Efficient Screening of Marine Extracts for Protease Inhibitors by Combining FRET Based Activity Assays and Surface Plasmon Resonance Spectroscopy Based Binding Assays


Abstract
:1. Introduction
2. Results and Discussion

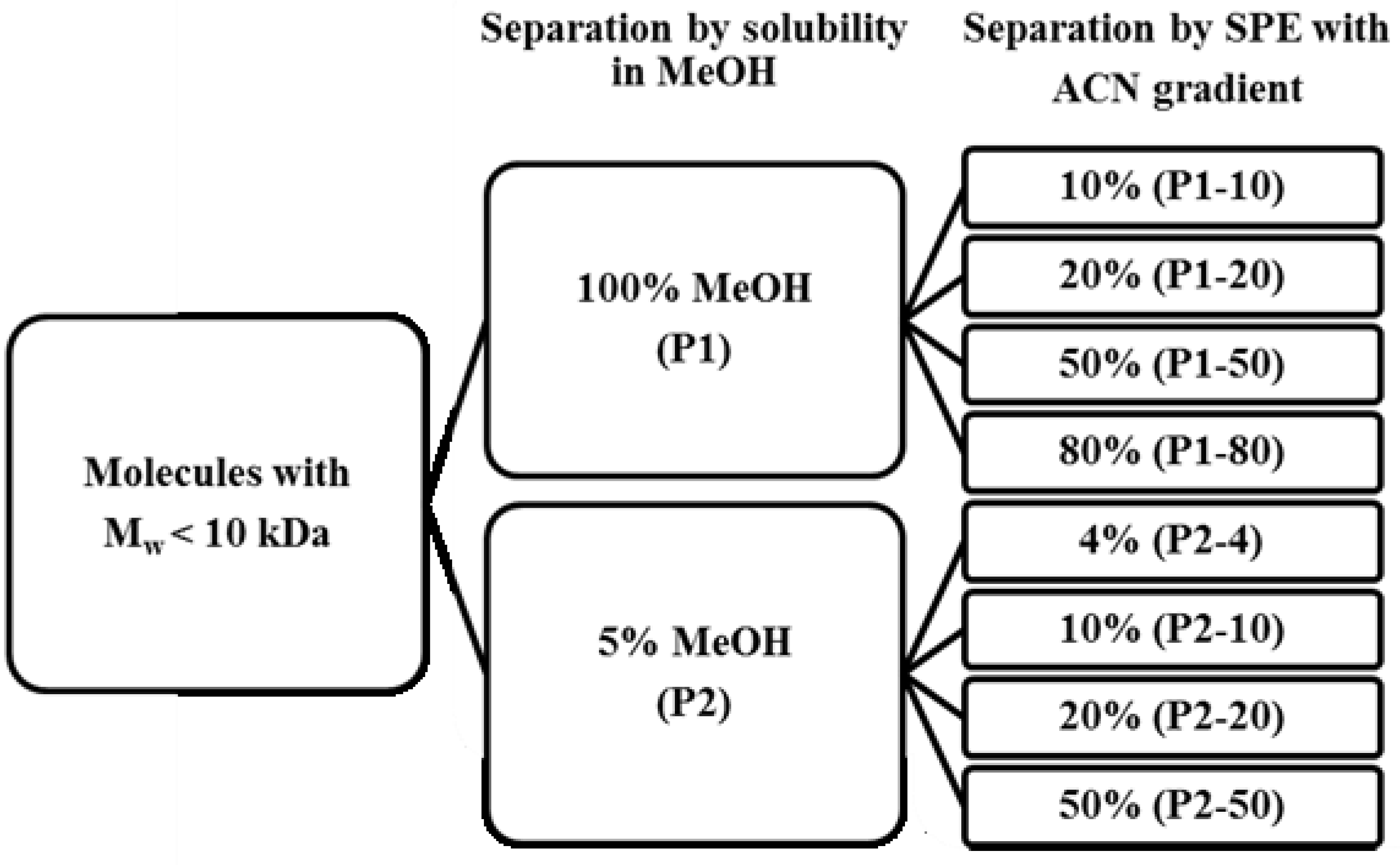
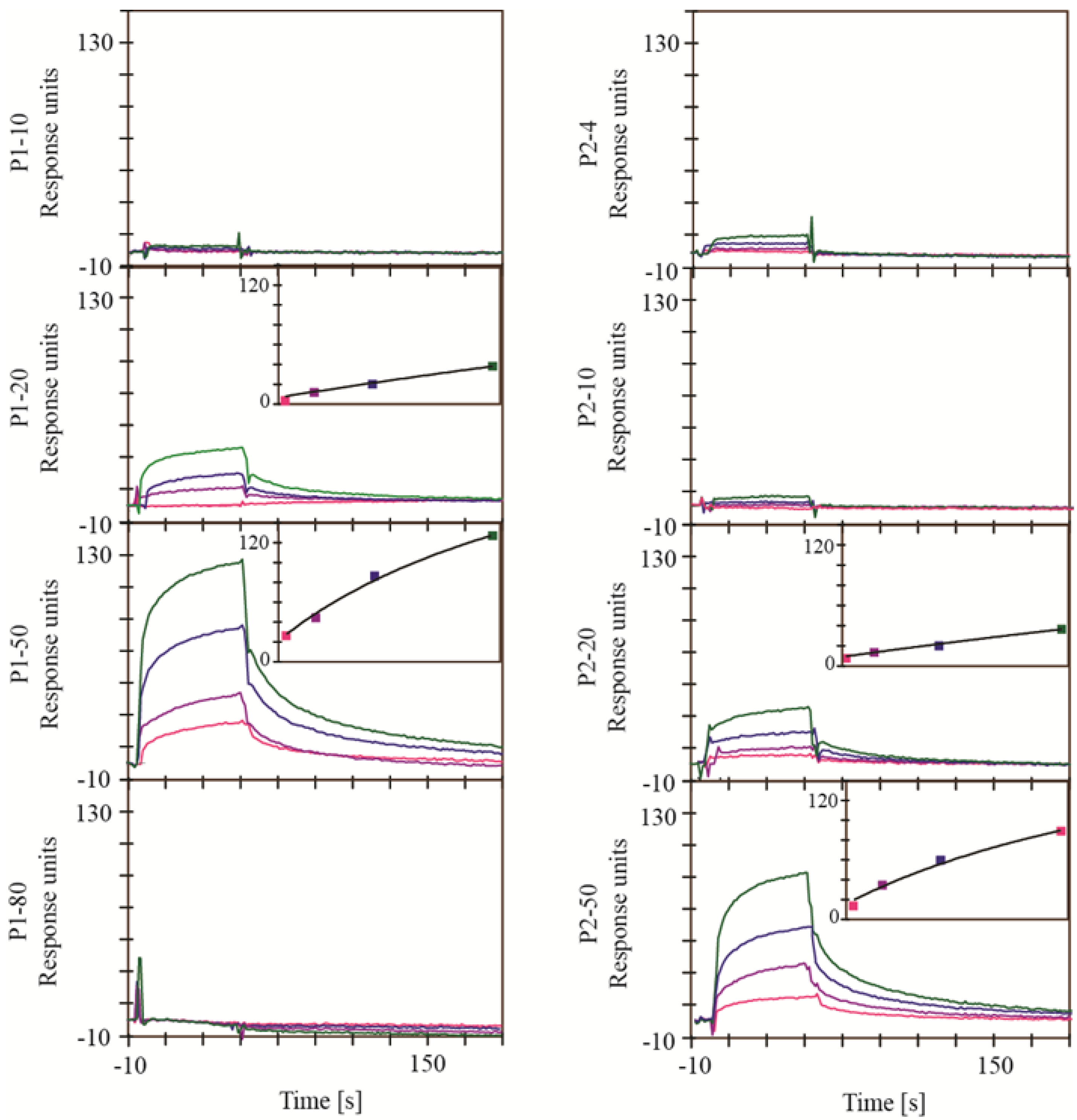
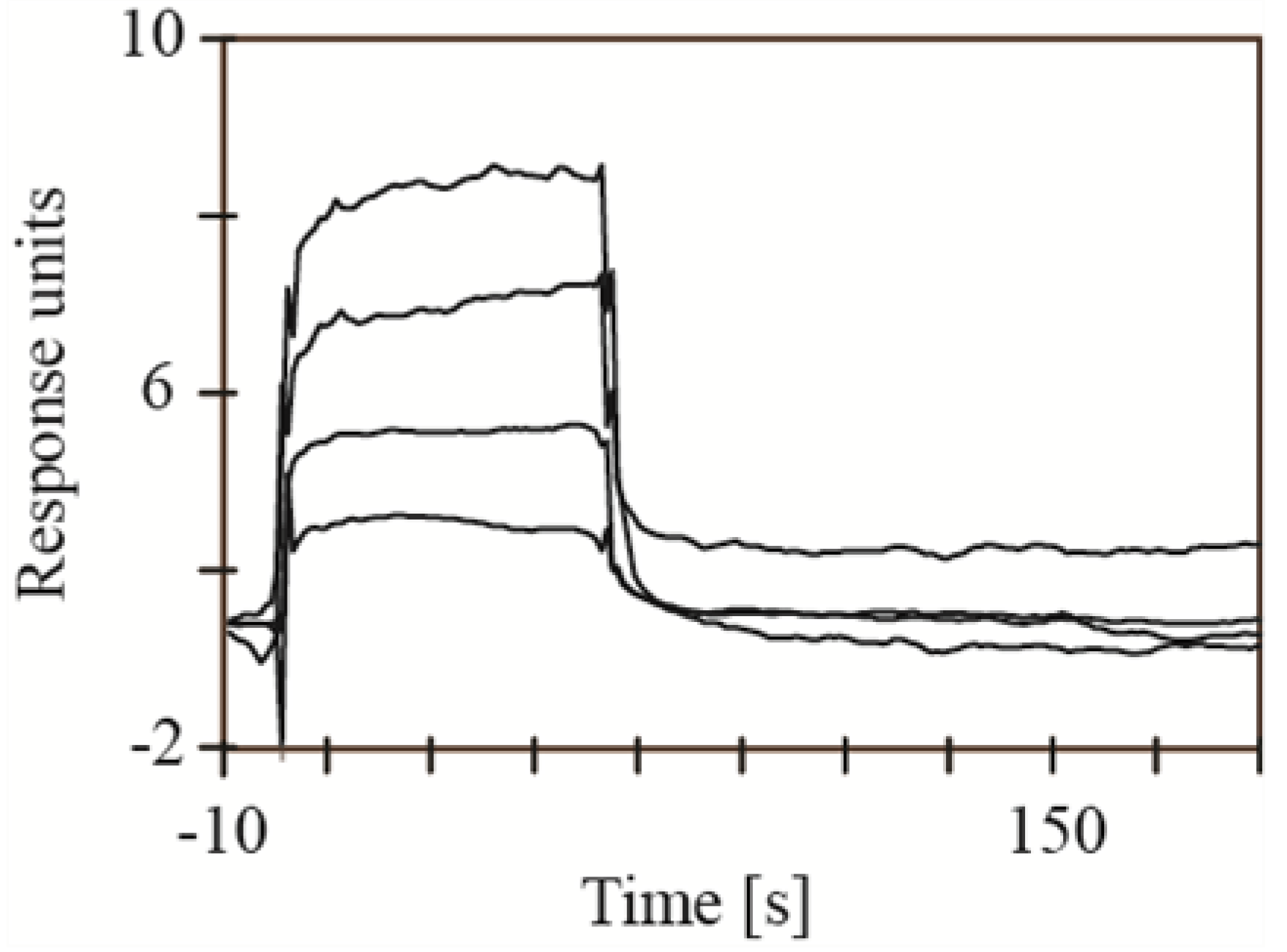
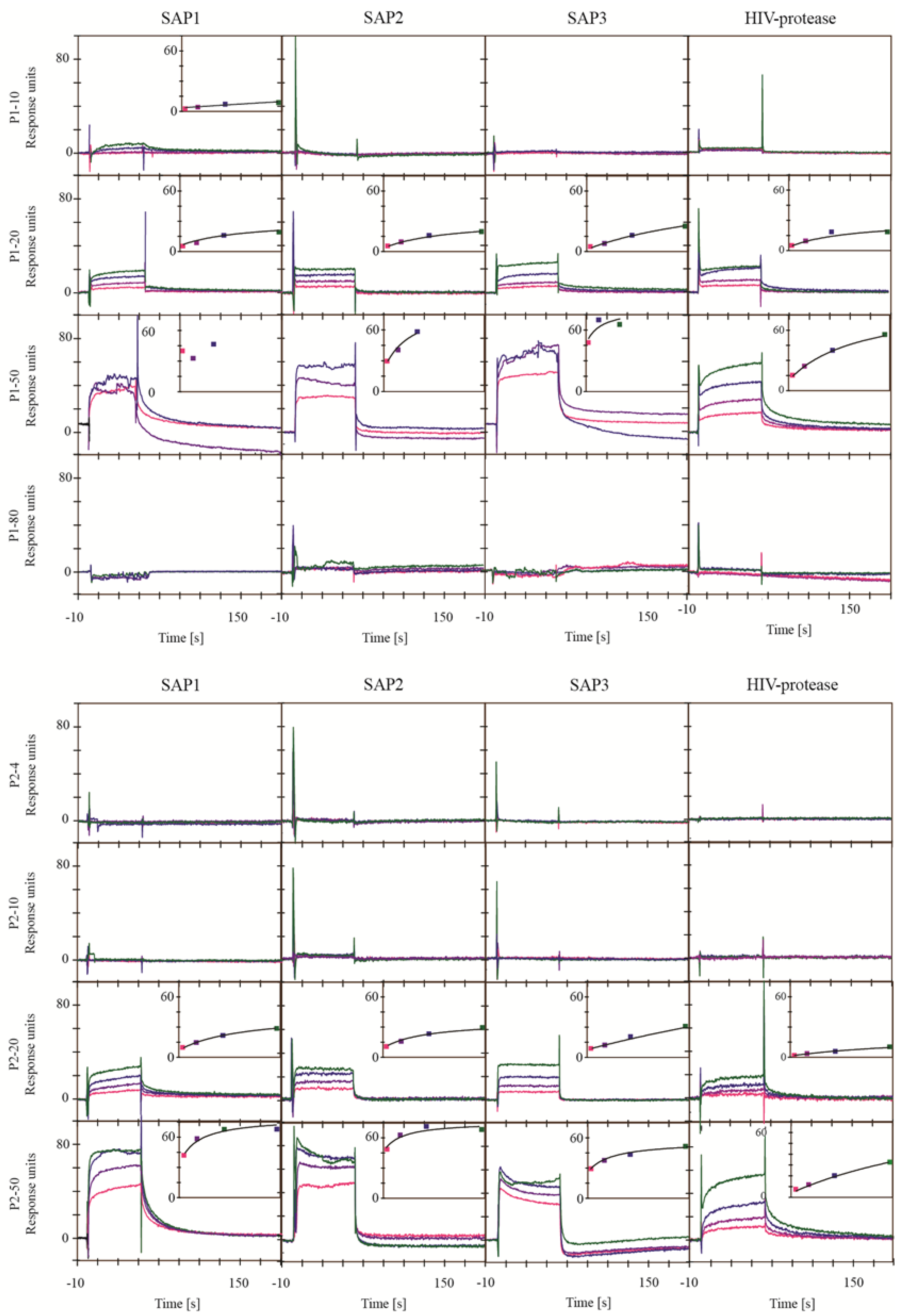
2.1. Screening for Inhibitors of HIV-1 Protease, SAP1, SAP2, SAP3 and Pepsin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % Inhibition | |||||||
|---|---|---|---|---|---|---|---|
| Extract | HIV-1 protease | SAP1 | SAP2 | SAP3 | Pepsin | BACE1 | HCMV Protease |
| P1-10 | 27 ± 1 | 11 ± 1 | −5 ± 6 | −6 ± 1 | 5 ± 2 | 7 ± 1 | 41 ± 4 |
| P1-20 | 70 ± 13 | 47 ± 1 | 36 ± 15 | 44 ± 1 | 34 ± 2 | 44 ± 3 | 71 ± 4 |
