Design of siRNA Therapeutics from the Molecular Scale

Abstract
:1. Introduction
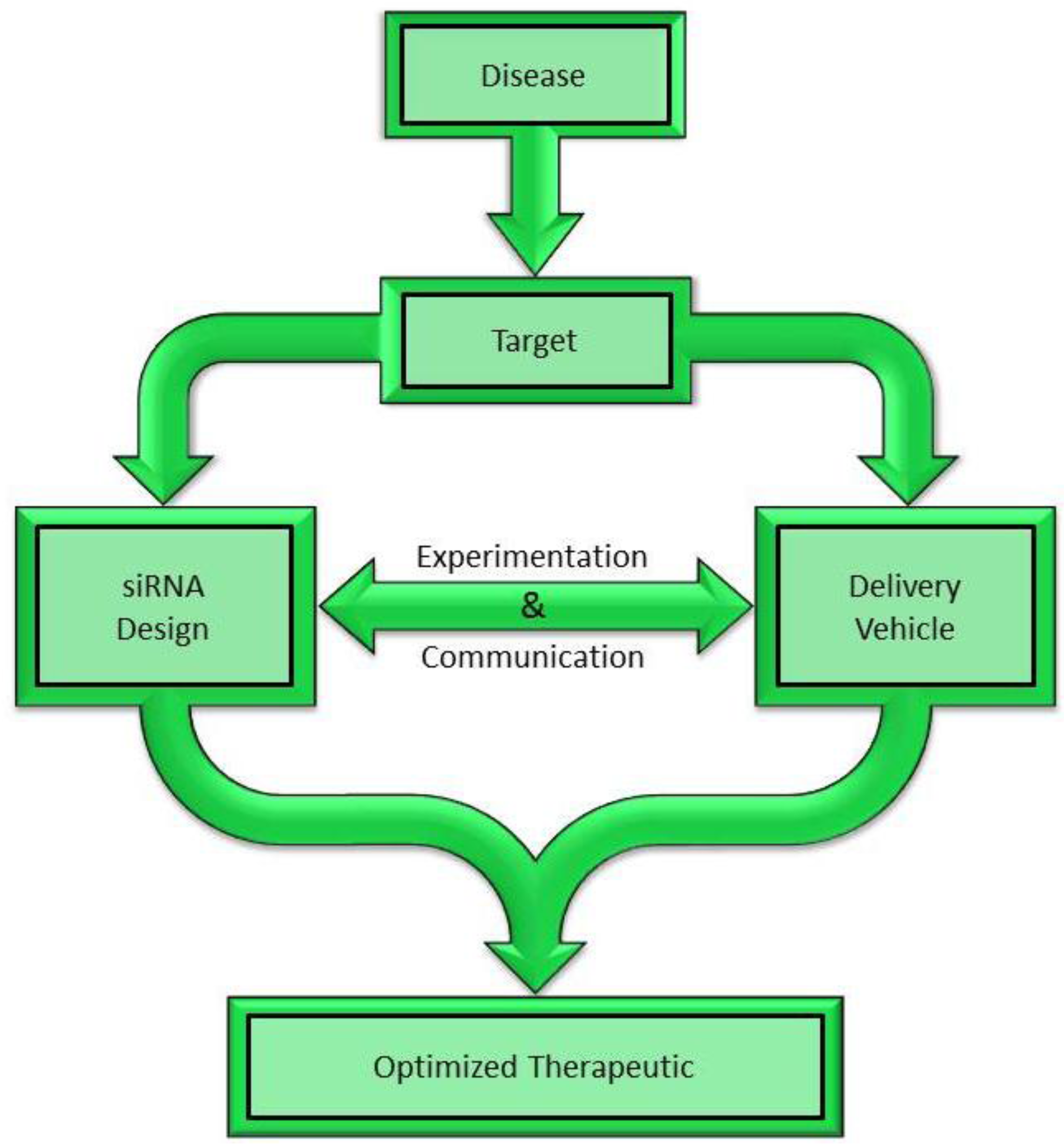
2. Choosing a Target for siRNA Mediated Silencing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Gene Function | Current Clinical Drugs Targeting | Protein Half-life | References |
|---|---|---|---|---|
| VEGFR/VEGFR2 | Angiogenesis | Sorafenib, Brivanib, Sunitinib, Cediranib, BIBF1120, Pazopanib, Regorafenib, E7080, TSU-68, Vandetanib, Ramucirumab, IMC-1121B | 70 min | [5,15,21] |
| EGFR | Signal Transduction | Erlotinib, Cetuximab, Gefitinib, Lapatinib, Vandetanib | 10 h | [5,16,21] |
| Bcr-ABL | Cell Proliferation | Destanib | 40 h | [5,10,17,21] |
| MEK | Signal Transduction | AZD6244 | 6 h | [5,18,21] |
| PDGFR | Angiogenesis/Signal Transduction | Sorafenib, Brivanib, Linifanib, BIBF1120, Pazopanib, TSU-68 | 3 h | [5,19,21] |
3. siRNA Design Considerations
3.1. Details of the RNAi Mechanism
3.2. Differential Terminal Hybridization Stability
3.3. 5’ Nucleotide Preference
3.4. mRNA Target Region
3.5. Immunogenicity
3.6. Non-Specific Effects

3.7. Other siRNA Design Criteria
3.8. Non-Canonical siRNA Structural Designs
3.9. Incorporation of Chemical Modifications
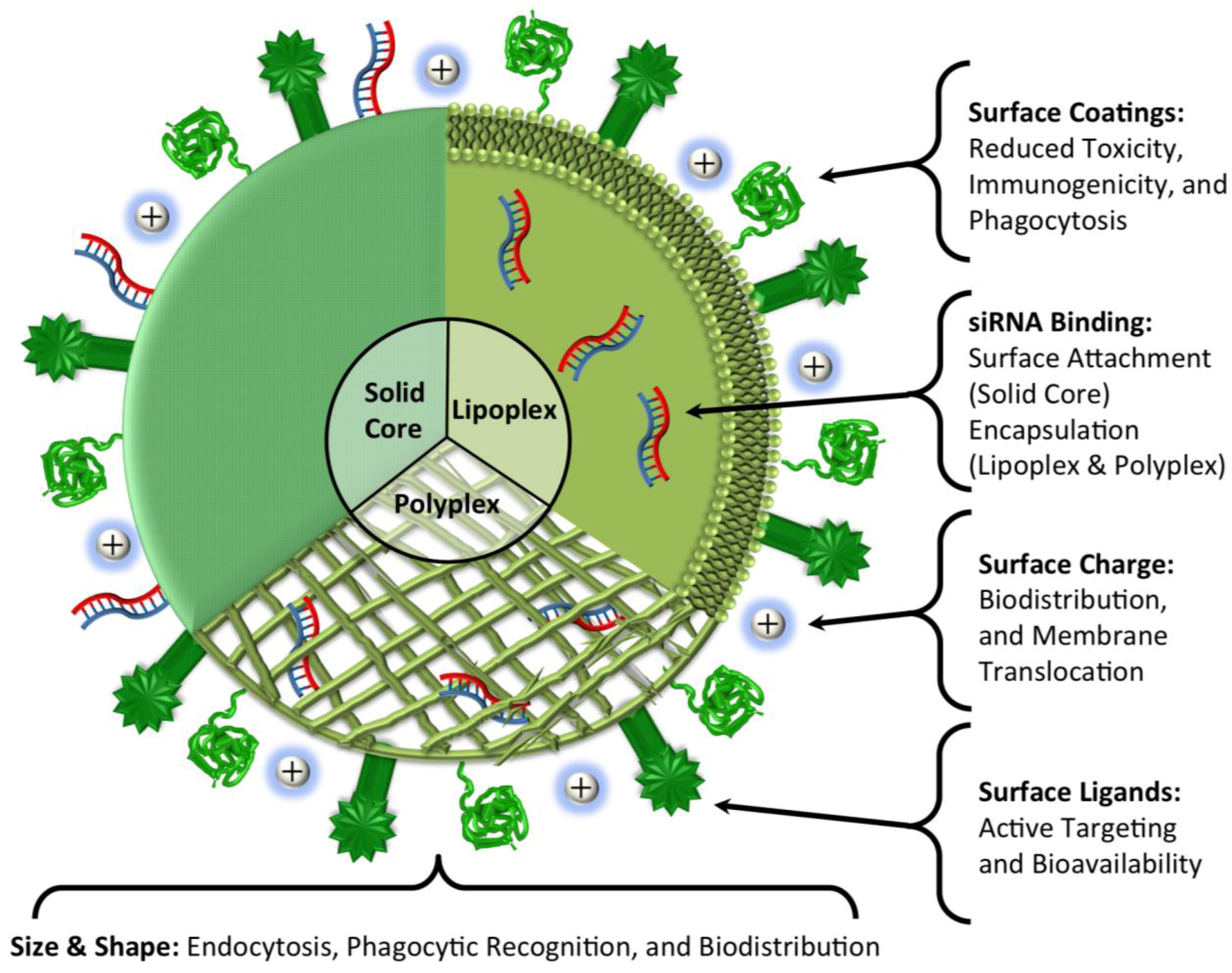
4. Delivery

4.1. Accessing the Cell Cytoplasm
4.2. Routes of Systemic Delivery
4.3. Delivery Specifically to Target Cells
4.4. Methods of Administration
4.5. Selection of a Specific Delivery Vehicle for Targeting HCCs
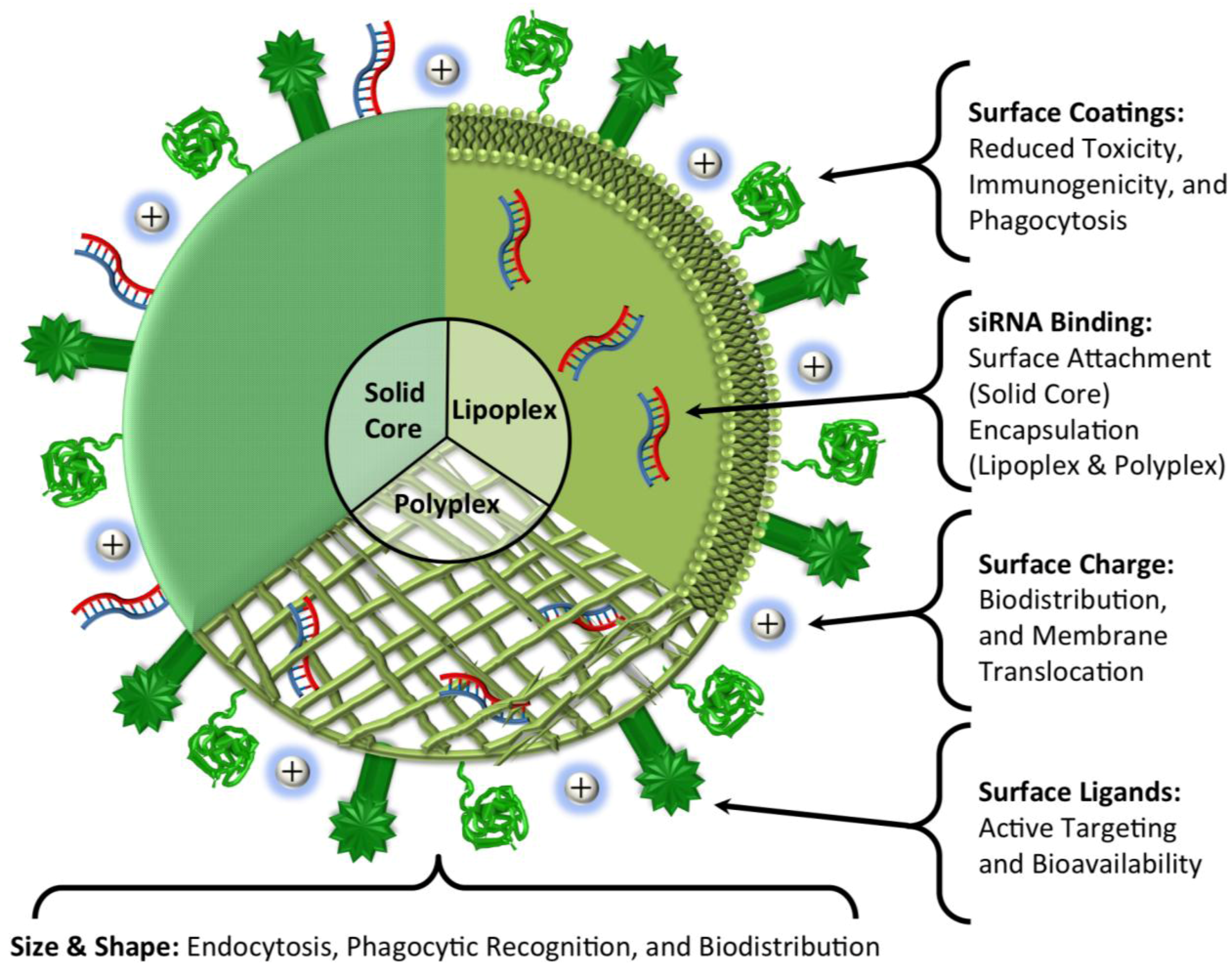
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Globocan 2008 v2.0, cancer incidence and mortality worldwide: Iarc cancerbase no. 10. Available online: http://globocan.iarc.fr/ (accessed on 26 November 2012).
- American cancer society. Cancer facts & figures 2012. Available online: http://www.cancer.org/research/cancerfactsfigures/cancerfactsfigures/cancer-facts-figures-2012/ (accessd on 26 November 2012).
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Zhu, A.X.; Duda, D.G.; Sahani, D.V.; Jain, R.K. Hcc and angiogenesis: Possible targets and future directions. Nat. Rev. Clin. Oncol. 2011, 8, 292–301. [Google Scholar] [CrossRef]
- Villanueva, A.; Llovet, J.M. Targeted therapies for hepatocellular carcinoma. Gastroenterology 2011, 140, 1410–1426. [Google Scholar]
- Burnett, J.C.; Rossi, J.J.; Tiemann, K. Current progress of sirna/shrna therapeutics in clinical trials. Biotechnol. J. 2011, 6, 1130–1146. [Google Scholar] [CrossRef]
- Haussecker, D. The business of rnai therapeutics in 2012. Mol. Ther. Nucleic Acids 2012, 1, e8. [Google Scholar] [CrossRef]
- Wei, J.; Jones, J.; Kang, J.; Card, A.; Krimm, M.; Hancock, P.; Pei, Y.; Ason, B.; Payson, E.; Dubinina, N.; et al. Rna-induced silencing complex-bound small interfering rna is a determinant of rna interference-mediated gene silencing in mice. Mol. Pharmacol. 2011, 79, 953–963. [Google Scholar] [CrossRef]
- Bartlett, D.W. Insights into the kinetics of sirna-mediated gene silencing from live-cell and live-animal bioluminescent imaging. Nucleic Acids Res. 2006, 34, 322–333. [Google Scholar] [CrossRef]
- Spiller, D.G.; Giles, R.V.; Broughton, C.M.; Grzybowski, J.; Ruddell, C.J.; Tidd, D.M.; Clark, R.E. The influence of target protein half-life on the effectiveness of antisense oligonucleotide analog-mediated biologic responses. Antisense Nucleic Acid Drug Dev. 1998, 8, 281–293. [Google Scholar] [CrossRef]
- Larsson, E.; Sander, C.; Marks, D. Mrna turnover rate limits sirna and microrna efficacy. Mol. Syst. Biol. 2010, 6, 1–9. [Google Scholar]
- Kennedy, S.; Wang, D.; Ruvkun, G. A conserved sirna-degrading rnase negatively regulates rna interference in c. Elegans. Nature 2004, 427, 645–649. [Google Scholar] [CrossRef]
- Bian, Y.; Zhou, W.; Zhao, Y.; Li, X.; Geng, W.; Hao, R.; Yang, Q.; Huang, W. High-dose sirnas upregulate mouse eri-1 at both transcription and posttranscription levels. PLoS ONE 2011, 6, e26466. [Google Scholar]
- Caffrey, D.R.; Zhao, J.; Song, Z.; Schaffer, M.E.; Haney, S.A.; Subramanian, R.R.; Seymour, A.B.; Hughes, J.D. Sirna off-target effects can be reduced at concentrations that match their individual potency. PLoS ONE 2011, 6, e21503. [Google Scholar] [CrossRef]
- Calera, M.R.; Venkatakrishnan, A.; Kazlauskas, A. Ve-cadherin increases the half-life of vegf receptor 2. Exp. Cell Res. 2004, 300, 248–256. [Google Scholar] [CrossRef]
- Schlessinger, J. The epidermal growth factor receptor as a multifunctional allosteric protein. Biochemistry 1988, 27, 3119–3123. [Google Scholar]
- Dhut, S.; Chaplin, T.; Young, B.D. Bcr-abl and bcr proteins: Biochemical characterization and localization. Leukemia 1990, 4, 745–750. [Google Scholar]
- Wang, P.Y.; Rao, J.N.; Zou, T.; Liu, L.; Xiao, L.; Yu, T.X.; Turner, D.J.; Gorospe, M.; Wang, J.Y. Post-transcriptional regulation of mek-1 by polyamines through the rna-binding protein hur modulating intestinal epithelial apoptosis. Biochem. J. 2010, 426, 293–306. [Google Scholar] [CrossRef]
- Keating, M.T.; Williams, L.T. Processing of the platelet-derived growth factor receptor. Biosynthetic and degradation studies using anti-receptor antibodies. J. Biol. Chem. 1987, 262, 7932–7937. [Google Scholar]
- Yang, E.; van Nimwegen, E.; Zavolan, M.; Rajewsky, N.; Schroeder, M.; Magnasco, M.; Darnell, J.E. Decay rates of human mrnas: Correlation with functional characteristics and sequence attributes. Genome Res. 2003, 13, 1863–1872. [Google Scholar]
- Llovet, J.M.; Bruix, J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008, 48, 1312–1327. [Google Scholar] [CrossRef]
- Reynolds, A.; Leake, D.; Boese, Q.; Scaringe, S.; Marshall, W.S.; Khvorova, A. Rational sirna design for rna interference. Nat. Biotechnol. 2004, 22, 326–330. [Google Scholar] [CrossRef]
- Jackson, A.L.; Bartz, S.R.; Schelter, J.; Kobayashi, S.V.; Burchard, J.; Mao, M.; Li, B.; Cavet, G.; Linsley, P.S. Expression profiling reveals off-target gene regulation by rnai. Nat. Biotechnol. 2003, 21, 635–637. [Google Scholar] [CrossRef]
- Naito, Y.; Ui-Tei, K. Sirna design software for a target gene-specific rna interference. Front. Genet. 2012, 3, 102. [Google Scholar]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of rna interference. Nature 2001, 409, 363–366. [Google Scholar]
- Zamore, P.D.; Tuschl, T.; Sharp, P.A.; Bartel, D.P. Rnai: Double-stranded rna directs the atp-dependent cleavage of mrna at 21 to 23 nucleotide intervals. Cell 2000, 101, 25–33. [Google Scholar] [CrossRef]
