RNAi Therapeutic Platforms for Lung Diseases

Abstract
:1. Introduction
2. Delivery of RNAi-Based Therapeutics to the Lungs
2.1. Pulmonary Delivery Approaches
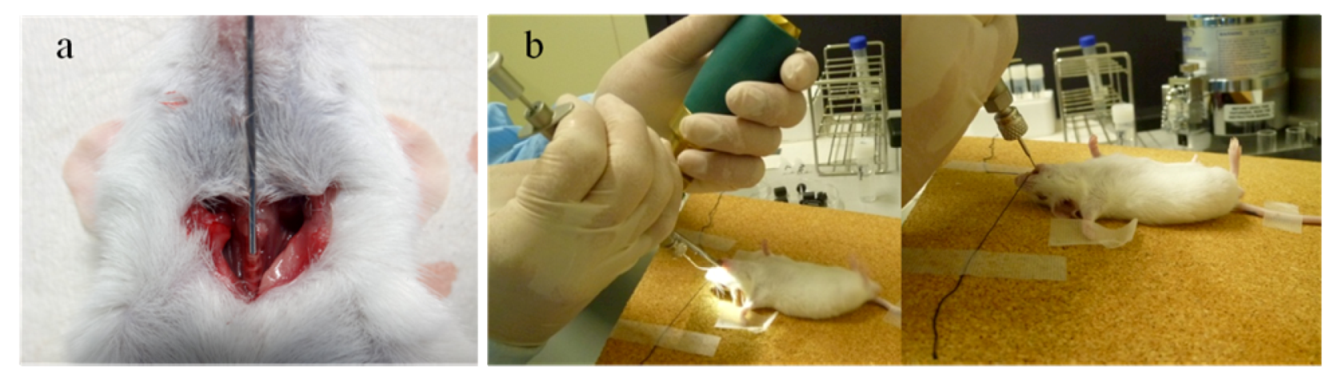
2.1.1. Intratracheal and Intranasal Delivery
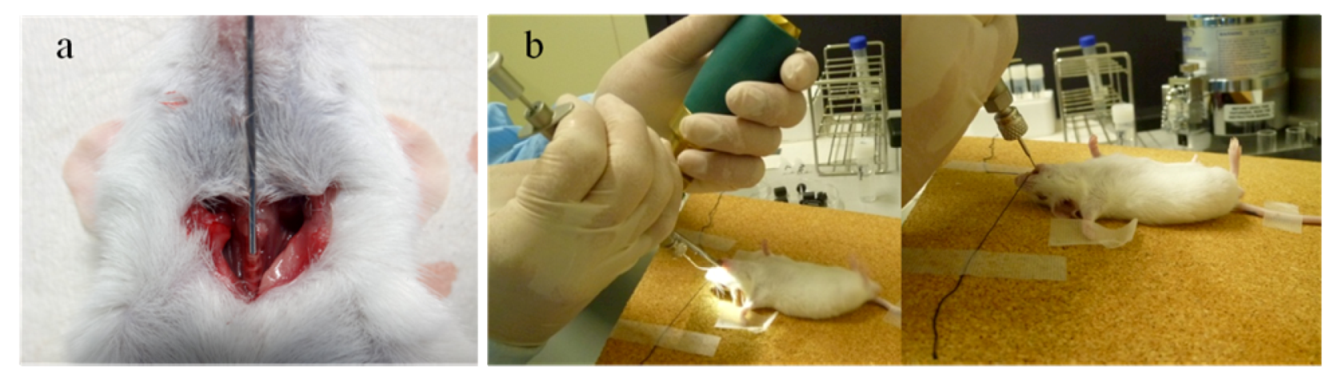
2.1.2. Inhalation Delivery
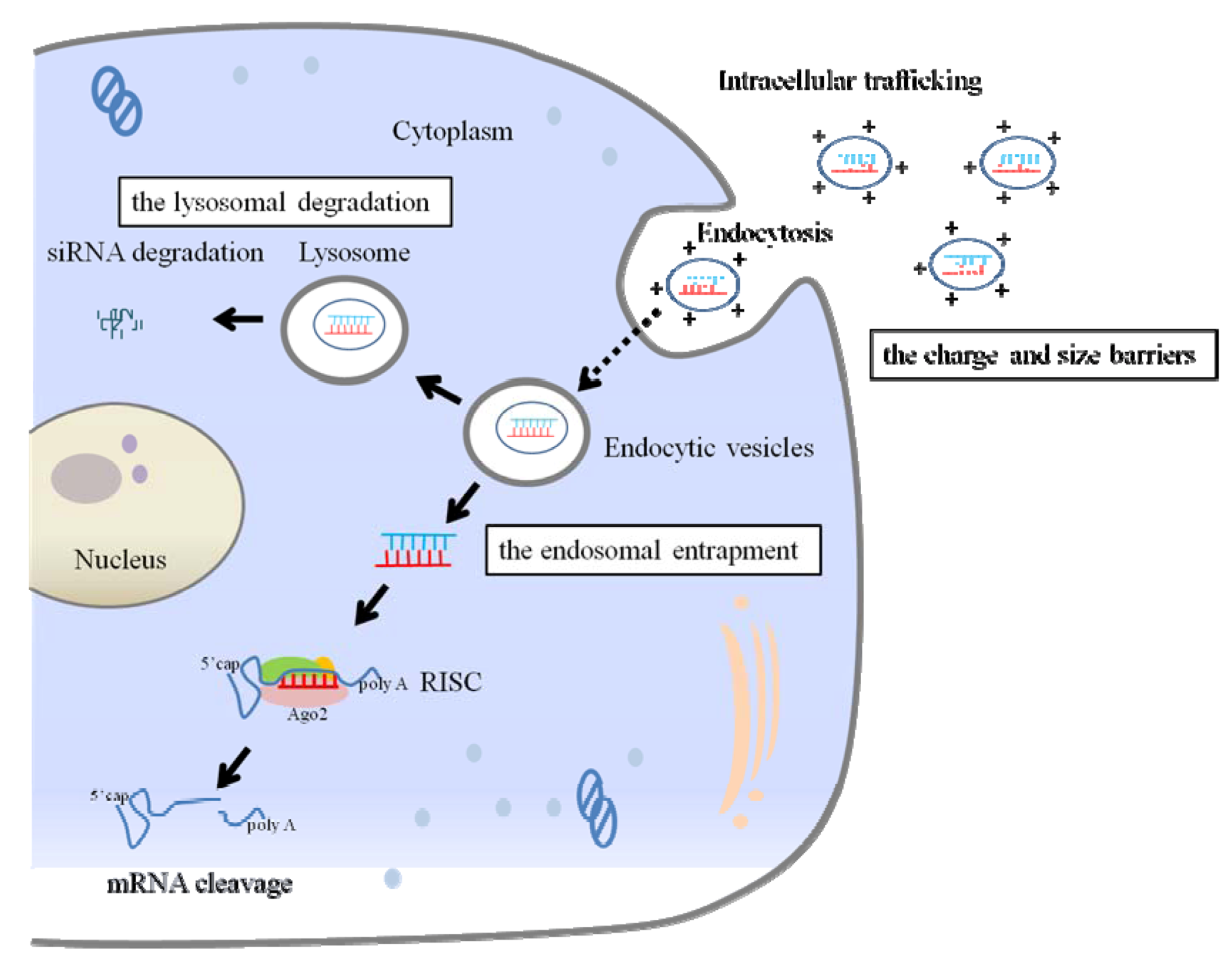
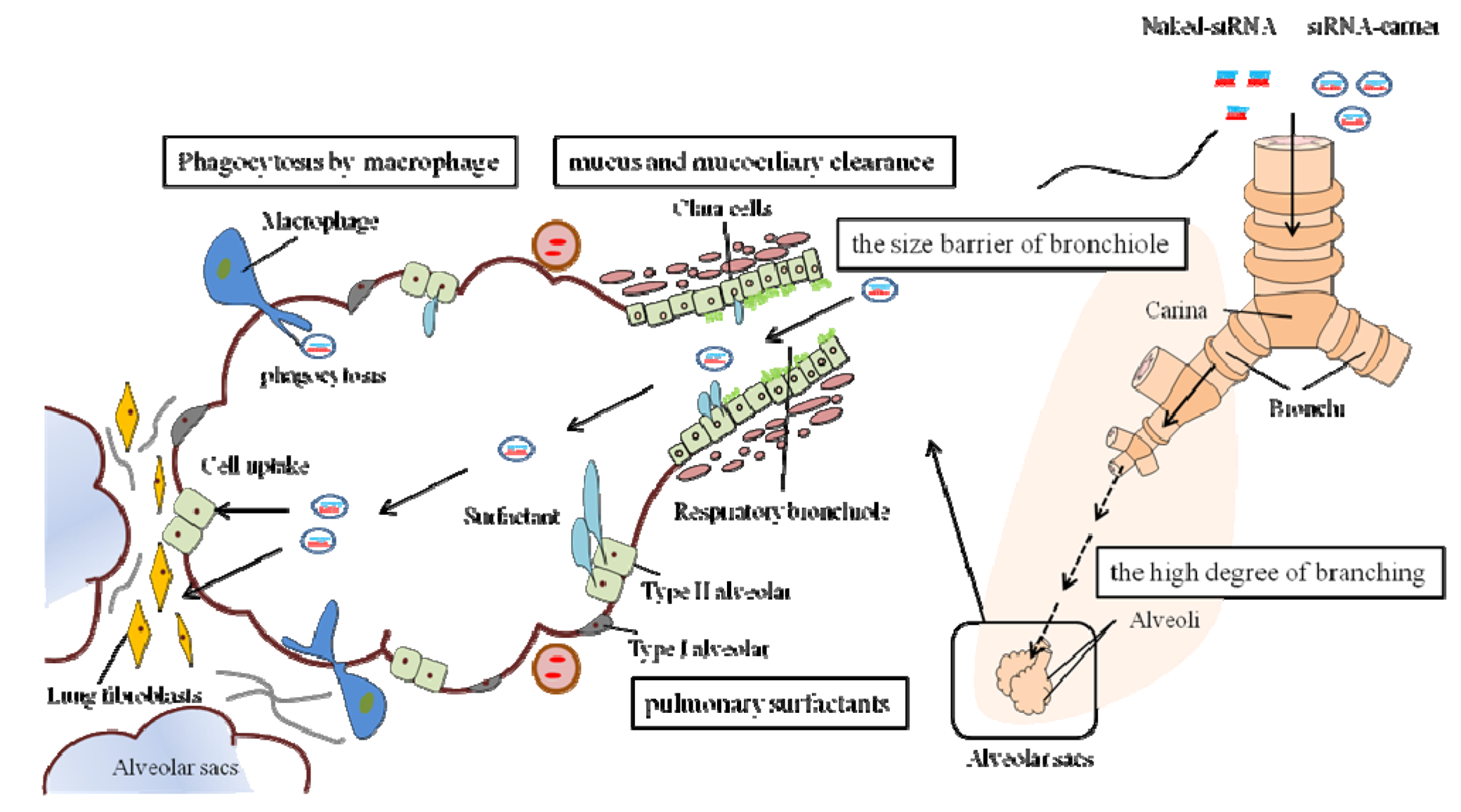
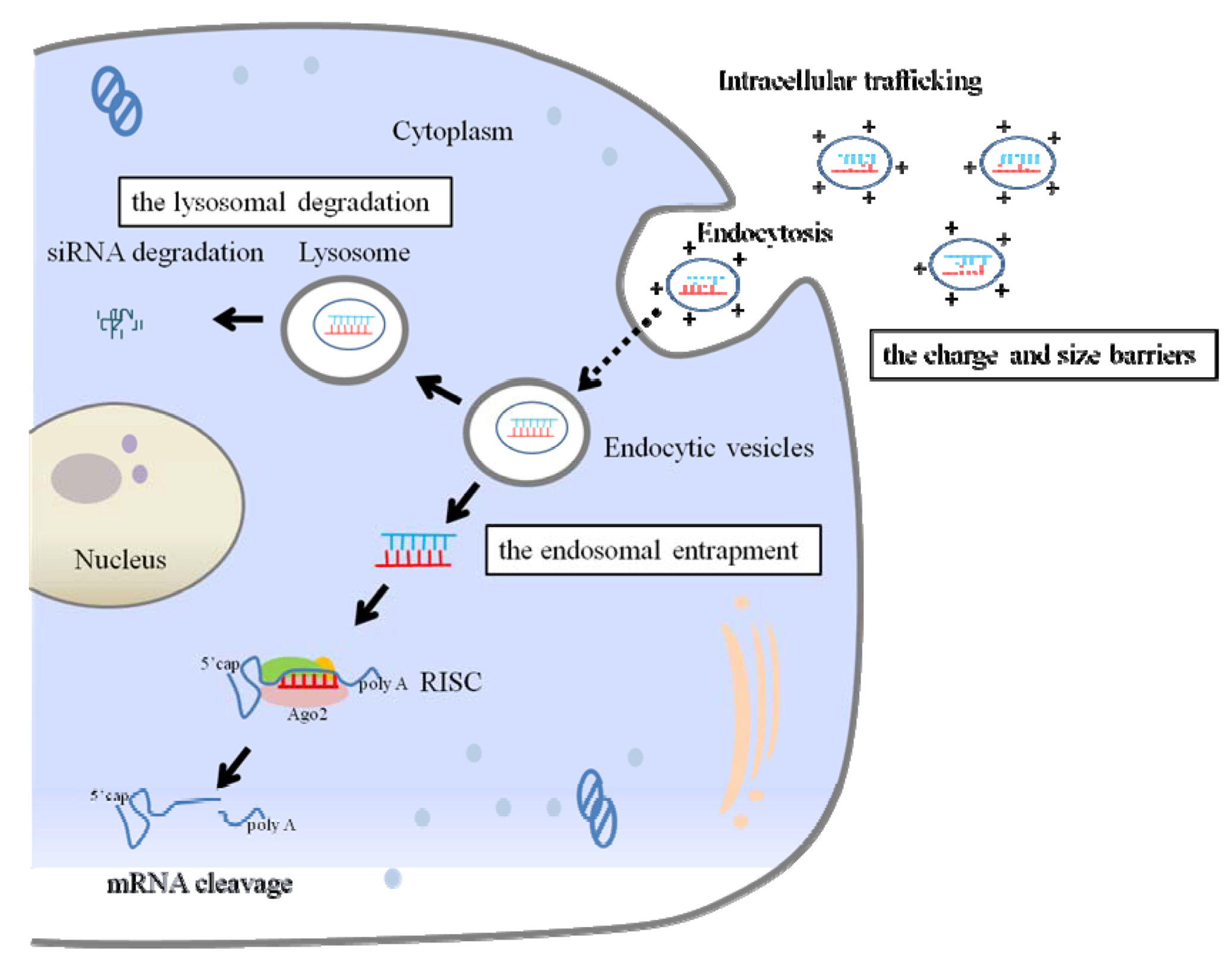
2.2. Extracellular and Intracellular Barriers to siRNA Delivery
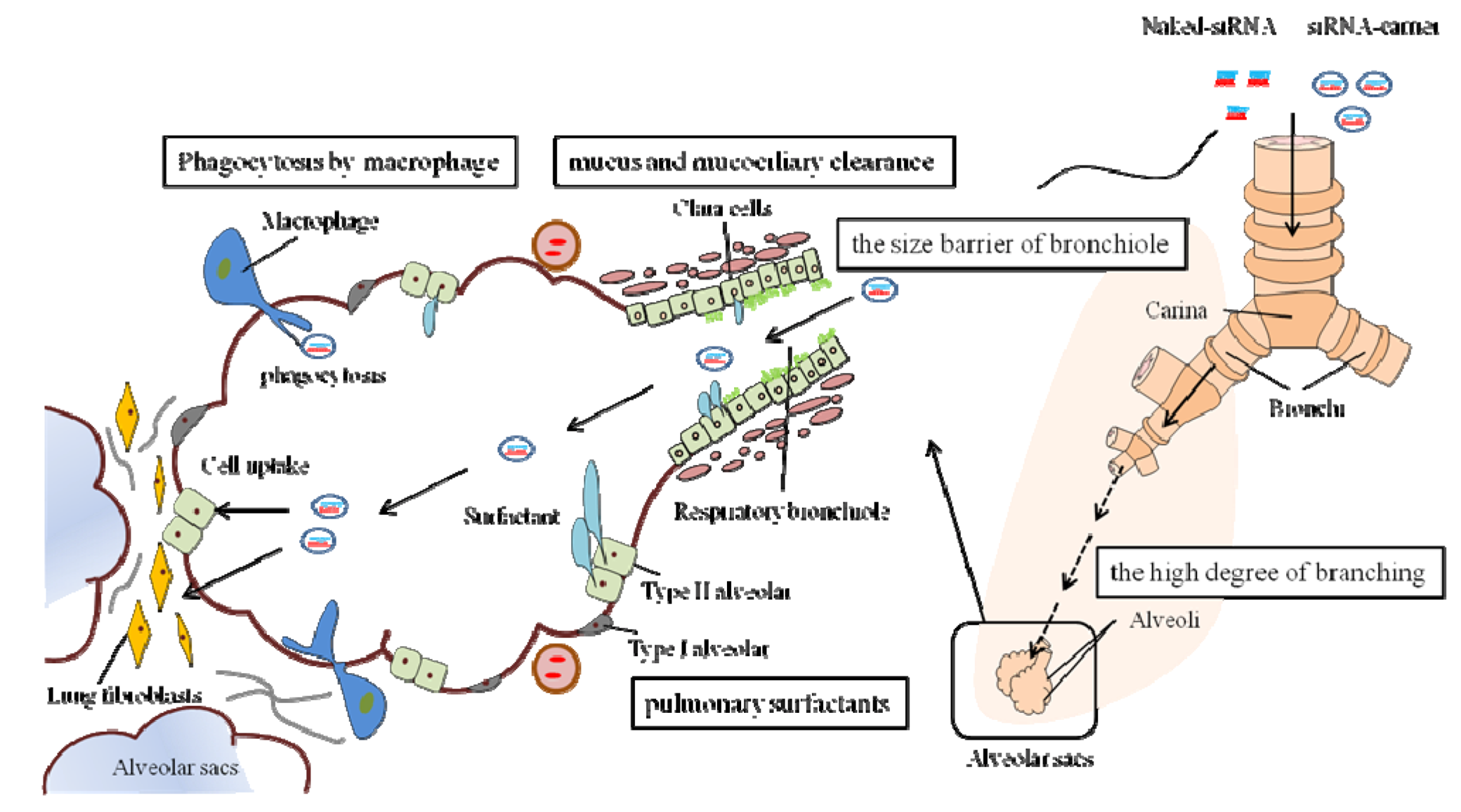
2.3. Delivery Method of siRNA to the Lungs
3. RNAi Medicine in Lung Diseases
3.1. Therapeutic siRNAs for Lung Diseases

{kind=link}
{kind=link}
{kind=link}
| Drug | Route of Administration | Delivery Agent | Disease | Target | Stage of Clinical Trial |
|---|---|---|---|---|---|
| ExcellairTM | Inhalation | Unknown | Asthma | Syk kinase | II |
| ALN-RSV01 | Intranasal spray | Naked siRNA | RSV infection | RSV nucleocapsid | IIb |
| Atu027 | IV | Lipid nanoparticles | Advanced solid cancer (Metastatic lung cancer) | PKN3 | I |
| TKM-ApoB | IV | Lipid nanoparticles | Hypercholesterolemia | ApoB | I |
| TKM-PLK1 | IV | Lipid nanoparticles | Cancer | Polo-like-kinase1 | I |
| ALN-VSP02 | IV | Lipid nanoparticles | Solid cancers with liver involvement | KSP and VEGF | I |
| ALN-TTR01 | IV | Lipid nanoparticles | Transthyretin-mediated amyloidosis (ATTR) | Transthyretin (TTR) | I |
| CALAA-01 | IV | Cyclodextrin nanoparticles | Solid tumor | RRM2 | I |
| siG12D LODER | EUS biopsy needle | Miniature biodegradable polymer matrix | Pancreatic ductal adenocarcinoma | KRAS | I |
| I5NP | IV | Naked siRNA | Acute kidney injury | p53 | I/II |
| QPI-1007 | IVT | Naked siRNA | Glaucoma and acute eye injury | Caspase-2 | I |
| TD101 | Intradermal injection | Naked siRNA | Pachyonychia congenita | Keratin 6a N171Kmutant mRNA | Ib |
| SYL040012 | Ophthalmic drops | Naked siRNA | Ocular hypertension and glaucoma | Adrenergic receptor beta-2 | I/II |
| AGN-745 | IVT | Naked siRNA | AMD | VEGF-receptor1 | II |
| PF-655 | IVT | Naked siRNA | AMD and diabetic macular edema | RTP801 (pro-angiogenic factor) | II |
| Bevasiranib | IVT | Naked siRNA | AMD | VEGF | III |
3.1.1. Pulmonary Viral Infections
3.1.2. Lung Cancer
3.1.3. Inflammatory Lung Diseases
3.2. Therapeutic microRNA/Anti-microRNA for Lung Diseases
| miRNA | Disease | Stage of clinical trial | Reference |
|---|---|---|---|
| miRNA replacement | |||
| let-7 | Lung cancer | Preclinical | [106] |
| miR-34 | Lung cancer, Prostate cancer | Preclinical | [107,108] |
| miR-29 | Cardiac fibrosis | Preclinical | [109] |
| miRNA antagonists | |||
| miR-122 | Hepatitis C virus | II | [110] |
| miR-208/499 | Chronic heart failure | Preclinical | [111] |
| miR-15/195 | Post-myocardial infarction remodeling | Preclinical | [112,113] |
| miR-206 | Amyotrophic lateral sclerosis | Preclinical | [114] |
| miR-451 | Myeloproliferative diseases | Preclinical | [115] |
3.2.1. Role of microRNA in Inflammatory Lung Diseases
| Lung diseases | Expression of specific miRNA | Reference | |
|---|---|---|---|
| COPD | miR-223/1274a | ↑ | [126] |
| let-7 | ↓ | [117] | |
| miR-1 | ↓ | [116] | |
| miR-146a | ↓ | [127] | |
| miR-150 | ↓ | [117] | |
| Asthma | miR-21 | ↑ | [123] |
| miR-126 | ↑ | [124] | |
| miR-155 | ↑ | [122] | |
| miR-133a | ↓ | [125] | |
| Pulmonary fibrosis | miR-21 | ↑ | [118] |
| miR-155 | ↑ | [128] | |
| let-7d | ↓ | [119] | |
| miR-29 | ↓ | [120] | |
| miR-200 | ↓ | [121] | |
| Smoking-related miRNA | let-7 | ↓ | [129] |
| miR-10a | ↓ | [129] | |
| miR-34 | ↓ | [129] | |
| miR-123 | ↓ | [129] | |
| miR-145 | ↓ | [129] | |
| miR-150 | ↓ | [117] | |
| miR-199b | ↓ | [130] | |
| miR-218 | ↓ | [130] | |
| miR-222 | ↓ | [117,129] | |
3.2.2. microRNA/Anti-microRNA Delivery for Lung Cancer Therapy
4. Conclusions and Prospects
Acknowledgements
Conflict of Interest
References
- Kim, D.H.; Rossi, J.J. Strategies for silencing human disease using RNA interference. Nat. Rev. Genet. 2007, 8, 173–184. [Google Scholar] [CrossRef]
- Bumcrot, D.; Manoharan, M.; Koteliansky, V.; Sah, D.W. RNAi therapeutics: A potential new class of pharmaceutical drugs. Nat. Chem. Biol. 2006, 2, 711–719. [Google Scholar] [CrossRef]
- Giladi, H.; Ketzinel-Gilad, M.; Rivkin, L.; Felig, Y.; Nussbaum, O.; Galun, E. Small interfering RNA inhibits hepatitis B virus replication in mice. Mol. Ther. 2003, 8, 769–776. [Google Scholar]
