J = 8.0, 2.0 Hz, H-9), 3.85 (3H, s, OMe), 3.84 (3H, s, OMe), 3.45 (2H, dt, J = 7.2, 7.2 Hz, H2-2), 2.94 (2H, t, J = 8.0 Hz, H2-12), 2.68 (2H, t, J = 7.2 Hz, H2-3), 2.42 (2H, t, J = 8.0 Hz, H2-11); 13C NMR (CDCl3, 100 MHz) δC 172.0 (C-10), 149.0 (C-6), 147.7 (C-7), 141.0 (C-13), 131.3 (C-4), 128.6 (C-15 and C-17), 128.3 (C-14 and C-18), 126.2 (C-16), 120.6 (C-9), 111.8 (C-5), 111.3 (C-8), 55.9 (OMe × 2), 40.6 (C-2), 38.5 (C-11), 35.2 (C-3), 31.7 (C-12); (+)-ESIMS m/z 314 [M + H]+; (+)-HRESIMS [M + H]+ 314.1748 (calcd. for C19H24NO3, 314.1751).
3.2.9. N-(3,4-Dimethoxyphenethyl)palmitamide (27)
To a cold (0 °C) solution of 2-(3,4-dimethoxyphenyl)ethylamine (21) (0.20 mL, 1.10 mmol) and Et3N (0.70 mL, 5.0 mmol) in THF (10 mL) was added palmitoyl chloride (1.18 mL, 3.86 mmol). The milky white solution was warmed to room temperature and then the solvents were removed in vacuo. CHCl3 (20 mL) was added, the solution washed with 10% aq. NaCO3 (50 mL), H2O (20 mL) and brine (20 mL) and then the organic phase was dried in vacuo. The residue was triturated with hexane (10 mL) and EtOAc (5 mL) to give 27 as a white solid (0.30 g, 65% yield).
Mp 94.0–95.1 °C; Rf (3 EtOAc:1 Hex) 0.73; IR νmax (ATR) 3301, 2955, 2918, 1705, 1638, 1591, 1232, 1140 cm−1; 1H NMR (CDCl3, 400 MHz) δH 6.80 (1H, d, J = 8.4 Hz, H-8), 6.73–6.71 (2H, m, H-5 and H-9), 5.43 (1H, s, NH-1), 3.86 (6H, s, OMe), 3.49 (2H, dt, J = 7.2, 7.2 Hz, H2-2), 2.75 (2H, t, J = 7.2 Hz, H2-3), 2.11 (2H, t, J = 7.6 Hz, H2-11), 1.58 (2H, br t, J = 7.2 Hz, H2-12), 1.24 (24H, br s, H2-13–H2-24), 0.87 (3H, t, J = 7.2 Hz, H3-25); 13C NMR (CDCl3, 100 MHz) δC 173.2 (C-10), 149.2 (C-6), 147.8 (C-7), 131.6 (C-4), 120.8 (C-9), 112.0 (C-5), 111.4 (C-8), 56.0 (OMe × 2), 40.7 (C-2), 37.0 (C-11), 35.5 (C-3), 32.1 (C-23), 29.8 (C-13–C-22), 25.9 (C-12), 22.8 (C-24), 14.3 (C-25); (+)-ESIMS m/z 420 [M + H]+; (+)-HRESIMS [M + H]+ 420.3460 (calcd. for C26H45NO3, 420.3472).
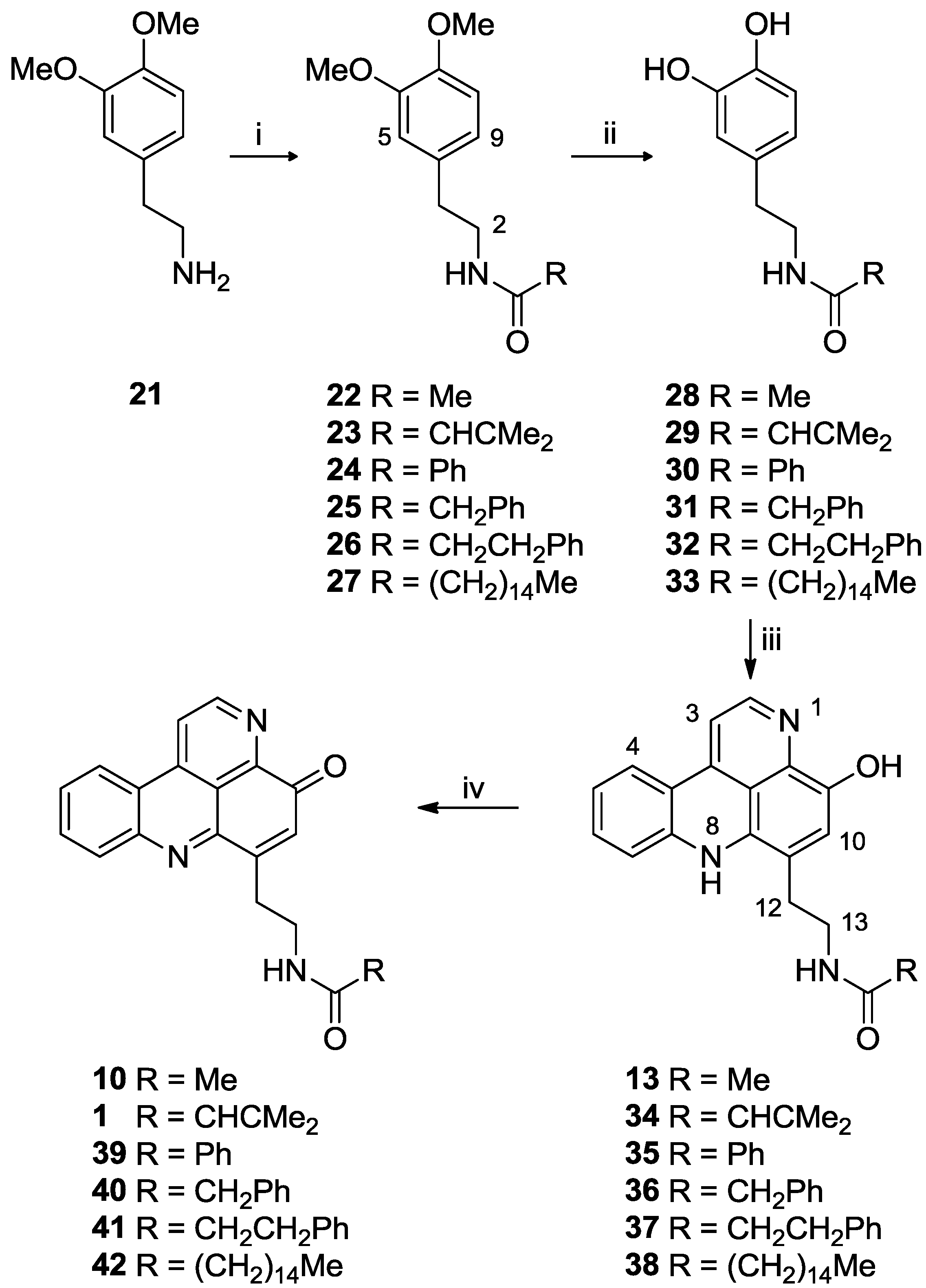
3.2.10. General Procedure for the Preparation of N-Acyl Dopamine Analogues 28–33
To a stirred solution of 3,4-dimethoxyphenethylamide 28–33 in dry CH2Cl2 (20 mL) in a salted ice bath, boron tribromide (10 equiv.) in dry CH2Cl2 (10 mL) was added dropwise. The solution turned from yellow to orange, and was stirred under N2 for 20 h with temperature rising to room temperature. MeOH (3 mL) and saturated brine (5 mL) were then added dropwise. EtOAc (30 mL) was added, the organic phase washed with H2O (30 mL), dried (MgSO4) and solvent removed in vacuo to afford the N-acyl dopamine analogue. The product was used in the subsequent reaction without further purification.
3.2.10.1. N-Acetyl Dopamine (28)
From N-(3,4-dimethoxyphenethyl)acetamide (22) (200 mg, 0.9 mmol) to afford 28 as a yellow oil (158 mg, 90% yield).
IR νmax (ATR) 3215, 1624, 1558, 1439, 1284 cm−1; 1H NMR (CD3OD, 400 MHz) δH 6.69 (1H, d, J = 8.0 Hz, H-8), 6.65 (1H, d, J = 2.0 Hz, H-5), 6.45 (1H, dd, J = 8.0, 2.0 Hz, H-9), 3.30 (2H, t, J = 7.2 Hz, H2-2), 2.60 (2H, t, J = 7.2 Hz, H2-3), 1.89 (3H, s, H3-11); 13C NMR (CD3OD, 100 MHz) δC 172.1 (C-10), 144.8 (C-6), 143.3 (C-7), 130.7 (C-4), 119.8 (C-9), 115.6 (C-8), 115.1 (C-5), 41.0 (C-2), 34.4 (C-3), 21.2 (C-11); (+)-ESIMS m/z 196 [M + H]+; (+)-HRESIMS [M + H]+ 196.0970 (calcd. for C10H14NO3, 196.0968).