- Nykänen, A.; Haley, B.; Zamore, P.D. Atp requirements and small interfering rna structure in the rna interference pathway. Cell 2001, 107, 309–321. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Martinez, J.; Patkaniowska, A.; Lendeckel, W.; Tuschl, T. Functional anatomy of sirnas for mediating efficient rnai in drosophila melanogaster embryo lysate. EMBO J. 2001, 20, 6877–6888. [Google Scholar] [CrossRef]
- Lima, W.F.; Murray, H.; Nichols, J.G.; Wu, H.; Sun, H.; Prakash, T.P.; Berdeja, A.R.; Gaus, H.J.; Crooke, S.T. Human dicer binds short single-strand and double-strand rna with high affinity and interacts with different regions of the nucleic acids. J. Biol. Chem. 2009, 284, 2535–2548. [Google Scholar]
- Sakurai, K.; Amarzguioui, M.; Kim, D.; Alluin, J.; Heale, B.; Song, M.; Gatignol, A.; Behlke, M.A.; Rossi, J.J. A role for human dicer in pre-risc loading of sirnas. Nucleic Acids Res. 2011, 39, 1510–1525. [Google Scholar] [CrossRef]
- Liu, Q.H.; Rand, T.A.; Kalidas, S.; Du, F.H.; Kim, H.E.; Smith, D.P.; Wang, X.D. R2d2, a bridge between the initiation and effector steps of the drosophila rnai pathway. Science 2003, 301, 1921–1925. [Google Scholar] [CrossRef]
- Tomari, Y.; Du, T.; Haley, B.; Schwarz, D.; Bennett, R.; Cook, H.; Koppetsch, B.; Theurkauf, W.; Zamore, P. Risc assembly defects in the drosophila rnai mutant armitage. Cell 2004, 116, 831–841. [Google Scholar] [CrossRef]
- Tomari, Y.; Matranga, C.; Haley, B.; Martinez, N.; Zamore, P.D. A protein sensor for sirna asymmetry. Science 2004, 306, 1377–1380. [Google Scholar] [CrossRef]
- Chendrimada, T.; Gregory, R.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. Trbp recruits the dicer complex to ago2 for microrna processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef]
- Hammond, S.M.; Bernstein, E.; Beach, D.; Hannon, G.J. An rna-directed nuclease mediates post-transcriptional gene silencing in drosophila cells. Nature 2000, 404, 293–296. [Google Scholar]
- MacRae, I.J.; Ma, E.; Zhou, M.; Robinson, C.V.; Doudna, J.A. In vitro reconstitution of the human risc-loading complex. Proc. Natl. Acad. Sci. USA 2008, 105, 512–517. [Google Scholar]
- Gredell, J.A.; Dittmer, M.J.; Wu, M.; Chan, C.; Walton, S.P. Recognition of sirna asymmetry by tar rna binding protein. Biochemistry 2010, 49, 3148–3155. [Google Scholar] [CrossRef]
- Noland, C.L.; Ma, E.; Doudna, J.A. Sirna repositioning for guide strand selection by human dicer complexes. Mol. Cell 2011, 43, 110–121. [Google Scholar] [CrossRef]
- Khvorova, A.; Reynolds, A.; Jayasena, S.D. Functional sirnas and mirnas exhibit strand bias. Cell 2003, 115, 209–216. [Google Scholar] [CrossRef]
- Schwarz, D.; Hutvagner, G.; Du, T.; Xu, Z.; Aronin, N.; Zamore, P. Asymmetry in the assembly of the rnai enzyme complex. Cell 2003, 115, 199–208. [Google Scholar] [CrossRef]
- Hamilton, A.; Baulcombe, D. A species of small antisense rna in posttranscriptional gene silencing in plants. Science 1999, 286, 950–952. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Lendeckel, W.; Tuschl, T. Rna interference is mediated by 21-and 22-nucleotide rnas. Genes Dev. 2001, 15, 188–200. [Google Scholar] [CrossRef]
- Rand, T.A.; Petersen, S.; Du, F.; Wang, X. Argonaute2 cleaves the anti-guide strand of sirna during risc activation. Cell 2005, 123, 621–629. [Google Scholar] [CrossRef]
- Leuschner, P.J.F.; Ameres, S.L.; Kueng, S.; Martinez, J. Cleavage of the sirna passenger strand during risc assembly in human cells. EMBO Rep. 2006, 7, 314–320. [Google Scholar] [CrossRef]
- Matranga, C.; Tomari, Y.; Shin, C.; Bartel, D.; Zamore, P. Passenger-strand cleavage facilitates assembly of sirna into ago2-containing rnai enzyme complexes. Cell 2005, 123, 607–620. [Google Scholar] [CrossRef]
- Yoda, M.; Kawamata, T.; Paroo, Z.; Ye, X.; Iwasaki, S.; Liu, Q.; Tomari, Y. Atp-dependent human risc assembly pathways. Nat. Struct. Mol. Biol. 2010, 17, 17–23. [Google Scholar] [CrossRef]
- Rivas, F.V.; Tolia, N.H.; Song, J.J.; Aragon, J.P.; Liu, J.D.; Hannon, G.J.; Joshua-Tor, L. Purified argonaute2 and an sirna form recombinant human risc. Nat. Struct. Mol. Biol. 2005, 12, 340–349. [Google Scholar] [CrossRef]
- Haley, B.; Zamore, P.D. Kinetic analysis of the rnai enzyme complex. Nat. Struct. Mol. Biol. 2004, 11, 599–606. [Google Scholar] [CrossRef]
- Liu, J.D.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 is the catalytic engine of mammalian rnai. Sci. Signal. 2004, 305, 1437–1437. [Google Scholar]
- Snead, N.M.; Rossi, J.J. Biogenesis and function of endogenous and exogenous sirnas. Wiley Interdiscip. Rev. RNA 2010, 1, 117–131. [Google Scholar]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of mirnas and sirnas. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef]
- Grimm, D.; Streetz, K.L.; Jopling, C.L.; Storm, T.A.; Pandey, K.; Davis, C.R.; Marion, P.; Salazar, F.; Kay, M.A. Fatality in mice due to oversaturation of cellular microrna/short hairpin rna pathways. Nature 2006, 441, 537–541. [Google Scholar] [CrossRef]
- Frank, F.; Sonenberg, N.; Nagar, B. Structural basis for 5'-nucleotide base-specific recognition of guide rna by human ago2. Nature 2010, 465, 818–822. [Google Scholar] [CrossRef]
- Walton, S.P.; Wu, M.; Gredell, J.A.; Chan, C. Designing highly active sirnas for therapeutic applications. FEBS J. 2010, 277, 4806–4813. [Google Scholar] [CrossRef]
- Betancur, J.G.; Tomari, Y. Dicer is dispensable for asymmetric risc loading in mammals. RNA 2012, 18, 24–30. [Google Scholar] [CrossRef]
- Hutvagner, G. Small rna asymmetry in rnai: Function in risc assembly and gene regulation. FEBS Lett. 2005, 579, 5850–5857. [Google Scholar] [CrossRef]
- Lu, Z.J.; Mathews, D.H. Efficient sirna selection using hybridization thermodynamics. Nucleic Acids Res. 2008, 36, 640–647. [Google Scholar]
- Ui-Tei, K.; Naito, Y.; Takahashi, F.; Haraguchi, T.; Ohki-Hamazaki, H.; Juni, A.; Ueda, R.; Saigo, K. Guidelines for the selection of highly effective sirna sequences for mammalian and chick rna interference. Nucleic Acids Res. 2004, 32, 936–948. [Google Scholar] [CrossRef]
- Jagla, B.; Aulner, N.; Kelly, P.D.; Song, D.; Volchuk, A.; Zatorski, A.; Shum, D.; Mayer, T.; de Angelis, D.A.; Ouerfelli, O.; et al. Sequence characteristics of functional sirnas. RNA 2005, 11, 864–872. [Google Scholar] [CrossRef]
- Huesken, D.; Lange, J.; Mickanin, C.; Weiler, J.; Asselbergs, F.; Warner, J.; Meloon, B.; Engel, S.; Rosenberg, A.; Cohen, D.; et al. Design of a genome-wide sirna library using an artificial neural network. Nat. Biotechnol. 2005, 23, 995–1001. [Google Scholar] [CrossRef]
- Ladunga, I. More complete gene silencing by fewer sirnas: Transparent optimized design and biophysical signature. Nucleic Acids Res. 2006, 35, 433–440. [Google Scholar] [CrossRef]