- Xia, H.; Mao, Q.; Eliason, S.L.; Harper, S.Q.; Martins, I.H.; Orr, H.T.; Paulson, H.L.; Yang, L.; Kotin, R.M.; Davidson, B.L. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat. Med. 2004, 10, 816–820. [Google Scholar] [CrossRef]
- Judge, A.D.; Robbins, M.; Tavakoli, I.; Levi, J.; Hu, L.; Fronda, A.; Ambegia, E.; McClintock, K.; MacLachlan, I. Confirming the RNAi-mediated mechanism of action of siRNA-based cancer therapeutics in mice. J. Clin. Invest. 2009, 119, 661–673. [Google Scholar] [CrossRef]
- Chernolovskaya, E.L.; Zenkova, M.A. Chemical modification of siRNA. Curr Opin Mol. Ther. 2010, 12, 158–167. [Google Scholar]
- Schlee, M.; Hornung, V.; Hartmann, G. siRNA and isRNA: Two edges of one sword. Mol. Ther. 2006, 14, 463–470. [Google Scholar] [CrossRef]
- Li, S.D.; Huang, L. Targeted delivery of antisense oligodeoxynucleotide and small interference RNA into lung cancer cells. Mol. Pharm. 2006, 3, 579–588. [Google Scholar] [CrossRef]
- Xu, C.X.; Jere, D.; Jin, H.; Chang, S.H.; Chung, Y.S.; Shin, J.Y.; Kim, J.E.; Park, S.J.; Lee, Y.H.; Chae, C.H.; et al. Poly(ester amine)-mediated, aerosol-delivered Akt1 small interfering RNA suppresses lung tumorigenesis. Am. J. Respir. Crit. Care Med. 2008, 178, 60–73. [Google Scholar] [CrossRef]
- Jere, D.; Xu, C.X.; Arote, R.; Yun, C.H.; Cho, M.H.; Cho, C.S. Poly(beta-amino ester) as a carrier for si/shRNA delivery in lung cancer cells. Biomaterials 2008, 29, 2535–2547. [Google Scholar] [CrossRef]
- Ren, X.L.; Xu, Y.M.; Bao, W.; Fu, H.J.; Wu, C.G.; Zhao, Y.; Li, Z.K.; Zhang, J.; Li, S.Q.; Chen, W.Q.; et al. Inhibition of non-small cell lung cancer cell proliferation and tumor growth by vector-based small interfering RNAs targeting HER2/neu. Cancer Lett. 2009, 281, 134–143. [Google Scholar] [CrossRef]
- Zamora-Avila, D.E.; Zapata-Benavides, P.; Franco-Molina, M.A.; Saavedra-Alonso, S.; Trejo-Avila, L.M.; Resendez-Perez, D.; Mendez-Vazquez, J.L.; Isaias-Badillo, J.; Rodriguez-Padilla, C. WT1 gene silencing by aerosol delivery of PEI-RNAi complexes inhibits B16-F10 lung metastases growth. Cancer Gene Ther. 2009, 16, 892–899. [Google Scholar] [CrossRef]
- Ge, Q.; Filip, L.; Bai, A.; Nguyen, T.; Eisen, H.N.; Chen, J. Inhibition of influenza virus production in virus-infected mice by RNA interference. Proc. Natl. Acad. Sci. USA 2004, 101, 8676–8681. [Google Scholar]
- Li, B.J.; Tang, Q.; Cheng, D.; Qin, C.; Xie, F.Y.; Wei, Q.; Xu, J.; Liu, Y.; Zheng, B.J.; Woodle, M.C.; et al. Using siRNA in prophylactic and therapeutic regimens against SARS coronavirus in Rhesus macaque. Nat. Med. 2005, 11, 944–951. [Google Scholar]
- Rosas-Taraco, A.G.; Higgins, D.M.; Sanchez-Campillo, J.; Lee, E.J.; Orme, I.M.; Gonzalez-Juarrero, M. Intrapulmonary delivery of XCL1-targeting small interfering RNA in mice chronically infected with Mycobacterium tuberculosis. Am. J. Respir. Cell Mol. Biol. 2009, 41, 136–145. [Google Scholar] [CrossRef]
- Fulton, A.; Peters, S.T.; Perkins, G.A.; Jarosinski, K.W.; Damiani, A.; Brosnahan, M.; Buckles, E.L.; Osterrieder, N.; Van de Walle, G.R. Effective treatment of respiratory alphaherpesvirus infection using RNA interference. PLoS One 2009, 4, 5. [Google Scholar]
- DeVincenzo, J.; Lambkin-Williams, R.; Wilkinson, T.; Cehelsky, J.; Nochur, S.; Walsh, E.; Meyers, R.; Gollob, J.; Vaishnaw, A. A randomized, double-blind, placebo-controlled study of an RNAi-based therapy directed against respiratory syncytial virus. Proc. Natl. Acad. Sci. USA 2010, 107, 8800–8805. [Google Scholar]
- Lee, C.C.; Chiang, B.L. RNA interference: new therapeutics in allergic diseases. Curr. Gene Ther. 2008, 8, 236–246. [Google Scholar] [CrossRef]
- Seguin, R.M.; Ferrari, N. Emerging oligonucleotide therapies for asthma and chronic obstructive pulmonary disease. Expert Opin. Investig. Drugs 2009, 18, 1505–1517. [Google Scholar]
- Senoo, T.; Hattori, N.; Tanimoto, T.; Furonaka, M.; Ishikawa, N.; Fujitaka, K.; Haruta, Y.; Murai, H.; Yokoyama, A.; Kohno, N. Suppression of plasminogen activator inhibitor-1 by RNA interference attenuates pulmonary fibrosis. Thorax 2010, 65, 334–340. [Google Scholar] [CrossRef]
- D'Alessandro-Gabazza, C.N.; Kobayashi, T.; Boveda-Ruiz, D.; Takagi, T.; Toda, M.; Gil-Bernabe, P.; Miyake, Y.; Yasukawa, A.; Matsuda, Y.; Suzuki, N.; et al. Development and preclinical efficacy of novel transforming growth factor-beta1 short interfering RNAs for pulmonary fibrosis. Am. J. Respir. Cell. Mol. Biol. 2012, 46, 397–406. [Google Scholar] [CrossRef]
