Amino Acid Prodrugs: An Approach to Improve the Absorption of HIV-1 Protease Inhibitor, Lopinavir

Abstract
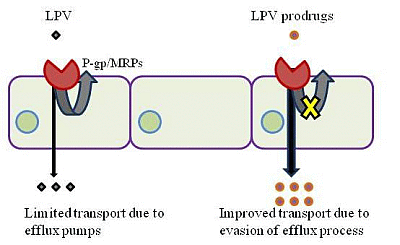
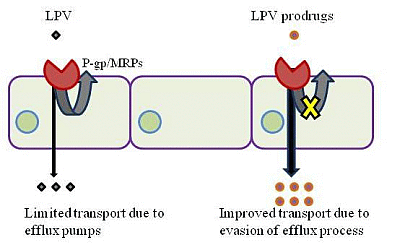
:
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Methods
2.2.1. Cell Culture
2.2.2. Solubility Study in Distilled Deionized Water (DDW)
2.2.3. Buffer Stability Studies
2.2.4. Cytotoxicity Studies
2.2.5. Uptake Studies
2.2.6. Transport Studies
2.2.7. Rat Microsomal Stability Studies
2.2.7.1. Time Dependent Stability Studies
2.2.7.2. Concentration Dependent Degradation Studies
2.3. Data Analysis
2.3.1. HPLC Analysis
2.3.2. Sample Preparation for LCMS/MS Analysis
2.3.3. LCMS/MS Analysis
2.3.4. Permeability Analysis
2.3.5. Statistical Analysis
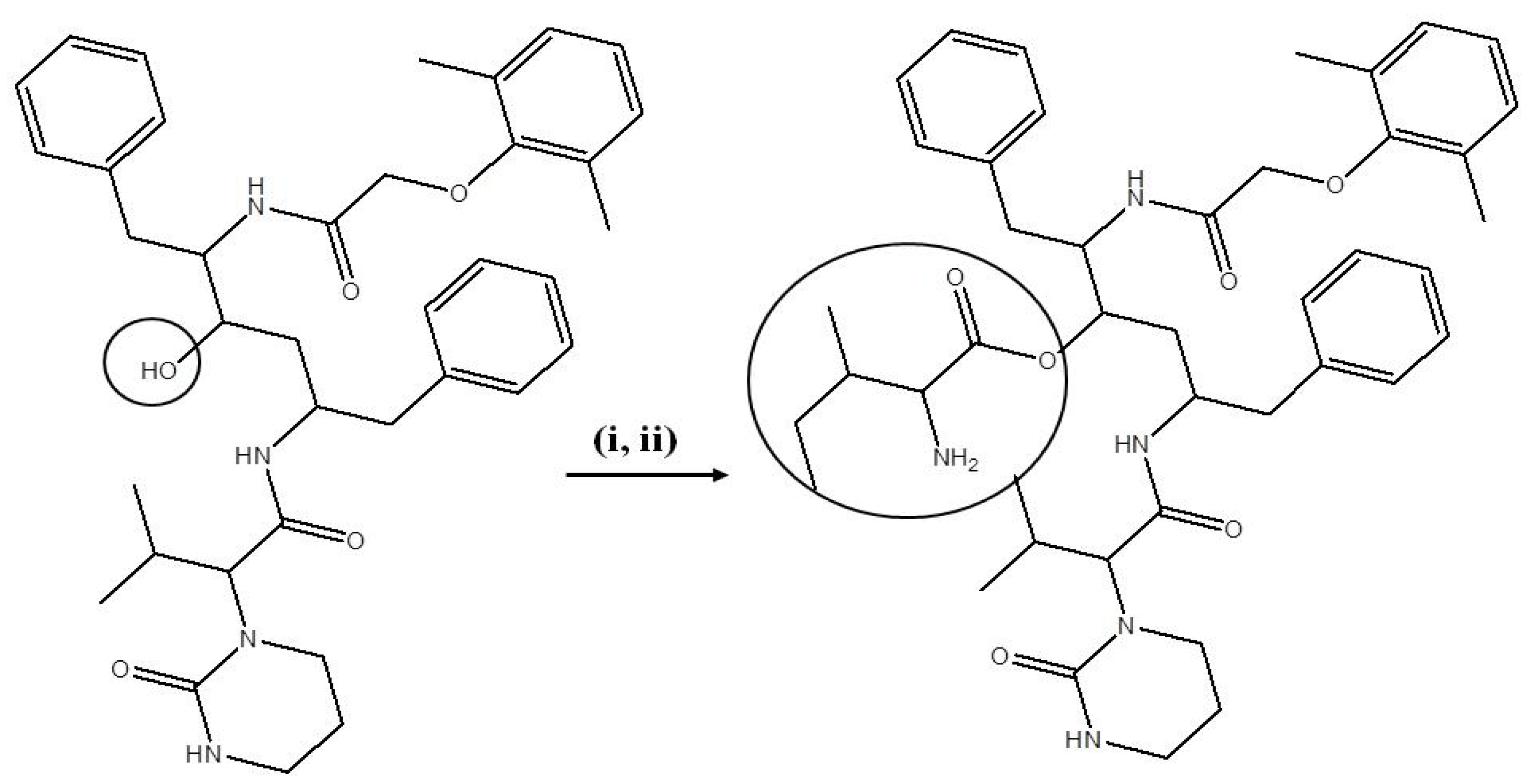
2.4. Synthesis and Identification of LPV Prodrugs
2.4.1. Synthesis of LPV Prodrugs
2.4.2. Deprotection of the N-Boc Group

2.5. Identification and Purity of Prodrugs
3. Results and Discussion
3.1. Solubility in DDW
3.2. Buffer Stability Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prodrugs | pH 4 | pH 5.5 | pH 7.4 |
|---|---|---|---|
| Ile-LPV | 101 ± 29 | 53 ± 10 | 6.4 ± 0.3 |
| Met-LPV | 119 ± 6 | 55 ± 6 | 6.2 ± 0.4 |