3.2.10.2. N-(3,4-Dihydroxyphenethyl)-3-methylbut-2-enamide (29)
From N-(3,4-dimethoxyphenethyl)-3-methylbut-2-enamide (23) (90 mg, 0.35 mmol) to afford 29 as a yellow oil (81 mg, 98% yield).
IR νmax (ATR) 3240, 2976, 1657, 1598, 1442, 1165 cm−1; 1H NMR (CD3OD, 400 MHz) δH 6.77 (1H, d, J = 8.0 Hz, H-8), 6.65 (1H, d, J = 2.0 Hz, H-5), 6.51 (1H, dd, J = 8.0, 2.0 Hz, H-9), 5.50 (1H, s, H-11), 3.30 (2H, t, J = 8.0 Hz, H2-2), 2.62 (2H, t, J = 8.0 Hz, H2-3), 2.06 (3H, s, H3-14), 1.80 (3H, s, H3-13); 13C NMR (CD3OD, 100 MHz) δC 169.7 (C-10), 151.4 (C-12), 146.2 (C-6), 144.7 (C-7), 132.2 (C-4), 121.0 (C-9), 119.6 (C-11), 116.9 (C-5), 116.3 (C-8), 42.1 (C-2), 36.1 (C-3), 27.2 (C-13), 20.0 (C-14); (+)-ESIMS m/z 236 [M + H]+; (+)-HRESIMS [M + H]+ 236.1275 (calcd. for C13H18NO3, 236.1281).
3.2.10.3. N-(3,4-Dihydroxyphenethyl)benzamide (30)
From N-(3,4-dimethoxyphenethyl)benzamide (24) (196 mg, 0.69 mmol) to afford 30 as a yellow oil (150 mg, 85% yield).
IR νmax (ATR) 3251, 2257, 1634, 1529, 1285 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 8.49 (1H, t, J = 5.6 Hz, NH-1), 7.83–7.80 (2H, m, H-12 and H-16), 7.51 (1H, tt, J = 7.2, 1.6 Hz, H-14), 7.45 (2H, td, J = 6.8, 1.6 Hz, H-13 and H-15), 6.65–6.62 (2H, m, H-5 and H-8), 6.47 (1H, dd, J = 8.0, 2.0 Hz, H-9), 3.39 (2H, dt, J = 8.0, 5.6 Hz, H2-2), 2.65 (2H, t, J = 8.0 Hz, H2-3); 13C NMR (DMSO-d6, 100 MHz) δC 166.0 (C-10), 145.0 (C-6), 143.4 (C-7), 134.6 (C-11), 130.9 (C-14), 130.2 (C-4), 128.2 (C-12 and C-16), 127.0 (C-13 and C-15), 119.2 (C-9), 115.9 (C-5), 115.4 (C-8), 41.2 (C-2), 34.6 (C-3); (+)-ESIMS m/z 258 [M + H]+; (+)-HRESIMS [M + H]+ 258.112 (calcd. for C15H16NO3, 258.1125).
3.2.10.4. N-(3,4-Dihydroxyphenethyl)-2-phenylacetamide (31)
From N-(3,4-dimethoxyphenethyl)-2-phenylacetamide (25) (200 mg, 0.67 mmol) to afford 31 as a yellow oil (142 mg, 79% yield).
IR ν
max (ATR) 3546, 3399, 3236, 1636, 1613, 1524, 1495, 1358, 1282, 1193 cm
−1;
1H NMR (CD
3OD, 400 MHz) δ
H 7.31–7.26 (2H, m, H-14, H-16), 7.24–7.30 (3H, m, H-13, H-15 and H-17), 6.65 (1H, d,
J = 8.0 Hz, H-8), 6.62 (1H, d,
J = 2.0 Hz, H-5), 6.45 (1H, dd,
J = 8.0, 2.0 Hz, H-9), 3.46 (2H, s, H
2-11), 3.36–3.33 (2H, m, H
2-2), 2.62 (2H, t,
J = 7.2 Hz, H
2-3);
13C NMR (CD
3OD, 100 MHz) δ
C 174.1 (C-10), 146.2 (C-6), 144.8 (C-7), 136.5 (C-12), 131.9 (C-4), 130.1 (C-13 and C-17), 129.6 (C-14 and C-16), 127.9 (C-15), 121.2 (C-9), 116.7 (C-5), 116.4 (C-8), 43.9 (C-11), 42.4 (C-2), 35.8 (C-3); (+)-ESIMS
m/z 272 [M + H]
+; (+)-HRESIMS [M + H]
+ 272.1272 (calcd. for C
16H
18NO
3, 272.1281).
1H NMR data agreed with literature [
25].
3.2.10.5. N-(3,4-Dihydroxyphenethyl)-3-phenylpropanamide (32)
From N-(3,4-dimethoxyphenethyl)-3-phenylpropanamide (26) (298 mg, 0.95 mmol) to afford 32 as a yellow oil (240 mg, 89% yield).
IR ν
max (ATR) 3214, 1604, 1521, 1446, 1281, 1190 cm
−1;
1H NMR (CD
3OD, 400 MHz) δ
H 7.15 (2H, dt,
J = 8.4, 0.8 Hz, H-15, H-17), 7.08–7.06 (3H, m, H-14, H-16 and H-18), 6.60 (1H, d,
J = 8.0 Hz, H-8), 6.56 (1H, d,
J = 2.4 Hz, H-5), 6.37 (1H, dd,
J = 8.0, 2.4 Hz, H-9), 3.22–3.20 (2H, m, H
2-2), 2.78 (2H, t,
J = 8.2 Hz, H
2-12), 2.48 (2H, t,
J = 7.6 Hz, H
2-3), 2.39 (2H, t,
J = 8.2 Hz, H
2-11);
13C NMR (CDCl
3, 100 MHz) δ
C 175.6 (C-10), 146.1 (C-6), 144.6 (C-7), 141.8 (C-13), 131.9 (C-4), 129.5 (C-14, C-15, C-17, and C-18), 127.3 (C-16), 121.1 (C-9), 116.8 (C-5), 116.4 (C-8), 42.5 (C-2), 38.7 (C-11), 35.7 (C-3), 33.0 (C-12); (+)-ESIMS
m/z 286 [M+H]
+; (+)-HRESIMS [M+H]
+ 286.1433 (calcd. for C
17H
20NO
3, 286.1438).
1H NMR data agreed with literature [
25].
3.2.10.6. N-(3,4-Dihydroxyphenethyl)palmitamide (33)
From N-(3,4-dimethoxyphenethyl)palmitamide (27) (200 mg, 0.48 mmol) to afford 33 as a colourless oil (150 mg, 75% yield).
IR ν
max (ATR) 3546, 3401, 3237, 2918, 2059, 1637, 1554, 1356, 1194, 1119 cm
−1;
1H NMR (DMSO-
d6, 400 MHz) δ
H 7.78 (1H, t,
J = 5.6 Hz, NH-1), 6.61 (1H, d,
J = 8.0 Hz, H-8), 6.55 (1H, d,
J = 2.5 Hz, H-5), 6.41 (1H, dd,
J = 8.0, 2.5 Hz, H-9), 3.14 (2H, dt,
J = 7.2, 5.6 Hz, H
2-2), 2.50 (2H, t,
J = 7.2 Hz, H
2-3), 2.01 (2H, t,
J = 7.6 Hz, H
2-11), 1.45 (2H, t,
J = 6.8 Hz, H
2-12), 1.23 (24H, br s, H
2-13–H
2-24), 0.87 (3H, t,
J = 7.2 Hz, H
3-25);
13C NMR (DMSO-
d6, 100 MHz) δ
C 171.8 (C-10), 144.9 (C-6), 143.4 (C-7), 130.2 (C-4), 119.1 (C-9), 115.8 (C-5), 115.3 (C-8), 40.4 (C-2), 35.4 (C-11), 34.7 (C-3), 31.2 (C-23), 28.7 (C-13–C-22), 25.2 (C-12), 22.0 (C-24), 13.9 (C-25); (+)-ESIMS
m/z 391 [M + H]
+; (+)-HRESIMS [M + H]
+ 392.3156 (calcd. for C
24H
42NO
3, 392.3159).
1H NMR data agreed with literature [
26].
