Application of HPLC-NMR in the Identification of Plocamenone and Isoplocamenone from the Marine Red Alga Plocamium angustum


Abstract
:1. Introduction
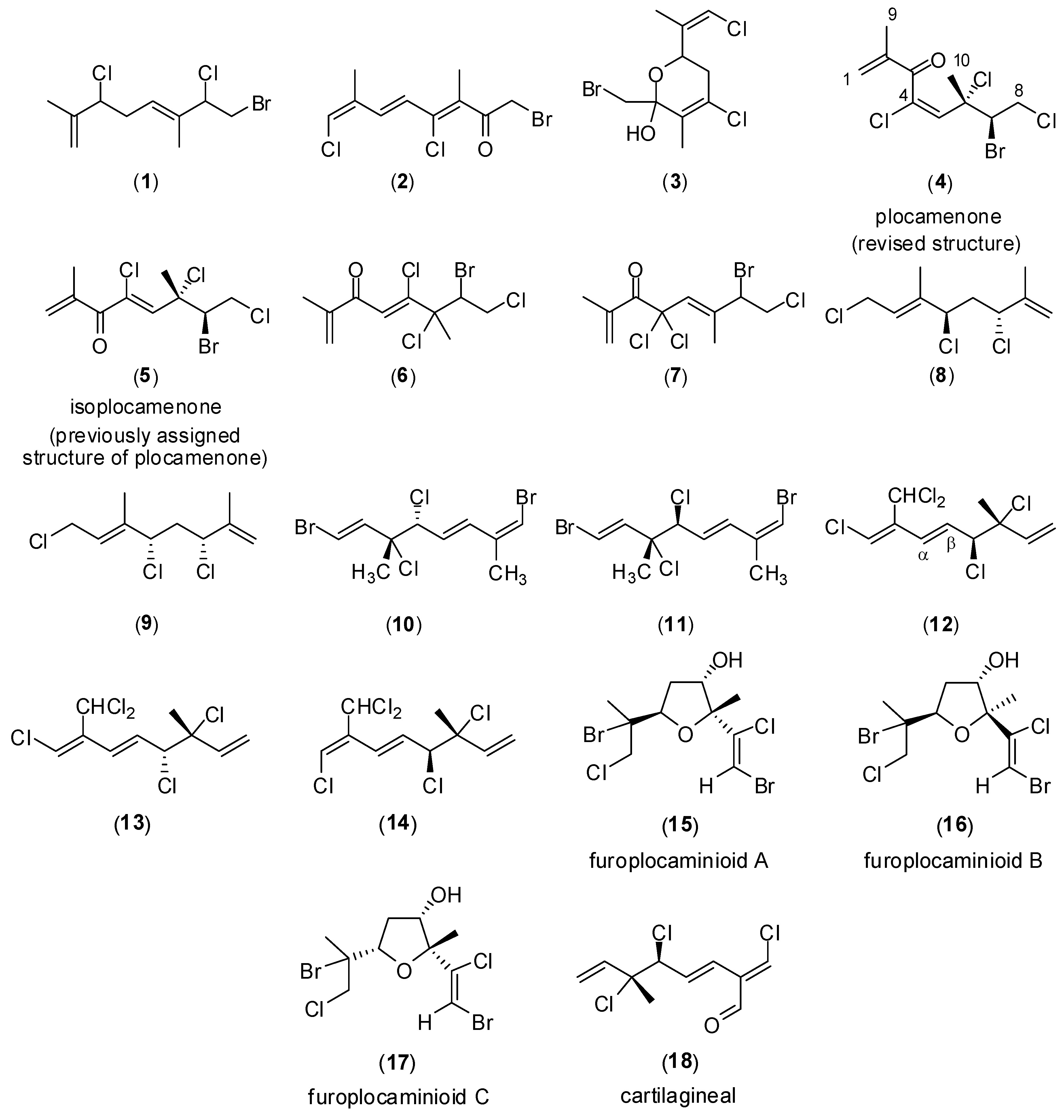
2. Results and Discussion
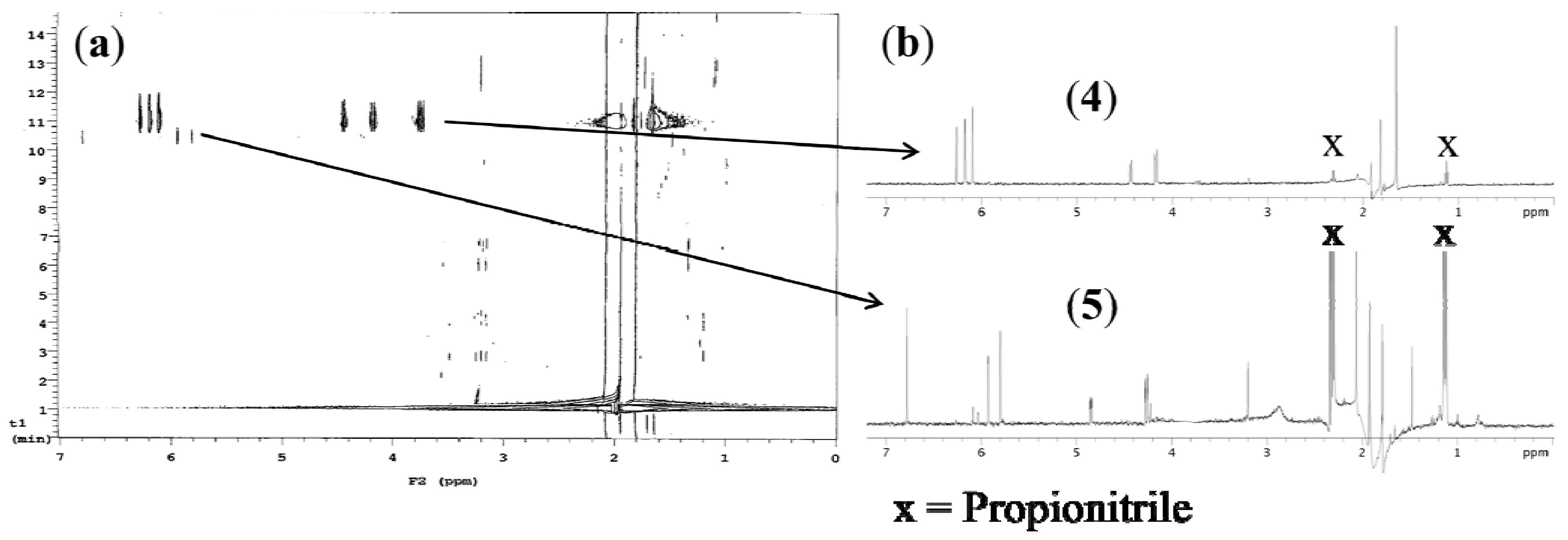
2.1. Secondary Metabolite Profiling of P. angustum Using On-Flow HPLC-NMR Analysis

2.2. Secondary Metabolite Profiling of P. angustum Using Stop-Flow HPLC-NMR Analysis
2.3. Off-Line Isolation and Purification of Secondary Metabolites from P. angustum

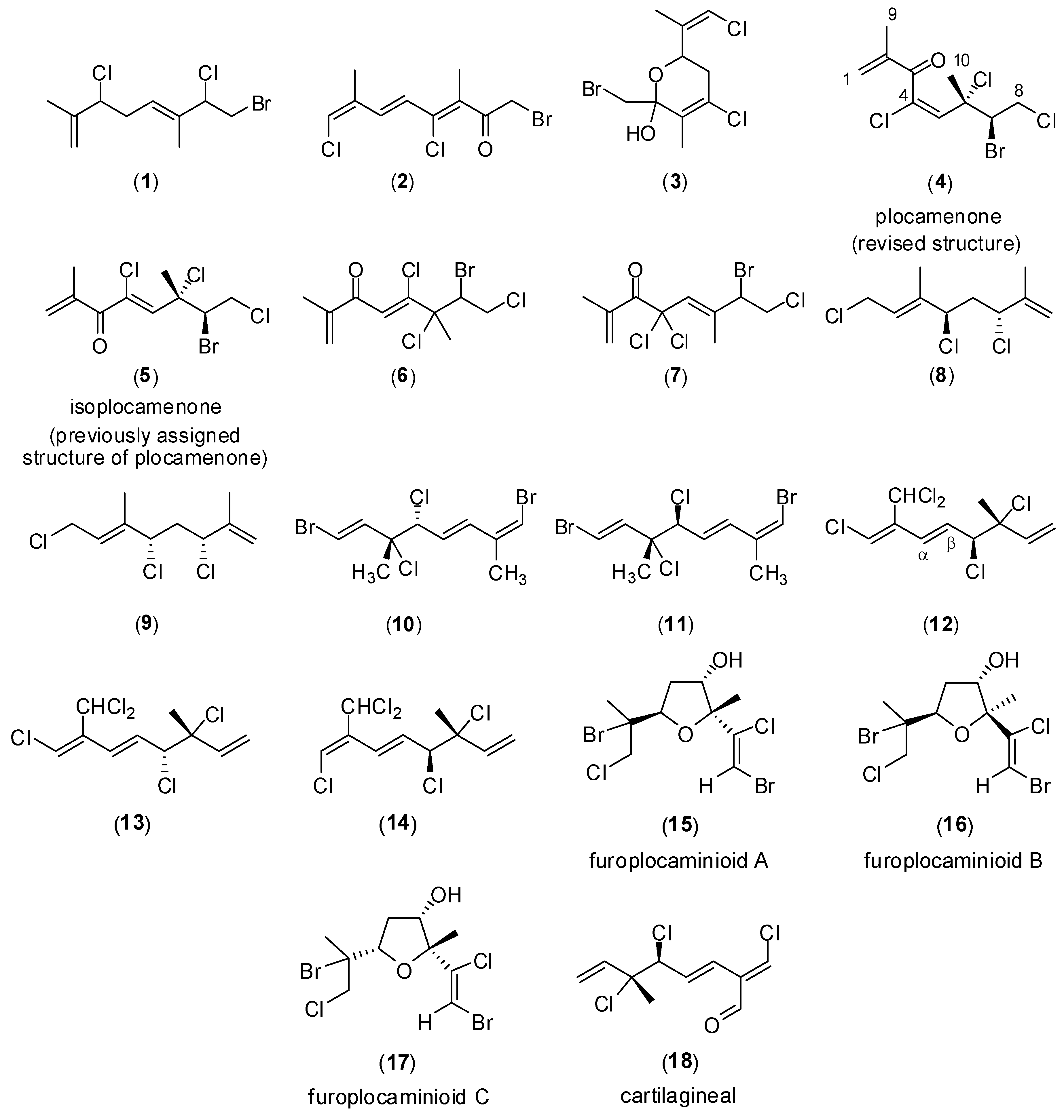{kind=link}
{kind=link}
| Plocamenone (4) | Isoplocamenone (5) | |||||||
|---|---|---|---|---|---|---|---|---|
| Position | δH (J in Hz) | δCa, mult | gCOSY | gHMBC | δH (J in Hz) | δCb, mult | gCOSY | gHMBC |
| 1a | 6.13, s | 131.2, CH2 | 9 | 2, 3, 9 | 5.91, s | 129.0, CH2 | 9 | 2, 3, 9 |
| 1b | 6.00, s | |||||||
| 2 | - | 141.7, C | - | - | - | 143.5, C | - | - |
| 3 | - | 191.5, C | - | - | - | 192.3, C | - | - |
| 4 | - | 129.9, C | - | - | 133.7, C | - | ||
| 5 | 6.20, s | 133.3, CH | - | 3, 4, 6, 7, 10 | 6.84, s | 140.8, CH | - | 3, 4, 6, 7, 10 |
| 6 | - | 69.0, C | - | - | - | 69.7, C | - | - |
| 7 | 4.36, dd, (3, 9) | 62.1, CH | 8a, 8b | 5, 6, 8, 10 | 4.97, dd, (2.5, 9) | 61.5, CH | 8a, 8b | 6, 8, 10 |
| 8a | 3.72, dd, (9, 12) | 46.3, CH2 | 7, 8b | 7 | 3.93, dd, (9, 13) | 47.1, CH2 | 7, 8b | 7 |
| 8b | 4.24, dd, (3, 12) | 7, 8a | 6 | 4.38, dd, (2.5, 13) | 7, 8a | 6 | ||
| 9 | 1.94, s | 16.5, CH3 | 1 | 1, 2, 3 | 1.97, s | 18.0, CH3 | 1b | 1, 2, 3 |
| 10 | 1.75, s | 26.8, CH3 | - | 5, 6, 7 | 2.05, s | 26.2, CH3 | - | 5, 6, 7 |
| plocamenone (4) | isoplocamenone (5) | Differences (ppm) | ||||
|---|---|---|---|---|---|---|