| Trp-LPV | 123 ± 14 | 50 ± 9 | 5.4 ± 0.3 |
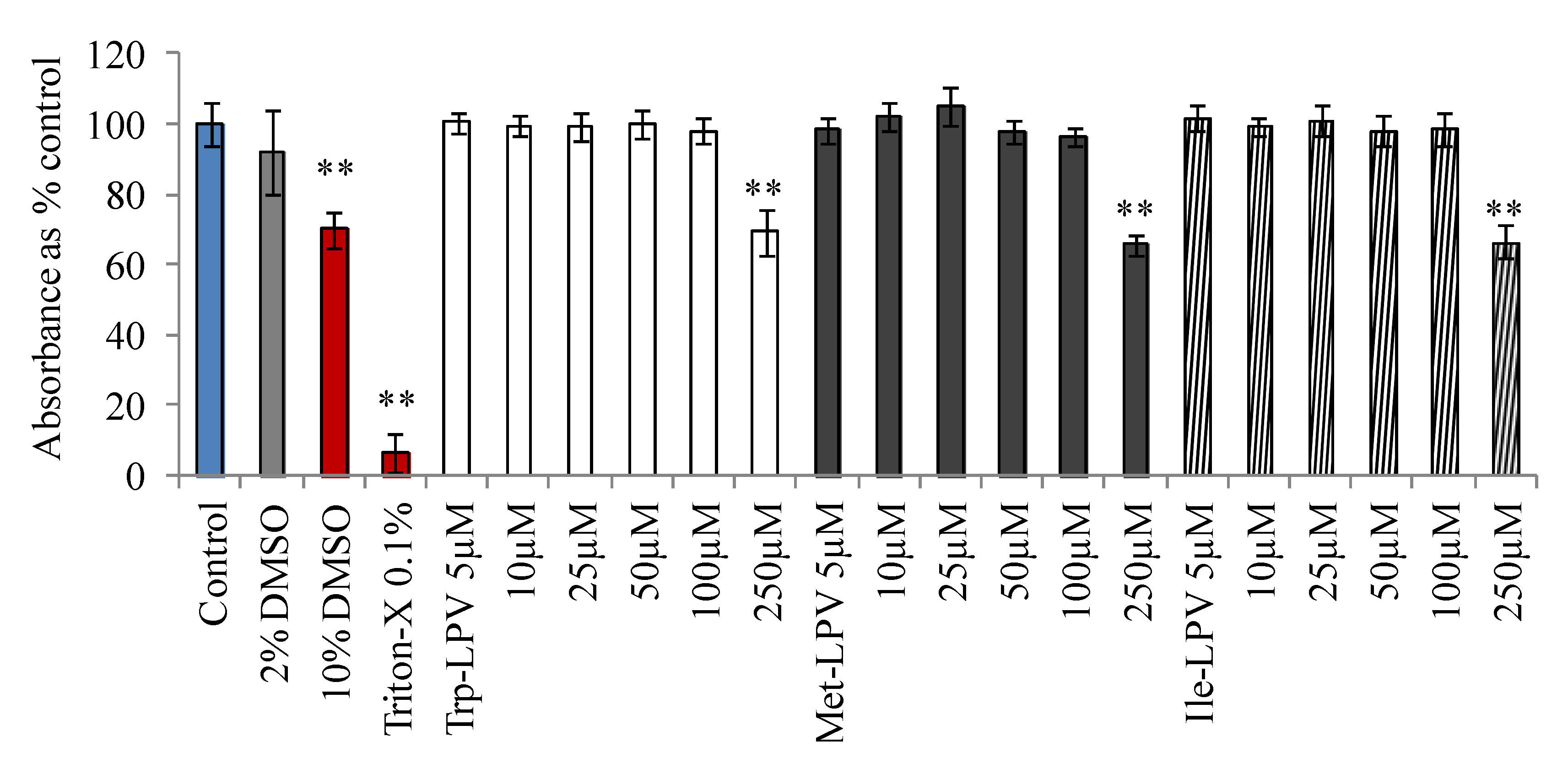
3.3. Cytotoxicity Studies
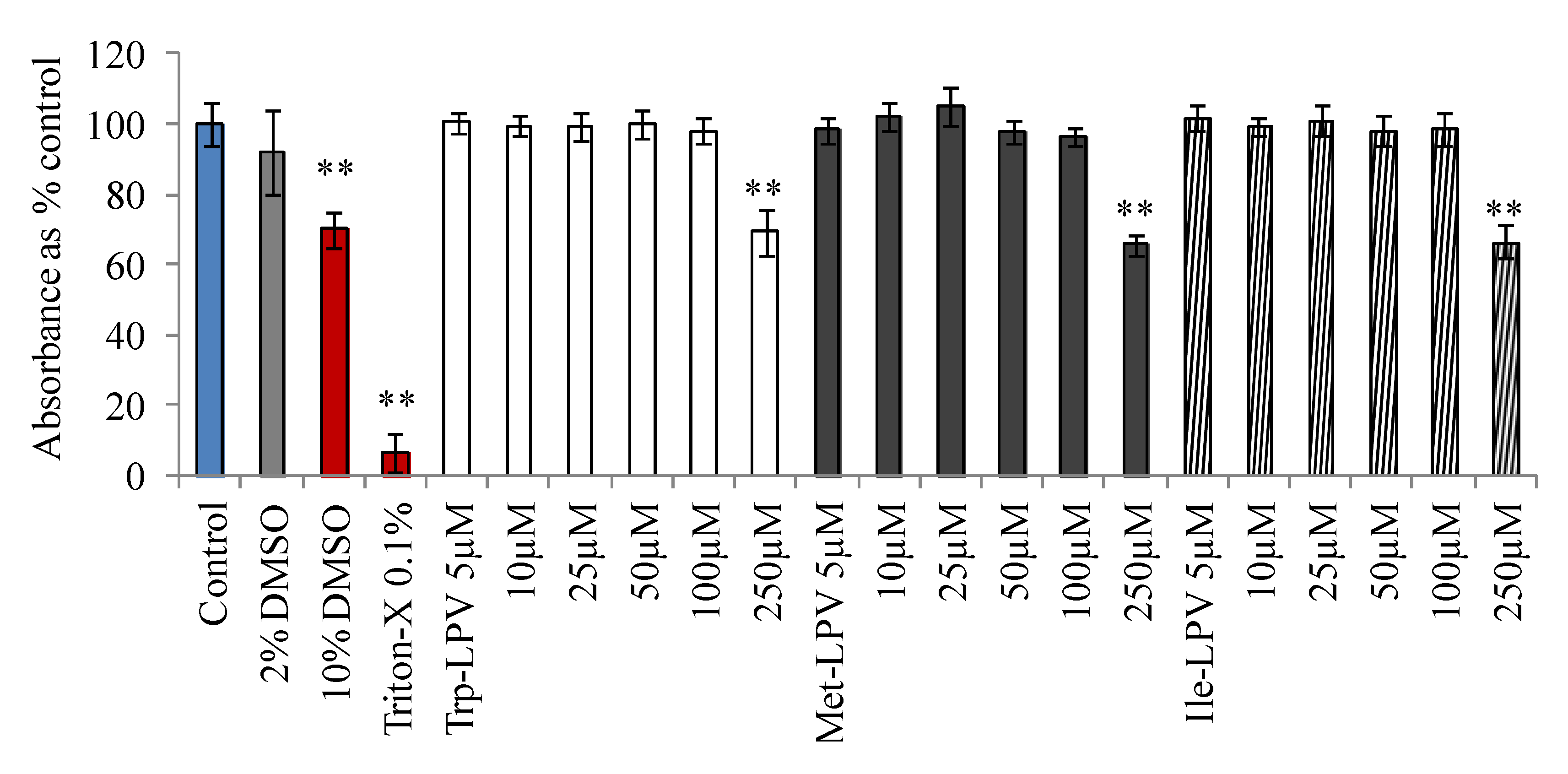
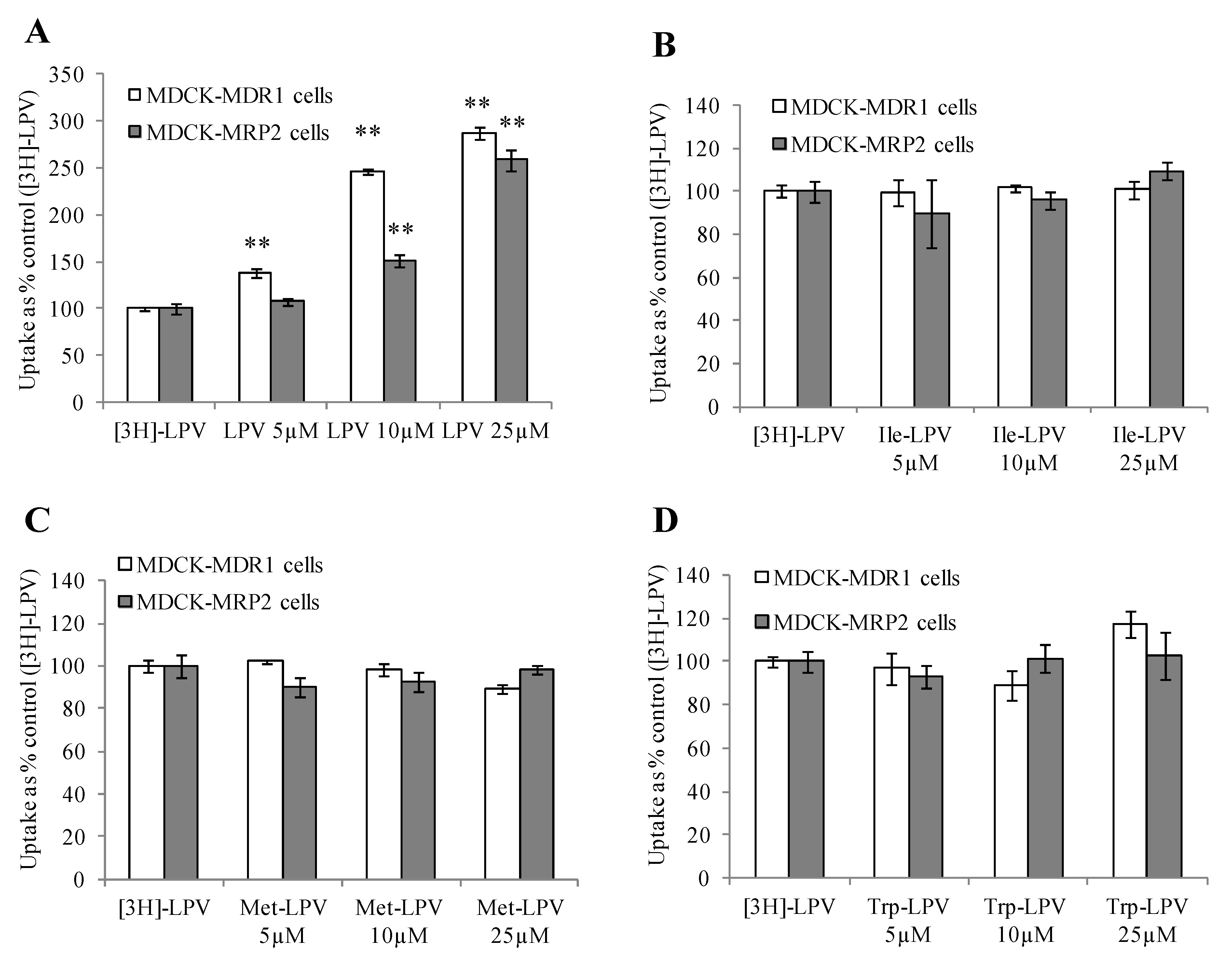
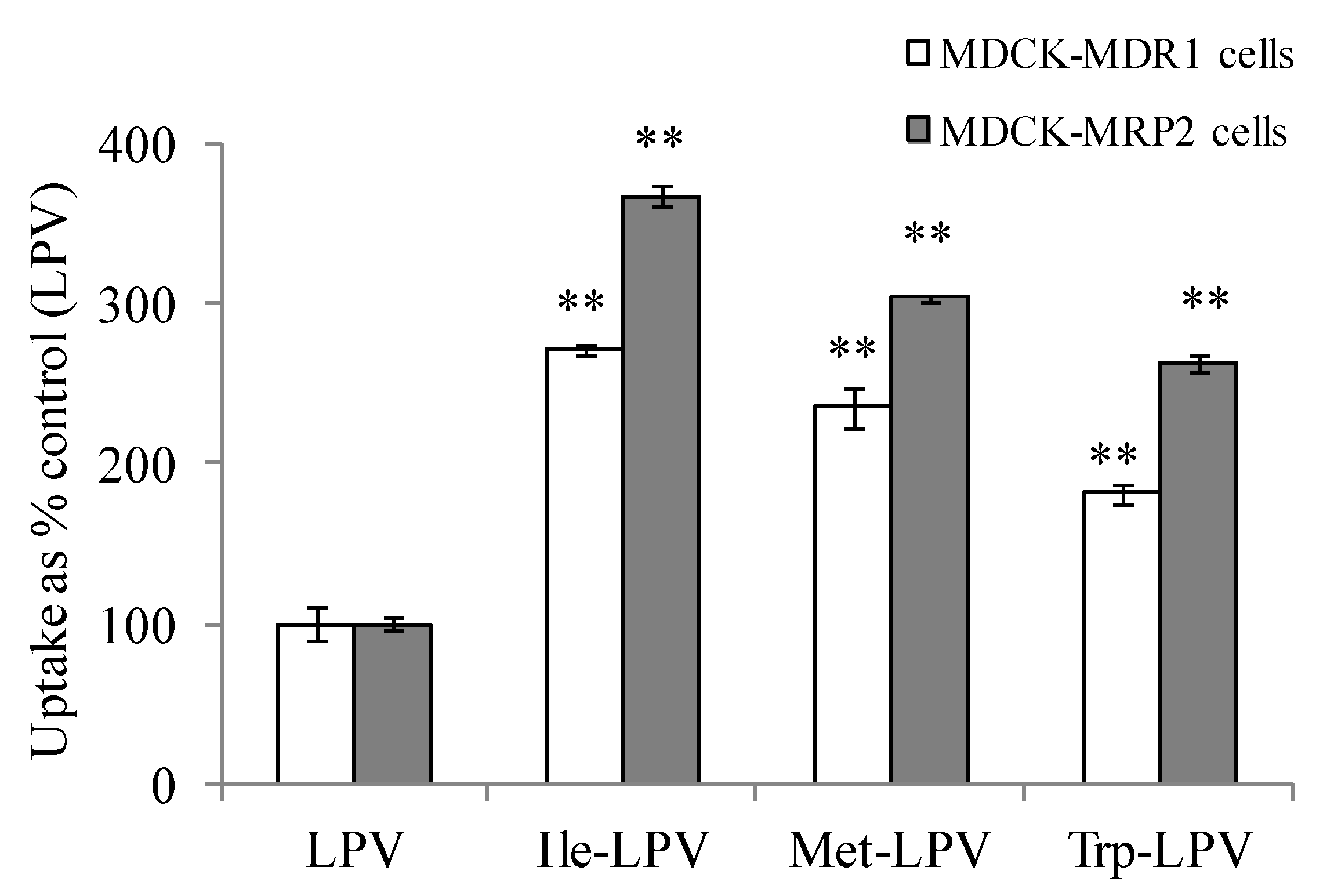
3.4. Cellular Uptake Studies
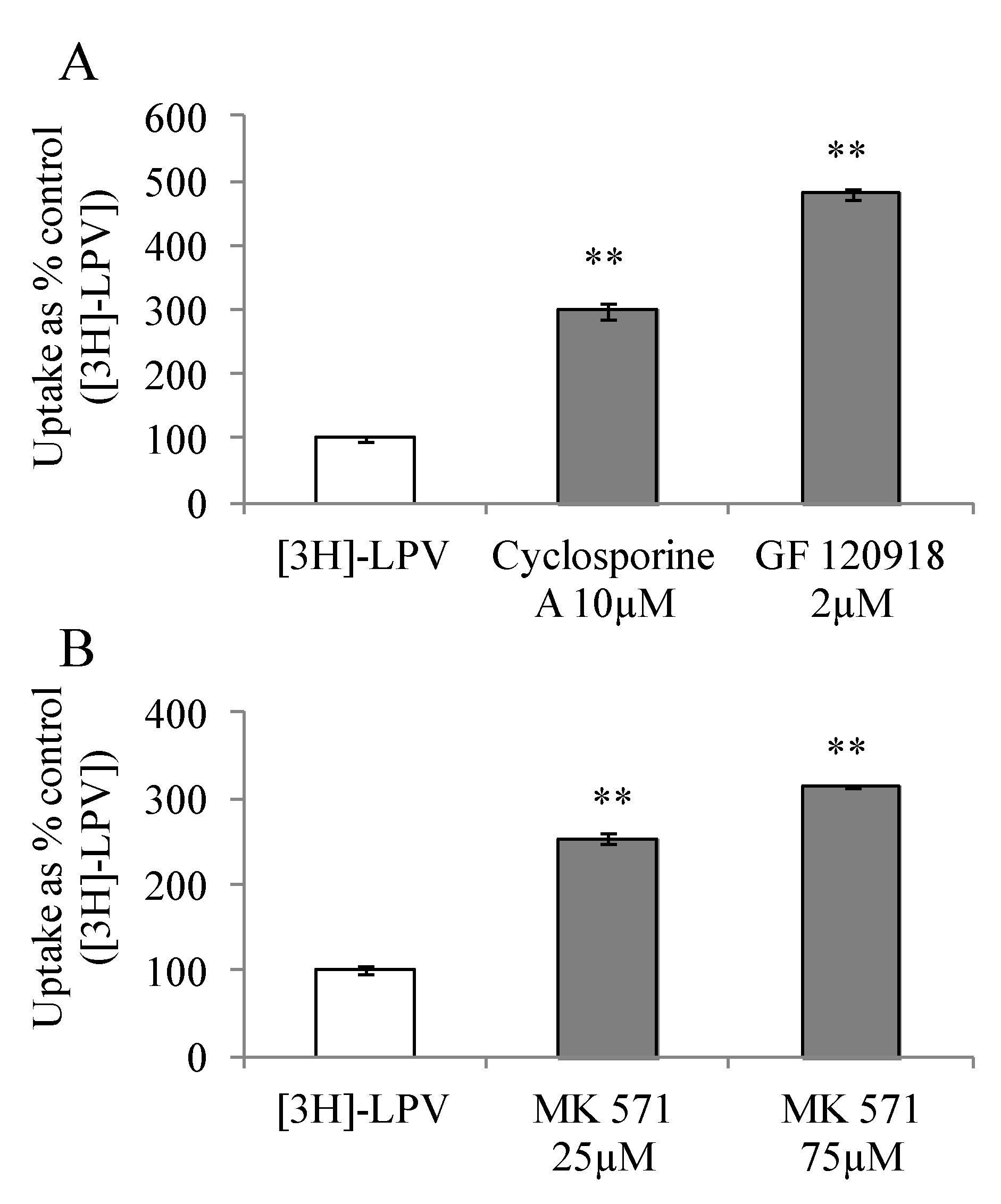
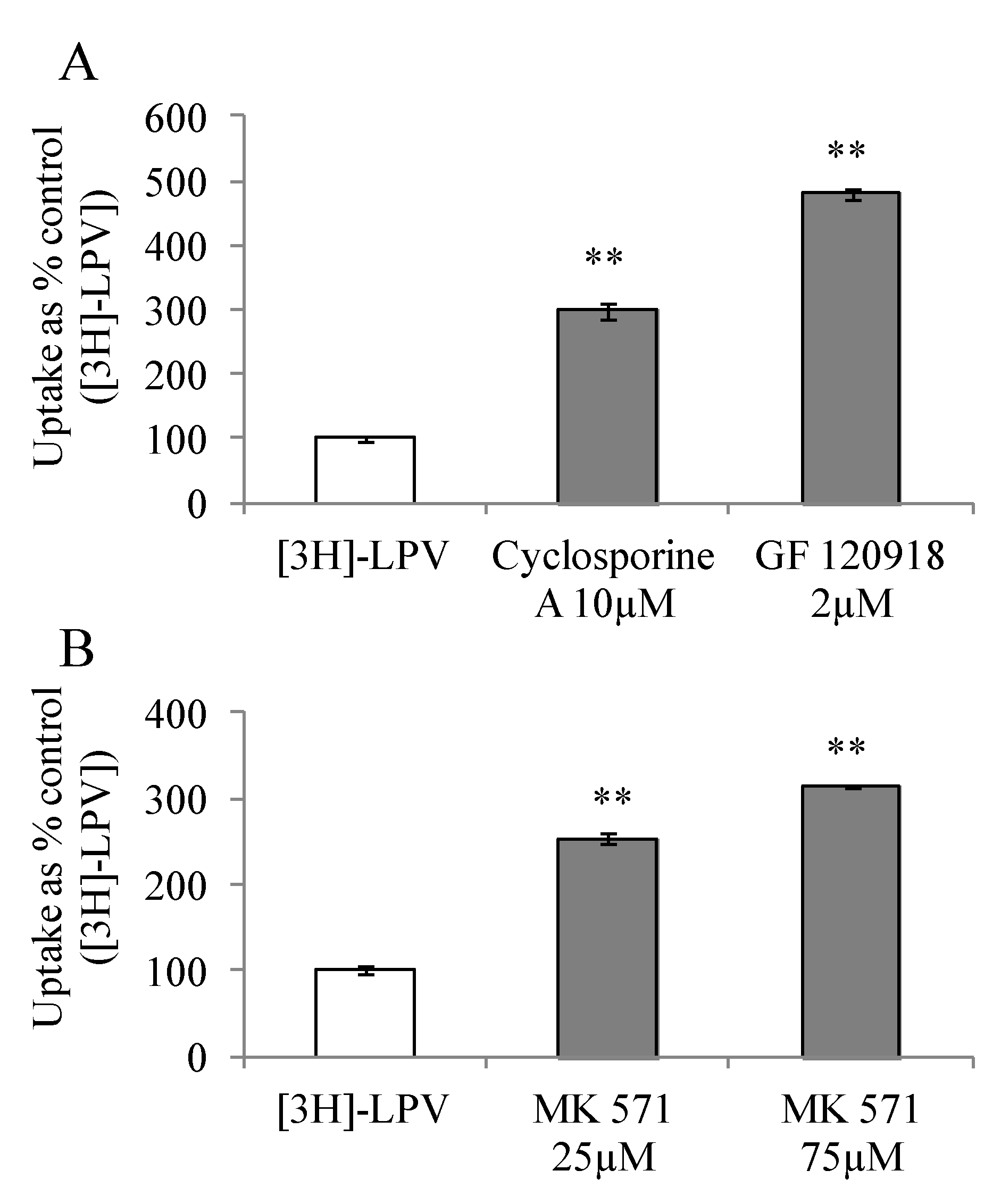
3.4.1. Uptake of [3H]-LPV in MDCK-MDR1 and MDCK-MRP2 Cells
3.4.2. Concentration Dependent Uptake Studies
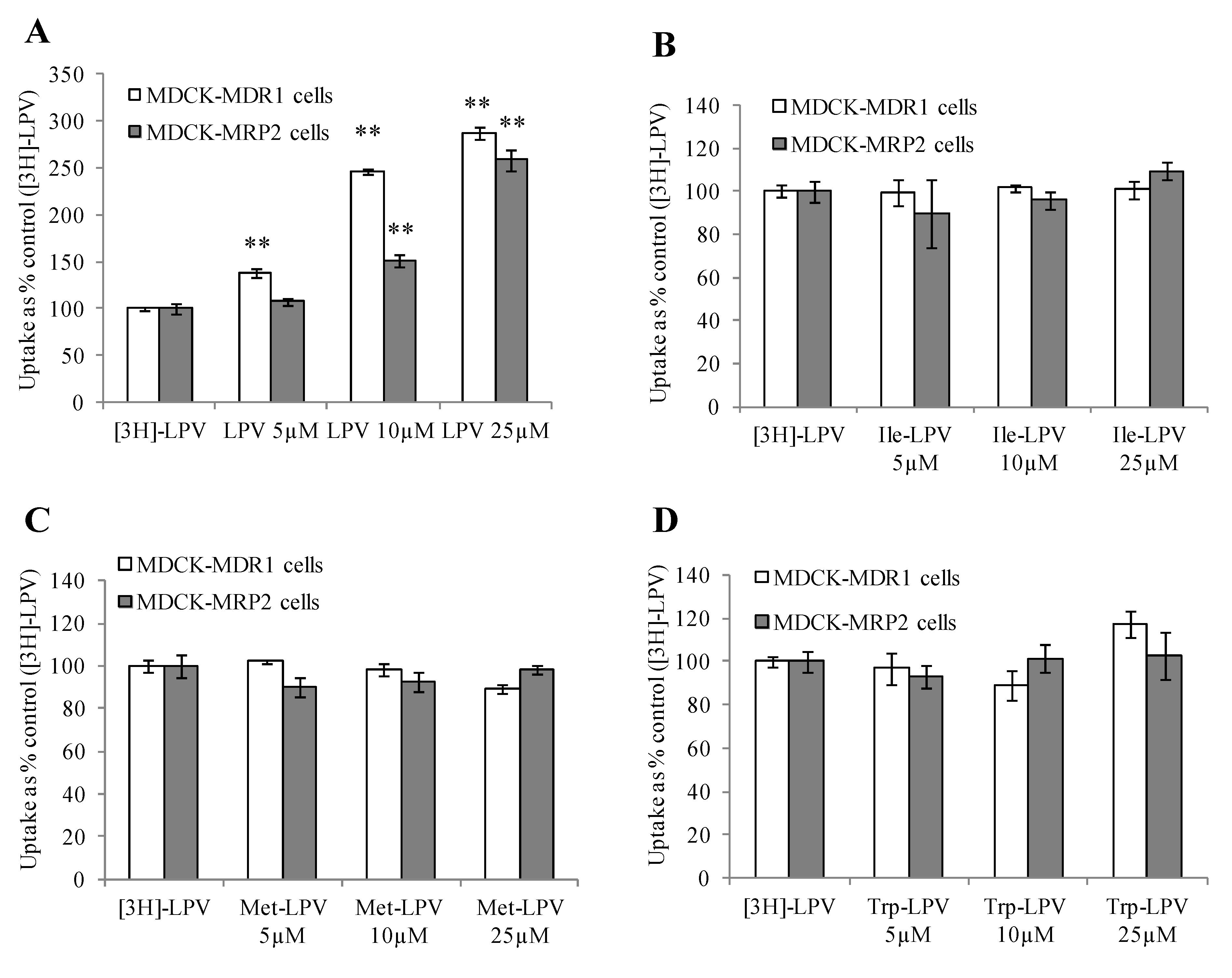
3.4.3. Cellular Uptake of Unlabelled LPV and Prodrugs
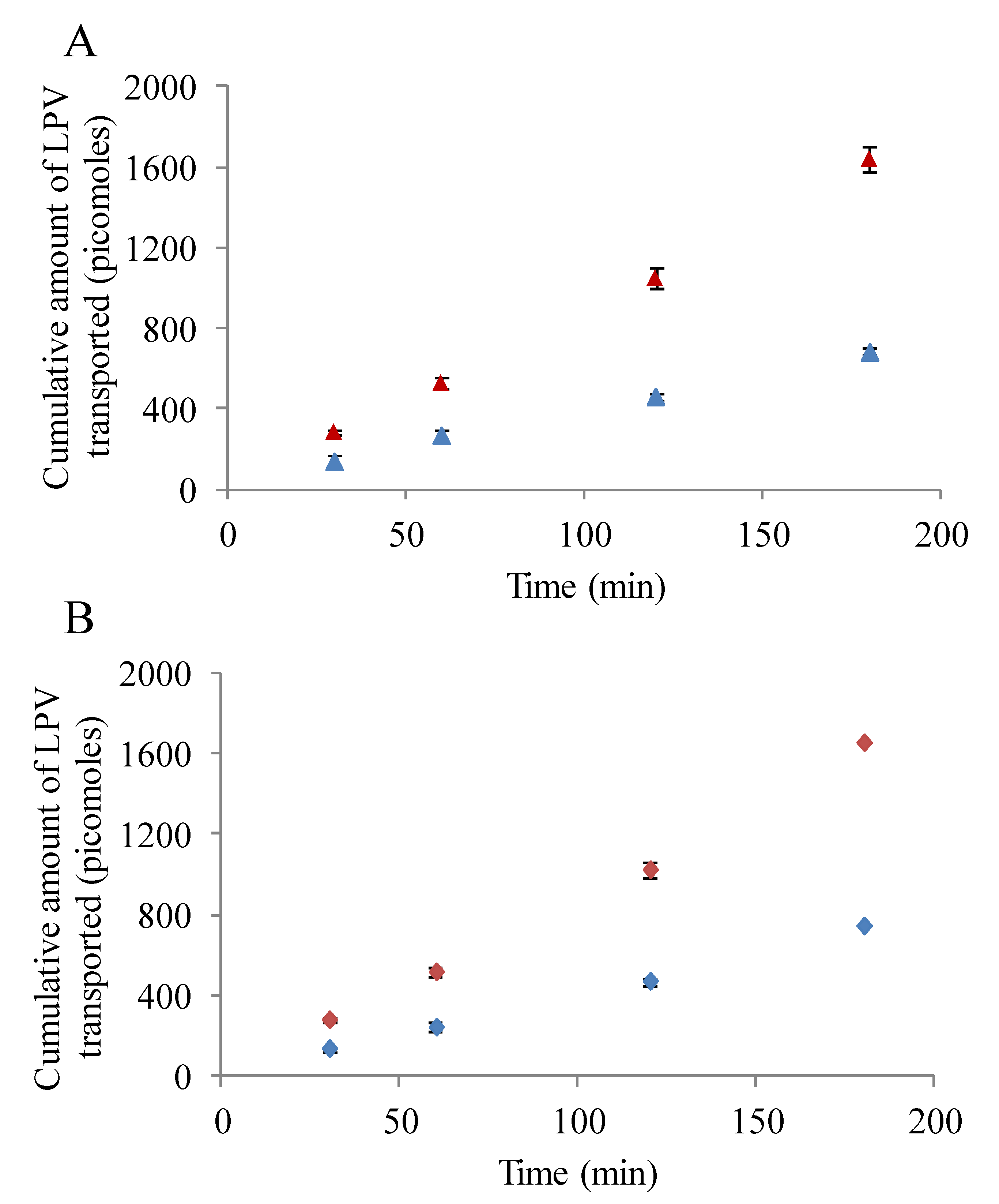
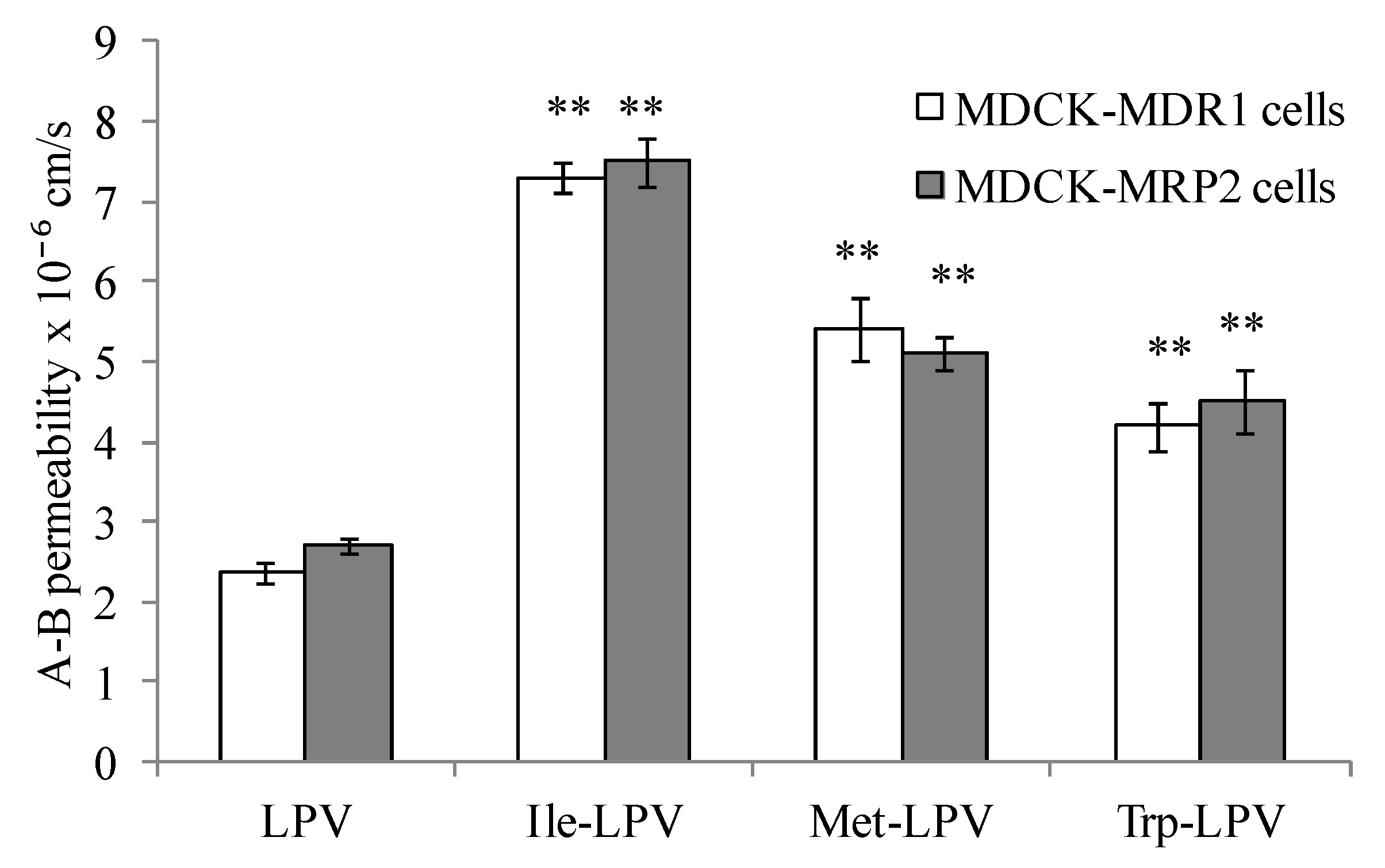
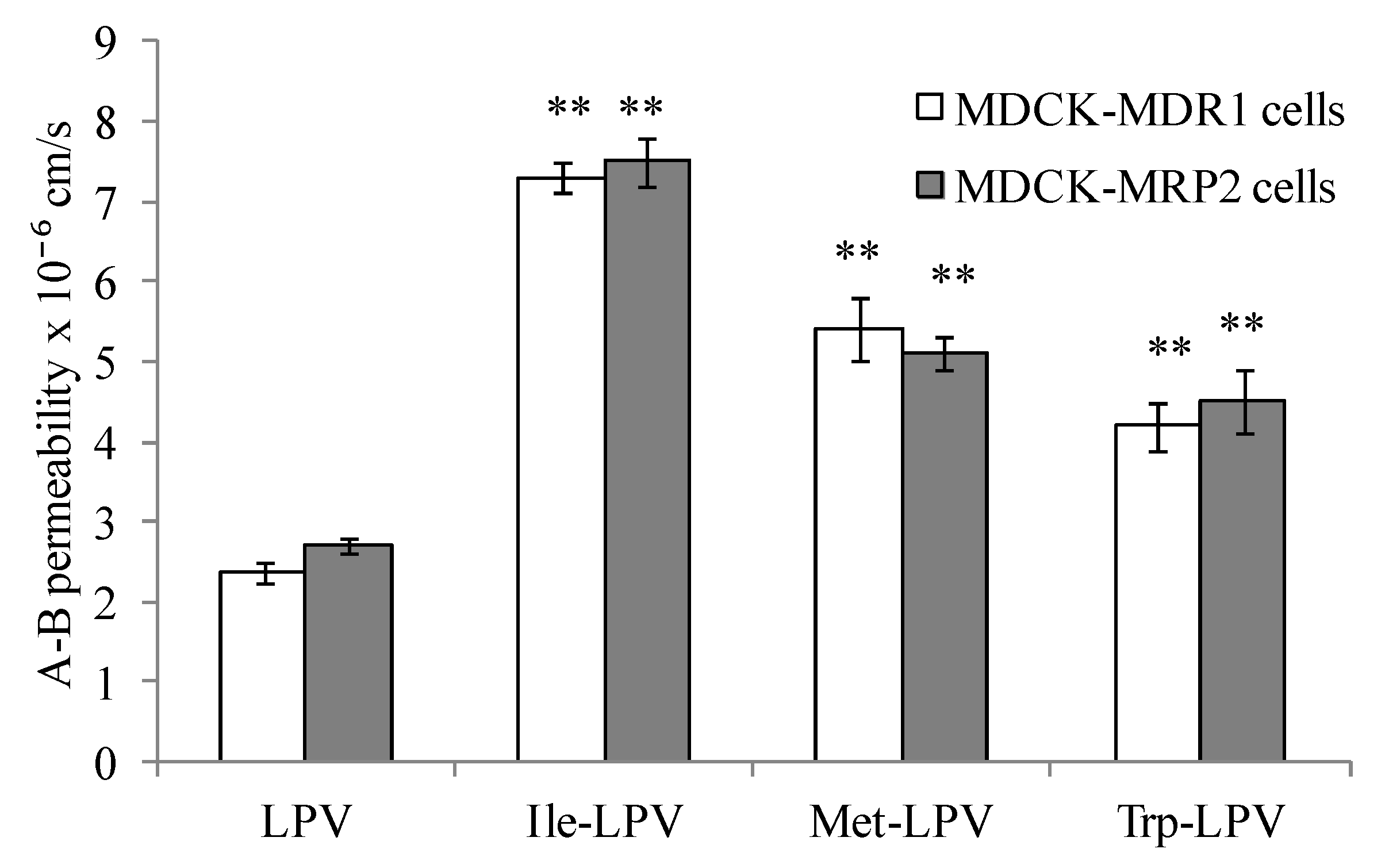
3.5. Transepithelial Transport Studies
3.5.1. LPV Transport across MDCK-MDR1 Cells
3.5.2. LPV Transport across MDCK-MRP2 Cells
3.5.3. Prodrug Transport across MDCK-MDR1 and MDCK-MRP2 Cells

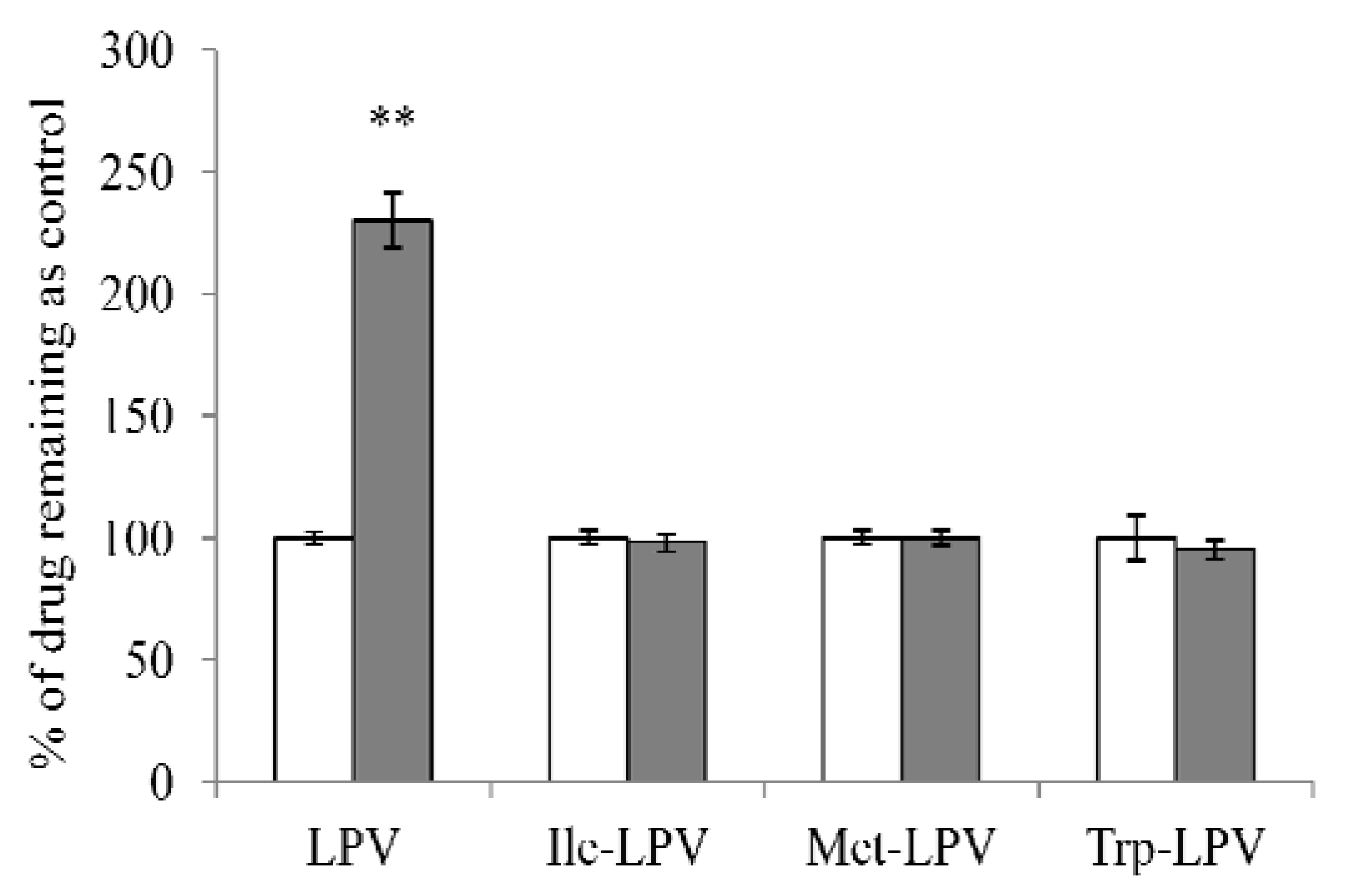
3.6. Liver Microsomal Stability Studies

3.6.1. Time Dependent Degradation Studies
3.6.2. Concentration Dependent Degradation Studies
4. Conclusions
Supplementary Files
Acknowledgements
Conflicts of Interest
References
- Agarwal, S.; Pal, D.; Mitra, A.K. Both P-gp and MRP2 mediate transport of Lopinavir, a protease inhibitor. Int. J. Pharm. 2007, 339, 139–147. [Google Scholar] [CrossRef]