| P1-50 | 56 ± 6 | 75 ± 11 | 68 ± 4 | 76 ± 3 | 47 ± 13 | 27 ± 3 | 68 ± 10 |
| P1-80 | −1 ± 1 | 29 ± 4 | 60 ± 1 | 51 ± 1 | 54 ± 14 | 2 ± 4 | 45 ± 5 |
| P2-4 | 11 ± 1 | 10 ± 7 | 4 ± 11 | 6 ± 4 | 11 ± 11 | 3 ± 3 | 43 ± 6 |
| P2-10 | 14 ± 3 | 21 ± 8 | −5 ± 4 | 8 ± 4 | 10 ± 5 | 11 ± 3 | 49 ± 2 |
| P2-20 | 28 ± 3 | −5 ± 15 | 7 ± 1 | −2 ± 7 | 12 ± 1 | 22 ± 4 | 30 ± 9 |
| P2-50 | −18 ± 4 | 8 ± 5 | 36 ± 13 | 14 ± 1 | 13 ± 6 | 9 ± 1 | 10 ± 3 |

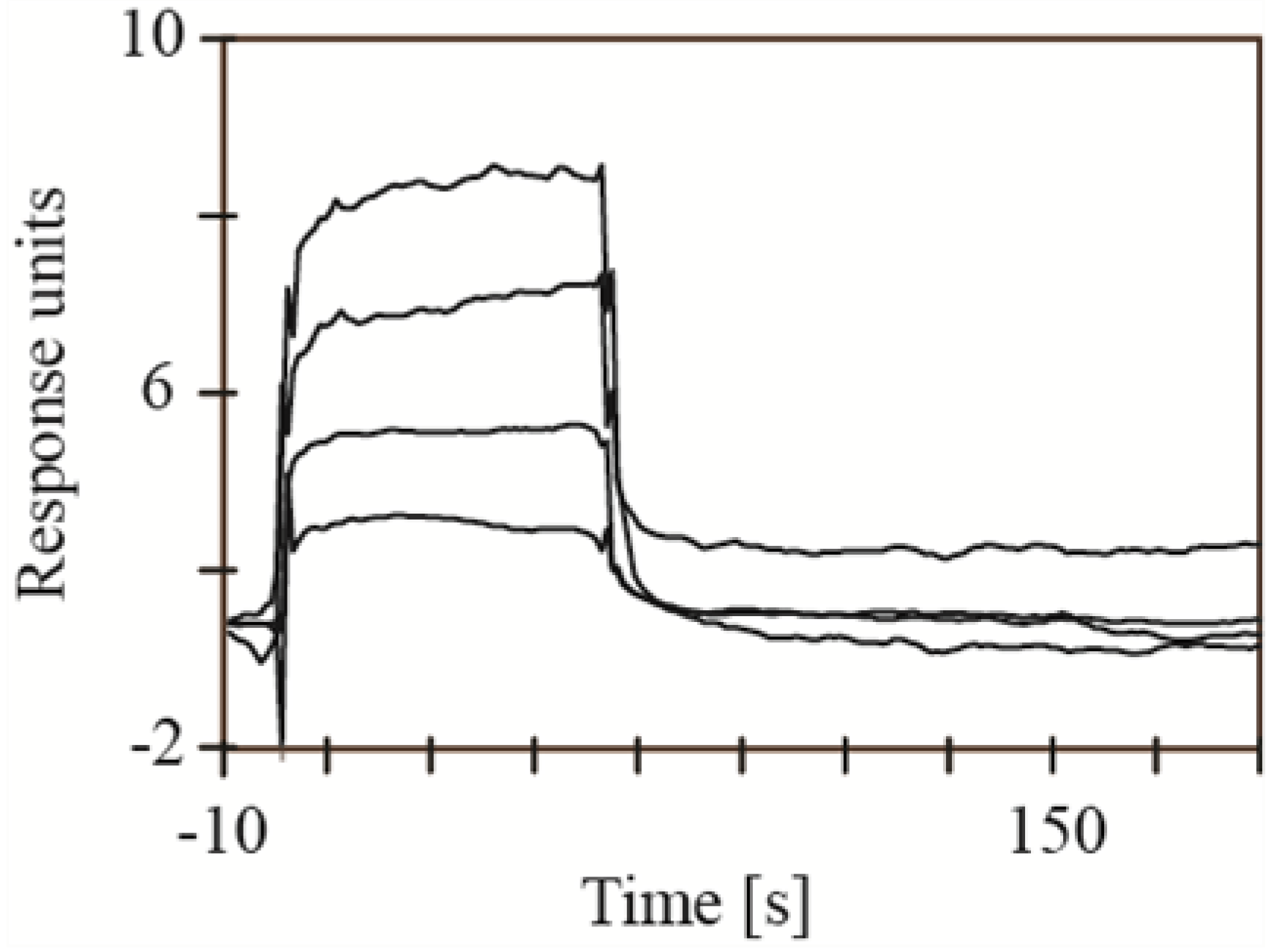
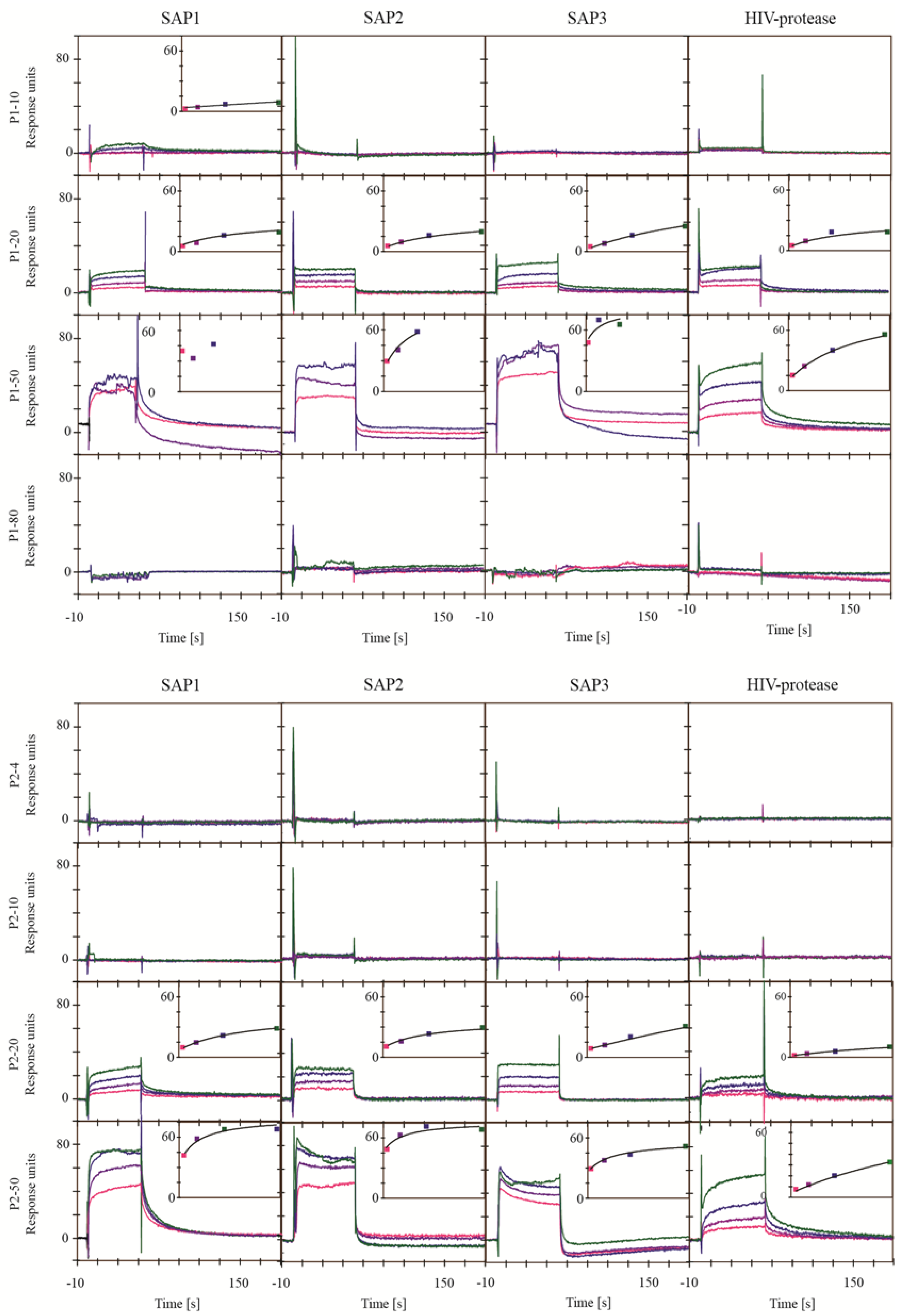
2.2. Screening for Inhibitors of BACE1
2.3. Screening for Inhibition of HCMV Protease
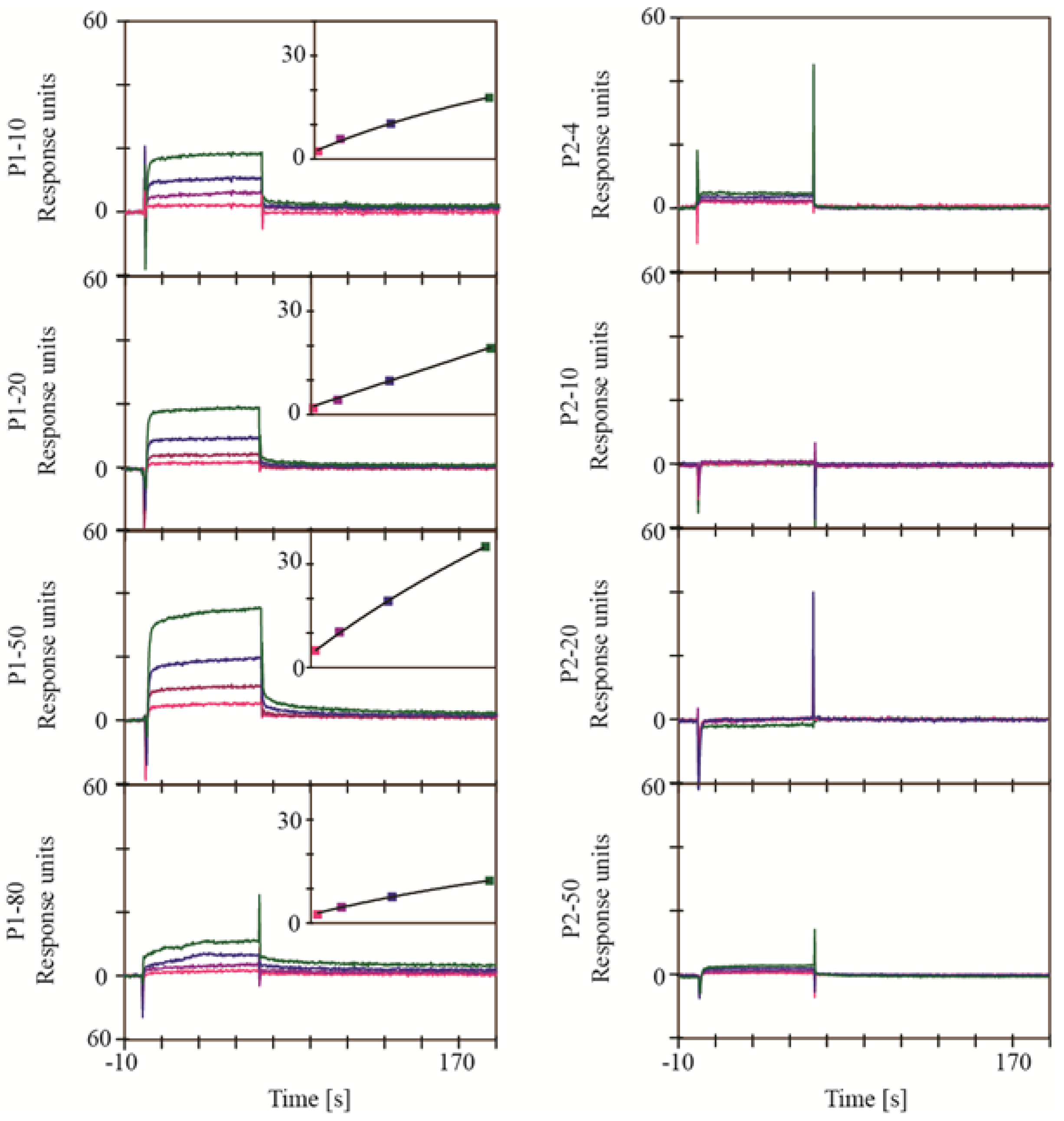
3. Experimental Section
3.1. Preparation of Extracts from Norwegian Spring Spawning Herring