| Position | δH | δCa | δH | δCa | ∆δH | ∆δC |
| 1a | 6.17, s | 132.3, (t) | 5.91, s | 129.0, (t) | +0.26 | +3.3 |
| 1b | 6.23, s | 6.00, s | +0.23 | |||
| 2 | - | 143.3, (s) | - | 143.5, (s) | −0.2 | |
| 3 | - | 193.2, (s) | - | 192.3, (s) | +0.9 | |
| 4 | - | 130.5, (s) | - | 133.7, (s) | −3.2 | |
| 5 | 6.39, s | 134.7, (d) | 6.84, s | 140.8, (d) | −0.45 | −6.1 |
| 6 | - | 70.7, (s) | - | 69.7, (s) | +1.0 | |
| 7 | 4.57, dd, (3, 9.5) | 63.5, (d) | 4.97, dd, (2.5, 9) | 61.5, (d) | −0.40 | +2.0 |
| 8a | 3.80, dd, (9.5, 12) | 47.6, (t) | 3.93, dd, (9, 13) | 47.1, (t) | −0.13 | +0.5 |
| 8b | 4.27, dd, (3, 12) | 4.37, dd (2.5, 13) | −0.10 | |||
| 9 | 1.93 | 16.6, (q) | 1.97, s | 18.0, (q) | −0.04 | −1.4 |
| 10 | 1.76 | 27.6, (q) | 2.05, s | 26.2, (q) | −0.29 | +1.4 |
| Specimen Voucher Code | P388 cytotoxiciy IC50 (ng/mL) |
|---|---|
| 2006-04 (crude extract) | <4,875 |
| 2009-14 (crude extract) | <4,875 |
| 2009-15 (crude extract) | 22,743 |
| plocamenone (4) (obtained from 2006-04) | 157.5 * |
| plocamenone (4) (obtained from 2009-14) | <97.5 |
| plocamenone (4) (obtained from 2009-15) | <97.5 |
| mixture of 4 and 5 (obtained from 2009-14) | <97.5 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. HPLC-NMR Experimental Procedure
3.3. Biological Evaluation and Details of Assays
3.4. Alga Collections and Identification
3.5. Extraction and Isolation
3.6. Off-Line Characterisation of 4 and 5
4. Conclusions
Supplementary Files
Acknowledgments
References
- Dias, D.; Urban, S. Phytochemical analysis of the Southern Australian marine alga, Plocamium mertensii using HPLC-NMR. Phytochem. Anal. 2008, 19, 453–470. [Google Scholar] [CrossRef]