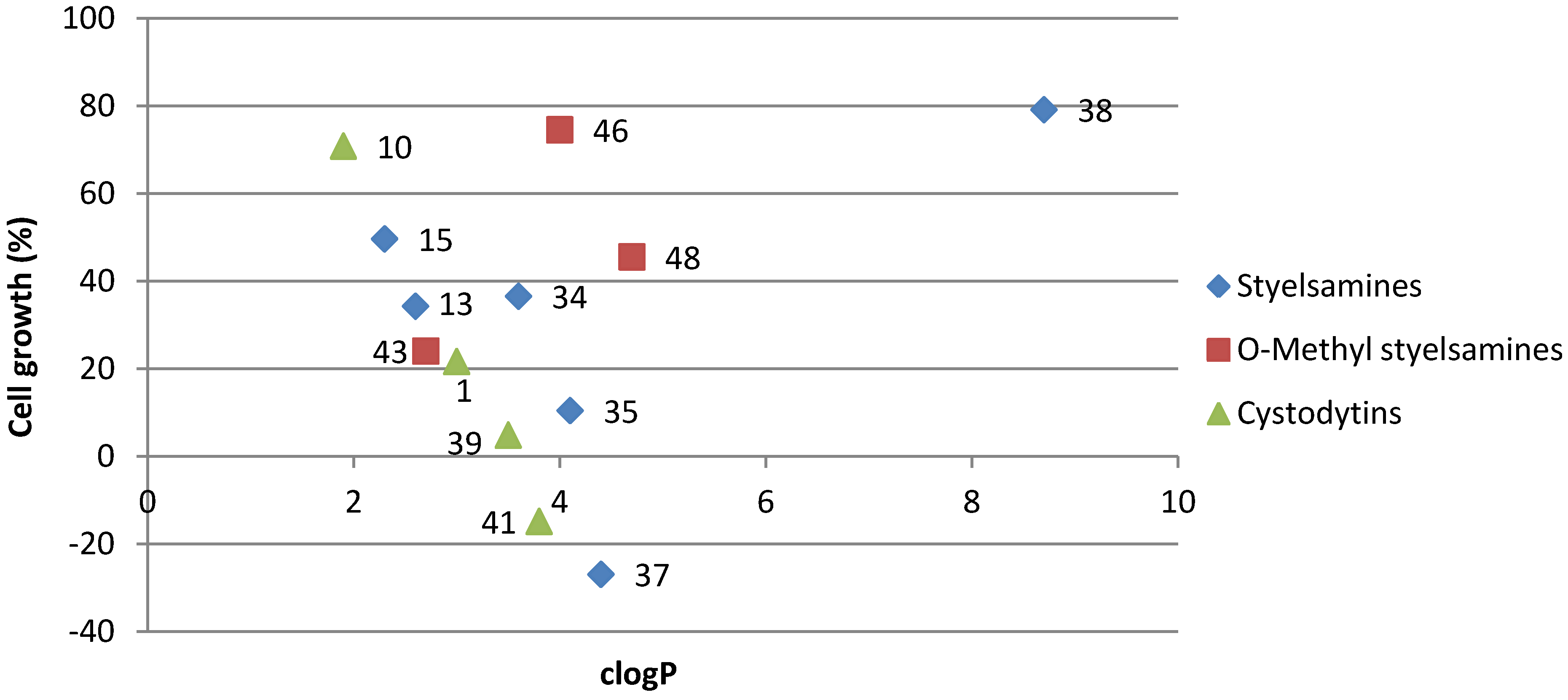
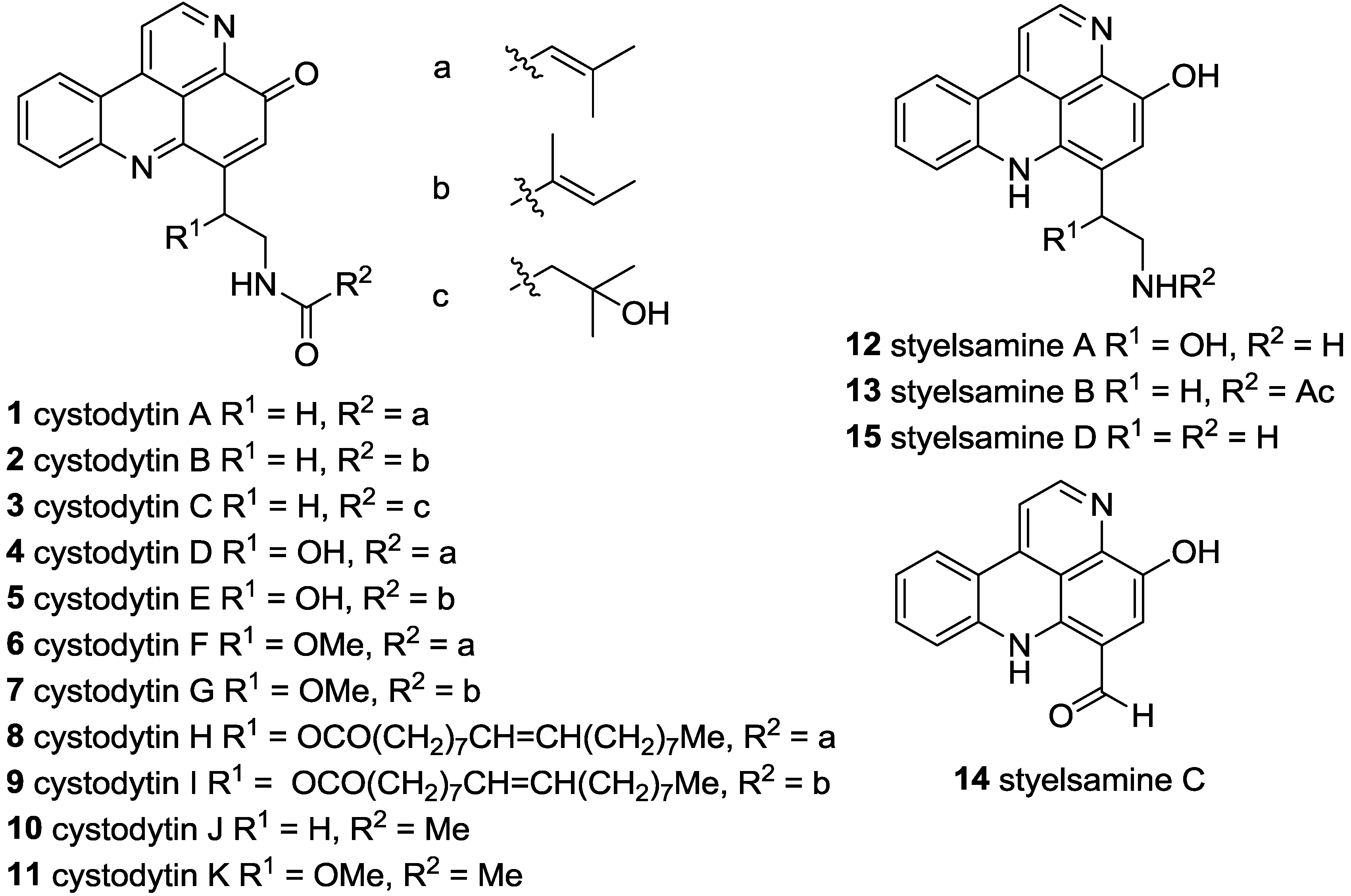
3.2.11. General Procedure for the Preparation of N-Acyl Styelsamine Analogues 13, 34–38
To a solution of N-acyl dopamine (1 equiv.) in degassed 2:1 MeOH:AcOH (6 mL) was added kynuramine dihydrobromide (1.05 equiv.) followed by CeCl3·7H2O (0.2 equiv.). To the stirred yellow solution under N2 was added Ag2O (2–4 equiv.) and the suspension warmed to 40 °C for 1.5 h. The yellow solution was filtered and added dropwise to stirring HCl (6 N, 15 mL) at 90 °C and heated for a further 30 min during which time the colour of the solution changed to purple. The solution was dried in vacuo and the residue purified by either RP-2 or RP-18 column chromatography using H2O (0.05% TFA)–MeOH solvent mixtures to afford the product as a purple oil.
3.2.11.1. Styelsamine B Trifluoroacetate (13)
Using the general procedure, reaction of N-acetyl dopamine (28) (52 mg, 0.27 mmol) with kynuramine dihydrobromide (92 mg, 0.28 mmol), CeCl3.7H2O (14.0 mg, 0.04 mmol) and Ag2O (123 mg, 0.53 mmol) afforded, after RP-2 column chromatography, 13 as a purple oil (22.0 mg, 19% yield).