- Kumar, G.N.; Jayanti, V.K.; Johnson, M.K.; Uchic, J.; Thomas, S.; Lee, R.D.; Grabowski, B.A.; Sham, H.L.; Kempf, D.J.; Denissen, J.F.; et al. Metabolism and disposition of the HIV-1 protease inhibitor lopinavir (ABT-378) given in combination with ritonavir in rats, dogs, and humans. Pharm. Res. 2004, 21, 1622–1630. [Google Scholar] [CrossRef]
- Kumar, G.N.; Dykstra, J.; Roberts, E.M.; Jayanti, V.K.; Hickman, D.; Uchic, J.; Yao, Y.; Surber, B.; Thomas, S.; Granneman, G.R. Potent inhibition of the cytochrome P-450 3A-mediated human liver microsomal metabolism of a novel HIV protease inhibitor by ritonavir: A positive drug-drug interaction. Drug Metab. Dispos. Biol. Fate Chem. 1999, 27, 902–908. [Google Scholar]
- Patel, M.; Mandava, N.K.; Vadlapatla, R.K.; Mitra, A.K. Recent patents and emerging therapeutics for HIV infections: A focus on protease inhibitors. Pharm. Patent Anal. 2013, 2, 513–538. [Google Scholar] [CrossRef]
- Gupta, D.; Varghese Gupta, S.; Dahan, A.; Tsume, Y.; Hilfinger, J.; Lee, K.D.; Amidon, G.L. Increasing oral absorption of polar neuraminidase inhibitors: A prodrug transporter approach applied to oseltamivir analogue. Mol. Pharm. 2013, 10, 512–522. [Google Scholar] [CrossRef]