- Dias, D.A.; Urban, S. Application of HPLC-NMR for the rapid chemical profiling of a Southern Australian sponge, Dactylospongia sp. J. Sep. Sci. 2009, 32, 542–548. [Google Scholar] [CrossRef]
- Dias, D.A.; White, J.M.; Urban, S. Laurencia filiformis: Phytochemical profiling by conventional and HPLC-NMR approaches. Nat. Prod. Commun. 2009, 4, 157–172. [Google Scholar]
- Timmers, M.; Urban, S. On-line (HPLC-NMR) and off-line phytochemical profiling of the australian plant, Lasiopetalum macrophyllum. Nat. Prod.Commun. 2011, 6, 1605–1616. [Google Scholar]
- Brkljaca, R.; Urban, S. Recent advancements in HPLC-NMR and applications for natural product profiling and identification. J. Liq. Chromatogr. Relat. Technol. 2011, 34, 1063–1076. [Google Scholar] [CrossRef]
- Urban, S.; Separovic, F. Developments in hyphenated spectroscopic methods in natural product profiling. Front. Drug Des. Discov. 2005, 1, 113–166. [Google Scholar]
- Mynderse, J.S.; Faulkner, D.J. Polyhalogenated monoterpenes from the red alga Plocamium cartilagineum. Tetrahedro. 1975, 31, 1963–1967. [Google Scholar] [CrossRef]
- Dunlop, R.W.; Murphy, P.T.; Wells, R.J. A new polyhalogenated monoterpene from the red alga Plocamium angustum. Aust. J. Chem. 1979, 32, 2735–2739. [Google Scholar] [CrossRef]
- Brownlee, R.T.C.; Hall, J.G.; Reiss, J.A. An application of the INEPT pulse sequence to the NMR assignment of halogenated marine natural products. Org. Magn. Reson. 1983, 21, 544–547. [Google Scholar] [CrossRef]
- Konig, G.M.; Wright, A.D.; de Nys, R. Halogenated Monoterpenes from Plocamium costatum and Their Biological Activity. J. Nat. Prod. 1999, 62, 383–385. [Google Scholar] [CrossRef]