- Shabalina, S.A.; Spiridonov, A.N.; Ogurtsov, A.Y. Computational models with thermodynamic and composition features improve sirna design. BMC Bioinformatics 2006, 7. [Google Scholar]
- Amarzguioui, M.; Prydz, H. An algorithm for selection of functional sirna sequences. Biochem. Biophys. Res. Commun. 2004, 316, 1050–1058. [Google Scholar] [CrossRef]
- Gong, W.; Ren, Y.; Xu, Q.; Wang, Y.; Lin, D.; Zhou, H.; Li, T. Integrated sirna design based on surveying of features associated with high rnai effectiveness. BMC Bioinformatics 2006, 7, 516. [Google Scholar] [CrossRef]
- Takasaki, S.; Kotani, S.; Konagaya, A. An effective method for selecting sirna target sequences in mammalian cells. Cell Cycle 2004, 3, 788–793. [Google Scholar] [CrossRef]
- Holen, T. Efficient prediction of sirnas with sirnarules 1.0: An open-source java approach to sirna algorithms. RNA 2006, 12, 1620–1625. [Google Scholar] [CrossRef]
- Takasaki, S. Selecting effective sirna target sequences by using bayes’ theorem. Comput. Biol. Chem. 2009, 33, 368–372. [Google Scholar] [CrossRef]
- Katoh, T.; Suzuki, T. Specific residues at every third position of sirna shape its efficient rnai activity. Nucleic Acids Res. 2007, 35, e27. [Google Scholar] [CrossRef]
- Seitz, H.; Tushir, J.S.; Zamore, P.D. A 5'-uridine amplifies mirna/mirna* asymmetry in drosophila by promoting rna-induced silencing complex formation. Silence 2011, 2, 4. [Google Scholar] [CrossRef]
- Brown, K.M.; Chu, C.Y.; Rana, T.M. Target accessibility dictates the potency of human risc. Nat. Struct. Mol. Biol. 2005, 12, 469–470. [Google Scholar] [CrossRef]
- Ameres, S.L.; Martinez, J.; Schroeder, R. Molecular basis for target rna recognition and cleavage by human risc. Cell 2007, 130, 101–112. [Google Scholar] [CrossRef]
- Mathews, D.; Sabina, J.; Zuker, M.; Turner, D. Expanded sequence dependence of thermodynamic parameters improves prediction of rna secondary structure. J. Mol. Biol. 1999, 288, 911–940. [Google Scholar] [CrossRef]
- Ding, Y.; Chan, C.Y.; Lawrence, C.E. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res. 2004, 32, W135–W141. [Google Scholar] [CrossRef]
- Tafer, H.; Ameres, S.L.; Obernosterer, G.; Gebeshuber, C.A.; Schroeder, R.; Martinez, J.; Hofacker, I.L. The impact of target site accessibility on the design of effective sirnas. Nat. Biotechnol. 2008, 26, 578–583. [Google Scholar] [CrossRef]
- Gredell, J.; Berger, A.; Walton, S. Impact of target mrna structure on sirna silencing efficiency: A large-scale study. Biotechnol. Bioeng. 2008, 100, 744–755. [Google Scholar] [CrossRef]
- Vickers, T.A.; Koo, S.; Bennett, C.F.; Crooke, S.T.; Dean, N.M.; Baker, B.F. Efficient reduction of target rnas by small interfering rna and rnase h-dependent antisense agents. A comparative analysis. J. Biol. Chem. 2003, 278, 7108–7118. [Google Scholar]
- Bohula, E.A.; Salisbury, A.J.; Sohail, M.; Playford, M.P.; Riedemann, J.; Southern, E.M.; Macaulay, V.M. The efficacy of small interfering rnas targeted to the type 1 insulin-like growth factor receptor (igf1r) is influenced by secondary structure in the igf1r transcript. J. Biol. Chem. 2003, 278, 15991–15997. [Google Scholar] [CrossRef]
- Overhoff, M.; Alken, M.; Far, R.K.-K.; Lemaitre, M.; Lebleu, B.; Sczakiel, G.; Robbins, I. Local rna target structure influences sirna efficacy: A systematic global analysis. J. Mol. Biol. 2005, 348, 871–881. [Google Scholar] [CrossRef]
- Schubert, S.; Grünweller, A.; Erdmann, V.A.; Kurreck, J. Local rna target structure influences sirna efficacy: Systematic analysis of intentionally designed binding regions. J. Mol. Biol. 2005, 348, 883–893. [Google Scholar] [CrossRef]
- Shao, Y.; Chan, C.Y.; Maliyekkel, A.; Lawrence, C.E.; Roninson, I.B.; Ding, Y. Effect of target secondary structure on rnai efficiency. RNA 2007, 13, 1631–1640. [Google Scholar] [CrossRef]
- Yoshinari, K.; Miyagishi, M.; Taira, K. Effects on rnai of the tight structure, sequence and position of the targeted region. Nucleic Acids Res. 2004, 32, 691–699. [Google Scholar] [CrossRef]
- Sledz, C.A.; Holko, M.; de Veer, M.J.; Silverman, R.H.; Williams, B.R.G. Activation of the interferon system by short-interfering rnas. Nat. Cell Biol. 2003, 5, 834–839. [Google Scholar] [CrossRef]
- Samuel-Abraham, S.; Leonard, J.N. Staying on message: Design principles for controlling nonspecific responses to sirna. FEBS J. 2010, 277, 4828–4836. [Google Scholar] [CrossRef]
- Jackson, A. Recognizing and avoiding sirna off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef]
- Robbins, M.; Judge, A.; Liang, L. 2'-o-methyl-modified rnas act as tlr7 antagonists. Mol. Ther. 2007, 15, 1663–1669. [Google Scholar] [CrossRef]
- Kodym, R.; Kodym, E.; Story, M.D. 2'-5'-oligoadenylate synthetase is activated by a specific rna sequence motif. Biochem. Biophys. Res. Commun. 2009, 388, 317–322. [Google Scholar] [CrossRef]
- Manche, L.; Green, S.R.; Schmedt, C.; Mathews, M.B. Interactions between double-stranded rna regulators and the protein kinase dai. Mol. Cell. Biol. 1992, 12, 5238–5248. [Google Scholar]
- Bevilacqua, P.C.; Cech, T.R. Minor-groove recognition of double-stranded rna by the double-stranded rna-binding domain from the rna-activated protein kinase pkr. Biochemistry 1996, 35, 9983–9994. [Google Scholar] [CrossRef]
- Marques, J.T.; Devosse, T.; Wang, D.; Zamanian-Daryoush, M.; Serbinowski, P.; Hartmann, R.; Fujita, T.; Behlke, M.A.; Williams, B.R. A structural basis for discriminating between self and nonself double-stranded rnas in mammalian cells. Nat. Biotechnol. 2006, 24, 559–565. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-i and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef]
- Gantier, M.P.; Williams, B.R.G. The response of mammalian cells to double-stranded rna. Cytokine Growth Factor Rev. 2007, 18, 363–371. [Google Scholar] [CrossRef]
- Nallagatla, S.R.; Hwang, J.; Toroney, R.; Zheng, X.; Cameron, C.E.; Bevilacqua, P.C. 5'-triphosphate-dependent activation of pkr by rnas with short stem-loops. Science 2007, 318, 1455–1458. [Google Scholar] [CrossRef]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef]