3.2. Protease Production and FRET Based Activity Assay
3.2.1. HIV-1 Protease
3.2.2. SAP1, SAP2 and SAP3
3.2.3. Pepsin
3.2.4. BACE1
3.2.5. HCMV Protease
3.3. SPR Based Binding Assays
3.3.1. HIV-1 Protease
3.3.2. SAP1, SAP2 and SAP3
3.3.3. BACE1
3.3.4. HCMV Protease
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2012, 29, 144–222. [Google Scholar] [CrossRef]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar]
- Bhatnagar, I.; Kim, S.K. Immense essence of excellence: Marine microbial bioactive compounds. Mar. Drugs 2010, 8, 2673–2701. [Google Scholar] [CrossRef]
- Seidel, V. Initial and bulk extraction of natural products isolation. Methods Mol. Biol. 2012, 864, 27–41. [Google Scholar] [CrossRef]
- Mishra, K.P.; Ganju, L.; Sairam, M.; Banerjee, P.K.; Sawhney, R.C. A review of high throughput technology for the screening of natural products. Biomed. Pharmacother. 2008, 62, 94–98. [Google Scholar]
- Harvey, A.L.; Cree, I.A. High-Throughput screening of natural products for cancer therapy. Planta Med. 2010, 76, 1080–1086. [Google Scholar] [CrossRef]
- Keseru, G.M.; Makara, G.M. Hit discovery and hit-to-lead approaches. Drug Discov. Today 2006, 11, 741–748. [Google Scholar] [CrossRef]
- Kim, G.B.; Kim, Y.P. Analysis of protease activity using quantum dots and resonance energy transfer. Theranostics 2012, 2, 127–138. [Google Scholar] [CrossRef]
- Gossas, T.; Danielson, U.H. Analysis of the ph-dependencies of the association and dissociation kinetics of hiv-1 protease inhibitors. J. Mol. Recognit. 2003, 16, 203–212. [Google Scholar] [CrossRef]