- Landowski, C.P.; Song, X.; Lorenzi, P.L.; Hilfinger, J.M.; Amidon, G.L. Floxuridine amino acid ester prodrugs: Enhancing Caco-2 permeability and resistance to glycosidic bond metabolism. Pharm. Res. 2005, 22, 1510–1518. [Google Scholar] [CrossRef]
- Gupta, S.V.; Gupta, D.; Sun, J.; Dahan, A.; Tsume, Y.; Hilfinger, J.; Lee, K.D.; Amidon, G.L. Enhancing the intestinal membrane permeability of zanamivir: A carrier mediated prodrug approach. Mol. Pharm. 2011, 8, 2358–2367. [Google Scholar] [CrossRef]
- Dias, C.; Nashed, Y.; Atluri, H.; Mitra, A. Ocular penetration of acyclovir and its peptide prodrugs valacyclovir and val-valacyclovir following systemic administration in rabbits: An evaluation using ocular microdialysis and LC-MS. Curr. Eye Res. 2002, 25, 243–252. [Google Scholar] [CrossRef]
- Majumdar, S.; Nashed, Y.E.; Patel, K.; Jain, R.; Itahashi, M.; Neumann, D.M.; Hill, J.M.; Mitra, A.K. Dipeptide monoester ganciclovir prodrugs for treating HSV-1-induced corneal epithelial and stromal keratitis: In vitro and in vivo evaluations. J. Ocular Pharm. Ther. Off. J. Assoc. Ocular Pharm. Ther. 2005, 21, 463–474. [Google Scholar]