- Weber, C.; Müller, C.; Podszuweit, A.; Montino, C.; Vollmer, J.; Forsbach, A. Toll-like receptor (tlr) 3 immune modulation by unformulated small interfering rna or DNA and the role of cd14 (in tlr-mediated effects). Immunology 2012, 136, 64–77. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded rna and activation of nf-kappab by toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Kariko, K.; Bhuyan, P.; Capodici, J.; Weissman, D. Small interfering rnas mediate sequence-independent gene suppression and induce immune activation by signaling through toll-like receptor 3. J. Immunol. 2004, 172, 6545–6549. [Google Scholar]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded rna via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef]
- Judge, A.D.; Sood, V.; Shaw, J.R.; Fang, D.; McClintock, K.; MacLachlan, I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic sirna. Nat. Biotechnol. 2005, 23, 457–462. [Google Scholar] [CrossRef]
- Jackson, A.L.; Burchard, J.; Leake, D.; Reynolds, A.; Schelter, J.; Guo, J.; Johnson, J.M.; Lim, L.; Karpilow, J.; Nichols, K.; et al. Position-specific chemical modification of sirnas reduces “off-target” transcript silencing. RNA 2006, 12, 1197–1205. [Google Scholar] [CrossRef]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of tlr7-mediated recognition of single-stranded rna. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef]
- Diebold, S.S.; Massacrier, C.; Akira, S.; Paturel, C.; Morel, Y.; Reis e Sousa, C. Nucleic acid agonists for toll-like receptor 7 are defined by the presence of uridine ribonucleotides. Eur. J. Immunol. 2006, 36, 3256–3267. [Google Scholar] [CrossRef]
- Goodchild, A.; Nopper, N.; King, A.; Doan, T.; Tanudji, M.; Arndt, G.M.; Poidinger, M.; Rivory, L.P.; Passioura, T. Sequence determinants of innate immune activation by short interfering rnas. BMC Immunol. 2009, 10. [Google Scholar]
- Kleinman, M.E.; Yamada, K.; Takeda, A.; Chandrasekaran, V.; Nozaki, M.; Baffi, J.Z.; Albuquerque, R.J.C.; Yamasaki, S.; Itaya, M.; Pan, Y.; Appukuttan, B.; Gibbs, D.; Yang, Z.; Karikó, K.; Ambati, B.K.; Wilgus, T.A.; DiPietro, L.A.; Sakurai, E.; Zhang, K.; Smith, J.R.; Taylor, E.W.; Ambati, J. Sequence- and target-independent angiogenesis suppression by sirna via tlr3. Nature 2008, 452, 591–597. [Google Scholar] [CrossRef]
- Reynolds, A.; Anderson, E.M.; Vermeulen, A.; Fedorov, Y.; Robinson, K.; Leake, D.; Karpilow, J.; Marshall, W.S.; Khvorova, A. Induction of the interferon response by sirna is cell type- and duplex length-dependent. RNA 2006, 12, 988–993. [Google Scholar] [CrossRef]
- Forsbach, A.; Nemorin, J.-G.; Montino, C.; Müller, C.; Samulowitz, U.; Vicari, A.P.; Jurk, M.; Mutwiri, G.K.; Krieg, A.M.; Lipford, G.B.; Vollmer, J. Identification of rna sequence motifs stimulating sequence-specific tlr8-dependent immune responses. J. Immunol. 2008, 180, 3729–3738. [Google Scholar]
- Sioud, M. Induction of inflammatory cytokines and interferon responses by double-stranded and single-stranded sirnas is sequence-dependent and requires endosomal localization. J. Mol. Biol. 2005, 348, 1079–1090. [Google Scholar] [CrossRef]
- Hornung, V.; Guenthner-Biller, M.; Bourquin, C.; Ablasser, A.; Schlee, M.; Uematsu, S.; Noronha, A.; Manoharan, M.; Akira, S.; de Fougerolles, A.; Endres, S.; Hartmann, G. Sequence-specific potent induction of ifn-α by short interfering rna in plasmacytoid dendritic cells through tlr7. Nat. Med. 2005, 11, 263–270. [Google Scholar] [CrossRef]
- Jurk, M.; Chikh, G.; Schulte, B.; Kritzler, A.; Richardt-Pargmann, D.; Lampron, C.; Luu, R.; Krieg, A.M.; Vicari, A.P.; Vollmer, J. Immunostimulatory potential of silencing rnas can be mediated by a non-uridine-rich toll-like receptor 7 motif. Nucleic Acid Ther. 2011, 21, 201–214. [Google Scholar]
- Fedorov, Y.; Anderson, E.M.; Birmingham, A.; Reynolds, A.; Karpilow, J.; Robinson, K.; Leake, D.; Marshall, W.S.; Khvorova, A. Off-target effects by sirna can induce toxic phenotype. RNA 2006, 12, 1188–1196. [Google Scholar] [CrossRef]
- Shafer, R.H.; Smirnov, I. Biological aspects of DNA/rna quadruplexes. Biopolymers 2000, 56, 209–227. [Google Scholar] [CrossRef]
- Doench, J.G.; Petersen, C.P.; Sharp, P.A. Sirnas can function as mirnas. Genes Dev. 2003, 17, 438–442. [Google Scholar] [CrossRef]
- Lambert, N.J.; Gu, S.G.; Zahler, A.M. The conformation of microrna seed regions in native micrornps is prearranged for presentation to mrna targets. Nucleic Acids Res. 2011, 39, 4827–4835. [Google Scholar] [CrossRef]
- Gu, S.; Jin, L.; Zhang, F.; Huang, Y.; Grimm, D.; Rossi, J.J.; Kay, M.A. Thermodynamic stability of small hairpin rnas highly influences the loading process of different mammalian argonautes. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 9208–9213. [Google Scholar]
- Bartel, D. Micrornas: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microrna targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Lin, X.; Ruan, X.; Anderson, M.G.; Mcdowell, J.A.; Kroeger, P.E.; Fesik, S.W.; Shen, Y. Sirna-mediated off-target gene silencing triggered by a 7 nt complementation. Nucleic Acids Res. 2005, 33, 4527. [Google Scholar] [CrossRef]
- Lai, E.C. Micro rnas are complementary to 3 ' utr sequence motifs that mediate negative post-transcriptional regulation. Nat. Genet. 2002, 30, 363–364. [Google Scholar] [CrossRef]
- Schultz, N.; Marenstein, D.R.; De Angelis, D.A.; Wang, W.-Q.; Nelander, S.; Jacobsen, A.; Marks, D.S.; Massagué, J.; Sander, C. Off-target effects dominate a large-scale rnai screen for modulators of the tgf-β pathway and reveal microrna regulation of tgfbr2. Silence 2011, 2, 3. [Google Scholar] [CrossRef]
- Didiano, D.; Hobert, O. Perfect seed pairing is not a generally reliable predictor for mirna-target interactions. Nat. Struct. Mol. Biol. 2006, 13, 849–851. [Google Scholar] [CrossRef]
- Doench, J.G.; Sharp, P.A. Specificity of microrna target selection in translational repression. Genes Dev. 2004, 18, 504–511. [Google Scholar] [CrossRef]
- Broderick, J.A.; Salomon, W.E.; Ryder, S.P.; Aronin, N.; Zamore, P.D. Argonaute protein identity and pairing geometry determine cooperativity in mammalian rna silencing. RNA 2011, 17, 1858–1869. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. Mirbase: Integrating microrna annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39, D152–D157. [Google Scholar] [CrossRef]
- Sigoillot, F.D.; Lyman, S.; Huckins, J.F.; Adamson, B.; Chung, E.; Quattrochi, B.; King, R.W. A bioinformatics method identifies prominent off-targeted transcripts in rnai screens. Nat. Methods 2012, 9, 363–366. [Google Scholar] [CrossRef]
- Sudbery, I.; Enright, A.J.; Fraser, A.G.; Dunham, I. Systematic analysis of off-target effects in an rnai screen reveals micrornas affecting sensitivity to trail-induced apoptosis. BMC Genomics 2010, 11, 175. [Google Scholar]