- Lam, J.K.; Liang, W.; Chan, H.K. Pulmonary delivery of therapeutic siRNA. Adv. Drug Deliv. Rev.. 2012, 64, 1–15. [Google Scholar]
- Agu, R.U.; Ugwoke, M.I.; Armand, M.; Kinget, R.; Verbeke, N. The lung as a route for systemic delivery of therapeutic proteins and peptides. Respir. Res. 2001, 2, 198–209. [Google Scholar]
- Jensen, D.M.; Cun, D.; Maltesen, M.J.; Frokjaer, S.; Nielsen, H.M.; Foged, C. Spray drying of siRNA-containing PLGA nanoparticles intended for inhalation. J. Control. Release 2010, 142, 138–145. [Google Scholar] [CrossRef]
- Jensen, D.K.; Jensen, L.B.; Koocheki, S.; Bengtson, L.; Cun, D.; Nielsen, H.M.; Foged, C. Design of an inhalable dry powder formulation of DOTAP-modified PLGA nanoparticles loaded with siRNA. J. Control. Release 2012, 157, 141–148. [Google Scholar] [CrossRef]
- Perl, M.; Chung, C.S.; Lomas-Neira, J.; Rachel, T.M.; Biffl, W.L.; Cioffi, W.G.; Ayala, A. Silencing of Fas, but not caspase-8, in lung epithelial cells ameliorates pulmonary apoptosis, inflammation, and neutrophil influx after hemorrhagic shock and sepsis. Am. J. Pathol. 2005, 167, 1545–1559. [Google Scholar] [CrossRef]
- Lomas-Neira, J.L.; Chung, C.S.; Wesche, D.E.; Perl, M.; Ayala, A. In vivo gene silencing (with siRNA) of pulmonary expression of MIP-2 versus KC results in divergent effects on hemorrhage-induced, neutrophil-mediated septic acute lung injury. J. Leukoc Biol 2005, 77, 846–853. [Google Scholar] [CrossRef]
- Merkel, O.M.; Beyerle, A.; Librizzi, D.; Pfestroff, A.; Behr, T.M.; Sproat, B.; Barth, P.J.; Kissel, T. Nonviral siRNA delivery to the lung: investigation of PEG-PEI polyplexes and their in vivo performance. Mol. Pharm. 2009, 6, 1246–1260. [Google Scholar] [CrossRef]
- Garbuzenko, O.B.; Saad, M.; Betigeri, S.; Zhang, M.; Vetcher, A.A.; Soldatenkov, V.A.; Reimer, D.C.; Pozharov, V.P.; Minko, T. Intratracheal versus intravenous liposomal delivery of siRNA, antisense oligonucleotides and anticancer drug. Pharm. Res. 2009, 26, 382–394. [Google Scholar] [CrossRef]
- Wang, J.C.; Lai, S.; Guo, X.; Zhang, X.; de Crombrugghe, B.; Sonnylal, S.; Arnett, F.C.; Zhou, X. Attenuation of fibrosis in vitro and in vivo with SPARC siRNA. Arthritis Res. Ther. 2010. [Google Scholar] [CrossRef]
- Gutbier, B.; Kube, S.M.; Reppe, K.; Santel, A.; Lange, C.; Kaufmann, J.; Suttorp, N.; Witzenrath, M. RNAi-mediated suppression of constitutive pulmonary gene expression by small interfering RNA in mice. Pulm. Pharmacol. Ther. 2010, 23, 334–344. [Google Scholar] [CrossRef]
- Driscoll, K.E.; Costa, D.L.; Hatch, G.; Henderson, R.; Oberdorster, G.; Salem, H.; Schlesinger, R.B. Intratracheal instillation as an exposure technique for the evaluation of respiratory tract toxicity: uses and limitations. Toxicol. Sci. 2000, 55, 24–35. [Google Scholar] [CrossRef]
- Bivas-Benita, M.; Zwier, R.; Junginger, H.E.; Borchard, G. Non-invasive pulmonary aerosol delivery in mice by the endotracheal route. Eur. J. Pharm. Biopharm. 2005, 61, 214–218. [Google Scholar] [CrossRef]
- Lu, D.; Hickey, A.J. Pulmonary vaccine delivery. Expert Rev. Vaccines 2007, 6, 213–226. [Google Scholar] [CrossRef]
- Bitko, V.; Musiyenko, A.; Shulyayeva, O.; Barik, S. Inhibition of respiratory viruses by nasally administered siRNA. Nat. Med. 2005, 11, 50–55. [Google Scholar] [CrossRef]
- Massaro, D.; Massaro, G.D.; Clerch, L.B. Noninvasive delivery of small inhibitory RNA and other reagents to pulmonary alveoli in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L1066–L1070. [Google Scholar] [CrossRef]
- Hickey, A.J.; Garcia-Contreras, L. Immunological and toxicological implications of short-term studies in animals of pharmaceutical aerosol delivery to the lungs: relevance to humans. Crit. Rev. Ther. Drug Carrier. Syst. 2001, 18, 387–431. [Google Scholar]
- DeVincenzo, J.; Cehelsky, J.E.; Alvarez, R.; Elbashir, S.; Harborth, J.; Toudjarska, I.; Nechev, L.; Murugaiah, V.; Van Vliet, A.; Vaishnaw, A.K.; et al. Evaluation of the safety, tolerability and pharmacokinetics of ALN-RSV01, a novel RNAi antiviral therapeutic directed against respiratory syncytial virus (RSV). Antiviral. Res. 2008, 77, 225–231. [Google Scholar] [CrossRef]
- Mastrandrea, L.D.; Quattrin, T. Clinical evaluation of inhaled insulin. Adv. Drug Deliv. Rev. 2006, 58, 1061–1075. [Google Scholar] [CrossRef]
- Bai, S.; Gupta, V.; Ahsan, F. Inhalable lactose-based dry powder formulations of low molecular weight heparin. J. Aerosol. Med. Pulm. Drug Deliv. 2010, 23, 97–104. [Google Scholar] [CrossRef]