- Patel, K.; Trivedi, S.; Luo, S.; Zhu, X.; Pal, D.; Kern, E.R.; Mitra, A.K. Synthesis, physicochemical properties and antiviral activities of ester prodrugs of ganciclovir. Int. J. Pharm. 2005, 305, 75–89. [Google Scholar] [CrossRef]
- Gunda, S.; Hariharan, S.; Mitra, A.K. Corneal absorption and anterior chamber pharmacokinetics of dipeptide monoester prodrugs of ganciclovir (GCV): In vivo comparative evaluation of these prodrugs with Val-GCV and GCV in rabbits. J. Ocular Pharm. Ther. Off. J. Assoc. Ocular Pharm. Ther. 2006, 22, 465–476. [Google Scholar] [CrossRef]
- Majumdar, S.; Kansara, V.; Mitra, A.K. Vitreal pharmacokinetics of dipeptide monoester prodrugs of ganciclovir. J. Ocular Pharm. Ther. Off. J. Assoc. Ocular Pharm. Ther. 2006, 22, 231–241. [Google Scholar] [CrossRef]
- Agarwal, S.; Boddu, S.H.; Jain, R.; Samanta, S.; Pal, D.; Mitra, A.K. Peptide prodrugs: Improved oral absorption of lopinavir, a HIV protease inhibitor. Int. J. Pharm. 2008, 359, 7–14. [Google Scholar] [CrossRef]
- Jain, R.; Duvvuri, S.; Kansara, V.; Mandava, N.K.; Mitra, A.K. Intestinal absorption of novel-dipeptide prodrugs of saquinavir in rats. Int. J. Pharm. 2007, 336, 233–240. [Google Scholar] [CrossRef]