- Anderson, E.M.; Birmingham, A.; Baskerville, S.; Reynolds, A.; Maksimova, E.; Leake, D.; Fedorov, Y.; Karpilow, J.; Khvorova, A. Experimental validation of the importance of seed complement frequency to sirna specificity. RNA 2008, 14, 853–861. [Google Scholar] [CrossRef]
- Snove, O.; Holen, T. Many commonly used sirnas risk off-target activity. Biochem. Biophys. Res. Commun. 2004, 319, 256–263. [Google Scholar] [CrossRef]
- Holen, T.; Moe, S.E.; Sorbo, J.G.; Meza, T.J.; Ottersen, O.P.; Klungland, A. Tolerated wobble mutations in sirnas decrease specificity, but can enhance activity in vivo. Nucleic Acids Res. 2005, 33, 4704–4710. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Ding, H.; Kennington, L.; Moore, J.T.; Schelter, J.; Burchard, J.; Linsley, P.S.; Aronin, N.; Xu, Z.; Zamore, P.D. Designing sirna that distinguish between genes that differ by a single nucleotide. PLoS Genet. 2006, 2, 1307–1318. [Google Scholar]
- Jackson, A.L.; Burchard, J.; Schelter, J.; Chau, B.N.; Cleary, M.; Lim, L.; Linsley, P.S. Widespread sirna “off-target” transcript silencing mediated by seed region sequence complementarity. RNA 2006, 12, 1179–1187. [Google Scholar] [CrossRef]
- Saxena, S.; Jónsson, Z.O.; Dutta, A. Small rnas with imperfect match to endogenous mrna repress translation. Implications for off-target activity of small inhibitory rna in mammalian cells. J. Biol. Chem. 2003, 278, 44312–44319. [Google Scholar] [CrossRef]
- Scacheri, P.C.; Rozenblatt-Rosen, O.; Caplen, N.J.; Wolfsberg, T.G.; Umayam, L.; Lee, J.C.; Hughes, C.M.; Shanmugam, K.S.; Bhattacharjee, A.; Meyerson, M. Short interfering rnas can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 1892–1897. [Google Scholar] [CrossRef]
- Aleman, L.M.; Doench, J.; Sharp, P.A. Comparison of sirna-induced off-target rna and protein effects. RNA 2007, 13, 385–395. [Google Scholar] [CrossRef]
- Liu, J.; Valencia-Sanchez, M.A.; Hannon, G.J.; Parker, R. Microrna-dependent localization of targeted mrnas to mammalian p-bodies. Nat. Cell Biol. 2005, 7, 719–723. [Google Scholar] [CrossRef]
- Teixeira, D.; Sheth, U.; Valencia-Sanchez, M.A.; Brengues, M.; Parker, R. Processing bodies require rna for assembly and contain nontranslating mrnas. RNA 2005, 11, 371–382. [Google Scholar] [CrossRef]
- Behm-Ansmant, I.; Rehwinkel, J.; Izaurralde, E. Micrornas silence gene expression by repressing protein expression and/or by promoting mrna decay. Cold Spring Harb. Symp. Quant. Biol. 2006, 71, 523–530. [Google Scholar] [CrossRef]
- Semizarov, D.; Frost, L.; Sarthy, A.; Kroeger, P.; Halbert, D.N.; Fesik, S.W. Specificity of short interfering rna determined through gene expression signatures. Proc. Natl. Acad. Sci. USA 2003, 100, 6347–6352. [Google Scholar] [CrossRef]
- Persengiev, S.P.; Zhu, X.; Green, M.R. Nonspecific, concentration-dependent stimulation and repression of mammalian gene expression by small interfering rnas (sirnas). RNA 2004, 10, 12–18. [Google Scholar] [CrossRef]
- Wilson, P.A.; Plucinski, M. A simple bayesian estimate of direct rnai gene regulation events from differential gene expression profiles. BMC Genomics 2011, 12, 250. [Google Scholar] [CrossRef]
- Sydor, J.R.; Nock, S. Protein expression profiling arrays: Tools for the multiplexed high-throughput analysis of proteins. Proteome Sci. 2003, 1, 3. [Google Scholar] [CrossRef]
- Pan, S.; Aebersold, R.; Chen, R.; Rush, J.; Goodlett, D.R.; McIntosh, M.W.; Zhang, J.; Brentnall, T.A. Mass spectrometry based targeted protein quantification: Methods and applications. J. Proteome Res. 2009, 8, 787–797. [Google Scholar] [CrossRef]
- Huang, H.; Qiao, R.; Zhao, D.; Zhang, T.; Li, Y.; Yi, F.; Lai, F.; Hong, J.; Ding, X.; Yang, Z.; et al. Profiling of mismatch discrimination in rnai enabled rational design of allele-specific sirnas. Nucleic Acids Res. 2009, 37, 7560–7569. [Google Scholar] [CrossRef]
- Kittler, R.; Surendranath, V.; Heninger, A.-K.; Slabicki, M.; Theis, M.; Putz, G.; Franke, K.; Caldarelli, A.; Grabner, H.; Kozak, K.; et al. Genome-wide resources of endoribonuclease-prepared short interfering rnas for specific loss-of-function studies. Nat. Methods 2007, 4, 337–344. [Google Scholar]
- Parker, J.S.; Parizotto, E.A.; Wang, M.; Roe, S.M.; Barford, D. Enhancement of the seed-target recognition step in rna silencing by a piwi/mid domain protein. Mol. Cell 2009, 33, 204–214. [Google Scholar] [CrossRef]
- Birmingham, A.; Anderson, E.M.; Reynolds, A.; Ilsley-Tyree, D.; Leake, D.; Fedorov, Y.; Baskerville, S.; Maksimova, E.; Robinson, K.; Karpilow, J.; Marshall, W.S.; Khvorova, A. 3' UTR seed matches, but not overall identity, are associated with rnai off-targets. Nat. Methods 2006, 3, 199–204. [Google Scholar] [CrossRef]
- Boland, A.; Tritschler, F.; Heimst Auml Dt, S.; Izaurralde, E.; Weichenrieder, O. Crystal structure and ligand binding of the mid domain of a eukaryotic argonaute protein. EMBO Rep. 2010, 11, 522–527. [Google Scholar] [CrossRef]
- Ma, J.-B.; Ye, K.; Patel, D.J. Structural basis for overhang-specific small interfering rna recognition by the paz domain. Nature 2004, 429, 318–322. [Google Scholar] [CrossRef]
- Lingel, A.; Simon, B.; Izaurralde, E.; Sattler, M. Nucleic acid 3'-end recognition by the argonaute2 paz domain. Nat. Struct. Mol. Biol. 2004, 11, 576–577. [Google Scholar] [CrossRef]
- Sashital, D.G.; Doudna, J.A. Structural insights into rna interference. Curr. Opin. Struct. Biol. 2010, 20, 90–97. [Google Scholar] [CrossRef]
- Vert, J.-P.; Foveau, N.; Lajaunie, C.; Vandenbrouck, Y. An accurate and interpretable model for sirna efficacy prediction. BMC Bioinformatics 2006, 7, 520. [Google Scholar] [CrossRef]
- Patzel, V.; Rutz, S.; Dietrich, I.; Köberle, C.; Scheffold, A.; Kaufmann, S.H.E. Design of sirnas producing unstructured guide-rnas results in improved rna interference efficiency. Nat. Biotechnol. 2005, 23, 1440–1444. [Google Scholar] [CrossRef]
- Köberle, C.; Kaufmann, S.H.E.; Patzel, V. Selecting effective sirnas based on guide rna structure. Nat. Protoc. 2006, 1, 1832–1839. [Google Scholar] [CrossRef]
- Hossbach, M.; Gruber, J.; Osborn, M.; Weber, K.; Tuschl, T. Gene silencing with sirna duplexes composed of target-mrna-complementary and partially palindromic or partially complementary single-stranded sirnas. RNA Biol. 2006, 3, 82–89. [Google Scholar] [CrossRef]
- Vermeulen, A.; Behlen, L.; Reynolds, A.; Wolfson, A.; Marshall, W.S.; Karpilow, J.; Khvorova, A. The contributions of dsrna structure to dicer specificity and efficiency. RNA 2005, 11, 674–682. [Google Scholar] [CrossRef]
- Sciabola, S.; Cao, Q.; Orozco, M.; Faustino, I.; Stanton, R.V. Improved nucleic acid descriptors for sirna efficacy prediction. Nucleic Acids Res. 2012. [Google Scholar]
