First Total Synthesis of a Naturally Occurring Iodinated 5′-Deoxyxylofuranosyl Marine Nucleoside


{kind=link}
{kind=link}
Abstract
:1. Introduction
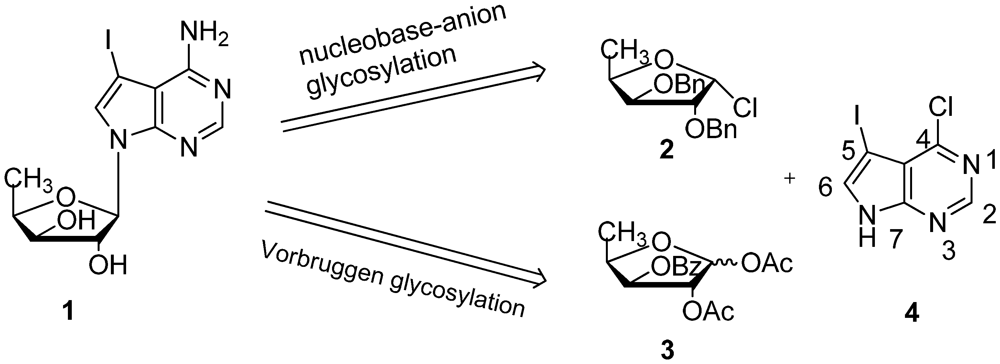
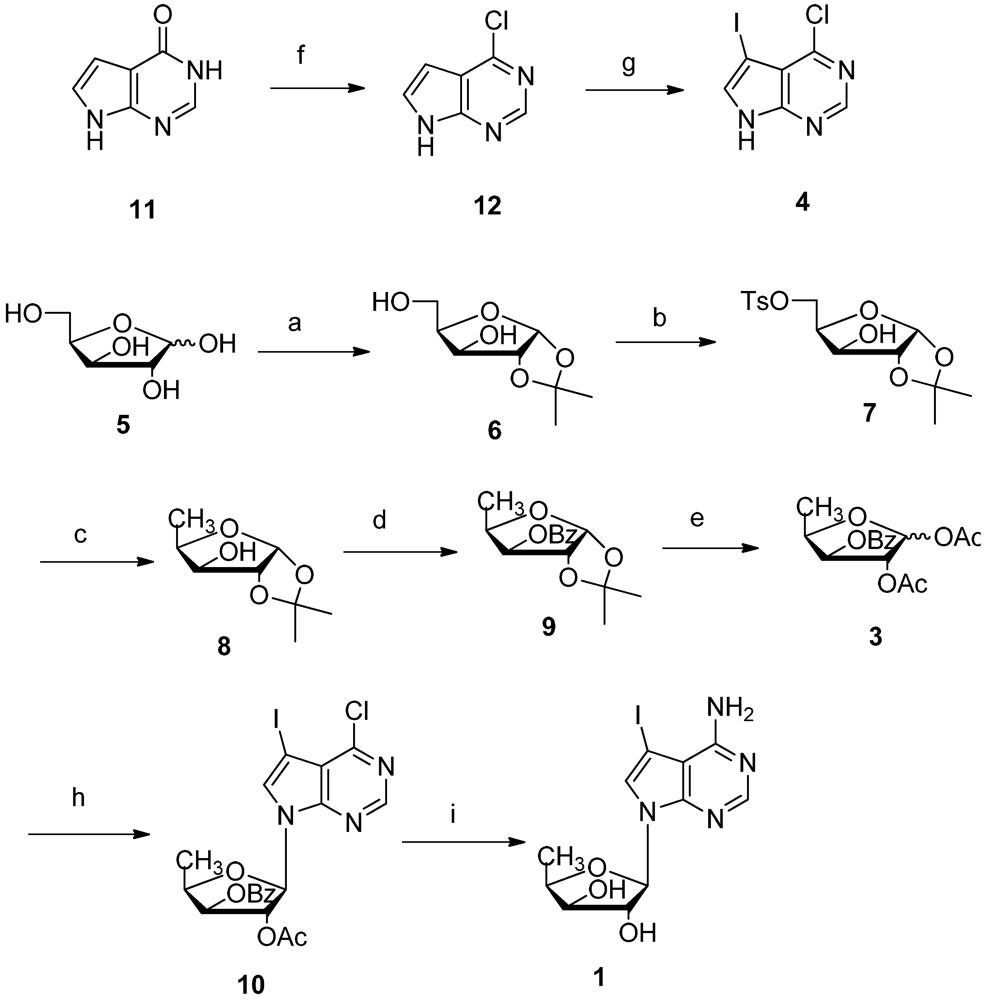
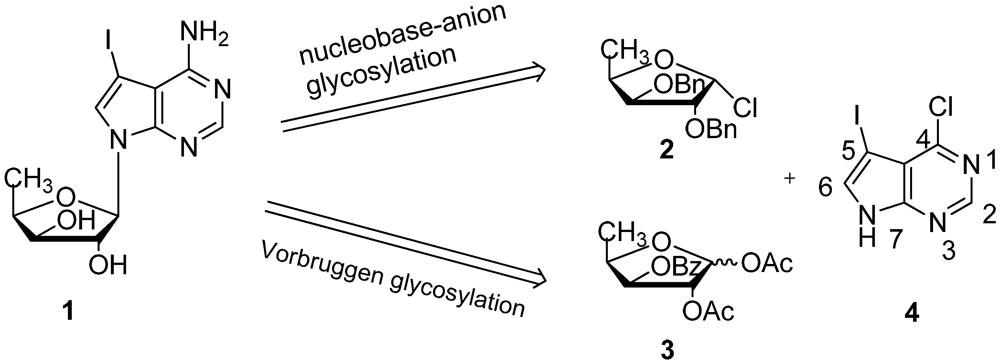
2. Results and Discussion

3. Experimental Section
3.1. General
3.2. Synthesis of 1,2-O-Isopropylidene-α-D-xylofuranose 6
3.3. Synthesis of 5-O-Tosylate-1,2-O-isopropylidene-α-D-xylofuranose 7
3.4. Synthesis of 5-Deoxy-1,2-O-isopropylidene-α-D-xylofuranose 8
3.5. Synthesis of 5-Deoxy-3-O-benzoyl-1,2-O-isopropylidene-α-D-xylofuranose 9
3.6. Synthesis of 5-Deoxy-3-O-benzoyl-1,2-O-diacetate-D-xylofuranose 3
3.7. Vorbrüggen Glycosylation of 5′-Deoxy-1,2,3-tri-O-acetyl-β-D-xylofuranose 5 and 5-Iodo-7H-pyrrolo[2,3-d]pyrimidine 10
3.8. Synthesis of 4-Amino-7-(5′-deoxy-β-D-xylofuranosyl)-5-iodo-pyrrolo[2,3-d]pyrimidine 1
4. Conclusions
Acknowledgments
- Sample Availability: Samples of the compound 1 are available from the authors.
References
- Duvall, L.R. Tubercidin. Cancer Chemother. Rep. 1963, 30, 61–62. [Google Scholar]
- Hayashi, K.; Karnio, S.; Oono, Y.; Townsend, L.B.; Nozaki, H. Toyocamycin specifically inhibits auxin signaling mediated by SCF(TIR1) pathway. Phytochemistry 2009, 70, 190–197. [Google Scholar]
- Stockwin, L.H.; Yu, S.X.; Stotler, H.; Hollingshead, M.G.; Newton, D.L. ARC (NSC 188491) has identical activity to Sangivamycin (NSC 65346) including inhibition of both P-TEFb and PKC. BMC Cancer 2009, 9, 63. [Google Scholar]
- Suzuki, H.; Kim, S.H.; Tahara, M.; Okazaki, K.; Okabe, T.; Wu, R.T.; Tanaka, N. Potentiation of cytotoxicity of 1-β-D-arabinofuranosylcytosine for K562 human-leukemic cells by cadeguomycin. Cancer Res. 1987, 47, 713–717. [Google Scholar]
- Jimeno, J.; Faircloth, G.; Sousa-Faro, J.; Scheuer, P.; Rinehart, K. New marine derived anticancer therapeutics—A journey from the sea to clinical trials. Mar. Drugs 2004, 2, 14–29. [Google Scholar]
- Seela, F.; Peng, X.H.; Budow, S. Advances in the synthesis of 7-deazapurine-pyrrolo[2,3-d]pyrimidine-2′-deoxyribonucleosides including D-and L-enantiomers, fluoro derivatives and 2′,3′-dideoxyribonucleosides. Curr. Org. Chem. 2007, 11, 427–462. [Google Scholar]
- Knapp, S. Synthesis of complex nucleoside antibiotics. Chem. Rev. 1995, 95, 1859–1876. [Google Scholar]