- Katragadda, S.; Talluri, R.S.; Mitra, A.K. Modulation of P-glycoprotein-mediated efflux by prodrug derivatization: An approach involving peptide transporter-mediated influx across rabbit cornea. J. Ocular Pharm. Ther. Off. J. Assoc. Ocular Pharm. Ther. 2006, 22, 110–120. [Google Scholar] [CrossRef]
- Patel, M.; Mandava, N.K.; Pal, D.; Mitra, A.K. Amino Acid Prodrug of Quinidine: An Approach to Circumvent P-glycoprotein Mediated Cellular Efflux. Int. J. Pharm. 2014, 464, 196–204. [Google Scholar] [CrossRef]
- Santos, C.; Morais, J.; Gouveia, L.; de Clercq, E.; Pannecouque, C.; Nielsen, C.U.; Steffansen, B.; Moreira, R.; Gomes, P. Dipeptide derivatives of AZT: Synthesis, chemical stability, activation in human plasma, hPEPT1 affinity, and antiviral activity. ChemMedChem 2008, 3, 970–978. [Google Scholar] [CrossRef]
- Kwatra, D.; Vadlapatla, R.K.; Vadlapudi, A.D.; Pal, D.; Mitra, A.K. Interaction of gatifloxacin with efflux transporters: A possible mechanism for drug resistance. Int. J. Pharm. 2010, 395, 114–121. [Google Scholar] [CrossRef]
- Luo, S.; Wang, Z.; Patel, M.; Khurana, V.; Zhu, X.; Pal, D.; Mitra, A.K. Targeting SVCT for enhanced drug absorption: Synthesis and in vitro evaluation of a novel vitamin C conjugated prodrug of saquinavir. Int. J. Pharm. 2011, 414, 77–85. [Google Scholar] [CrossRef]