- Konig, G.M.; Wright, A.D.; Sticher, O. A new polyhalogenated monoterpene from the red alga Plocamium cartilagineum. J. Nat. Prod. 1990, 53, 1615–1618. [Google Scholar] [CrossRef]
- Ankisetty, S.; Nandiraju, S.; Win, H.; Park, Y.C.; Amsler, C.D.; McClintock, J.B.; Baker, J.A.; Diyabalanage, T.K.; Pasaribu, A.; Singh, M.P.; et al. Chemical investigation of predator-deterred macroalgae from the Antarctic Peninsula. J. Nat. Prod. 2004, 67, 1295–1302. [Google Scholar] [CrossRef]
- Konig, G.M.; Wright, A.D.; Linden, A. Plocamium hamatum and its monoterpenes: Chemical and biological investigations of the tropical marine red alga. Phytochemistry 1999, 52, 1047–1053. [Google Scholar] [CrossRef]
- Knott, M.G.; Mkwananzi, H.; Arendse, C.E.; Hendricks, D.T.; Bolton, J.J.; Beukes, D.R. Plocoralides A-C, polyhalogenated monoterpenes from the marine alga Plocamium corallorhiza. Phytochemistr. 2005, 66, 1108–1112. [Google Scholar] [CrossRef]
- Argandona, V.H.; Rovirosa, J.; San-Martin, A.; Riquelme, A.; Diaz-Marrero, A.R.; Cueto, M.; Darias, J.; Santana, O.; Guadano, A.; Gonzalez-Coloma, A. Antifeedant effects of marine halogenated monoterpenes. J. Agric. Food Chem. 2002, 50, 7029–7033. [Google Scholar] [CrossRef]
- Afolayan, A.F.; Mann, M.G.A.; Lategan, C.A.; Smith, P.J.; Bolton, J.J.; Beukes, D.R. Antiplasmodial halogenated monoterpenes from the marine red alga Plocamium cornutum. Phytochemistr. 2009, 70, 597–600. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Yoon, M.-Y.; Cha, M.-R.; Hwang, J.-H.; Park, E.; Choi, S.-U.; Park, H.-R.; Hwang, Y.-I. Methanolic Extracts of Plocamium telfairiae Induce Cytotoxicity and Caspase-Dependent Apoptosis in HT-29 Human Colon Carcinoma Cells. J. Med. Foo. 2007, 10, 587–593. [Google Scholar] [CrossRef]
