Screening Mangrove Endophytic Fungi for Antimalarial Natural Products



Abstract
:1. Introduction
2. Results and Discussion
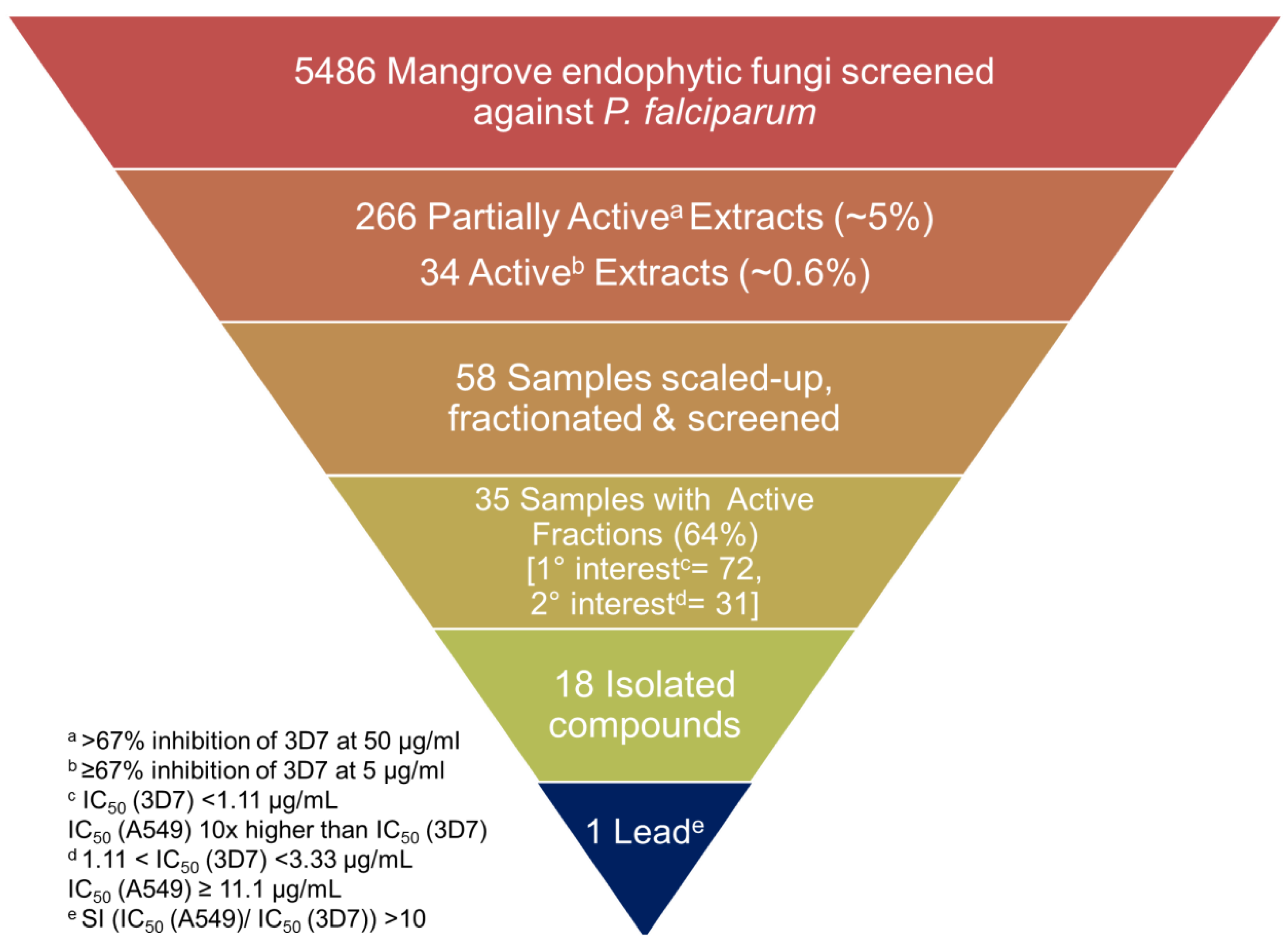
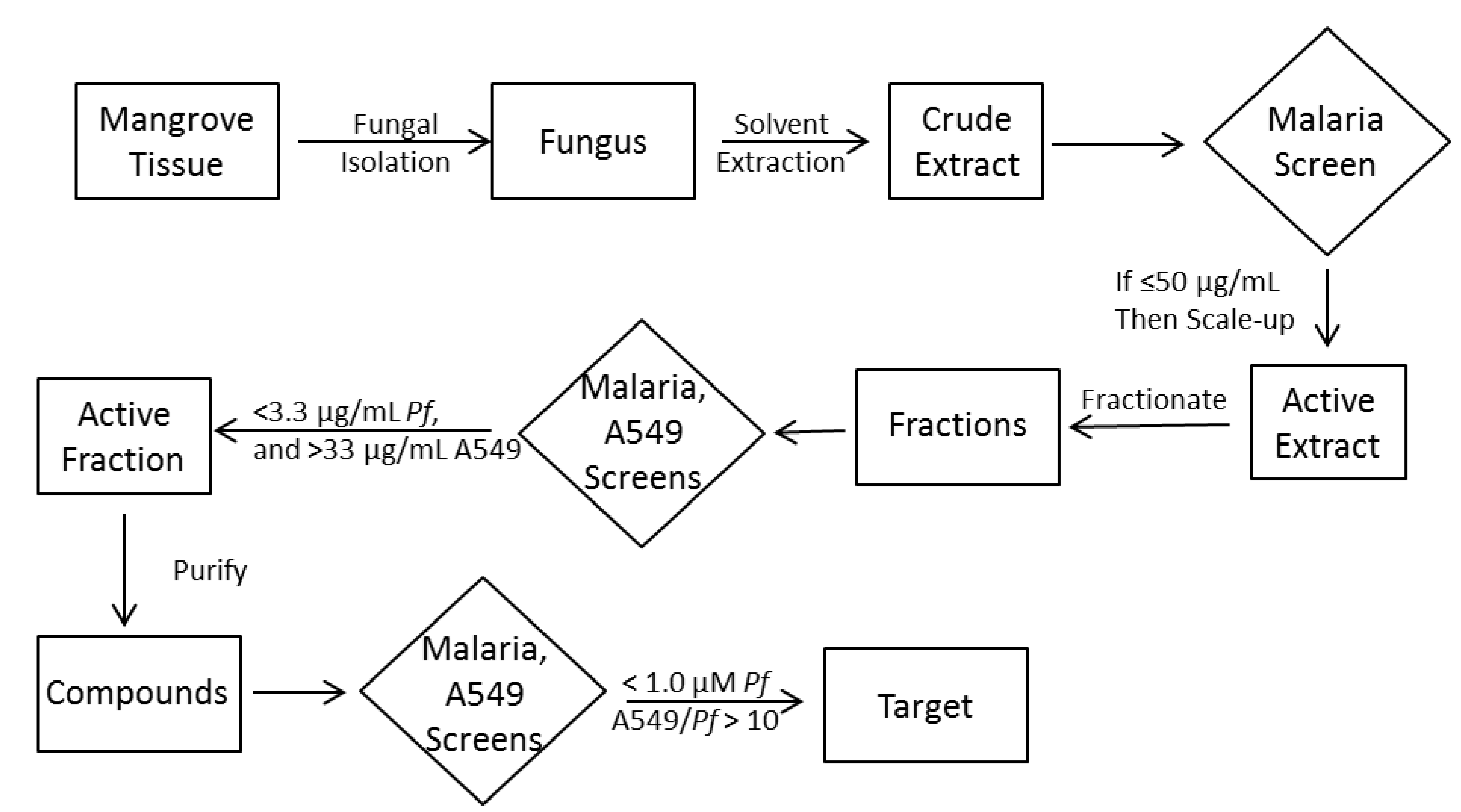
2.1. Strategy

2.2. Fungal Isolation and Fermentation
2.3. Extraction, Plating and Screening of Miniaturized Cultures
2.4. Chromatographic Separation, Screening and Structure Elucidation
2.5. Fungal Chemistry and its Bioactivity
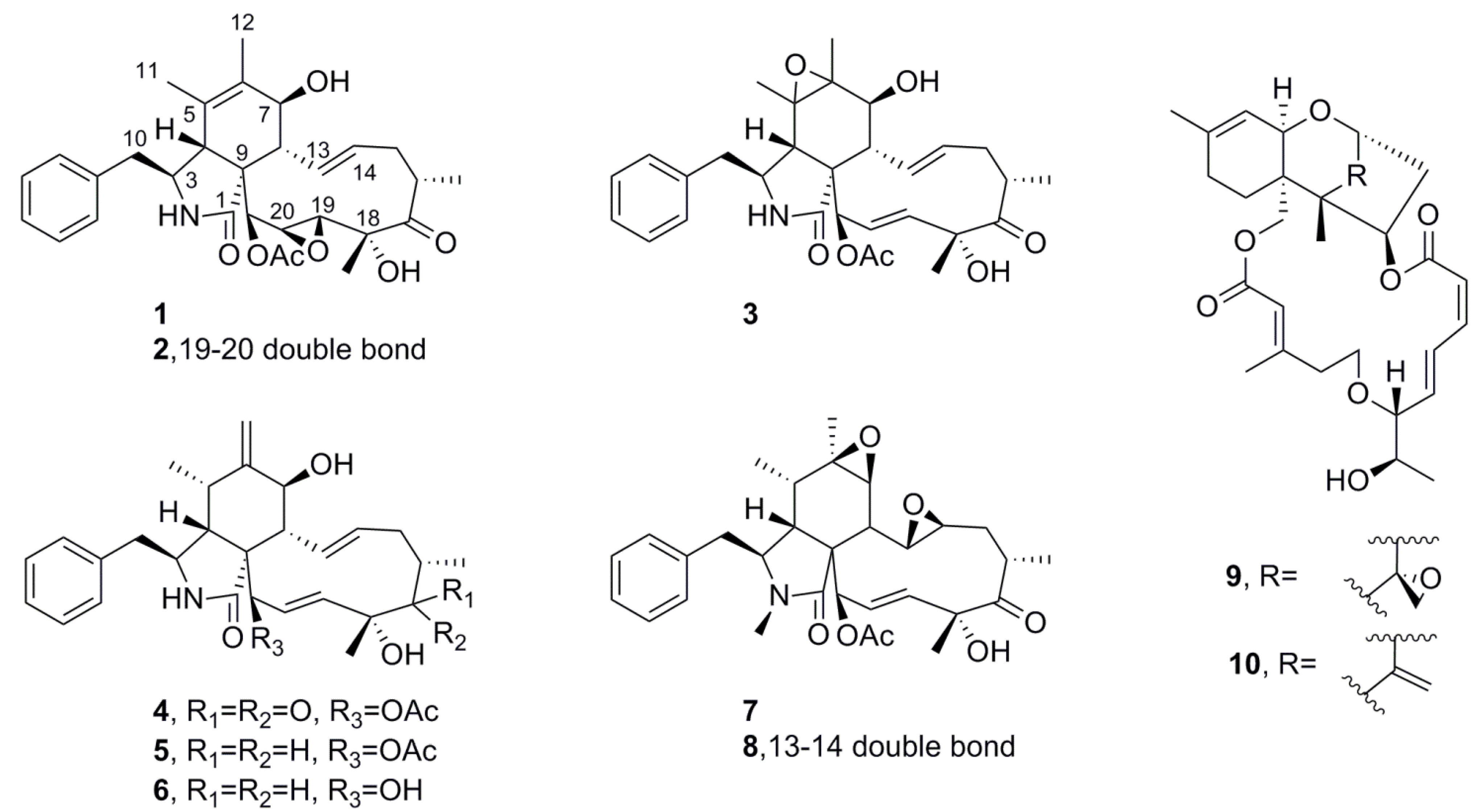
2.5.1. Mycotoxins



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Cpd | IC50 (nM) | S.I. | Source (Taxonomic Identification) | |
|---|---|---|---|---|---|
| 3D7 | A549 | A549/3D7 | |||
| Mycotoxin | |||||
| Cytochalasin | 1 | <20 | ND * | − | CY-5286 (Diaporthe sp.) |
| 2 | 136 | ND * | − | CY-5331 (Xylaria sp.) | |
| 3 | <20 | ND * | − | CY-5368 (Verticillium sp.) | |
| 4 | 25.8 | ND * | − | CY-5286 (Diaporthe sp.), CY-6884 (Xylaria sp.), NTOU-3332 (Xylaria sp.), NTOU-1430 (Xylaria sp.) | |
| 5 | <20 | ND * | − | CY-5286 (Diaporthe sp.) | |
| 6 | <20 | ND * | − | CY-5286 (Diaporthe sp.) | |
| 7 | 290 | ND * | − | CY-5331 (Xylaria sp.) | |
| 8 | 26 | ND * | − | CY-5331 (Xylaria sp.) | |
| Trichothecene | 9 | <20 | <200 | − | CY-3923 † |
| 10 | <20 | <200 | − | CY-3923 † | |
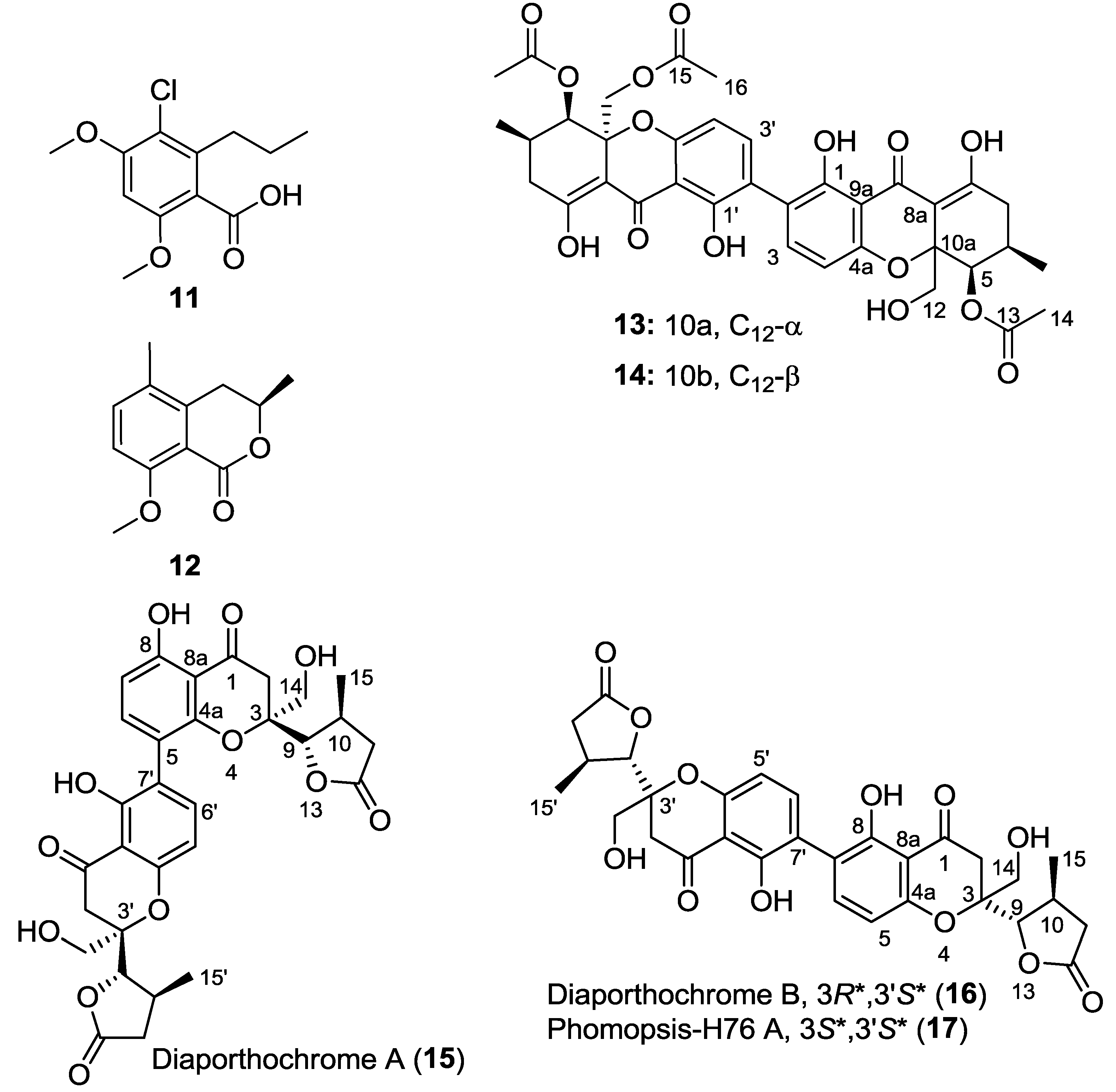
| Polyketide | 11 | 36,000 | >38,700 | − | NTOU-1455 (Xylaria sp.) |
| 12 | >48,500 | >48,500 | − | NTOU-2009 (Phomopsis sp.) | |