- Snead, N.M.; Rossi, J.J. Rna interference trigger variants: Getting the most out of rna for rna interference-based therapeutics. Nucleic Acid Ther. 2012, 22, 139–146. [Google Scholar]
- Martinez, J.; Patkaniowska, A.; Urlaub, H.; Lührmann, R.; Tuschl, T. Single-stranded antisense sirnas guide target rna cleavage in rnai. Cell 2002, 110, 563–574. [Google Scholar] [CrossRef]
- Holen, T.; Amarzguioui, M.; Babaie, E.; Prydz, H. Similar behaviour of single-strand and double-strand sirnas suggests they act through a common rnai pathway. Nucleic Acids Res. 2003, 31, 2401–2407. [Google Scholar] [CrossRef]
- Lima, W.F.; Prakash, T.P.; Murray, H.M.; Kinberger, G.A.; Li, W.; Chappell, A.E.; Li, C.S.; Murray, S.F.; Gaus, H.; Seth, P.P.; et al. Single-stranded sirnas activate rnai in animals. Cell 2012, 150, 883–894. [Google Scholar] [CrossRef]
- Haringsma, H.J.; Li, J.J.; Soriano, F.; Kenski, D.M.; Flanagan, W.M.; Willingham, A.T. Mrna knockdown by single strand rna is improved by chemical modifications. Nucleic Acids Res. 2012, 40, 4125–4136. [Google Scholar] [CrossRef]
- Chu, C.-Y.; Rana, T.M. Potent rnai by short rna triggers. RNA 2008, 14, 1714–1719. [Google Scholar] [CrossRef]
- Sun, X.; Rogoff, H.A.; Li, C.J. Asymmetric rna duplexes mediate rna interference in mammalian cells. Nat. Biotechnol. 2008, 26, 1379–1382. [Google Scholar] [CrossRef]
- Chang, C.I.; Yoo, J.W.; Hong, S.W.; Lee, S.E.; Kang, H.S.; Sun, X.; Rogoff, H.A.; Ban, C.; Kim, S.; Li, C.J.; et al. Asymmetric shorter-duplex sirna structures trigger efficient gene silencing with reduced nonspecific effects. Mol. Ther. 2009, 17, 725–732. [Google Scholar] [CrossRef]
- Hohjoh, H. Enhancement of rnai activity by improved sirna duplexes. FEBS Lett. 2004, 557, 193–198. [Google Scholar] [CrossRef]
- Bramsen, J.B.; Laursen, M.B.; Damgaard, C.K.; Lena, S.W.; Babu, B.R.; Wengel, J.; Kjems, J. Improved silencing properties using small internally segmented interfering rnas. Nucleic Acids Res. 2007, 35, 5886–5897. [Google Scholar] [CrossRef]
- Dua, P.; Yoo, J.W.; Kim, S.; Lee, D.-K. Modified sirna structure with a single nucleotide bulge overcomes conventional sirna-mediated off-target silencing. Mol. Ther. 2011, 19, 1676–1687. [Google Scholar] [CrossRef]
- Amarzguioui, M.; Lundberg, P.; Cantin, E.; Hagstrom, J.; Behlke, M.A.; Rossi, J.J. Rational design and in vitro and in vivo delivery of dicer substrate sirna. Nat. Protoc. 2006, 1, 508–517. [Google Scholar] [CrossRef]
- Collingwood, M.A.; Rose, S.D.; Huang, L.; Hillier, C.; Amarzguioui, M.; Wiiger, M.T.; Soifer, H.S.; Rossi, J.J.; Behlke, M.A. Chemical modification patterns compatible with high potency dicer-substrate small interfering rnas. Oligonucleotides 2008, 18, 187–200. [Google Scholar] [CrossRef]
- Tanudji, M.; Machalek, D.; Arndt, G.M.; Rivory, L. Competition between sirna duplexes: Impact of rna-induced silencing complex loading efficiency and comparison between conventional-21 bp and dicer-substrate sirnas. Oligonucleotides 2010, 20, 27–32. [Google Scholar] [CrossRef]
- Foster, D.J.; Barros, S.; Duncan, R.; Shaikh, S.; Cantley, W.; Dell, A.; Bulgakova, E.; O’Shea, J.; Taneja, N.; Kuchimanchi, S.; et al. Comprehensive evaluation of canonical versus dicer-substrate sirna in vitro and in vivo. RNA 2012, 18, 557–568. [Google Scholar] [CrossRef]
- Turner, J.J.; Jones, S.W.; Moschos, S.A.; Lindsay, M.A.; Gait, M.J. Maldi-tof mass spectral analysis of sirna degradation in serum confirms an rnase a-like activity. Mol. Biosyst. 2006, 3, 43–50. [Google Scholar]
- Volkov, A.A.; Kruglova, N.y.S.; Meschaninova, M.I.; Venyaminova, A.G.; Zenkova, M.A.; Vlassov, V.V.; Chernolovskaya, E.L. Selective protection of nuclease-sensitive sites in sirna prolongs silencing effect. Oligonucleotides 2009, 19, 191–202. [Google Scholar] [CrossRef]
- Hong, J.; Huang, Y.; Li, J.; Yi, F.; Zheng, J.; Huang, H.; Wei, N.; Shan, Y.; An, M.; Zhang, H.; et al. Comprehensive analysis of sequence-specific stability of sirna. FASEB J. 2010, 24, 4844–4855. [Google Scholar] [CrossRef]
- Allerson, C.R.; Sioufi, N.; Jarres, R.; Prakash, T.P.; Naik, N.; Berdeja, A.; Wanders, L.; Griffey, R.H.; Swayze, E.E.; Bhat, B. Fully 2'-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering rna. J. Med. Chem. 2005, 48, 901–904. [Google Scholar] [CrossRef]
- Bramsen, J.B.; Laursen, M.B.; Nielsen, A.F.; Hansen, T.B.; Bus, C.; Langkjaer, N.; Babu, B.R.; Hojland, T.; Abramov, M.; Van Aerschot, A.; Odadzic, D.; et al. A large-scale chemical modification screen identifies design rules to generate sirnas with high activity, high stability and low toxicity. Nucleic Acids Res. 2009, 37, 2867–2881. [Google Scholar] [CrossRef]
- Kenski, D.M.; Butora, G.; Willingham, A.T.; Cooper, A.J.; Fu, W.; Qi, N.; Soriano, F.; Davies, I.W.; Flanagan, W.M. Sirna-optimized modifications for enhanced in vivo activity. Mol. Ther. Nucleic Acids 2012, 1, e5. [Google Scholar] [CrossRef]
- Braasch, D.A.; Jensen, S.; Liu, Y.; Kaur, K.; Arar, K.; White, M.A.; Corey, D.R. Rna interference in mammalian cells by chemically-modified rna. Biochemistry 2003, 42, 7967–7975. [Google Scholar] [CrossRef]
- Chiu, Y.-L.; Rana, T.M. Sirna function in rnai: A chemical modification analysis. RNA 2003, 9, 1034–1048. [Google Scholar] [CrossRef]
- Manoharan, M.; Akinc, A.; Pandey, R.K.; Qin, J.; Hadwiger, P.; John, M.; Mills, K.; Charisse, K.; Maier, M.A.; Nechev, L.; et al. Unique gene-silencing and structural properties of 2'-fluoro-modified sirnas. Angew. Chem. Int. Ed. Engl. 2011, 50, 2284–2288. [Google Scholar]
- Cekaite, L.; Furset, G.; Hovig, E.; Sioud, M. Gene expression analysis in blood cells in response to unmodified and 2-modified sirnas reveals tlr-dependent and independent effects. J. Mol. Biol. 2007, 365, 90–108. [Google Scholar] [CrossRef]
- Tluk, S.; Jurk, M.; Forsbach, A.; Weeratna, R.; Samulowitz, U.; Krieg, A.M.; Bauer, S.; Vollmer, J. Sequences derived from self-rna containing certain natural modifications act as suppressors of rna-mediated inflammatory immune responses. Int. Immunol. 2009, 21, 607–619. [Google Scholar] [CrossRef]
- Fucini, R.V.; Haringsma, H.J.; Deng, P.; Flanagan, W.M.; Willingham, A.T. Adenosine modification may be preferred for reducing sirna immune stimulation. Nucleic Acid Ther. 2012, 22, 205–210. [Google Scholar]
- Haupenthal, J.; Baehr, C.; Kiermayer, S.; Zeuzem, S.; Piiper, A. Inhibition of rnase a family enzymes prevents degradation and loss of silencing activity of sirnas in serum. Biochem. Pharmacol. 2006, 71, 702–710. [Google Scholar] [CrossRef]