- Sanders, N.; Rudolph, C.; Braeckmans, K.; De Smedt, S.C.; Demeester, J. Extracellular barriers in respiratory gene therapy. Adv. Drug Deliv. Rev. 2009, 61, 115–127. [Google Scholar] [CrossRef]
- Roy, I.; Vij, N. Nanodelivery in airway diseases: Challenges and therapeutic applications. Nanomedicine 2010, 6, 237–244. [Google Scholar]
- Groneberg, D.A.; Eynott, P.R.; Lim, S.; Oates, T.; Wu, R.; Carlstedt, I.; Roberts, P.; McCann, B.; Nicholson, A.G.; Harrison, B.D.; et al. Expression of respiratory mucins in fatal status asthmaticus and mild asthma. Histopathology 2002, 40, 367–373. [Google Scholar] [CrossRef]
- Jeffery, P.K. Remodeling in asthma and chronic obstructive lung disease. Am. J. Respir. Crit. Care Med. 2001, 164, S28–S38. [Google Scholar]
- Lebhardt, T.; Roesler, S.; Beck-Broichsitter, M.; Kissel, T. Polymeric nanocarriers for drug delivery to the lung. J. Drug Deliv. Sci. Technol. 2010, 20, 171–180. [Google Scholar]
- Akhtar, S.; Benter, I.F. Nonviral delivery of synthetic siRNAs in vivo. J. Clin. Invest. 2007, 117, 3623–3632. [Google Scholar] [CrossRef]
- Goula, D.; Becker, N.; Lemkine, G.F.; Normandie, P.; Rodrigues, J.; Mantero, S.; Levi, G.; Demeneix, B.A. Rapid crossing of the pulmonary endothelial barrier by polyethylenimine/DNA complexes. Gene Ther. 2000, 7, 499–504. [Google Scholar] [CrossRef]
- Wang, J.; Lu, Z.; Wientjes, M.G.; Au, J.L. Delivery of siRNA therapeutics: Barriers and carriers. AAPS J. 2010, 12, 492–503. [Google Scholar] [CrossRef]
- Endoh, T.; Ohtsuki, T. Cellular siRNA delivery using cell-penetrating peptides modified for endosomal escape. Adv. Drug Deliv. Rev. 2009, 61, 704–709. [Google Scholar]
- Novobrantseva, T.I.; Akinc, A.; Borodovsky, A.; de Fougerolles, A. Delivering silence: advancements in developing siRNA therapeutics. Curr. Opin. Drug Discov. Devel. 2008, 11, 217–224. [Google Scholar]
- Shen, C.; Buck, A.K.; Liu, X.; Winkler, M.; Reske, S.N. Gene silencing by adenovirus-delivered siRNA. FEBS Lett. 2003, 539, 111–114. [Google Scholar] [CrossRef]
- Narvaiza, I.; Aparicio, O.; Vera, M.; Razquin, N.; Bortolanza, S.; Prieto, J.; Fortes, P. Effect of adenovirus-mediated RNA interference on endogenous microRNAs in a mouse model of multidrug resistance protein 2 gene silencing. J. Virol. 2006, 80, 12236–12247. [Google Scholar]
- Guo, X.; Wang, W.; Hu, J.; Feng, K.; Pan, Y.; Zhang, L.; Feng, Y. Lentivirus-mediated RNAi knockdown of NUPR1 inhibits human nonsmall cell lung cancer growth in vitro and in vivo. Anat. Rec. 2012, 295, 2114–2121. [Google Scholar] [CrossRef]
- Scanlon, K.J. Cancer gene therapy: Challenges and opportunities. Anticancer Res. 2004, 24, 501–504. [Google Scholar]
- Teichler Zallen, D. US gene therapy in crisis. Trends Genet. 2000, 16, 272–275. [Google Scholar] [CrossRef]
- Musiyenko, A.; Bitko, V.; Barik, S. RNAi-dependent and -independent antiviral phenotypes of chromosomally integrated shRNA clones: role of VASP in respiratory syncytial virus growth. J. Mol. Med. 2007, 85, 745–752. [Google Scholar] [CrossRef]
- Thomas, M.; Lu, J.J.; Chen, J.; Klibanov, A.M. Non-viral siRNA delivery to the lung. Adv. Drug Deliv. Rev. 2007, 59, 124–133. [Google Scholar] [CrossRef]
- Tseng, Y.C.; Mozumdar, S.; Huang, L. Lipid-based systemic delivery of siRNA. Adv. Drug Deliv. Rev. 2009, 61, 721–731. [Google Scholar] [CrossRef]
- Tompkins, S.M.; Lo, C.Y.; Tumpey, T.M.; Epstein, S.L. Protection against lethal influenza virus challenge by RNA interference in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 8682–8686. [Google Scholar]
- Han, S.W.; Roman, J. Fibronectin induces cell proliferation and inhibits apoptosis in human bronchial epithelial cells: pro-oncogenic effects mediated by PI3-kinase and NF-kappa B. Oncogene 2006, 25, 4341–4349. [Google Scholar] [CrossRef]
- Thomas, M.; Lu, J.J.; Ge, Q.; Zhang, C.; Chen, J.; Klibanov, A.M. Full deacylation of polyethylenimine dramatically boosts its gene delivery efficiency and specificity to mouse lung. Proc. Natl. Acad. Sci. USA 2005, 102, 5679–5684. [Google Scholar]
- Sioud, M.; Sorensen, D.R. Cationic liposome-mediated delivery of siRNAs in adult mice. Biochem. Biophys. Res. Commun. 2003, 312, 1220–1225. [Google Scholar]
- Wu, S.Y.; McMillan, N.A. Lipidic systems for in vivo siRNA delivery. AAPS J. 2009, 11, 639–652. [Google Scholar] [CrossRef]