- Isono, K. Nucleoside antibiotics-structure, biological-acitvity and biosynthesis. J. Antibiot. 1988, 41, 1711–1739. [Google Scholar]
- Li, K.; Li, Q.-L.; Ji, N.-Y.; Liu, B.; Zhang, W.; Cao, X.-P. Deoxyuridines from the marine sponge associated actinomycete streptomyces microflavus. Mar. Drugs 2011, 9, 690–695. [Google Scholar]
- Sagar, S.; Kaur, M.; Minneman, K.P. Antiviral lead compounds from marine sponges. Mar. Drugs 2010, 8, 2619–2638. [Google Scholar]
- Bhakuni, D.S.; Rawat, D.S. Bioactive Marine Natural Products; Co-published by Springer with Anamaya Publishers: New Delhi, India, 2005; pp. 208–234. [Google Scholar]
- Margiastuti, P.; Ogi, T.; Teruya, T.; Taira, J.; Suenaga, K.; Ueda, K. An unusual iodinated 5′-deoxyxylofuranosyl nucleoside from an Okinawan ascidian, Diplosoma sp. Chem. Lett. 2008, 37, 448–449. [Google Scholar]
- Vorbruggen, H.; Krolikiewicz, K.; Bennua, B. Nucleoside synthesis. 22. Nucleoside synthesis with trimethylsilyl triflate and perchlorate as catalysis. Chem. Ber. Recl. 1981, 114, 1234–1255. [Google Scholar] [CrossRef]
- Niedballa, U.; Vorbruggen, H. General synthesis of N-glycosides. 6. Mechanism of stannic chloride catalyzed silyl Hibert–Johnson reaction. J. Org. Chem. 1976, 41, 2084–2086. [Google Scholar] [CrossRef]
- Vorbrüggen, H.; Ruh-Pohlenz, C. Handbook of Nucleoside Synthesis; John Wiley Sons: New York, NY, USA, 2001. [Google Scholar]
- Bio, M.M.; Xu, F.; Waters, M.; Williams, J.M.; Savary, K.A.; Cowden, C.J.; Yang, C.H.; Buck, E.; Song, Z.G.J.; Tschaen, D.M.; et al. Practical synthesis of a potent hepatitis C virus RNA replication inhibitor. J. Org. Chem. 2004, 69, 6257–6266. [Google Scholar]
- Dempcy, R.O.; Skibo, E.B. Regioselective synthesis of imidazo [4,5-g]quinazoline quinone nucleosides and quinazoline amino nucleosides. Studies of their xanthine-oxidase and purine nucleoside phosphorylase substrate activity. J. Org. Chem. 1991, 56, 776–785. [Google Scholar] [CrossRef]
- Ramasamy, K.; Imamura, N.; Robins, R.K.; Revankar, G.R. A facile synthesis of tubercidin and related 7-deazapurine nucleosides via the stereospecific sodium-salt glycosylation procedure. Tetrahedron Lett. 1987, 28, 5107–5110. [Google Scholar]