| 13 | >25,000 | >25,000 | − | CY-5188 (Diaporthe sp.) | |
| 14 | 600 | 7800 | 13 | CY-5188 (Diaporthe sp.) | |
| 15 | >25,000 | >25,000 | − | CY-5286 (Diaporthe sp.) | |
| 16 | >25,000 | >25,000 | − | CY-5286 (Diaporthe sp.) | |
| Lipid | 18 | >25,000 | >25,000 | − | CY-5331(Xylaria sp.) |
| 19 | >25,000 | >25,000 | − | CY-6884 (Xylaria sp.) | |
| Control | CQ | 4.5 | >25,000 | − | |
| ATO | 0.3 | >25,000 | − | ||
| DHA | 0.8 | >25,000 | − | ||
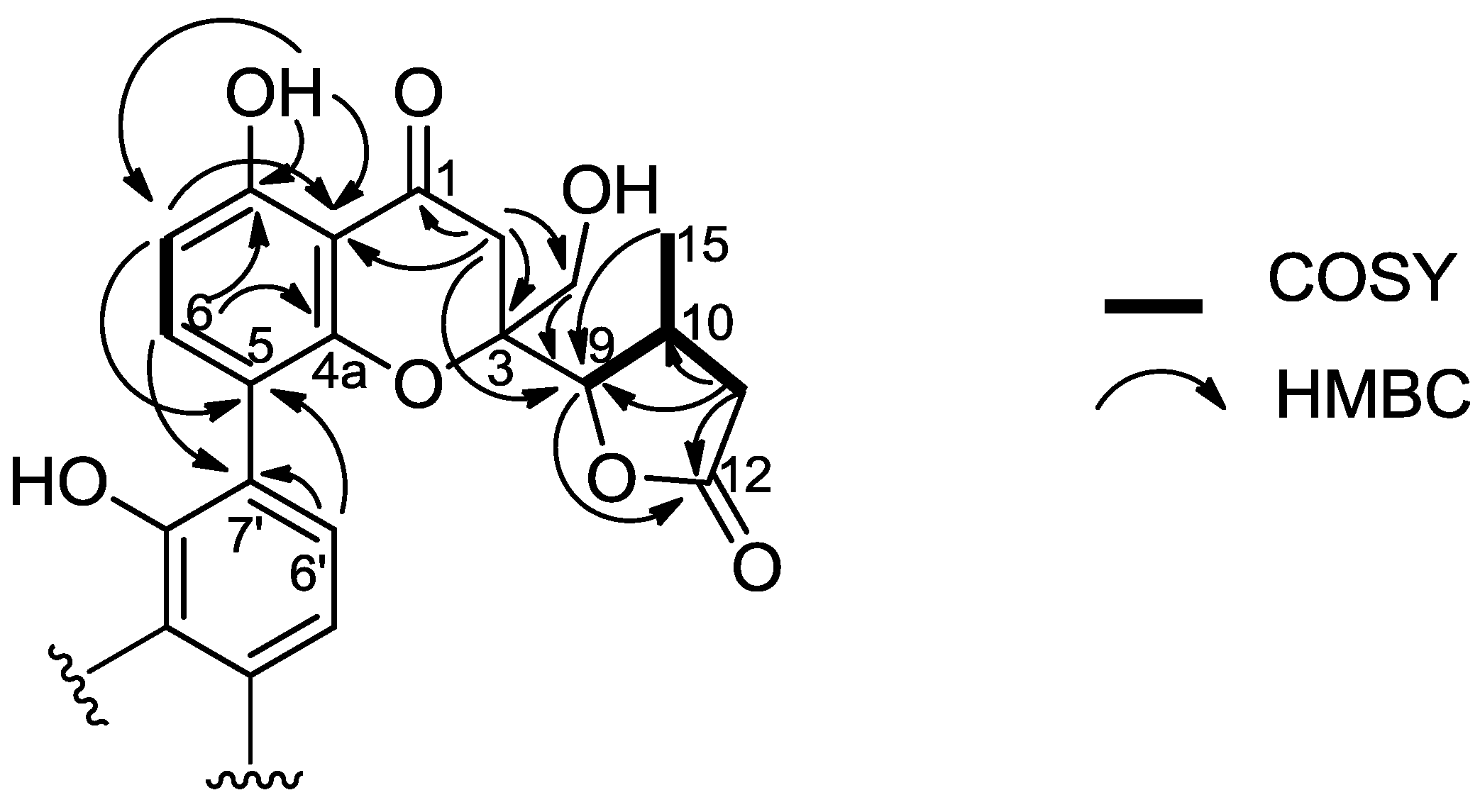
2.5.2. Polyketides

| 15 a | 16 a | 17 b | ||||
|---|---|---|---|---|---|---|
| Position | δH (J in Hz) | δC | δH (int., J in Hz) | δC | δH | δC |
| 1 | 196.9 | 195.9 | 200.2 | |||
| 2 | 3.22 (1H, d, 17.5) 2.86 (1H, d, 17.6) | 38.3 | 3.05 (1H, dd, 17.6, 0.8) 2.90 (1H, brd, 15.1) | 37.9 | 3.11 2.93 | 39.9 |
| 3 | 83.5 | 82.9 * | 86.6 | |||
| 4a | 155.6 | 158.1 | 161.5 | |||
| 5 | 115.2 | 6.45 * (1H, d, 8.6) | 106.9 | 6.48 | 109.4 | |
| 6 | 7.25 (1H, d, 8.8) | 140.2 | 7.43 * (1H, d, 8.5) | 140.8 | 7.38 | 142.7 |
| 7 | 6.59 (1H, d, 8.8) | 109.9 | 117.2 | 118.8 | ||
| 8 | 161.7 | 159.0 | 160.6 | |||
| 8a | 107.0 | 107.2 * | 109.6 | |||
| 9 | 4.22 (1H, d, 3.9) | 86.9 | 4.40 (1H, dd, 1.5,4.4) | 86.5 * | 4.37 | 89.3 |
| 10 | 2.67 (1H, m) | 29.6 | 2.84 (1H, m) | 29.9 | 2.79 | 31.8 |
| 11 | 2.34 (1H, dd, 17.8, 9.0) 2.00 (1H, dd, 18.1, 5.4) | 36.2 | 2.26 (1H, m) 2.23 (1H, m) | 36.5 | 2.75 2.22 | 38.6 |
| 12 | 176.0 | 175.6 | 178.7 | |||