- Jackson, A.L.; Bartz, S.R.; Schelter, J.; Kobayashi, S.V.; Burchard, J.; Mao, M.; Li, B.; Cavet, G.; Linsley, P.S. Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 2003, 21, 635–637. [Google Scholar]
- Bridge, A.J.; Pebernard, S.; Ducraux, A.; Nicoulaz, A.L.; Iggo, R. Induction of an interferon response by RNAi vectors in mammalian cells. Nat. Genet. 2003, 34, 263–264. [Google Scholar] [CrossRef]
- Heidel, J.D.; Hu, S.; Liu, X.F.; Triche, T.J.; Davis, M.E. Lack of interferon response in animals to naked siRNAs. Nat. Biotechnol. 2004, 22, 1579–1582. [Google Scholar] [CrossRef]
- Urban-Klein, B.; Werth, S.; Abuharbeid, S.; Czubayko, F.; Aigner, A. RNAi-mediated gene-targeting through systemic application of polyethylenimine (PEI)-complexed siRNA in vivo. Gene Ther. 2005, 12, 461–466. [Google Scholar] [CrossRef]
- Pal, A.; Ahmad, A.; Khan, S.; Sakabe, I.; Zhang, C.; Kasid, U.N.; Ahmad, I. Systemic delivery of RafsiRNA using cationic cardiolipin liposomes silences RAF-1 expression and inhibits tumor growth in xenograft model of human prostate cancer. Int. J. Oncol. 2005, 26, 1087–1091. [Google Scholar]
- Nielsen, E.J.; Nielsen, J.M.; Becker, D.; Karlas, A.; Prakash, H.; Glud, S.Z.; Merrison, J.; Besenbacher, F.; Meyer, T.F.; Kjems, J.; Howard, K.A. Pulmonary gene silencing in transgenic EGFP mice using aerosolised chitosan/siRNA nanoparticles. Pharm. Res. 2010, 27, 2520–2527. [Google Scholar] [CrossRef]
- Akinc, A.; Zumbuehl, A.; Goldberg, M.; Leshchiner, E.S.; Busini, V.; Hossain, N.; Bacallado, S.A.; Nguyen, D.N.; Fuller, J.; Alvarez, R.; et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat. Biotechnol. 2008, 26, 561–569. [Google Scholar]
- Cosio, M.G.; Saetta, M.; Agusti, A. Immunologic aspects of chronic obstructive pulmonary disease. New Engl. J. Med. 2009, 360, 2445–2454. [Google Scholar]
- Xu, L.; Anchordoquy, T. Drug delivery trends in clinical trials and translational medicine: challenges and opportunities in the delivery of nucleic acid-based therapeutics. J. Pharm. Sci. 2011, 100, 38–52. [Google Scholar] [CrossRef]
- Shen, J.; Samul, R.; Silva, R.L.; Akiyama, H.; Liu, H.; Saishin, Y.; Hackett, S.F.; Zinnen, S.; Kossen, K.; Fosnaugh, K.; et al. Suppression of ocular neovascularization with siRNA targeting VEGF receptor 1. Gene Ther. 2006, 13, 225–234. [Google Scholar] [CrossRef]
- The US National Institutes of Health. ClinicalTrials.gov. Available online: http://www.clinicaltrials.gov/ (accessed on 30 November 2012).
- Falsey, A.R.; Hennessey, P.A.; Formica, M.A.; Cox, C.; Walsh, E.E. Respiratory syncytial virus infection in elderly and high-risk adults. New Engl. J. Med. 2005, 352, 1749–1759. [Google Scholar] [CrossRef]
- Zamora, M.R.; Budev, M.; Rolfe, M.; Gottlieb, J.; Humar, A.; Devincenzo, J.; Vaishnaw, A.; Cehelsky, J.; Albert, G.; Nochur, S.; et al. RNA interference therapy in lung transplant patients infected with respiratory syncytial virus. Am. J. Respir. Crit. Care Med. 2011, 183, 531–538. [Google Scholar] [CrossRef]
- Dong, A.Q.; Kong, M.J.; Ma, Z.Y.; Qian, J.F.; Xu, X.H. Down-regulation of IGF-IR using small, interfering, hairpin RNA (siRNA) inhibits growth of human lung cancer cell line A549 in vitro and in nude mice. Cell Biol. Int. 2007, 31, 500–507. [Google Scholar] [CrossRef]
- Xia, H.; Yu, C.H.; Zhang, Y.; Yu, J.; Li, J.; Zhang, W.; Zhang, B.; Li, Y.; Guo, N. EZH2 silencing with RNAi enhances irradiation-induced inhibition of human lung cancer growth in vitro and in vivo. Oncol. Lett. 2012, 4, 135–140. [Google Scholar]
- Aleku, M.; Schulz, P.; Keil, O.; Santel, A.; Schaeper, U.; Dieckhoff, B.; Janke, O.; Endruschat, J.; Durieux, B.; Roder, N.; et al. Atu027, a liposomal small interfering RNA formulation targeting protein kinase N3, inhibits cancer progression. Cancer Res. 2008, 68, 9788–9798. [Google Scholar]
- Leenders, F.; Mopert, K.; Schmiedeknecht, A.; Santel, A.; Czauderna, F.; Aleku, M.; Penschuck, S.; Dames, S.; Sternberger, M.; Rohl, T.; et al. PKN3 is required for malignant prostate cell growth downstream of activated PI 3-kinase. EMBO J. 2004, 23, 3303–3313. [Google Scholar] [CrossRef]
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 615–675. [Google Scholar] [CrossRef]
- Hao, S.; Baltimore, D. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol. 2009, 10, 281–288. [Google Scholar]
- Falk, J.A.; Minai, O.A.; Mosenifar, Z. Inhaled and systemic corticosteroids in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2008, 5, 506–512. [Google Scholar] [CrossRef]