- Patel, M.; Vadlapatla, R.K.; Pal, D.; Mitra, A.K. Molecular and functional characterization of riboflavin specific transport system in rat brain capillary endothelial cells. Brain Res. 2012, 1468, 1–10. [Google Scholar]
- Patel, M.; Vadlapatla, R.K.; Shah, S.; Mitra, A.K. Molecular expression and functional activity of sodium dependent multivitamin transporter in human prostate cancer cells. Int. J. Pharm. 2012, 436, 324–331. [Google Scholar] [CrossRef]
- Patel, M.; Dalvi, P.; Gokulgandhi, M.; Kesh, S.; Kohli, T.; Pal, D.; Mitra, A.K. Functional characterization and molecular expression of large neutral amino acid transporter (LAT1) in human prostate cancer cells. Int. J. Pharm. 2013, 443, 245–253. [Google Scholar] [CrossRef]
- Feierman, D.E.; Melinkov, Z.; Nanji, A.A. Induction of CYP3A by ethanol in multiple in vitro and in vivo models. Alcohol. Clin. Exp. Res. 2003, 27, 981–988. [Google Scholar] [CrossRef]
- Tang, F.; Horie, K.; Borchardt, R.T. Are MDCK cells transfected with the human MDR1 gene a good model of the human intestinal mucosa? Pharm. Res. 2002, 19, 765–772. [Google Scholar] [CrossRef]