| 14 | 3.95 (1H, dd, 5.6, 11.6) 3.89 (1H, dd,5.5, 12.3) | 62.9 | 3.94 * (2H, m) | 62.4 | 3.68 | 65.0 |
| 15 | 1.10 (3H, d, 6.8) | 20.5 | 1.27 (3H, d, 7.0) | 20.8 | 1.15 | 22.7 |
| 1′ | 197.0 | 196.7 | 200.2 | |||
| 2′ | 3.24 (1H, d, 17.6) 3.10 (1H, d, 17.5) | 38.8 | 3.22 (1H, d, 17.6) 2.96 (1H, d, 17.6, 1.5) | 38.2 | 3.11 2.93 | 39.9 |
| 3′ | 83.3 | 82.9 * | 86.6 | |||
| 4a′ | 158.5 | 158.1 | 161.5 | |||
| 5′ | 6.50 (1H, d, 8.3) | 107.4 | 6.46 * (1H, dd, 1.5, 8.5) | 106.9 | 6.48 | 109.4 |
| 6′ | 7.29 (1H, d, 8.3) | 140.1 | 7.44 * (1H, dd, 1.5, 8.5) | 140.8 | 7.38 | 142.7 |
| 7′ | 118.6 | 117.2 | 118.8 | |||
| 8′ | 158.4 | 159.0 | 160.6 | |||
| 8a′ | 106.8 | 107.0 * | 109.6 | |||
| 9′ | 4.33 (1H, d, 3.9) | 87.6 | 4.35 (1H, dd, 1.5, 4.3) | 86.4 * | 4.37 | 89.3 |
| 10′ | 2.95 (1H, m) | 29.2 | 2.90 (1H, m) | 29.4 | 2.79 | 31.8 |
| 11′ | 2.88 (1H, dd, 18.0, 10) 2.22 (1H, dd, 17.6, 4.9) | 36.4 | 2.90 (2H, m) | 36.6 | 2.75 2.22 | 38.6 |
| 12′ | 175.3 | 175.8 | 178.7 | |||
| 14′ | 3.82 (2H, m) | 63.2 | 3.94 * (1H, m) 3.86 (1H, brd, 11.7) | 62.8 | 3.68 | 65.0 |
| 15′ | 1.30 (3H, d, 6.8) | 20.8 | 1.30 (3H, d, 6.4) | 20.6 | 1.15 | 22.7 |
| 8-OH | 11.73 (1H, s) | 12.01 * (1H, s) | 11.97 | |||
| 8′-OH | 12.28 (1H, brs) | 12.00 * (1H, d, 1.5) | 11.97 | |||

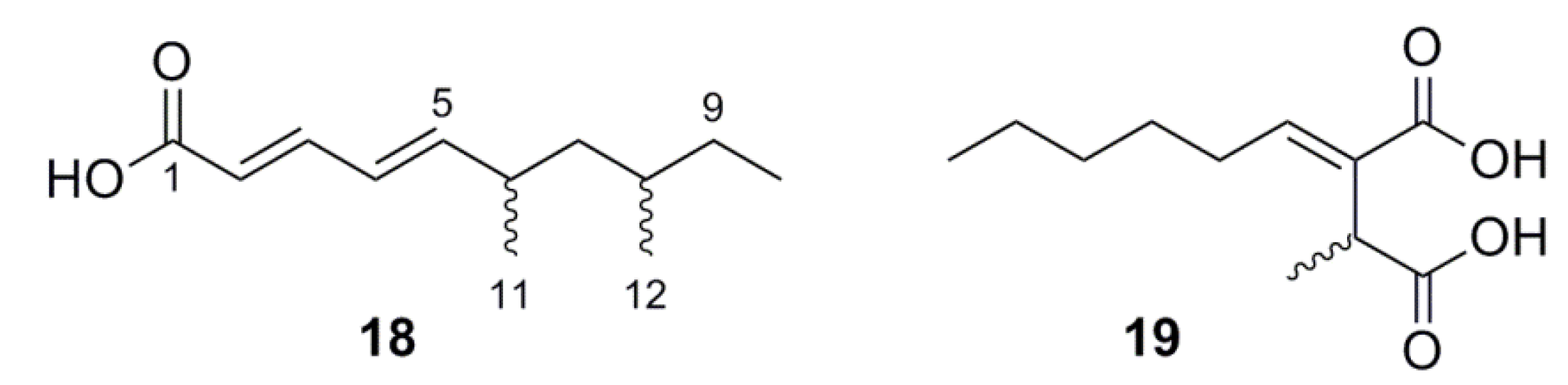
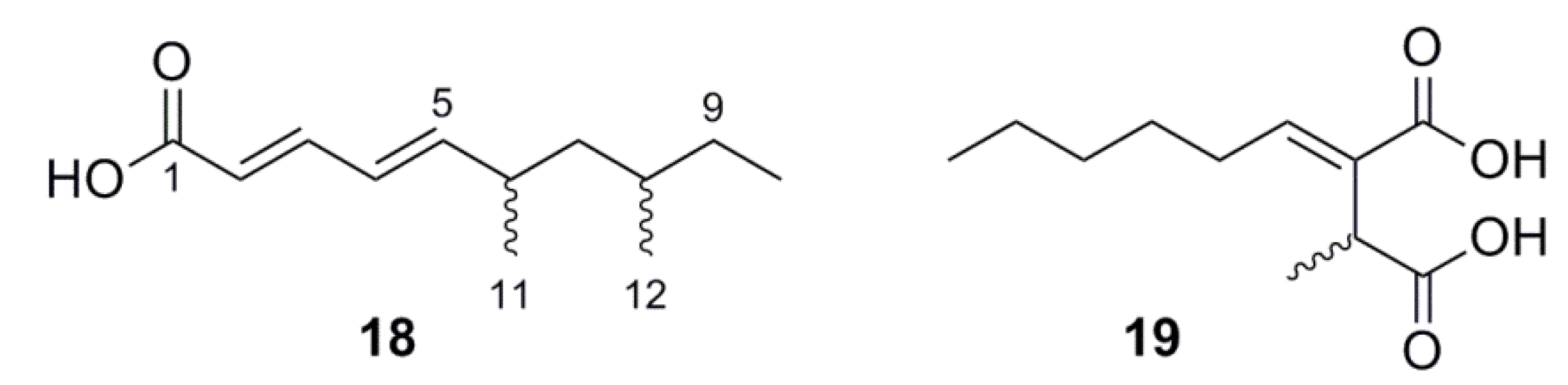
2.5.3. Lipids
3. Experimental Section
3.1. General Experimental Procedures