- Crews, P.; Kho-Wiseman, E. Acyclic polyhalogenated monoterpenes from the red algae Plocamium violaceum. J. Org. Chem. 1977, 42, 2812–2815. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2010, 27, 165–237. [Google Scholar] [CrossRef]
- Stierle, D.B.; Wing, R.M.; Sims, J.J. Marine natural products. XVI. Polyhalogenated acyclic monoterpenes from the red alga Plocamium of Antarctica. Tetrahedron 1979, 35, 2855–2859. [Google Scholar] [CrossRef]
- Leary, J.V.; Kfir, R.; Sims, J.J.; Fulbright, D.W. The mutagenicity of natural products from marine algae. Mutat. Res. 1979, 68, 301–305. [Google Scholar] [CrossRef]
- Naylor, S.; Manes, L.V.; Crews, P. Carbon-13 substituent effects in multifunctional marine natural products. J. Nat. Prod. 1985, 48, 72–75. [Google Scholar] [CrossRef]
- Stierle, D.B.; Sims, J.J. Plocamenone, a unique halogenated monoterpene from the red alga, Plocamium. Tetrahedron Lett. 1984, 25, 153–156. [Google Scholar] [CrossRef]
- Kesternich, V.; Martinez, R.; Gutierrez, E.; Ballesteros, K.; Mansilla, H. Antibacterial activity of some compounds isolated from Ceramium rubrum against gram negative bacteria. Bol. Soc. Chil. Quim. 1997, 42, 105–108. [Google Scholar]
- Gribble, G.W. Naturally Occurring Organohalogen Compounds—A Comprehensive Update; Series Fortschritte der Chemie organischer Naturstoffe Progress in the Chemistry of Organic Natural Products; Springer-Verlag/Wien: New York, NY, USA, 2010; Volume 91, pp. 9–348. [Google Scholar]
- Jongaramruong, J.; Blackman, A.J. Polyhalogenated monoterpenes from a Tasmanian collection of the red seaweed Plocamium cartilagineum. J. Nat. Prod. 2000, 63, 272–275. [Google Scholar] [CrossRef]