IR ν
max (ATR) 3389, 3075, 1679, 1205, 1138 cm
−1;
Rt = 5.99 min;
1H NMR (CD
3OD, 400 MHz) δ
H 7.93 (1H, d,
J = 5.8 Hz, H-2), 7.81 (1H, d,
J = 7.8 Hz, H-4), 7.52 (1H, t,
J = 8.0 Hz, H-6), 7.46 (1H, d,
J = 8.0 Hz, H-7), 7.20 (1H, s, H-10), 7.14 (1H, d,
J = 5.8 Hz, H-3), 7.10 (1H, t,
J = 7.8 Hz, H-5), 3.20 (2H, t,
J = 6.8 Hz, H
2-13), 2.79 (2H, t,
J = 6.8 Hz, H
2-12), 2.04 (3H, s, H
3-16);
13C NMR (CD
3OD, 100 MHz) δ
C 174.7 (C-15), 150.8 (C-3a), 143.4 (C-2), 142.3 (C-7a), 138.0 (C-11), 136.2 (C-6), 129.7 (C-8a), 127.2 (C-11a), 126.0 (C-4), 123.9 (C-5), 122.6 (C-10), 121.6 (C-11b), 118.9 (C-7), 117.3 (C-9), 115.2 (C-3b), 105.6 (C-3), 39.3 (C-13), 31.9 (C-12), 22.5 (C-16); (+)-ESIMS
m/z 320 [M + H]
+; (+)-HRESIMS [M + H]
+ 320.1391 (calcd. for C
19H
18N
3O
2, 320.1394).
1H and
13C NMR data agreed with literature [
6].
3.2.11.2. Styelsamine-N14-3-methyl-but-2-enamide (34)
Using the general procedure, reaction of N-(3,4-dihydroxyphenethyl)-3-methylbut-2-enamide (29) (98 mg, 0.42 mmol) with kynuramine dihydrobromide (142 mg, 0.44 mmol), CeCl3·7H2O (22 mg, 0.06 mmol) and Ag2O (360 mg, 1.57 mmol) afforded, after RP-2 column chromatography, 34 as a purple oil (12.0 mg, 6% yield).
IR νmax (ATR) 3374, 3069, 1699, 1684, 1499, 1205 cm−1; Rt = 9.53 min; 1H NMR (CD3OD, 400 MHz) δH 7.89 (1H, d, J = 5.8 Hz, H-2), 7.78 (1H, d, J = 8.0 Hz, H-4), 7.47 (2H, br s, H-6 and H-7), 7.14 (1H, s, H-10), 7.11–7.05 (2H, m, H-3 and H-5), 5.68 (1H, s, H-16), 3.15 (2H, t, J = 8.4 Hz, H2-13), 2.75 (2H, t, J = 7.2 Hz, H2-12), 2.22 (3H, s, H3-19), 1.87 (3H, s, H3-18); 13C NMR (CD3OD, 100 MHz) δC 170.9 (C-15), 153.4 (C-17), 151.0 (C-3a), 143.3 (C-2), 142.5 (C-7a), 137.9 (C-11), 136.1 (C-6), 130.6 (C-8a), 127.0 (C-11a), 126.0 (C-4), 123.8 (C-5), 122.7 (C-11b and C-10), 118.9 (C-16), 118.8 (C-7), 117.5 (C-9), 115.4 (C-3b), 105.4 (C-3), 39.2 (C-13), 32.5 (C-12), 27.5 (C-18), 20.3 (C-19); (+)-ESIMS m/z 360 [M + H]+; (+)-HRESIMS [M + H]+ 360.1723 (calcd. for C22H22N3O2, 360.1707).
3.2.11.3. Styelsamine-N14-benzamide (35)
Using the general procedure, reaction of N-(3,4-dihydroxyphenethyl)benzamide (30) (43.0 mg, 0.17 mmol) with kynuramine dihydrobromide (57.0 mg, 0.18 mmol), CeCl3·7H2O (9.0 mg, 0.03 mmol) and Ag2O (97 mg, 0.42 mmol) afforded, after RP-2 column chromatography, 35 as a purple oil (13.0 mg, 15% yield).
IR νmax (ATR) 3327, 3052, 1654, 1582, 1205 cm−1; Rt = 9.17 min; 1H NMR (CD3OD, 400 MHz) δH 11.66 (1H, s, NH-1), 8.97 (1H, t, J = 5.4 Hz, NH-14), 8.05 (1H, d, J = 8.0 Hz, H-2), 8.00 (1H, d, J = 7.8 Hz, H-4), 7.88 (2H, d, J = 7.6 Hz, H-17 and H-21), 7.70 (1H, d, J = 8.0 Hz, H-7), 7.63 (1H, t, J = 8.0 Hz, H-6), 7.56 (1H, dt, J = 6.4, 0.8 Hz, H-19), 7.47 (2H, t, J = 8.0 Hz, H-18 and H-20), 7.35–7.31 (2H, m, H-3 and H-10), 7.19 (1H, t, J = 7.8 Hz, H-5), 3.49 (2H, t, J = 7.6 Hz, H2-13), 3.05 (2H, t, J = 7.6 Hz, H2-12); 13C NMR (CD3OD, 100 MHz) δC 171.3 (C-15), 151.4 (C-3a), 143.5 (C-2), 142.7 (C-7a), 138.1 (C-11), 136.4 (C-6), 134.9 (C-16), 133.1 (C-19), 130.2 (C-11b), 129.7 (C-18 and C-20), 128.4 (C-17 and C-21), 127.6 (C-11a), 126.2 (C-4), 124.0 (C-5), 122.8 (C-10), 122.0 (C-8a), 119.2 (C-7), 117.5 (C-9), 115.5 (C-3b), 105.7 (C-3), 39.9 (C-13), 32.2 (C-12); (+)-ESIMS m/z 382 [M + H]+; (+)-HRESIMS [M + H]+ 382.1538 (calcd. for C24H20N3O2, 382.1550).
3.2.11.4. Styelsamine-N14-2-phenylacetamide (36)
Using the general procedure, reaction of N-(3,4-dihydroxyphenethyl)-2-phenylacetamide (31) (142 mg, 0.52 mmol) with kynuramine dihydrobromide (178 mg, 0.55 mmol), CeCl3·7H2O (28.0 mg, 0.08 mmol) and Ag2O (360 mg, 1.57 mmol) afforded, after RP-2 column chromatography, 36 as a purple oil (28.0 mg, 11% yield).
IR νmax (ATR) 3283, 3068, 1661, 1583 cm−1; Rt = 8.25 min; 1H NMR (CD3OD, 400 MHz) δH 8.05 (1H, d, J = 6.8 Hz, H-2), 8.00 (1H, d, J = 8.4 Hz, H-4), 7.62 (1H, br d, J = 8.2 Hz, H-6), 7.57 (1H, d, J = 8.2 Hz, H-7), 7.35–7.33 (1H, m, H-3), 7.33 (1H, s, H-10), 7.30–7.28 (3H, m, H-5, H-19 and H-21), 7.25–7.17 (3H, m, H-18, H-20 and H-22), 3.56 (2H, s, H2-16), 3.35 (2H, t, J = 8.0 Hz, H2-13), 2.94 (2H, t, J = 7.6 Hz, H2-12); 13C NMR (CD3OD, 100 MHz) δC 175.4 (C-15), 151.4 (C-3a), 143.5 (C-2), 142.7 (C-7a), 138.1 (C-11), 136.7 (C-17), 136.3 (C-6), 130.2 (C-18 and C-22), 129.7 (C-8a, C-19 and C-21), 128.1 (C-11a), 127.5 (C-20), 126.2 (C-4), 124.0 (C-5), 122.8 (C-10), 122.0 (C-11b), 119.1 (C-7), 117.3 (C-9), 115.5 (C-3b), 105.7 (C-3), 43.8 (C-16), 39.5 (C-13), 31.7 (C-12); (+)-ESIMS m/z 396 [M + H]+; (+)-HRESIMS [M + H]+ 396.1701 (calcd. for C25H22N3O2, 396.1707).
3.2.11.5. Styelsamine-N14-3-phenylpropanamide (37)
Using the general procedure, reaction of N-(3,4-dihydroxyphenethyl)-3-phenylpropanamide (32) (80.0 mg, 0.28 mmol) with kynuramine dihydrobromide (96.0 mg, 0.30 mmol), CeCl3·7H2O (15.0 mg, 0.04 mmol) and Ag2O (163 mg, 0.70 mmol) afforded, after RP-2 column chromatography, 37 as a purple oil (29.0 mg, 20% yield).