- Tang, F.; Horie, K.; Borchardt, R.T. Are MDCK cells transfected with the human MRP2 gene a good model of the human intestinal mucosa? Pharm. Res. 2002, 19, 773–779. [Google Scholar] [CrossRef]
- Wang, Z.; Pal, D.; Mitra, A.K. Stereoselective evasion of P-glycoprotein, cytochrome P450 3A, and hydrolases by peptide prodrug modification of saquinavir. J. Pharm. Sci. 2012, 101, 3199–3213. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Patel, M.; Mandava, N.; Gokulgandhi, M.; Pal, D.; Mitra, A.K. Amino Acid Prodrugs: An Approach to Improve the Absorption of HIV-1 Protease Inhibitor, Lopinavir. Pharmaceuticals 2014, 7, 433-452. https://doi.org/10.3390/ph7040433
Patel M, Mandava N, Gokulgandhi M, Pal D, Mitra AK. Amino Acid Prodrugs: An Approach to Improve the Absorption of HIV-1 Protease Inhibitor, Lopinavir. Pharmaceuticals. 2014; 7(4):433-452. https://doi.org/10.3390/ph7040433
Chicago/Turabian StylePatel, Mitesh, Nanda Mandava, Mitan Gokulgandhi, Dhananjay Pal, and Ashim K. Mitra. 2014. "Amino Acid Prodrugs: An Approach to Improve the Absorption of HIV-1 Protease Inhibitor, Lopinavir" Pharmaceuticals 7, no. 4: 433-452. https://doi.org/10.3390/ph7040433