- Backman, D.; Monod, M.; Danielson, U.H. Biosensor-Based screening and characterization of hiv-1 inhibitor interactions with sap 1, sap 2, and sap 3 from candida albicans. J. Biomol. Screen. 2006, 11, 165–175. [Google Scholar] [CrossRef]
- Christopeit, T.; Stenberg, G.; Gossas, T.; Nystrom, S.; Baraznenok, V.; Lindstrom, E.; Danielson, U.H. A surface plasmon resonance-based biosensor with full-length bace1 in a reconstituted membrane. Anal. Biochem. 2011, 414, 14–22. [Google Scholar]
- Danielson, U.H. Fragment library screening and lead characterization using spr biosensors. Curr. Top. Med. Chem. 2009, 9, 1725–1735. [Google Scholar] [CrossRef]
- Borch, J.; Roepstorff, P. Screening for enzyme inhibitors by surface plasmon resonance combined with mass spectrometry. Anal. Chem. 2004, 76, 5243–5248. [Google Scholar] [CrossRef]
- Beutler, J.A. Natural products as a foundation for drug discovery. Curr. Protoc. Pharmacol. 2009, 46, 9.11.1–9.11.21. [Google Scholar]
- Li, J.W.; Vederas, J.C. Drug discovery and natural products: End of an era or an endless frontier? Science 2009, 325, 161–165. [Google Scholar] [CrossRef]
- Drag, M.; Salvesen, G.S. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 2010, 9, 690–701. [Google Scholar] [CrossRef]
- Braga-Silva, L.A.; Santos, A.L. Aspartic protease inhibitors as potential anti-candida albicans drugs: Impacts on fungal biology, virulence and pathogenesis. Curr. Med. Chem. 2011, 18, 2401–2419. [Google Scholar] [CrossRef]
- Holwerda, B.C. Herpesvirus proteases: Targets for novel antiviral drugs. Antivir. Res. 1997, 35, 1–21. [Google Scholar] [CrossRef]
- Evin, G.; Lessene, G.; Wilkins, S. Bace inhibitors as potential drugs for the treatment of Alzheimer’s disease: Focus on bioactivity. Recent Pat. CNS Drug Discov. 2011, 6, 91–106. [Google Scholar] [CrossRef]
- Calugi, C.; Guarna, A.; Trabocchi, A. Insight into the structural similarity between hiv protease and secreted aspartic protease-2 and binding mode analysis of hiv-candida albicans inhibitors. J. Enzyme Inhib. Med. Chem. 2012, 28, 936–943. [Google Scholar] [CrossRef]
- Hoegl, L.; Korting, H.C.; Klebe, G. Inhibitors of aspartic proteases in human diseases: Molecular modeling comes of age. Die Pharm. 1999, 54, 319–329. [Google Scholar]
- Biacore™ Assay Handbook, Edition AA ed; GE Healthcare Bio-Sciences AB: Uppsala, Sweden, 2012.