IR νmax (ATR) 3321, 1661, 1584, 1202, 1130, 1015 cm−1; Rt = 10.60 min; 1H NMR (DMSO-d6, 400 MHz) δH 13.49 (1H, br s, NH-1), 11.48 (1H, br s, OH), 10.80 (1H, br s, NH-8), 8.46 (1H, t, J = 5.6 Hz, NH-14), 8.26 (1H, d, J = 6.4 Hz, H-2), 8.22 (1H, d, J = 8.0 Hz, H-4), 7.71 (2H, d, J = 4.0 Hz, H-6 and H-7), 7.55 (1H, d, J = 6.4 Hz, H-3), 7.45 (1H, s, H-10), 7.28-7.14 (6H, m, H-5, H-19, H-20, H-21, H-22 and H-23), 3.28 (2H, dt, J = 6.8, 5.6 Hz, H2-13), 2.96 (2H, t, J = 6.8 Hz, H2-12), 2.85 (2H, t, J = 7.6 Hz, H2-17), 2.45 (2H, t, J = 7.6 Hz, H2-16); 13C NMR (DMSO-d6, 100 MHz) δC 173.2 (C-15), 149.2 (C-3a), 143.4 (C-2), 141.1 (C-7a and C-18), 136.7 (C-11), 135.0 (C-6), 128.3 (C-11a), 128.4 (C-8a), 128.2 (C-19, C-20, C-22 and C-23), 126.0 (C-21), 125.5 (C-4), 122.4 (C-5), 121.7 (C-10), 120.4 (C-11b), 117.7 (C-7), 116.2 (C-9), 113.9 (C-3b), 105.0 (C-3), 37.7 (C-13), 36.8 (C-16), 31.1 (C-17), 30.3 (C-12); (+)-ESIMS m/z 410 [M + H]+; (+)-HRESIMS [M + H]+ 410.1875 (calcd. for C26H24N3O2, 410.1863).
3.2.11.6. Styelsamine-N14-palmitamide (38)
Using the general procedure, reaction of N-(3,4-dihydroxyphenethyl)palmitamide (33) (150.0 mg, 0.38 mmol) with kynuramine dihydrobromide (122 mg, 0.38 mmol), CeCl3.7H2O (2.0 mg, 0.05 mmol) and Ag2O (210 mg, 0.89 mmol) afforded, after RP-18 column chromatography, 38 as a purple oil (40 mg, 16% yield).
IR νmax (ATR) 3286, 3074, 2917, 1685, 1560, 1511, 1467, 1200 cm−1; 1H NMR (CD3OD, 400 MHz) δH 8.13 (2H, d, J = 6.4 Hz, H-2 and H-4), 7.74–7.67 (2H, m, H-6 and H-7), 7.47 (1H, d, J = 6.8 Hz, H-3), 7.42 (1H, s, H-10), 7.27 (1H, t, J = 7.4 Hz, H-5), 3.37 (2H, t, J = 7.6 Hz, H2-13), 3.06 (2H, t, J = 7.6 Hz, H2-12), 2.26–2.24 (2H, m, H2-16), 1.65 (2H, t, J = 7.2 Hz, H2-17), 1.28 (24H, br s, H2-18–H2-29), 0.87 (3H, J = 7.2 Hz, H3-30); 13C NMR (CD3OD, 100 MHz) δC 177.8 (C-15), 151.7 (C-3a), 143.7 (C-2), 142.9 (C-7a), 138.2 (C-11), 136.4 (C-6), 130.4 (C-8a), 127.8 (C-11a), 126.3 (C-4), 124.1 (C-5), 122.9 (C-10), 122.3 (C-11b), 119.2 (C-7), 117.6 (C-9), 115.7 (C-3b), 105.8 (C-3), 39.4 (C-13), 37.0 (C-16), 33.1 (C-28), 32.1 (C-12), 30.8 (C-18–C-27), 26.9 (C-17), 23.8 (C-29), 14.5 (C-30); (+)-ESIMS m/z 516 [M + H]+; (+)-HRESIMS [M + H]+ 516.3589 (calcd. for C33H46N3O2, 516.3585).
3.2.12. General Procedure for the Preparation of N-Acyl Cystodytin Analogues 1, 10, 39–42
To a stirring solution of styelsamine analogue (1 equiv.) in MeOH (1.0 mL) was added Ag2O (1.5 equiv.) followed by sat. NaHCO3 (3 mL) dropwise. The purple mixture turned to red/orange then to yellow/green. The mixture was filtered, H2O (1.0 mL) and EtOAc (5.0 mL) added and the organic phase separated and dried in vacuo to afford the product as a yellow oil or solid.
3.2.12.1. Cystodytin J (10)
Using the general procedure, reaction of styelsamine B (
13) (7.0 mg, 0.016 mmol) with Ag
2O (5.0 mg, 0.022 mmol) afforded
10 as a yellow oil (4.0 mg, 79% yield) [
10].
1H NMR (CDCl
3, 400 MHz) δ
H 8.94 (1H, d,
J = 5.3 Hz, H-2), 8.59 (1H, d,
J = 8.1 Hz, H-4), 8.55 (1H, d,
J = 5.3 Hz, H-3), 8.32 (1H, dd,
J = 8.4, 1.1 Hz, H-7), 7.91 (1H, dt,
J = 8.4, 1.4 Hz, H-6), 7.83 (1H, dt,
J = 8.1, 1.1 Hz, H-5), 6.95 (1H, s, H-10), 3.78 (2H, dt,
J = 6.4, 6.4 Hz, H
2-13), 3.32 (2H, t,
J = 6.4 Hz, H
2-12), 1.59 (3H, s, H
3-16); (+)-ESIMS
m/z 318 [M + H]
+; (+)-HRESIMS [M + H]
+ 318.1230 (calcd. for C
19H
16N
3O
2, 318.1237).
1H NMR data agreed with literature [
4].
3.2.12.2. Cystodytin A (1)
Using the general procedure, reaction of styelsamine analogue 34 (15.0 mg, 0.032 mmol) with Ag2O (8.0 mg, 0.042 mmol) afforded 1 as a yellow oil (7.0 mg, 62% yield).
1H NMR (CDCl
3, 400 MHz) δ
H 9.24 (1H, d,
J = 5.6 Hz, H-2), 8.60 (1H, dd,
J = 7.8, 1.4 Hz, H-4), 8.57 (2H, d,
J = 5.6 Hz, H-3), 8.32 (1H, dd,
J = 7.8, 1.0 Hz, H-7), 7.94 (1H, dt,
J = 7.8, 1.4 Hz, H-6), 7.85 (1H, dt,
J = 7.8, 1.0 Hz, H-5), 6.95 (1H, s, H-10), 5.85 (1H, br s, NH-14), 5.50 (1H, s, H-16), 3.81 (2H, dt,
J = 6.4, 6.4 Hz, H
2-13), 3.36 (2H, t,
J = 6.4 Hz, H
2-12), 2.10 (3H, s, H
3-19), 1.79 (3H, s, H
3-18); (+)-ESIMS
m/z 358 [M + H]
+; (+)-HRESIMS [M + H]
+ 358.1544 (calcd. for C
22H
20N
3O
2, 358.1550).
1H NMR data agreed with literature [
2].
3.2.12.3. Cystodytin-N14-benzamide (39)
Using the general procedure, reaction of styelsamine analogue 35 (20.0 mg, 0.040 mmol) with Ag2O (12.0 mg, 0.053 mmol) afforded 39 as a yellow oil (8.00 mg, 52% yield).
Rf (5% MeOH/CH2Cl2) 0.37; IR νmax (smear) 3298, 1653, 1550, 1432, 1202, 766 cm−1; 1H NMR (CDCl3, 400 MHz) δH 9.24 (1H, d, J = 5.6 Hz, H-2), 8.60 (1H, d, J = 7.6 Hz, H-4), 8.58 (1H, d, J = 5.6 Hz, H-3), 8.30 (1H, d, J = 8.2 Hz, H-7), 7.93 (1H, dt, J = 8.2, 1.2 Hz, H-6), 7.86 (1H, t, J = 7.6 Hz, H-5), 7.65 (2H, d, J = 7.7 Hz, H-17 and H-21), 7.42 (1H, t, J = 1.1 Hz, H-19), 7.31 (2H, t, J = 7.7 Hz, H-18 and H-20), 7.01 (1H, s, H-10), 6.86 (1H, br s, NH-14), 3.98 (2H, dt, J = 6.4, 6.4 Hz, H2-13), 3.44 (2H, t, J = 6.4 Hz, H2-12); 13C NMR (CDCl3, 100 MHz) δC 181.8 (C-11), 173.8 (C-15), 152.7 (C-9), 151.6 (C-8a), 149.7 (C-2), 147.1 (C-11a), 145.3 (C-7a), 137.5 (C-3a), 134.4 (C-16), 132.1 (C-10), 131.9 (C-7), 131.3 (C-6), 131.2 (C-18 and C-20), 129.8 (C-5), 128.5 (C-19), 126.5 (C-17 and C-21), 123.0 (C-4), 121.4 (C-3b), 119.3 (C-3), 118.4 (C-11b), 40.0 (C-13), 31.0 (C-12); (+)-ESIMS m/z 380 [M + H]+; (+)-HRESIMS [M + H]+ 380.1411 (calcd. for C24H18N3O2 380.1394).
3.2.12.4. Cystodytin-N14-2-phenylacetamide (40)
Using the general procedure, reaction of styelsamine analogue 36 (21.0 mg, 0.041 mmol) with Ag2O (12.0 mg, 0.053 mmol) afforded 40 as a yellow oil (2.1 mg, 13% yield).