- Rezanka, T.; Dembitsky, V.M. Polyhalogenated homosesquiterpenic fatty acids from Plocamium cartilagineum. Phytochemistr. 2001, 57, 607–611. [Google Scholar] [CrossRef]
- Crews, P. Monoterpene halogenation by the red alga Plocamium oregonum. J. Org. Chem. 1977, 42, 2634–2636. [Google Scholar] [CrossRef]
- Abreu, P.M.; Galindro, J.M. Polyhalogenated monoterpenes from Plocamium cartilagineum from the Portuguese coast. J. Nat. Prod. 1996, 59, 1159–1162. [Google Scholar] [CrossRef]
- Crews, P.; Naylor, S.; Hanke, F.J.; Hogue, E.R.; Kho, E.; Braslau, R. Halogen regiochemistry and substituent stereochemistry determination in marine monoterpenes by carbon-13 NMR. J. Org. Chem. 1984, 49, 1371–1377. [Google Scholar] [CrossRef]
- Hevesi, L.; Nagy, J.B.; Krief, A.; Derouane, E.G. Proton and carbon-13 studies of alkenes, epoxides, and cyclic thionocarbonates. Org. Magn. Reson. 1977, 10, 14–19. [Google Scholar] [CrossRef]
- Chukovskaya, E.C.; Dostovalova, V.I.; Vasileva, T.T.; Freidlina, R.K. Carbon-13 NMR spectra of some polychloroalkenes. Org. Magn. Reson. 1976, 8, 229–232. [Google Scholar]
- Kimpenhaus, W.; Auf der Heyde, W. Structure determination of highly chlorinated butenoic acids by carbon-13 NMR spectroscopy. Liebigs Ann. Chem. 1983, 378–392. [Google Scholar] [CrossRef]
- Darias, J.; Rovirosa, J.; San, M.A.; Diaz, A.R.; Dorta, E.; Cueto, M. Furoplocamioids A-C, novel polyhalogenated furanoid monoterpenes from Plocamium cartilagineum. J. Nat. Prod. 2001, 64, 1383–1387. [Google Scholar] [CrossRef]
- ACD/Labs, version 8.0; Advanced Chemistry Development, Inc.: Toronto, Canada, 2012. Available online: http://www.acdlabs.com (accessed on 4 May 2012).