- Kazimierczuk, Z.; Revankar, G.R.; Robins, R.K. Total synthesis of certain 2-monosubstituted-tubercidin, 6-mono-substituted-tubercidin and 2,6-disubstituted-tubercidin derivatives-synthesis of tubercidin via the sodium-salt glycosylation procedure. Nucleic Acids Res. 1984, 12, 1179–1192. [Google Scholar]
- Seela, F.; Westermann, B.; Bindig, U. Liquid–liquid and solid–liquid phase-transfer glycosylation of pyrrolo[2,3-d]pyrimidines-stereospecific synthesis of 2-deoxy-β-D-ribofuranosides related to 2′-deoxy-7-carbaguanosine. J. Chem. Soc. Perkin Trans. 1 1988, 697–702. [Google Scholar]
- Seela, F.; Muth, H.P.; Bindig, U. Synthesis of 6-substituted 7-carbapurine 2′,3′-dideoxynucleosides-solid-liquid phase-transfer glycosylation of 4-chloropyrrolo[2,3-d]pyrimidine and deoxygenation of its 2′-deoxyribofuranoside. Synthesis 1988, 670–674. [Google Scholar]
- Lupke, U.; Seela, F. Ribosidation of 7H-pyrrolo[2,3-d] pyrimidin-4(3H)-one at N-3. Chem. Ber. Recl. 1979, 112, 3526–3529. [Google Scholar]
- Wilcox, C.S.; Otoski, R.M. Stereoselective preparations of ribofuranosyl chlorides and ribofuranosyl acetates-solvent effects and stereoselectivity in the reaction of ribofuranosyl acetates with trimethylallylsilane. Tetrahedron Lett. 1986, 27, 1011–1014. [Google Scholar]
- Kane, P.D.; Mann, J. The preparation and utility of ethyl 2-(5′-O-tertbutyldimethylsilyl-2′,3′-O-isopropylidene-β-D-ribofuranosyl)propenoate as a key intermediate for C-nucleoside synthesis. J. Chem. Soc. Perkin Trans. 1 1984, 657–660. [Google Scholar]
- Song, Y.; Ding, H.X.; Dou, Y.H.; Yang, R.C.; Sun, Q.; Xiao, Q.; Ju, Y. Efficient and practical synthesis of 5′-deoxytubercidin and its analogues via vorbruggen glycosylation. Synthesis 2011, 1442–1446. [Google Scholar]
- Seela, F.; Ming, X. 7-Functionalized 7-deazapurine β-D- and β-L-ribonucleosides related to tubercidin and 7-deazainosine: glycosylation of pyrrolo 2,3-d pyrimidines with 1-O-acetyl-2,3,5-tri-O-benzoyl-β-D or β-L-ribofuranose. Tetrahedron 2007, 63, 9850–9861. [Google Scholar]
- Seela, F.; Peng, X.H. Pyrrolo[2,3-d]pyrimidine β-L-nucleosides containing 7-deazaadenine, 2-amino-7-deazaadenine, 7-deazaguanine, 7-deazaisoguanine, and 7-deazaxanthine. Collect. Czech. Chem. Commun. 2006, 71, 956–977. [Google Scholar]