Rf (5% MeOH/CH2Cl2) 0.25; IR νmax (smear) 3290, 2924, 1657, 1585, 1432, 1201, 722 cm−1; 1H NMR (CDCl3, 400 MHz) δH 9.23 (1H, d, J = 5.3 Hz, H-2), 8.58 (1H, d, J = 7.8 Hz, H-4), 8.55 (1H, d, J = 6.6 Hz, H-3), 8.25 (1H, d, J = 7.9 Hz, H-7), 7.92 (1H, t, J = 7.9 Hz, H-6), 7.84 (1H, t, J = 6.6 Hz, H-5), 7.21–7.18 (2H, m, H-19 and H-21), 7.16–7.11 (3H, m, H-18, H-20 and H-22), 6.81 (1H, s, H-10), 5.74 (1H, s, NH-14), 3.78–3.72 (2H, m, H2-13), 3.52 (2H, s, H2-16), 3.25 (2H, t, J = 6.3 Hz, H2-12); 13C NMR (CDCl3, 100 MHz) δC 183.4 (C-11), 171.2 (C-15), 151.8 (C-9), 150.3 (C-8a), 150.0 (C-2), 147.2 (C-11a), 145.4 (C-7a), 137.8 (C-3a), 134.6 (C-17), 132.8 (C-10), 132.0 (C-7), 131.7 (C-6), 129.9 (C-5), 129.3 (C-19 and C-21), 129.0 (C-18 and C-22), 127.3 (C-20), 122.9 (C-4), 121.9 (C-3b), 119.3 (C-3), 118.5 (C-11b), 43.8 (C-16), 39.3 (C-13), 31.3 (C-12); (+)-ESIMS m/z 394 [M + H]+; (+)-HRESIMS [M + H]+ 394.1552 (calcd. for C25H20N3O2 394.1550).
3.2.12.5. Cystodytin-N14-3-phenylpropanamide (41)
Using the general procedure, reaction of styelsamine analogue 37 (9.0 mg, 0.017 mmol) with Ag2O (5.0 mg, 0.022 mmol) afforded 41 as a yellow oil (5.0 mg, 71% yield).
Rf (5% MeOH/CH2Cl2) 0.13; IR νmax (smear) 3285, 3072, 2922, 1647, 1551, 1588, 1332 cm−1; 1H NMR (CDCl3, 400 MHz) δH 9.20 (1H, d, J = 5.5 Hz, H-2), 8.59 (1H, dd, J = 7.6, 1.4 Hz, H-4), 8.51 (1H, d, J = 5.5 Hz, H-3), 8.28 (1H, dd, J = 7.6, 1.1 Hz, H-7), 7.94 (1H, dt, J = 7.6, 1.4 Hz, H-6), 7.85 (1H, t, J = 7.6, 1.1 Hz, H-5), 7.13 (2H, td, J = 7.5, 1.5 Hz, H-20 and H-22), 7.09–7.07 (2H, m, H-19 and H-23), 7.03 (1H, tt, J = 7.1, 1.4 Hz, H-21), 6.86 (1H, s, H-10), 6.05 (1H, br s, NH-14), 3.75 (2H, dt, J = 6.2, 6.2, H2-13), 3.24 (2H, t, J = 6.2 Hz, H2-12), 2.89 (2H, t, J = 7.6 Hz, H2-17), 2.44 (2H, t, J = 7.6 Hz, H2-16); 13C NMR (CDCl3, 100 MHz) δC 184.0 (C-11), 172.4 (C-15), 152.2 (C-9), 150.7 (C-8a), 150.2 (C-2), 147.0 (C-11a), 145.5 (C-7a), 140.9 (C-18), 137.4 (C-3a), 133.0 (C-10), 132.0 (C-6 and C-7), 130.1 (C-5), 128.5 (C-20 and C-22), 128.4 (C-19 and C-23), 126.3 (C-21), 123.1 (C-4), 122.1 (C-3b), 119.4 (C-3), 118.3 (C-11b), 39.5 (C-13), 38.7 (C-16), 31.9 (C-17), 31.6 (C-12); (+)-ESIMS m/z 407 [M + H]+; (+)-HRESIMS [M + H]+ 408.1716 (calcd. for C26H22N3O2, 408.1707).
3.2.12.6. Cystodytin-N14-palmitamide (42)
Using the general procedure, reaction of styelsamine analogue 38 (37.0 mg, 0.059 mmol) with Ag2O (17.0 mg, 0.072 mmol) afforded 42 as a yellow oil (5.2 mg, 17% yield).
Rf (5% MeOH/CH2Cl2) 0.31; IR νmax (smear) 3303, 2914, 2849, 1656, 1553, 1470, 774 cm−1; 1H NMR (CDCl3, 400 MHz) δH 9.25 (1H, d, J = 5.5 Hz, H-2), 8.61 (1H, dd, J = 8.1, 1.4 Hz, H-4), 8.58 (1H, d, J = 5.5 Hz, H-3), 8.32 (1H, dd, J = 7.5, 1.3 Hz, H-7), 7.95 (1H, td, J = 7.5, 1.4 Hz, H-6), 7.84 (1H, td, J = 8.1, 1.3 Hz, H-5), 6.95 (1H, s, H-10), 6.01 (1H, s, NH-14), 3.79 (2H, dt, J = 6.4, 6.4 Hz, H2-13), 3.32 (2H, t, J = 6.4 Hz, H2-12), 2.11 (2H, d, J = 7.4 Hz, H2-16), 1.55 (2H, br s, H2-17), 1.25 (24H, br s, H2-18–H2-29), 0.88 (3H, t, J = 7.1 Hz, H3-30); 13C NMR (CDCl3, 100 MHz) δC 184.7 (C-11), 173.4 (C-15), 151.9 (C-9), 150.6 (C-8a), 150.2 (C-2), 146.9 (C-11a), 145.5 (C-7a), 137.5 (C-3a), 133.0 (C-10), 132.0 (C-6 and C-7), 130.1 (C-5), 123.3 (C-4), 122.0 (C-3b), 119.5 (C-3), 117.2 (C-11b), 39.5 (C-13), 37.0 (C-16), 32.1 (C-28), 31.8 (C-12), 29.6 (C-18–C-27), 25.9 (C-17), 22.8 (C-29), 14.3 (C-30); (+)-ESIMS m/z 514 [M + H]+; (+)-HRESIMS [M + H]+ 514.3410 (calcd. for C33H44N3O2 514.3428).