- ChemDraw Ultra, version 6.0; PerkinElmer Inc.: Cambridge, MA, USA, 2012. Available online: http://www.cambridgesoft.com (accessed on 4 May 2012).
- Crews, P.; Kho-Wiseman, E. Cartilagineal. Unusual monoterpene aldehyde from marine alga. J. Org. Chem. 1974, 39, 3303–3304. [Google Scholar] [CrossRef]
- Tobey, S.W. Structure assignments in polysubstituted ethylenes by nuclear magnetic resonance. J. Org. Chem. 1969, 34, 1281–1298. [Google Scholar] [CrossRef]
- Pascual, C.; Meier, J.; Simon, W. Rule for the estimation of the chemical proton shift in double bonds. Helv. Chim. Act. 1966, 49, 164–168. [Google Scholar] [CrossRef]
- Crews, P.; Kho-Wiseman, E. Stereochemical assignments in marine natural products by carbon-13 NMR γ effects. Tetrahedron Lett. 1978, 19, 2483–2486. [Google Scholar] [CrossRef]
- Lopez, A.; Gerwick, W.H. Two new icosapentaenoic acids from the temperate red seaweed Ptilota filicina J. Agardh. Lipids 1987, 22, 190–194. [Google Scholar] [CrossRef]
- Itokawa, H.; Morris-Natschke, S.L.; Akiyama, T.; Lee, K.-H. Plant-derived natural product research aimed at new drug discovery. J. Nat. Med. 2008, 62, 263–280. [Google Scholar] [CrossRef]
- Dias, D.; Urban, S. Chemical constituents of the lichen, Candelaria concolor: A complete NMR and chemical degradative investigation. Nat. Prod. Res. 2009, 23, 925–939. [Google Scholar] [CrossRef]
- Samples Availability: Available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Timmers, M.A.; Dias, D.A.; Urban, S. Application of HPLC-NMR in the Identification of Plocamenone and Isoplocamenone from the Marine Red Alga Plocamium angustum. Mar. Drugs 2012, 10, 2089-2102. https://doi.org/10.3390/md10092089
Timmers MA, Dias DA, Urban S. Application of HPLC-NMR in the Identification of Plocamenone and Isoplocamenone from the Marine Red Alga Plocamium angustum. Marine Drugs. 2012; 10(9):2089-2102. https://doi.org/10.3390/md10092089
Chicago/Turabian StyleTimmers, Michael Anthony, Daniel Anthony Dias, and Sylvia Urban. 2012. "Application of HPLC-NMR in the Identification of Plocamenone and Isoplocamenone from the Marine Red Alga Plocamium angustum" Marine Drugs 10, no. 9: 2089-2102. https://doi.org/10.3390/md10092089
APA StyleTimmers, M. A., Dias, D. A., & Urban, S. (2012). Application of HPLC-NMR in the Identification of Plocamenone and Isoplocamenone from the Marine Red Alga Plocamium angustum. Marine Drugs, 10(9), 2089-2102. https://doi.org/10.3390/md10092089