- Amarzguioui, M. Tolerance for mutations and chemical modifications in a sirna. Nucleic Acids Res. 2003, 31, 589–595. [Google Scholar] [CrossRef]
- Yang, X.; Sierant, M.; Janicka, M.; Peczek, L.; Martinez, C.; Hassell, T.; Li, N.; Li, X.; Wang, T.; Nawrot, B. Gene silencing activity of sirna molecules containing phosphorodithioate substitutions. ACS Chem. Biol. 2012, 7, 1214–1220. [Google Scholar] [CrossRef]
- Bramsen, J.B.; Kjems, J. Development of therapeutic-grade small interfering rnas by chemical engineering. Front. Genet. 2012, 3, 154–154. [Google Scholar]
- Wang, J.; Byrne, J.D.; Napier, M.E.; DeSimone, J.M. More effective nanomedicines through particle design. Small 2011, 7, 1919–1931. [Google Scholar] [CrossRef]
- Elsabahy, M.; Wooley, K.L. Design of polymeric nanoparticles for biomedical delivery applications. Chem. Soc. Rev. 2012, 41, 2545–2561. [Google Scholar] [CrossRef]
- Siegwart, D.J.; Whitehead, K.A.; Nuhn, L.; Sahay, G.; Cheng, H.; Jiang, S.; Ma, M.; Lytton-Jean, A.; Vegas, A.; Fenton, P.; et al. Combinatorial synthesis of chemically diverse core-shell nanoparticles for intracellular delivery. Proc. Natl. Acad. Sci. USA 2011, 108, 12996–13001. [Google Scholar] [CrossRef]
- Ulery, B.D.; Nair, L.S.; Laurencin, C.T. Biomedical applications of biodegradable polymers. J. Polym. Sci. B Polym. Phys. 2011, 49, 832–864. [Google Scholar] [CrossRef]
- Whitehead, K.A.; Sahay, G.; Li, G.Z.; Love, K.T.; Alabi, C.A.; Ma, M.; Zurenko, C.; Querbes, W.; Langer, R.S.; Anderson, D.G. Synergistic silencing: Combinations of lipid-like materials for efficacious sirna delivery. Mol. Ther. 2011, 19, 1688–1694. [Google Scholar] [CrossRef]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.Y.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for sirna delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef]
- Malam, Y.; Loizidou, M.; Seifalian, A.M. Liposomes and nanoparticles: Nanosized vehicles for drug delivery in cancer. Trends Pharmacol. Sci. 2009, 30, 592–599. [Google Scholar] [CrossRef]
- Mutlu, G.M.; Budinger, G.R.S.; Green, A.A.; Urich, D.; Soberanes, S.; Chiarella, S.E.; Alheid, G.F.; McCrimmon, D.R.; Szleifer, I.; Hersam, M.C. Biocompatible nanoscale dispersion of single-walled carbon nanotubes minimizes in vivo pulmonary toxicity. Nano Lett. 2010, 10, 1664–1670. [Google Scholar] [CrossRef]
- Cheung, W.; Pontoriero, F.; Taratula, O.; Chen, A.M.; He, H.X. DNA and carbon nanotubes as medicine. Adv. Drug Deliv. Rev. 2010, 62, 633–649. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; Aggarwal, P.; Hall, J.B.; McNeil, S.E. Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol. Pharm. 2008, 5, 487–495. [Google Scholar] [CrossRef]
- Li, Y.; Sun, L.; Jin, M.; Du, Z.; Liu, X.; Guo, C.; Li, Y.; Huang, P.; Sun, Z. Size-dependent cytotoxicity of amorphous silica nanoparticles in human hepatoma hepg2 cells. Toxicol. In Vitro 2011, 25, 1343–1352. [Google Scholar] [CrossRef]
- Slowing, I.I.; Trewyn, B.G.; Giri, S.; Lin, V.S.Y. Mesoporous silica nanoparticles for drug delivery and biosensing applications. Adv. Funct. Mater. 2007, 17, 1225–1236. [Google Scholar] [CrossRef]
- Asefa, T.; Tao, Z. Biocompatibility of mesoporous silica nanoparticles. Chem. Res. Toxicol. 2012, 25, 2265–2284. [Google Scholar] [CrossRef]
- Colombo, M.; Carregal-Romero, S.; Casula, M.F.; Gutierrez, L.; Morales, M.P.; Bohm, I.B.; Heverhagen, J.T.; Prosperi, D.; Parak, W.J. Biological applications of magnetic nanoparticles. Chem. Soc. Rev. 2012, 41, 4306–4334. [Google Scholar] [CrossRef]
- Prijic, S.; Sersa, G. Magnetic nanoparticles as targeted delivery systems in oncology. Radiol. Oncol. 2011, 45, 1–16. [Google Scholar] [CrossRef]
- Khlebtsov, N.; Dykman, L. Biodistribution and toxicity of engineered gold nanoparticles: A review of in vitro and in vivo studies. Chem. Soc. Rev. 2011, 40, 1647–1671. [Google Scholar] [CrossRef]
- Cho, E.C.; Zhang, Q.; Xia, Y. The effect of sedimentation and diffusion on cellular uptake of gold nanoparticles. Nat. Nanotechnol. 2011, 6, 385–391. [Google Scholar]
- Kong, W.H.; Bae, K.H.; Jo, S.D.; Kim, J.S.; Park, T.G. Cationic lipid-coated gold nanoparticles as efficient and non-cytotoxic intracellular sirna delivery vehicles. Pharm. Res. 2012, 29, 362–374. [Google Scholar] [CrossRef]
- Daka, A.; Peer, D. Rnai-based nanomedicines for targeted personalized therapy. Adv. Drug Deliv. Rev. 2012, 64, 1508–1521. [Google Scholar] [CrossRef]
- Parveen, S.; Misra, R.; Sahoo, S.K. Nanoparticles: A boon to drug delivery, therapeutics, diagnostics and imagin. Nanomedicine 2012, 8, 147–166. [Google Scholar] [CrossRef]
- Burnett, J.C.; Rossi, J.J. Rna-based therapeutics: Current progress and future prospects. Chem. Biol. 2012, 19, 60–71. [Google Scholar]
- De Ilarduya, C.T.; Sun, Y.; Duezguenes, N. Gene delivery by lipoplexes and polyplexes. Eur. J. Pharm. Sci. 2010, 40, 159–170. [Google Scholar]
- De Planque, M.R.R.; Aghdaei, S.; Roose, T.; Morgan, H. Electrophysiological characterization of membrane disruption by nanoparticles. ACS Nano 2011, 5, 3599–3606. [Google Scholar] [CrossRef]
- Khalil, I.A.; Kogure, K.; Akita, H.; Harashima, H. Uptake pathways and subsequent intracellular trafficking in nonviral gene delivery. Pharmacol. Rev. 2006, 58, 32–45. [Google Scholar] [CrossRef]
- Ding, H.M.; Tian, W.D.; Ma, Y.Q. Designing nanoparticle translocation through membranes by computer simulations. ACS Nano 2012, 6, 1230–1238. [Google Scholar] [CrossRef]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef]
- Kerr, M.C.; Teasdale, R.D. Defining macropinocytosis. Traffic 2009, 10, 364–371. [Google Scholar] [CrossRef]
- Mayor, S.; Pagano, R.E. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 603–612. [Google Scholar] [CrossRef]
- Rejman, J.; Bragonzi, A.; Conese, M. Role of clathrin- and caveolae-mediated endocytosis in gene transfer mediated by lipo- and polyplexes. Mol. Ther. 2005, 12, 468–474. [Google Scholar] [CrossRef]
- Frohlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomedicine 2012, 7, 5577–5591. [Google Scholar] [CrossRef]
- Goncalves, C.; Mennesson, E.; Fuchs, R.; Gorvel, J.P.; Midoux, P.; Pichon, C. Macropinocytosis of polyplexes and recycling of plasmid via the clathrin-dependent pathway impair the transfection efficiency of human hepatocarcinoma cells. Mol. Ther. 2004, 10, 373–385. [Google Scholar] [CrossRef]
