Two New Tryptamine Derivatives, Leptoclinidamide and (-)-Leptoclinidamine B, from an Indonesian Ascidian Leptoclinides dubius

Abstract

1. Introduction
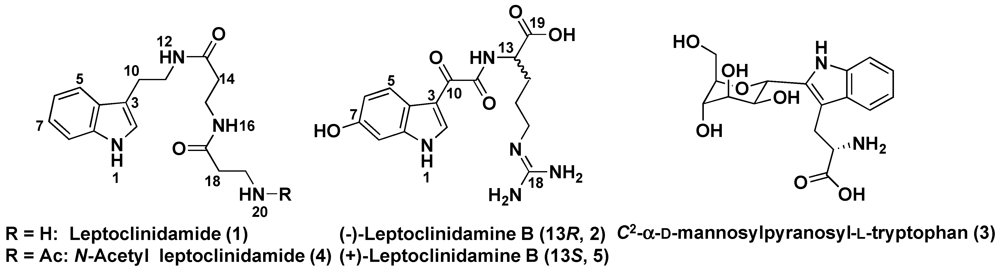
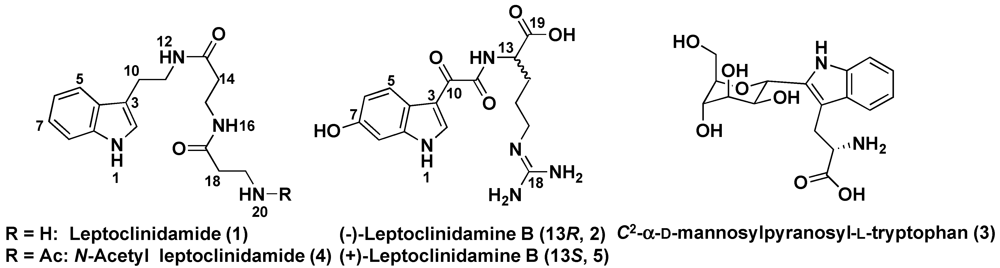
2. Results and Discussion