3.2. Biological Materials
3.3. Dicerandrol D (14)
3.4. Diaporthochromones (15 and 16)
3.5. (2E,4E)-6,8-Dimethyldeca-2,4-dienoic Acid (18)
3.6. Malaria Assay
3.7. In Vitro Toxicity Assay
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar]
- Xu, J. Biomolecules produced by mangrove-associated microbes. Curr. Med. Chem. 2011, 18, 5224–5266. [Google Scholar] [CrossRef]
- Wu, J.; Xiao, Q.; Xu, J.; Li, M.Y.; Pan, J.Y.; Yang, M.H. Natural products from true mangrove flora: Source, chemistry and bioactivities. Nat. Prod. Rep. 2008, 25, 955–981. [Google Scholar] [CrossRef]
- Omar, S.; Godard, K.; Ingham, A.; Hussain, H.; Wongpanich, V.; Pezzuto, J.; Durst, T.; Eklu, C.; Gbeassor, M.; Sanchez-Vindas, P.; et al. Antimalarial activities of gedunin and 7-methoxygedunin and synergistic activity with dillapiol. Ann. Appl. Biol. 2003, 143, 135–141. [Google Scholar] [CrossRef]
- Castillo, U.; Harper, J.K.; Strobel, G.A.; Sears, J.; Alesi, K.; Ford, E.; Lin, J.; Hunter, M.; Maranta, M.; Ge, H.; et al. Kakadumycins, novel antibiotics from Streptomyces sp. NRRL, 30566, an endophyte of Grevillea pteridifolia. FEMS Microbiol. Lett. 2003, 224, 183–190. [Google Scholar] [CrossRef]
- Isaka, M.; Suyarnsestakorn, C.; Tanticharoen, M.; Kongsaeree, P.; Thebtaranonth, Y. Aigialomycins A–E, new resorcylic macrolides from the marine mangrove fungus Aigialus parvus. J. Org. Chem. 2002, 67, 1561–1566. [Google Scholar] [CrossRef]