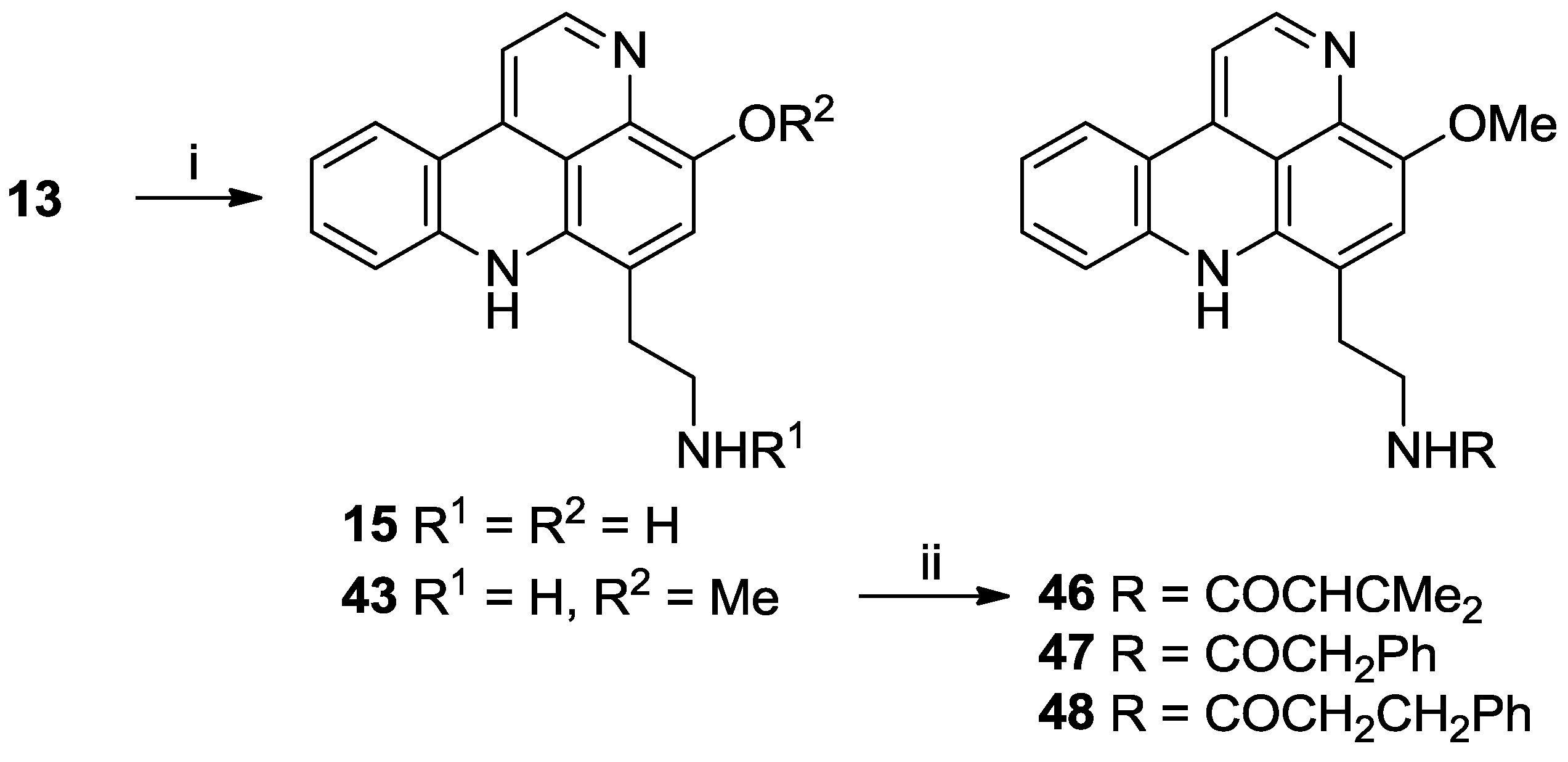
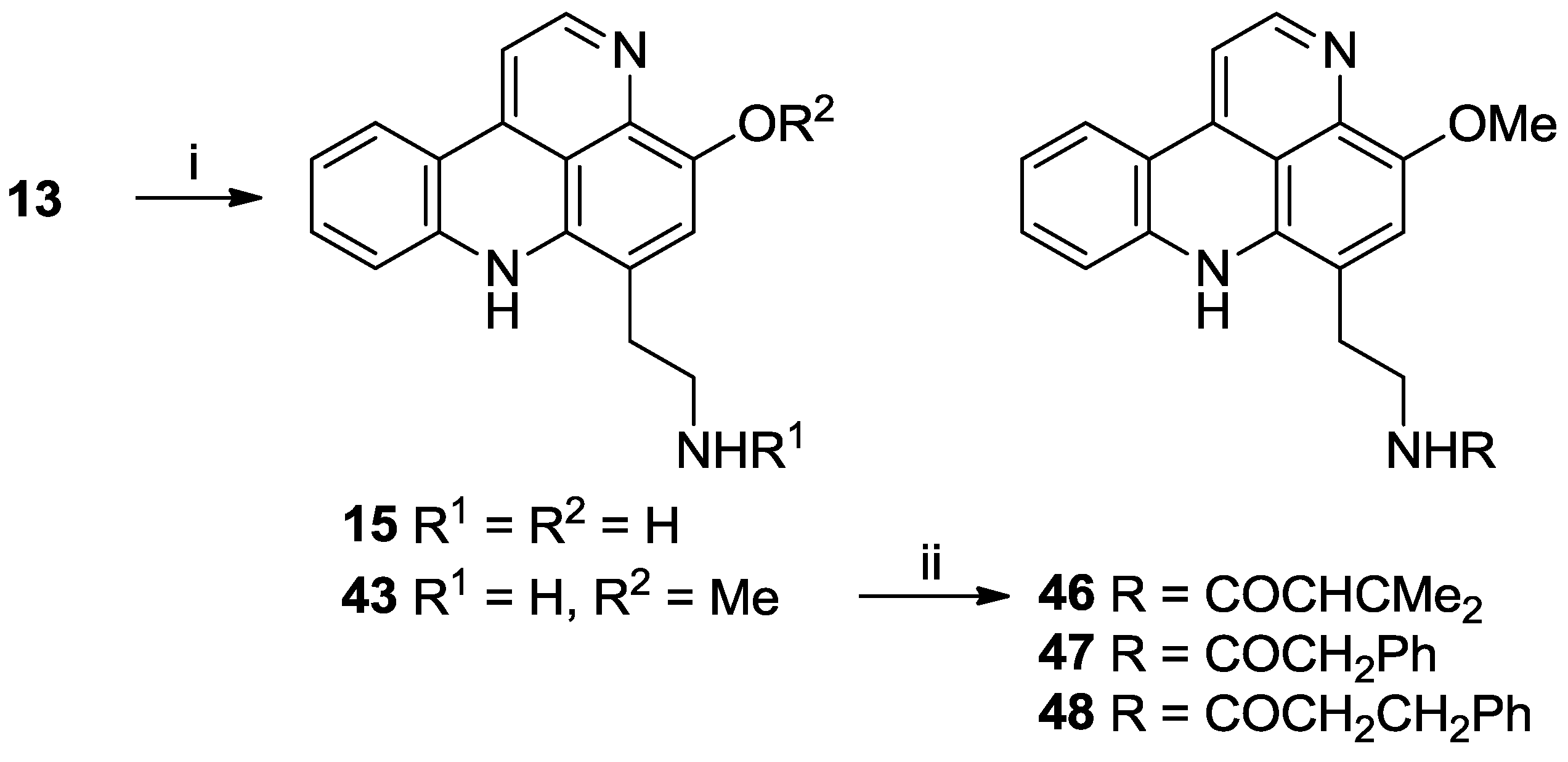
3.2.13. Styelsamine D Ditrifluoroacetate (15)
Styelsamine B (13) (35.1 mg, 0.081 mmol) was dissolved in 1:1 MeOH/4N HCl (10 mL) and heated to 80 °C. After 24 h, the solvents were removed in vacuo and the product purified by RP-18 column chromatography (H2O (0.05% TFA):MeOH (0.05% TFA) (100:0 to 85:15)) to afford 15 as a purple oil (30.5 mg, 75% yield).
IR ν
max (ATR) 3412, 1678, 1434, 1203, 1180, 1129, 765 cm
−1;
Rt = 4.68 min;
1H NMR (DMSO-
d6, 400 MHz) δ
H 13.85 (1H, s, NH-1), 10.93 (1H, br s, OH), 8.32 (1H, d,
J = 6.8 Hz, H-2), 8.25 (1H, d,
J = 7.4 Hz, H-4), 8.03 (3H, br s, NH
3-15), 7.75–7.68 (2H, m, H-6 and H-7), 7.62 (1H, d,
J = 6.8 Hz, H-3), 7.51 (1H, s, H-10), 7.25 (1H, dt,
J = 7.4, 1.5 Hz, H-5), 3.23 (2H, br t,
J = 7.5 Hz, H
2-12), 3.12–3.11 (2H, m, H
2-13);
13C NMR (DMSO-
d6, 100 MHz) δ
C 149.5 (C-3a), 143.6 (C-2), 141.5 (C-7a), 137.4 (C-11), 134.9 (C-6), 128.6 (C-8a), 126.8 (C-11a), 125.5 (C-4), 122.2 (C-5), 121.3 (C-10), 120.8 (C-11b), 118.1 (C-7), 114.2 (C-3b), 113.3 (C-9), 105.4 (C-3), 38.2 (C-13), 28.6 (C-12); (+)-ESIMS
m/z 278 [M + H]
+; (+)-HRESIMS [M + H]
+ 278.1298 (calcd. for C
17H
16N
3O, 278.1288).
1H and
13C NMR data agreed with literature [
6].
3.2.14. O-Methyl Styelsamine D Ditrifluoroacetate (43)
Styelsamine B (13) (19.0 mg, 0.044 mmol) was dissolved in 1:1 MeOH/4N HCl (10 mL) and heated to 80 °C. After 48 h, solvents were removed in vacuo and the residue purified by RP-18 column chromatography to afford 15 as a purple oil (13.4 mg, 60% yield) and 43 as a purple oil (10.3 mg, 45% yield).
IR νmax (ATR) 3382, 1675, 1429, 1178, 1138, 765 cm−1; Rt = 4.90 min; 1H NMR (DMSO-d6, 400 MHz) δH 13.85 (1H, br s, NH-1), 11.37 (1H, br s, NH-8), 8.38 (1H, d, J = 6.5 Hz, H-2), 8.28 (1H, d, J = 8.0 Hz, H-4), 8.18 (3H, br s, NH3-14), 8.04 (1H, d, J = 8.3 Hz, H-7), 7.79 (1H, s, H-10), 7.73–7.70 (2H, m, H-3 and H-6), 7.27 (1H, t, J = 8.0 Hz, H-5), 4.06 (3H, s, OMe), 3.38 (2H, t, J = 7.2 Hz, H2-12), 3.14–3.12 (2H, m, H2-13); 13C NMR (DMSO-d6, 100 MHz) δC 149.1 (C-3a), 143.5 (C-2), 141.2 (C-7a), 138.7 (C-11), 135.0 (C-6), 129.6 (C-8a), 127.3 (C-11a), 125.5 (C-4), 122.7 (C-5), 120.1 (C-11b), 119.5 (C-10), 118.0 (C-7), 117.9 (C-9), 113.8 (C-3b), 106.0 (C-3), 56.9 (OMe), 37.9 (C-13), 28.4 (C-12); (+)-ESIMS m/z 292 [M + H]+; (+)-HRESIMS [M + H]+ 292.1448 (calcd. for C18H18N3O 292.1444).