{kind=link}
{kind=link}
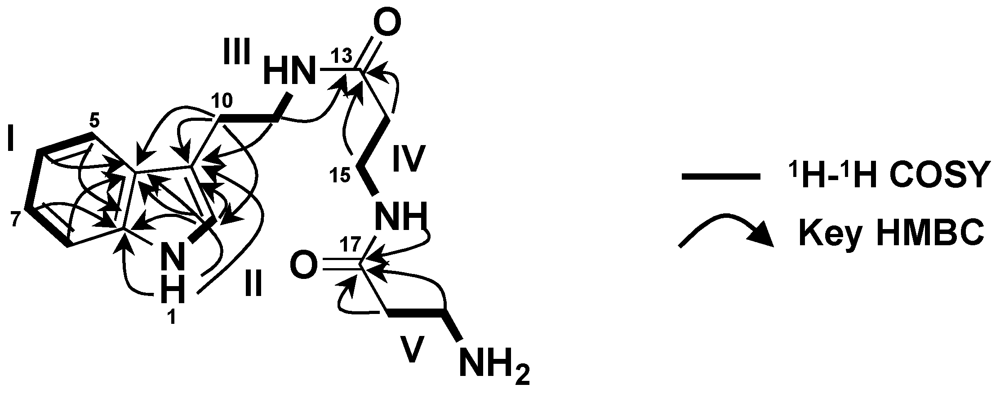
{kind=link}
{kind=link}
| No. | δC | δH ( J in Hz) | HMBC |
|---|---|---|---|
| 1-NH | - | 10.8 brs | 3, 4, 9 |
| 2 | 122.5 | 7.09 d (2.0) | 3, 4, 9 |
| 3 | 111.8 | - | |
| 4 | 127.2 | - | |
| 5 | 118.1 | 7.48 d (8.0) | 9 |
| 6 | 118.1 | 6.93 t (8.0) | 4 |
| 7 | 120.8 | 7.02 t (8.0) | 9 |
| 8 | 111.3 | 7.29 d (8.0) | 4 |
| 9 | 136.2 | - | |
| 10 | 25.1 | 2.77 brt (7.4) | 2, 3, 4 |
| 11 | 39.2 | 3.29 brt (7.4) | 3, 13 |
| 12-NH | - | 7.93 brt (5.8) | |
| 13 | 170.0 | - | |
| 14 | 35.2 | 2.24 brt (7.4) | 13 |
| 15 | 35.3 | 3.23 brt (7.4) | 13 |
| 16-NH | - | 8.07 brt (5.8) | 17 |
| 17 | 169.2 | - | |
| 18 | 31.9 | 2.38 brt (7.4) | 17 |
| 19 | 35.3 | 2.93 brt (7.4) | 17 |
| 20-NH2 | - | 7.66 brs |
3. Experimental Section
3.1. General
3.2. Materials
3.3. Ascidian
3.4. Extraction and Isolation
3.5. Synthesis of N-Acetyl Derivative (4) of Compound 1
3.6. Acid Hydrolysis of Compound 2
3.7. Chiral HPLC of Peptide Hydrolysate
3.8.Marfey’s Analysis
3.9. Antimicrobial Assay
3.10. Cytotoxicity Assay
4. Conclusions
Acknowledgments
References
- Faulkner, D.J. Marine natural products. Nat. Prod. Rep. 2002, 19, 1–48, and previous reports in this series. [Google Scholar]
- Blunt, J.W.; Copp, B.R.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2011, 28, 196–268, and previous reports in this series. [Google Scholar] [CrossRef]
- Wang, W.F.; Namikoshi, M. Bioactive nitrogenous metabolites from ascidians. Heterocycles 2007, 74, 53–88. [Google Scholar]
- De Beer, T.; Vliegenthart, J.F.G.; Loffler, A.; Hofsteenge, J. The hexopyranosyl residue that is C-glycosidically linked to the side chain of tryptophan-7 in human RNase Us is α-mannopyranose. Biochemistry 1995, 34, 11785–11789. [Google Scholar]
- Gutsche, B.; Grun, C.; Scheutzow, D.; Herderich, M. Tryptophan glycoconjugates in food and human urine. Biochem. J. 1999, 343, 11–19. [Google Scholar]
- Manabe, S.; Ito, Y. Total synthesis of novel subclass of glyco-amino acid structure motif: C2-α-L-C-mannosylpyranosyl-L-tryptophan. J. Am. Chem. Soc. 1999, 121, 9754–9755. [Google Scholar]
- Nishikawa, T.; Ishikawa, M.; Isobe, M. Synthesis of a α-C-mannosyltryptophan derivative, naturally occurring C-glycosyl amino acid found in human ribonuclease. Synlett 1999, 123–125. [Google Scholar]
- Garcia, A.; Lenis, L.A.; Jimenez, C.; Debitus, C.; Quinoa, E.; Riguera, R. The occurrence of the human glycoconjugate C2-α-D-mannosylpyranosyl-L-tryptophan in marine ascidians. Org. Lett. 2000, 2, 2765–2767. [Google Scholar]
- Carroll, A.R.; Avery, V.M. Leptoclinidamines A-C, indole alkaloids from the Australian ascidian Leptoclinides durus. J. Nat. Prod. 2009, 72, 696–699. [Google Scholar]
- Marfey, P. Determination of D-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlsberg Res. Commun. 1984, 49, 591–596. [Google Scholar] [CrossRef]
- Kuo, H.K.; Kai, K.; Akiyama, K.; Hayashi, H. Novel bioactive peptides, PF1171F and PF1171G, from unidentified ascomycete OK-128. Tetrahedron Lett. 2012, 53, 429–431. [Google Scholar]
- Li, J.L.; Han, S.C.; Yoo, E.S.; Shin, S.; Hong, J.; Cui, Z.; Li, H.; Jung, J.H. Anti-inflammatory amino acid derivatives from the ascidian Herdmania momus. J. Nat. Prod. 2011, 74, 1792–1797. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar]
- Samples Availability: Not available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yamazaki, H.; Wewengkang, D.S.; Nishikawa, T.; Rotinsulu, H.; Mangindaan, R.E.P.; Namikoshi, M. Two New Tryptamine Derivatives, Leptoclinidamide and (-)-Leptoclinidamine B, from an Indonesian Ascidian Leptoclinides dubius. Mar. Drugs 2012, 10, 349-357. https://doi.org/10.3390/md10020349
Yamazaki H, Wewengkang DS, Nishikawa T, Rotinsulu H, Mangindaan REP, Namikoshi M. Two New Tryptamine Derivatives, Leptoclinidamide and (-)-Leptoclinidamine B, from an Indonesian Ascidian Leptoclinides dubius. Marine Drugs. 2012; 10(2):349-357. https://doi.org/10.3390/md10020349
Chicago/Turabian StyleYamazaki, Hiroyuki, Defny S. Wewengkang, Teruaki Nishikawa, Henki Rotinsulu, Remy E. P. Mangindaan, and Michio Namikoshi. 2012. "Two New Tryptamine Derivatives, Leptoclinidamide and (-)-Leptoclinidamine B, from an Indonesian Ascidian Leptoclinides dubius" Marine Drugs 10, no. 2: 349-357. https://doi.org/10.3390/md10020349
APA StyleYamazaki, H., Wewengkang, D. S., Nishikawa, T., Rotinsulu, H., Mangindaan, R. E. P., & Namikoshi, M. (2012). Two New Tryptamine Derivatives, Leptoclinidamide and (-)-Leptoclinidamine B, from an Indonesian Ascidian Leptoclinides dubius. Marine Drugs, 10(2), 349-357. https://doi.org/10.3390/md10020349