- Feller, I.C.; Lovelock, C.E.; Berger, U.; McKee, K.L.; Joye, S.B.; Ball, M.C. Biocomplexity in mangrove ecosystems. Annu. Rev. Mar. Sci. 2010, 2, 395–417. [Google Scholar] [CrossRef]
- Mangrove Forest Threats. Available online: http://wwf.panda.org/about_our_earth/blue_planet/coasts/mangroves/mangrove_threats/ (accessed on 8 September 2013).
- Alongi, D.M. Present state of future and the world’s mangrove forests. Environ. Conserv. J. 2002, 29, 331–349. [Google Scholar]
- Valiela, I.; Bowen, J.L.; York, J.K. Mangrove forests: one of the world’s threatened major tropical environments. BioScience 2001, 51, 807–915. [Google Scholar] [CrossRef]
- Polidoro, B.A.; Carpenter, K.E.; Collins, L.; Duke, N.C.; Ellison, A.M.; Farnsworth, E.J.; Fernando, E.S.; Kathiresan, K.; Koedam, N.E.; Livingstone, S.R.; et al. The loss of species: Mangrove extinction risk and geographic areas of global concern. PLoS One 2010, 5, e10095. [Google Scholar] [CrossRef]
- Mace, G.M.; Norris, K.; Fitter, A.H. Biodiversity and ecosystem services: A multilayered relationship. Trends Ecol. Evol. 2011, 27, 19–26. [Google Scholar] [CrossRef]
- Lebar, M.D.; Hahn, K.N.; Mutka, T.; Maignan, P.; van Olphen, A.; Kyle, D.E.; McClintock, J.B.; Amsler, C.D.; Baker, B.J. CNS and antimalarial activity of synthetic meridianin and psammopemmin analogs. Bioorg. Med. Chem. 2011, 19, 5756–5762. [Google Scholar] [CrossRef]