3.2.15. O-Methyl-styelsamine-N14-3-methylbut-2-enamide Trifluoroacetate (46)
To a solution of O-methyl styelsamine D (43) (11.0 mg, 0.021 mmol) in dry DMF (3 mL) was added 3,3-dimethylacrylic acid (5.96 mg, 0.060 mmol) and PyBOP (31.0 mg, 0.060 mmol) followed by Et3N (22 µL, 0.16 mmol). The solution was stirred under N2 at room temperature for 25 h. CH2Cl2 (20 mL) was added, washed with H2O (15 mL) and the organic phase dried in vacuo to give a purple/red oil. Purification using RP-2 column chromatography (H2O (0.05% TFA):MeOH (0.05% TFA) (100:0 to 40:60)) afforded 46 as a purple oil (9.1 mg, 88% yield).
IR ν
max (ATR) 3396, 1675, 1586, 1433, 1182, 1134, 765 cm
−1;
Rt = 7.58 min;
1H NMR (DMSO-
d6, 400 MHz) δ
H 13.78 (1H, br s, NH-1), 11.85 (1H, s, NH-8), 8.51 (1H, t,
J = 5.6 Hz, NH-14), 8.34 (1H, d,
J = 6.5 Hz, H-2), 8.28 (1H, d,
J = 8.2 Hz, H-4), 7.79 (1H, d,
J = 7.9 Hz, H-7), 7.76–7.72 (2H, m, H-6 and H-10), 7.68 (1H, d,
J = 6.5 Hz, H-3), 7.27 (1H, td,
J = 8.2, 1.0 Hz, H-5), 5.72 (1H, s, H-16), 4.03 (3H, s, OMe), 3.34 (2H, dt,
J = 7.2, 5.6 Hz, H
2-13), 3.08 (2H, t,
J = 7.2 Hz, H
2-12), 2.16 (3H, s, H
3-19), 1.81 (3H, s, H
3-18);
13C NMR (DMSO-
d6, 100 MHz) δ
C 167.8 (C-15), 150.1 (C-17), 149.4 (C-3a), 143.5 (C-2), 141.2 (C-7a), 138.5 (C-11), 135.2 (C-6), 129.5 (C-8a), 126.7 (C-11a), 125.7 (C-4), 122.7 (C-5), 119.9 (C-11b), 119.1 (C-10), 118.3 (C-16), 118.1 (C-7), 117.7 (C-9), 114.1 (C-3b), 105.7 (C-3), 56.9 (OMe), 37.4 (C-13), 30.6 (C-12), 26.9 (C-18), 19.5 (C-19); (+)-ESIMS
m/z 374 [M + H]
+; (+)-HRESIMS [M + H]
+ 374.1875 (calcd. for C
23H
24N
3O
2, 374.1876).
1H NMR data agreed with literature [
2].
3.2.16. O-Methyl-styelsamine-N14-2-phenylacetamide Trifluoroacetate (47)
To a solution of O-methyl styelsamine D (43) (11.0 mg, 0.021 mmol) in dry DMF (2 mL) was added phenylacetic acid (5.1 mg, 0.038 mmol) and PyBOP (19.0 mg, 0.038 mmol) followed by Et3N (16 µL, 0.11 mmol). The solution was stirred under N2 at room temperature for 1 h. CH2Cl2 (10 mL) was added, washed with H2O (10 mL) and the organic phase dried in vacuo to give a purple/red oil. Purification using RP-2 column chromatography (H2O (0.05% TFA):MeOH (0.05% TFA) (100:0 to 50:50)) afforded 47 as a purple oil (5.28 mg, 48% yield).
IR νmax (ATR) 3394, 1679, 1584, 1489, 1140, 1052, 708 cm−1; Rt = 7.33 min; 1H NMR (DMSO-d6, 400 MHz) δH 13.74 (1H, s, NH-1), 11.39 (1H, s, NH-8), 8.56 (1H, t, J = 5.6 Hz, NH-14), 8.35 (1H, d, J = 6.5 Hz, H-2), 8.27 (1H, d, J = 8.2 Hz, H-4), 7.76–7.74 (1H, m, H-7), 7.73–7.71 (2H, m, H-6 and H-10), 7.68 (1H, d, J = 6.5 Hz, H-3), 7.29–7.26 (1H, m, H-5), 7.29–7.23 (2H, m, H-18 and H-22), 7.23–7.19 (3H, m , H-19, H-20 and H-21), 4.00 (3H, s, OMe), 3.46 (2H, s, H2-16), 3.40 (2H, dt, J = 6.9, 5.6 Hz, H2-13), 3.08 (2H, t, J = 6.9 Hz, H2-12); 13C NMR (DMSO-d6, 100 MHz) δC 171.6 (C-15), 149.2 (C-3a), 143.5 (C-2), 141.0 (C-7a), 138.5 (C-11), 136.0 (C-17), 135.2 (C-6), 129.4 (C-8a), 128.8 (C-18 and C-22), 128.2 (C-19 and C-21), 126.7 (C-11a), 126.4 (C-20), 125.6 (C-4), 122.7 (C-5), 119.9 (C-11b), 119.0 (C-10), 118.2 (C-7), 116.0 (C-9), 114.0 (C-3b), 105.7 (C-3), 56.9 (OMe), 42.2 (C-16), 37.6 (C-13), 30.1 (C-12); (+)-ESIMS m/z 410 [M + H]+; (+)-HRESIMS [M + H]+ 410.1855 (calcd. for C26H24N3O2, 410.1863).
3.2.17. O-Methyl-styelsamine-N14-3-phenylpropanamide Trifluoroacetate (48)
To a cold (0 °C) solution of O-methyl styelsamine D (43) (7.0 mg, 0.013 mmol) in dry THF (2 mL) was added dihydrocinnamoyl chloride (5.64 µL, 0.038 mmol) followed by Et3N (7.1 µL, 0.051 mmol). The solution was stirred under N2 at room temperature for 30 min. Solvents were removed in vacuo to give a purple/red oil. Purification using RP-2 (H2O (0.05% TFA):MeOH (0.05% TFA) (100:0 to 50:50)) and LH-20 column chromatography (MeOH (0.05% TFA)) afforded 48 as a purple oil (3.2 mg, 43% yield).
IR νmax (ATR) 3407, 1679, 1584, 1489, 1140, 1052, 701 cm−1; Rt = 7.85 min; 1H NMR (DMSO-d6, 400 MHz) δH 13.69 (1H, s, NH-1), 11.63 (1H, s, NH-8), 8.49 (1H, t, J = 5.5 Hz, NH-14), 8.36 (1H, d, J = 6.4 Hz, H-2), 8.31 (1H, d, J = 8.3 Hz, H-4), 7.80–7.74 (2H, m, H-6 and H-7), 7.76 (1H, s, H-10), 7.70 (1H, d, J = 6.4 Hz, H-3), 7.29 (1H, dt, J = 8.3, 2.2 Hz, H-5), 7.24 (2H, d, J = 7.4 Hz, H-19 and H-23), 7.21–7.15 (3H, m, H-20, H-21 and 22), 4.04 (3H, s, OMe), 3.33 (2H, dt, J = 7.2, 5.5 Hz, H2-13), 3.03 (2H, t, J = 7.2 Hz, H2-12), 2.86 (2H, t, J = 7.7 Hz, H2-17), 2.47–2.45 (2H, m, H2-16); 13C NMR (DMSO-d6, 100 MHz) δC 173.3 (C-15), 149.2 (C-3a), 143.5 (C-2), 141.0 (C-7a and C-18), 138.4 (C-11), 135.3 (C-6), 129.5 (C-8a), 128.2 (C-19, C-20, C-22 and C-23), 125.9 (C-11a and C-21), 125.7 (C-4), 122.7 (C-5), 119.9 (C-11b), 119.1 (C-10), 117.8 (C-7), 116.0 (C-9), 114.1 (C-3b), 105.7 (C-3), 56.9 (OMe), 37.5 (C-13), 36.8 (C-16), 31.0 (C-17), 30.3 (C-12); (+)-ESIMS m/z 424 [M + H]+; (+)-HRESIMS [M + H]+ 424.2005 (calcd. for C27H26N3O2, 424.2020).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}