- Tsuji, T.; Aoshiba, K.; Nagai, A. Cigarette smoke induces senescence in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 31, 643–649. [Google Scholar] [CrossRef]
- Tuder, R.M.; Kern, J.A.; Miller, Y.E. Senescence in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2012, 9, 62–63. [Google Scholar] [CrossRef]
- Molfino, N.A. Genetics of COPD. Chest 2004, 125, 1929–1940. [Google Scholar]
- Chen, Z.H.; Kim, H.P.; Ryter, S.W.; Choi, A.M. Identifying targets for COPD treatment through gene expression analyses. Int. J. Chron. Obstruct. Pulmon. Dis. 2008, 3, 359–370. [Google Scholar]
- Edwards, M.R.; Bartlett, N.W.; Clarke, D.; Birrell, M.; Belvisi, M.; Johnston, S.L. Targeting the NF-kappaB pathway in asthma and chronic obstructive pulmonary disease. Pharmacol. Ther. 2009, 121, 1–13. [Google Scholar] [CrossRef]
- Shapiro, S.D.; Goldstein, N.M.; Houghton, A.M.; Kobayashi, D.K.; Kelley, D.; Belaaouaj, A. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am. J. Pathol. 2003, 163, 2329–2335. [Google Scholar] [CrossRef]
- Yoshida, T.; Tuder, R.M. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol. Rev. 2007, 87, 1047–1082. [Google Scholar] [CrossRef]
- Huang, S.L.; Su, C.H.; Chang, S.C. Tumor necrosis factor-alpha gene polymorphism in chronic bronchitis. Am. J. Respir. Crit. Care Med. 1997, 156, 1436–1439. [Google Scholar]
- Stevenson, C.S.; Coote, K.; Webster, R.; Johnston, H.; Atherton, H.C.; Nicholls, A.; Giddings, J.; Sugar, R.; Jackson, A.; Press, N.J.; et al. Characterization of cigarette smoke-induced inflammatory and mucus hypersecretory changes in rat lung and the role of CXCR2 ligands in mediating this effect. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, 29. [Google Scholar]
- Stevenson, C.S.; Docx, C.; Webster, R.; Battram, C.; Hynx, D.; Giddings, J.; Cooper, P.R.; Chakravarty, P.; Rahman, I.; Marwick, J.A.; et al. Comprehensive gene expression profiling of rat lung reveals distinct acute and chronic responses to cigarette smoke inhalation. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L1183–L1193. [Google Scholar] [CrossRef]
- Aoshiba, K.; Yokohori, N.; Nagai, A. Alveolar wall apoptosis causes lung destruction and emphysematous changes. Am. J. Respir. Cell Mol. Biol. 2003, 28, 555–562. [Google Scholar]
- Kasahara, Y.; Tuder, R.M.; Taraseviciene-Stewart, L.; Le Cras, T.D.; Abman, S.; Hirth, P.K.; Waltenberger, J.; Voelkel, N.F. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J. Clin. Invest. 2000, 106, 1311–1319. [Google Scholar]
- Barnes, P.J.; Stockley, R.A. COPD: current therapeutic interventions and future approaches. Eur. Respir. J. 2005, 25, 1084–1106. [Google Scholar] [CrossRef]
- Banerjee, A.; Koziol-White, C.; Panettieri, R., Jr. p38 MAPK inhibitors, IKK2 inhibitors, and TNFalpha inhibitors in COPD. Curr. Opin. Pharmacol. 2012, 12, 287–292. [Google Scholar]
- World Health Organization. Chronic respiratory disease—Asthma. Available online: http://www.who.int/respiratory/asthma/en/ (accessed on 30 November 2012).
- Huang, H.Y.; Chiang, B.L. siRNA as a therapy for asthma. Curr. Opin. Mol. Ther. 2009, 11, 652–663. [Google Scholar]
- Meinicke, H.; Darcan, Y.; Hamelmann, E. Targeting allergic airway diseases by siRNA: An option for the future? Curr. Mol. Med. 2009, 9, 483–494. [Google Scholar] [CrossRef]
- Popescu, F.D. Antisense- and RNA interference-based therapeutic strategies in allergy. J. Cell Mol. Med. 2005, 9, 840–853. [Google Scholar]
- Stenton, G.R.; Kim, M.K.; Nohara, O.; Chen, C.F.; Hirji, N.; Wills, F.L.; Gilchrist, M.; Hwang, P.H.; Park, J.G.; Finlay, W.; et al. Aerosolized Syk antisense suppresses Syk expression, mediator release from macrophages, and pulmonary inflammation. J. Immunol. 2000, 164, 3790–3797. [Google Scholar]
- Ulanova, M.; Puttagunta, L.; Marcet-Palacios, M.; Duszyk, M.; Steinhoff, U.; Duta, F.; Kim, M.K.; Indik, Z.K.; Schreiber, A.D.; Befus, A.D. Syk tyrosine kinase participates in beta1-integrin signaling and inflammatory responses in airway epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, 19. [Google Scholar]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell. Biol 2009, 11, 228–234. [Google Scholar] [CrossRef]
- Pagdin, T.; Lavender, P. MicroRNAs in lung diseases. Thorax 2012, 67, 183–184. [Google Scholar]
- Trang, P.; Medina, P.P.; Wiggins, J.F.; Ruffino, L.; Kelnar, K.; Omotola, M.; Homer, R.; Brown, D.; Bader, A.G.; Weidhaas, J.B.; et al. Regression of murine lung tumors by the let-7 microRNA. Oncogene 2010, 29, 1580–1587. [Google Scholar]