- Kowalski, T.; Kehr, R.D. Endophytic fungal colonization of branch bases in several forest tree species. Sydowia 1992, 44, 137–168. [Google Scholar]
- Pang, K.L.; Vrijmoed, L.L.P.; Goh, T.K.; Plaingam, N.; Jones, G.E.B. Fungal endophytes associated with Kandelia candel (Rhizophoraceae) in Mai Po Nature Reserve, Hong Kong. Bot. Mar. 2008, 51, 171–178. [Google Scholar]
- Edwards, R.L.; Maitland, J.; Whalley, A.J.S. Metabolites of the higher fungi. Part 24. Cytochalasin N, O, P, Q, and R. New cytochalasins from the fungus Hypoxylon terricola Mill. J. Chem. Soc. Perkin Trans. I 1989, 57–65. [Google Scholar]
- Liu, J.; Jianwen, T.; Dong, Z.; Ding, Z.; Wang, X.; Liu, P. Neoengleromycin, a novel compound from Engleromyces goetzii. Helv. Chem. Acta 2002, 85, 1439–1442. [Google Scholar] [CrossRef]
- Espada, A.; Rivera-Sagredo, A.; de la Fuente, J.M.; Hueso-Rodriguez, J.A.; Elson, S.W. New cytochalasins from the fungus Xylaria hypoxylon. Tetrahedron 1997, 53, 6485–5492. [Google Scholar] [CrossRef]
- Konig, G.M.; Wright, A.D.; Angerhofer, C.K. Antimalarial diterpene isonitriles, isothiocyanates and isocyanates from the tropical marine sponge Cymbastela hooperi. J. Org. Chem. 1996, 61, 3259–3267. [Google Scholar] [CrossRef]
- Namikoshi, M.; Akano, K.; Meguro, S.; Kasuga, I.; Mine, Y.; Takahashi, T.; Kobayashi, H. A new macrocyclic trichothecene, 12,13-deoxyroridin E, produced by the marine-derived fungus Myrothecium roridum collected in Palau. J. Nat. Prod. 2001, 64, 396–398. [Google Scholar] [CrossRef]