- Seela, F.; Peng, X.H. 7-Functionalized 7-deazapurine ribonucleosides related to 2-aminoadenosine, guanosine, and xanthosine: Glycosylation of pyrrolo 2,3-d pyrimidines with 1-O-acetyl-2,3,5-tri-O-benzoyl-D-ribofuranose. J. Org. Chem. 2006, 71, 81–90. [Google Scholar]
- Moravcova, J.; Capkova, J.; Stanek, J. One-pot synthesis of 1,2-O-isopropylidene-α-D-xylofuranose. Carbohydr. Res. 1994, 263, 61–66. [Google Scholar]
- Hildebrandt, B.; Nakamura, Y.; Ogawa, S. Practical synthesis of optically pure 3,4-epoxy-5-methyldihydro-2(3H)-furanones from D-xylose by regioselective and stereoselective functionalization. Carbohydr. Res. 1991, 214, 87–93. [Google Scholar]
- Mathe, C.; Imbach, J.L.; Gosselin, G. 1,2-Di-O-acetyl-5-O-benzoyl-3-deoxy-L-erythro-pentofuran ose, a convenient precursor for the stereospecific synthesis of nucleoside analogues with the unnatural β-L-configuration. Carbohydr. Res. 2000, 323, 226–229. [Google Scholar]
- Reigan, P.; Gbaj, A.; Stratford, I.J.; Bryce, R.A.; Freeman, S. Xanthine oxidase-activated prodrugs of thymidine phosphorylase inhibitors. Eur. J. Med. Chem. 2008, 43, 1248–1260. [Google Scholar]
- Reigan, P.; Gbaj, A.; Chinje, E.; Stratford, I.J.; Douglas, K.T.; Freeman, S. Synthesis enzymatic evaluation of xanthine oxidase-activated prodrugs based on inhibitors of thymidine phosphorylase. Biorg. Med. Chem. Lett. 2004, 14, 5247–5250. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sun, J.; Dou, Y.; Ding, H.; Yang, R.; Sun, Q.; Xiao, Q. First Total Synthesis of a Naturally Occurring Iodinated 5′-Deoxyxylofuranosyl Marine Nucleoside. Mar. Drugs 2012, 10, 881-889. https://doi.org/10.3390/md10040881
Sun J, Dou Y, Ding H, Yang R, Sun Q, Xiao Q. First Total Synthesis of a Naturally Occurring Iodinated 5′-Deoxyxylofuranosyl Marine Nucleoside. Marine Drugs. 2012; 10(4):881-889. https://doi.org/10.3390/md10040881
Chicago/Turabian StyleSun, Jianyun, Yanhui Dou, Haixin Ding, Ruchun Yang, Qi Sun, and Qiang Xiao. 2012. "First Total Synthesis of a Naturally Occurring Iodinated 5′-Deoxyxylofuranosyl Marine Nucleoside" Marine Drugs 10, no. 4: 881-889. https://doi.org/10.3390/md10040881
APA StyleSun, J., Dou, Y., Ding, H., Yang, R., Sun, Q., & Xiao, Q. (2012). First Total Synthesis of a Naturally Occurring Iodinated 5′-Deoxyxylofuranosyl Marine Nucleoside. Marine Drugs, 10(4), 881-889. https://doi.org/10.3390/md10040881