- Wiggins, J.F.; Ruffino, L.; Kelnar, K.; Omotola, M.; Patrawala, L.; Brown, D.; Bader, A.G. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 2010, 70, 5923–5930. [Google Scholar] [CrossRef]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Orum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar]
- van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 2007, 316, 575–579. [Google Scholar]
- Porrello, E.R.; Johnson, B.A.; Aurora, A.B.; Simpson, E.; Nam, Y.J.; Matkovich, S.J.; Dorn, G.W., II.; van Rooij, E.; Olson, E.N. MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ. Res. 2011, 109, 670–679. [Google Scholar] [CrossRef]
- Zhu, H.; Yang, Y.; Wang, Y.; Li, J.; Schiller, P.W.; Peng, T. MicroRNA-195 promotes palmitate-induced apoptosis in cardiomyocytes by down-regulating Sirt1. Cardiovasc. Res. 2011, 92, 75–84. [Google Scholar] [CrossRef]
- Williams, A.H.; Valdez, G.; Moresi, V.; Qi, X.; McAnally, J.; Elliott, J.L.; Bassel-Duby, R.; Sanes, J.R.; Olson, E.N. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 2009, 326, 1549–1554. [Google Scholar] [CrossRef]
- Patrick, D.M.; Zhang, C.C.; Tao, Y.; Yao, H.; Qi, X.; Schwartz, R.J.; Jun-Shen Huang, L.; Olson, E.N. Defective erythroid differentiation in miR-451 mutant mice mediated by 14–3-3zeta. Genes Dev. 2010, 24, 1614–1619. [Google Scholar]
- Lewis, A.; Riddoch-Contreras, J.; Natanek, S.A.; Donaldson, A.; Man, W.D.; Moxham, J.; Hopkinson, N.S.; Polkey, M.I.; Kemp, P.R. Downregulation of the serum response factor/miR-1 axis in the quadriceps of patients with COPD. Thorax 2012, 67, 26–34. [Google Scholar] [CrossRef]
- Pottelberge, G.R.; Mestdagh, P.; Bracke, K.R.; Thas, O.; Durme, Y.M.; Joos, G.F.; Vandesompele, J.; Brusselle, G.G. MicroRNA expression in induced sputum of smokers and patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 183, 898–906. [Google Scholar] [CrossRef]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar]
- Pandit, K.V.; Corcoran, D.; Yousef, H.; Yarlagadda, M.; Tzouvelekis, A.; Gibson, K.F.; Konishi, K.; Yousem, S.A.; Singh, M.; Handley, D.; et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 220–229. [Google Scholar] [CrossRef]
- Cushing, L.; Kuang, P.P.; Qian, J.; Shao, F.; Wu, J.; Little, F.; Thannickal, V.J.; Cardoso, W.V.; Lu, J. miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 45, 287–294. [Google Scholar] [CrossRef]
- Yang, S.; Banerjee, S.; de Freitas, A.; Sanders, Y.Y.; Ding, Q.; Matalon, S.; Thannickal, V.J.; Abraham, E.; Liu, G. Participation of miR-200 in pulmonary fibrosis. Am. J. Pathol. 2012, 180, 484–493. [Google Scholar] [CrossRef]
- Rodriguez, A.; Vigorito, E.; Clare, S.; Warren, M.V.; Couttet, P.; Soond, D.R.; van Dongen, S.; Grocock, R.J.; Das, P.P.; Miska, E.A.; et al. Requirement of bic/microRNA-155 for normal immune function. Science 2007, 316, 608–611. [Google Scholar]
- Lu, T.X.; Munitz, A.; Rothenberg, M.E. MicroRNA-21 is up-regulated in allergic airway inflammation and regulates IL-12p35 expression. J. Immunol. 2009, 182, 4994–5002. [Google Scholar] [CrossRef]
- Mattes, J.; Collison, A.; Plank, M.; Phipps, S.; Foster, P.S. Antagonism of microRNA-126 suppresses the effector function of TH2 cells and the development of allergic airways disease. Proc. Natl. Acad. Sci. USA 2009, 106, 18704–18709. [Google Scholar]
- Chiba, Y.; Tanabe, M.; Goto, K.; Sakai, H.; Misawa, M. Down-regulation of miR-133a contributes to up-regulation of Rhoa in bronchial smooth muscle cells. Am. J. Respir. Crit. Care Med. 2009, 180, 713–719. [Google Scholar] [CrossRef]
- Ezzie, M.E.; Crawford, M.; Cho, J.H.; Orellana, R.; Zhang, S.; Gelinas, R.; Batte, K.; Yu, L.; Nuovo, G.; Galas, D.; et al. Gene expression networks in COPD: microRNA and mRNA regulation. Thorax 2012, 67, 122–131. [Google Scholar] [CrossRef]
- Sato, T.; Liu, X.; Nelson, A.; Nakanishi, M.; Kanaji, N.; Wang, X.; Kim, M.; Li, Y.; Sun, J.; Michalski, J.; et al. Reduced miR-146a increases prostaglandin E(2)in chronic obstructive pulmonary disease fibroblasts. Am. J. Respir. Crit. Care Med. 2010, 182, 1020–1029. [Google Scholar] [CrossRef]
- Pottier, N.; Maurin, T.; Chevalier, B.; Puissegur, M.P.; Lebrigand, K.; Robbe-Sermesant, K.; Bertero, T.; Lino Cardenas, C.L.; Courcot, E.; Rios, G.; et al. Identification of keratinocyte growth factor as a target of microRNA-155 in lung fibroblasts: Implication in epithelial-mesenchymal interactions. PLoS One 2009. [Google Scholar] [CrossRef]