- Pontius, A.; Mohamed, I.; Krick, A.; Kehraus, S.; Konig, G.M. Aromatic polyketides from marine algicolous fungi. J. Nat. Prod. 2008, 71, 272–274. [Google Scholar]
- Kokubun, T.; Bridge, P.D.; Simmonds, M.S.J. Dihydroisocoumarins and a tetralone from Cytospora eucalypticola. Phytochemistry 2003, 62, 779–782. [Google Scholar] [CrossRef]
- Kamisuki, S.; Ishimaru, C.; Onoda, K.; Kuriyama, I.; Ida, N.; Sugawara, F.; Yoshida, H.; Mizushina, Y. Nodulisporol and nodulisporone, novel specific inhibitors of human DNA polymerase lambda from a fungus, Nodulisporium sp. Bioorg. Med. Chem. 2007, 15, 3109–3114. [Google Scholar] [CrossRef]
- Ma, W.S. Natural Product Drug Discovery against Tropical Diseases. Ph.D. Thesis, Department of Chemistry, College of Arts and Sciences, University of South Florida, Tampa, FL, USA, 11 November 2011. [Google Scholar]
- Wagenaar, M.M.; Clardy, J. Dicerandrols, new antibiotic and cytotoxic dimers produced by the fungus Phomopsis longicolla isolated from an endangered mint. J. Nat. Prod. 2001, 64, 1006–1009. [Google Scholar] [CrossRef]
- Yang, J.F.; Xu, F.; Huang, C.H.; Li, J.; She, Z.G.; Pei, Z.; Lin, Y.C. Metabolites from the mangrove endophytic fungus Phomopsis sp. (#zsu-H76). Eur. J. Org. Chem. 2010, 2010, 3692–3695. [Google Scholar] [CrossRef]