- Giannetti, A.M.; Koch, B.D.; Browner, M.F. Surface plasmon resonance based assay for the detection and characterization of promiscuous inhibitors. J. Med. Chem. 2008, 51, 574–580. [Google Scholar] [CrossRef]
- Markgren, P.O.; Hamalainen, M.; Danielson, U.H. Kinetic analysis of the interaction between hiv-1 protease and inhibitors using optical biosensor technology. Anal. Biochem. 2000, 279, 71–78. [Google Scholar] [CrossRef]
- Batra, R.; Gupta, M.N. Enhancement of enzyme-activity in aqueous-organic solvent mixtures. Biotechnol. Lett. 1994, 16, 1059–1064. [Google Scholar]
- Vassar, R. Beta-secretase (bace) as a drug target for Alzheimer’s disease. Adv. Drug Deliv. Rev. 2002, 54, 1589–1602. [Google Scholar]
- Hong, L.; Koelsch, G.; Lin, X.; Wu, S.; Terzyan, S.; Ghosh, A.K.; Zhang, X.C.; Tang, J. Structure of the protease domain of memapsin 2 (beta-secretase) complexed with inhibitor. Science 2000, 290, 150–153. [Google Scholar] [CrossRef]
- Backman, D.; Danielson, U.H. Kinetic and mechanistic analysis of the association and dissociation of inhibitors interacting with secreted aspartic acid proteases 1 and 2 from candida albicans. Biochim. Biophys. Acta 2003, 1646, 184–195. [Google Scholar] [CrossRef]
- Geitmann, M.; Danielson, U.H. Studies of substrate-induced conformational changes in human cytomegalovirus protease using optical biosensor technology. Anal. Biochem. 2004, 332, 203–214. [Google Scholar]
- Burck, P.J.; Berg, D.H.; Luk, T.P.; Sassmannshausen, L.M.; Wakulchik, M.; Smith, D.P.; Hsiung, H.M.; Becker, G.W.; Gibson, W.; Villarreal, E.C. Human cytomegalovirus maturational proteinase: Expression in escherichia coli, purification, and enzymatic characterization by using peptide substrate mimics of natural cleavage sites. J. Virol. 1994, 68, 2937–2946. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Christopeit, T.; Øverbø, K.; Danielson, U.H.; Nilsen, I.W. Efficient Screening of Marine Extracts for Protease Inhibitors by Combining FRET Based Activity Assays and Surface Plasmon Resonance Spectroscopy Based Binding Assays. Mar. Drugs 2013, 11, 4279-4293. https://doi.org/10.3390/md11114279
Christopeit T, Øverbø K, Danielson UH, Nilsen IW. Efficient Screening of Marine Extracts for Protease Inhibitors by Combining FRET Based Activity Assays and Surface Plasmon Resonance Spectroscopy Based Binding Assays. Marine Drugs. 2013; 11(11):4279-4293. https://doi.org/10.3390/md11114279
Chicago/Turabian StyleChristopeit, Tony, Kersti Øverbø, U. Helena Danielson, and Inge W. Nilsen. 2013. "Efficient Screening of Marine Extracts for Protease Inhibitors by Combining FRET Based Activity Assays and Surface Plasmon Resonance Spectroscopy Based Binding Assays" Marine Drugs 11, no. 11: 4279-4293. https://doi.org/10.3390/md11114279