- Brigger, I.; Dubernet, C.; Couvreur, P. Nanoparticles in cancer therapy and diagnosis. Adv. Drug Deliv. Rev. 2002, 54, 631–651. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Hunter, A.C.; Andresen, T.L. Factors controlling nanoparticle pharmacokinetics: An integrated analysis and perspective. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 481–503. [Google Scholar] [CrossRef]
- Venturoli, D.; Rippe, B. Ficoll and dextran vs. Globular proteins as probes for testing glomerular permselectivity: Effects of molecular size, shape, charge, and deformability. Am. J. Physiol. Renal Physiol. 2005, 288, F605–F613. [Google Scholar] [CrossRef]
- Petros, R.A.; DeSimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Drug delivery systems: Entering the mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef]
- Vonarbourg, A.; Passirani, C.; Saulnier, P.; Benoit, J.P. Parameters influencing the stealthiness of colloidal drug delivery systems. Biomaterials 2006, 27, 4356–4373. [Google Scholar] [CrossRef]
- Doshi, N.; Mitragotri, S. Macrophages recognize size and shape of their targets. PLoS ONE 2010, 5, e10051. [Google Scholar] [CrossRef]
- Decuzzi, P.; Pasqualini, R.; Arap, W.; Ferrari, M. Intravascular delivery of particulate systems: Does geometry really matter? Pharm. Res. 2009, 26, 235–243. [Google Scholar] [CrossRef]
- Champion, J.A.; Mitragotri, S. Role of target geometry in phagocytosis. Proc. Natl. Acad. Sci. USA 2006, 103, 4930–4934. [Google Scholar] [CrossRef]
- He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31, 3657–3666. [Google Scholar] [CrossRef]
- Galvin, P.; Thompson, D.; Ryan, K.B.; McCarthy, A.; Moore, A.C.; Burke, C.S.; Dyson, M.; MacCraith, B.D.; Gun’ko, Y.K.; Byrne, M.T.; et al. Nanoparticle-based drug delivery: Case studies for cancer and cardiovascular applications. Cell. Mol. Life Sci. 2012, 69, 389–404. [Google Scholar] [CrossRef]
- Schipper, M.L.; Iyer, G.; Koh, A.L.; Cheng, Z.; Ebenstein, Y.; Aharoni, A.; Keren, S.; Bentolila, L.A.; Li, J.Q.; Rao, J.H.; et al. Particle size, surface coating, and pegylation influence the biodistribution of quantum dots in living mice. Small 2009, 5, 126–134. [Google Scholar] [CrossRef]
- Lemarchand, C.; Gref, R.; Couvreur, P. Polysaccharide-decorated nanoparticles. Eur. J. Pharm. Biopharm. 2004, 58, 327–341. [Google Scholar] [CrossRef]
- Dufort, S.; Sancey, L.; Coll, J.L. Physico-chemical parameters that govern nanoparticles fate also dictate rules for their molecular evolution. Adv. Drug Deliv. Rev. 2012, 64, 179–189. [Google Scholar] [CrossRef]
- Noguchi, Y.; Wu, J.; Duncan, R.; Strohalm, J.; Ulbrich, K.; Akaike, T.; Maeda, H. Early phase tumor accumulation of macromolecules: A great difference in clearance rate between tumor and normal tissues. Jpn. J. Cancer Res. 1998, 89, 307–314. [Google Scholar] [CrossRef]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the epr effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef]
- Singh, S.; Sharma, A.; Robertson, G.P. Realizing the clinical potential of cancer nanotechnology by minimizing toxicologic and targeted delivery concerns. Cancer Res. 2012, 72, 5663–5668. [Google Scholar] [CrossRef]
- Lo, A.; Lin, C.-T.; Wu, H.-C. Hepatocellular carcinoma cell-specific peptide ligand for targeted drug delivery. Mol. Cancer Ther. 2008, 7, 579–589. [Google Scholar] [CrossRef]
- Yan, H.B.; Tram, K. Glycotargeting to improve cellular delivery efficiency of nucleic acids. Glycoconj. J. 2007, 24, 107–123. [Google Scholar] [CrossRef]
- Vaishnaw, A.K.; Gollob, J.; Gamba-Vitalo, C.; Hutabarat, R.; Sah, D.; Meyers, R.; de Fougerolles, T.; Maraganore, J. A status report on rnai therapeutics. Silence 2010, 1, 14. [Google Scholar] [CrossRef]
- De Fougerolles, A.R. Delivery vehicles for small interfering rna in vivo. Hum. Gene Ther. 2008, 19, 125–132. [Google Scholar] [CrossRef]
- Wu, Y.; Ho, Y.P.; Mao, Y.C.; Wang, X.M.; Yu, B.; Leong, K.W.; Lee, L.J. Uptake and intracellular fate of multifunctional nanoparticles: A comparison between lipoplexes and polyplexes via quantum dot mediated forster resonance energy transfer. Mol. Pharm. 2011, 8, 1662–1668. [Google Scholar] [CrossRef]
- Strumberg, D.; Schultheis, B.; Traugott, U.; Vank, C.; Santel, A.; Keil, O.; Giese, K.; Kaufmann, J.; Drevs, J. Phase i clinical development of atu027, a sirna formulation targeting pkn3 in patients with advanced solid tumors. Int. J. Clin. Pharmacol. Ther. 2012, 50, 76–78. [Google Scholar]
- Lonez, C.; Vandenbranden, M.; Ruysschaert, J.M. Cationic liposomal lipids: From gene carriers to cell signaling. Prog. Lipid Res. 2008, 47, 340–347. [Google Scholar] [CrossRef]
- Barros, S.A.; Gollob, J.A. Safety profile of rnai nanomedicines. Adv. Drug Deliv. Rev. 2012, 64, 1730–1737. [Google Scholar] [CrossRef]
- Davis, M.E. The first targeted delivery of sirna in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: From concept to clinic. Mol. Pharm. 2009, 6, 659–668. [Google Scholar] [CrossRef]
- Heidel, J.D.; Davis, M.E. Clinical developments in nanotechnology for cancer therapy. Pharm. Res. 2011, 28, 187–199. [Google Scholar] [CrossRef]
- Rozema, D.B.; Lewis, D.L.; Wakefield, D.H.; Wong, S.C.; Klein, J.J.; Roesch, P.L.; Bertin, S.L.; Reppen, T.W.; Chu, Q.; Blokhin, A.V.; et al. Dynamic polyconjugates for targetedin vivo delivery of sirna to hepatocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 12982–12987. [Google Scholar]
- Panyam, J.; Labhasetwar, V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv. Drug Deliv. Rev. 2003, 55, 329–347. [Google Scholar] [CrossRef]
- Reilly, M.J.; Larsen, J.D.; Sullivan, M.O. Histone h3 tail peptides and poly(ethylenimine) have synergistic effects for gene delivery. Mol. Pharm. 2012, 9, 1031–1040. [Google Scholar]
- Zhou, J.; Liu, J.; Cheng, C.J.; Patel, T.R.; Weller, C.E.; Piepmeier, J.M.; Jiang, Z.; Saltzman, W.M. Biodegradable poly(amine-co-ester) terpolymers for targeted gene delivery. Nat. Mater. 2012, 11, 82–90. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Angart, P.; Vocelle, D.; Chan, C.; Walton, S.P. Design of siRNA Therapeutics from the Molecular Scale. Pharmaceuticals 2013, 6, 440-468. https://doi.org/10.3390/ph6040440
Angart P, Vocelle D, Chan C, Walton SP. Design of siRNA Therapeutics from the Molecular Scale. Pharmaceuticals. 2013; 6(4):440-468. https://doi.org/10.3390/ph6040440
Chicago/Turabian StyleAngart, Phillip, Daniel Vocelle, Christina Chan, and S. Patrick Walton. 2013. "Design of siRNA Therapeutics from the Molecular Scale" Pharmaceuticals 6, no. 4: 440-468. https://doi.org/10.3390/ph6040440
APA StyleAngart, P., Vocelle, D., Chan, C., & Walton, S. P. (2013). Design of siRNA Therapeutics from the Molecular Scale. Pharmaceuticals, 6(4), 440-468. https://doi.org/10.3390/ph6040440