- Anderson, J.R.; Edward, R.L.; Whalley, A.C.J. Metabolites of the higher fungi. Part 22. 2-Butyl-3-methylsuccinic acid and 2-hexylidene-3-methylsuccinic acid from xylariaceous fungi. J. Chem. Soc. Perkin Trans. I 1985, 1481–1485. [Google Scholar] [CrossRef]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Calcul, L.; Waterman, C.; Ma, W.S.; Lebar, M.D.; Harter, C.; Mutka, T.; Morton, L.; Maignan, P.; Olphen, A.V.; Kyle, D.E.; et al. Screening Mangrove Endophytic Fungi for Antimalarial Natural Products. Mar. Drugs 2013, 11, 5036-5050. https://doi.org/10.3390/md11125036
Calcul L, Waterman C, Ma WS, Lebar MD, Harter C, Mutka T, Morton L, Maignan P, Olphen AV, Kyle DE, et al. Screening Mangrove Endophytic Fungi for Antimalarial Natural Products. Marine Drugs. 2013; 11(12):5036-5050. https://doi.org/10.3390/md11125036
Chicago/Turabian StyleCalcul, Laurent, Carrie Waterman, Wai Sheung Ma, Matthew D. Lebar, Charles Harter, Tina Mutka, Lindsay Morton, Patrick Maignan, Alberto Van Olphen, Dennis E. Kyle, and et al. 2013. "Screening Mangrove Endophytic Fungi for Antimalarial Natural Products" Marine Drugs 11, no. 12: 5036-5050. https://doi.org/10.3390/md11125036
APA StyleCalcul, L., Waterman, C., Ma, W. S., Lebar, M. D., Harter, C., Mutka, T., Morton, L., Maignan, P., Olphen, A. V., Kyle, D. E., Vrijmoed, L., Pang, K.-L., Pearce, C., & Baker, B. J. (2013). Screening Mangrove Endophytic Fungi for Antimalarial Natural Products. Marine Drugs, 11(12), 5036-5050. https://doi.org/10.3390/md11125036