- Izzotti, A.; Calin, G.A.; Arrigo, P.; Steele, V.E.; Croce, C.M.; de Flora, S. Downregulation of microRNA expression in the lungs of rats exposed to cigarette smoke. FASEB J. 2009, 23, 806–812. [Google Scholar]
- Schembri, F.; Sridhar, S.; Perdomo, C.; Gustafson, A.M.; Zhang, X.; Ergun, A.; Lu, J.; Liu, G.; Zhang, X.; Bowers, J.; et al. MicroRNAs as modulators of smoking-induced gene expression changes in human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 2319–2324. [Google Scholar]
- Decramer, M.; Rennard, S.; Troosters, T.; Mapel, D.W.; Giardino, N.; Mannino, D.; Wouters, E.; Sethi, S.; Cooper, C.B. COPD as a lung disease with systemic consequences--clinical impact, mechanisms, and potential for early intervention. COPD 2008, 5, 235–256. [Google Scholar] [CrossRef]
- Gower, A.C.; Steiling, K.; Brothers, J.F., II.; Lenburg, M.E.; Spira, A. Transcriptomic studies of the airway field of injury associated with smoking-related lung disease. Proc. Am. Thorac. Soc. 2011, 8, 173–179. [Google Scholar] [CrossRef]
- Agusti, A.G. COPD, a multicomponent disease: implications for management. Respir. Med. 2005, 99, 670–682. [Google Scholar] [CrossRef]
- Locksley, R.M. Asthma and allergic inflammation. Cell 2010, 140, 777–783. [Google Scholar] [CrossRef]
- Williams, A.E.; Larner-Svensson, H.; Perry, M.M.; Campbell, G.A.; Herrick, S.E.; Adcock, I.M.; Erjefalt, J.S.; Chung, K.F.; Lindsay, M.A. MicroRNA expression profiling in mild asthmatic human airways and effect of corticosteroid therapy. PLoS One 2009. [Google Scholar] [CrossRef] [Green Version]
- Oglesby, I.K.; McElvaney, N.G.; Greene, C.M. MicroRNAs in inflammatory lung disease--master regulators or target practice? Respir. Res. 2010, 11, 148. [Google Scholar] [CrossRef]
- Boudreau, R.L.; Martins, I.; Davidson, B.L. Artificial microRNAs as siRNA shuttles: improved safety as compared to shRNAs in vitro and in vivo. Mol. Ther. 2009, 17, 169–175. [Google Scholar] [CrossRef]
- Bader, A.G.; Brown, D.; Winkler, M. The promise of microRNA replacement therapy. Cancer Res. 2010, 70, 7027–7030. [Google Scholar]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef]
- He, X.Y.; Chen, J.X.; Zhang, Z.; Li, C.L.; Peng, Q.L.; Peng, H.M. The let-7a microRNA protects from growth of lung carcinoma by suppression of k-Ras and c-Myc in nude mice. J. Cancer Res. Clin. Oncol. 2010, 136, 1023–1028. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Trang, P.; Wiggins, J.F.; Patrawala, L.; Cheng, A.; Ford, L.; Weidhaas, J.B.; Brown, D.; Bader, A.G.; Slack, F.J. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle 2008, 7, 759–764. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, X.; Zhang, X.; Liu, B.; Huang, L. Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Mol. Ther. 2010, 18, 1650–1656. [Google Scholar] [CrossRef]
- Ling, B.; Wang, G.X.; Long, G.; Qiu, J.H.; Hu, Z.L. Tumor suppressor miR-22 suppresses lung cancer cell progression through post-transcriptional regulation of ErbB3. J. Cancer Res. Clin. Oncol. 2012, 138, 1355–1361. [Google Scholar]
- Krutzfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with “antagomirs”. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef]
- Elmen, J.; Lindow, M.; Schutz, S.; Lawrence, M.; Petri, A.; Obad, S.; Lindholm, M.; Hedtjarn, M.; Hansen, H.F.; Berger, U.; et al. LNA-mediated microRNA silencing in non-human primates. Nature 2008, 452, 896–899. [Google Scholar] [CrossRef]
- Li, Y.J.; Zhang, Y.X.; Wang, P.Y.; Chi, Y.L.; Zhang, C.; Ma, Y.; Lv, C.J.; Xie, S.Y. Regression of A549 lung cancer tumors by anti-miR-150 vector. Oncol. Rep. 2012, 27, 129–134. [Google Scholar]
- Zheng, T.; Wang, J.; Chen, X.; Liu, L. Role of microRNA in anticancer drug resistance. Int. J. Cancer 2010, 126, 2–10. [Google Scholar] [CrossRef]
- Ji, Q.; Hao, X.; Zhang, M.; Tang, W.; Yang, M.; Li, L.; Xiang, D.; Desano, J.T.; Bommer, G.T.; Fan, D.; et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One 2009, 4. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fujita, Y.; Takeshita, F.; Kuwano, K.; Ochiya, T. RNAi Therapeutic Platforms for Lung Diseases. Pharmaceuticals 2013, 6, 223-250. https://doi.org/10.3390/ph6020223
Fujita Y, Takeshita F, Kuwano K, Ochiya T. RNAi Therapeutic Platforms for Lung Diseases. Pharmaceuticals. 2013; 6(2):223-250. https://doi.org/10.3390/ph6020223
Chicago/Turabian StyleFujita, Yu, Fumitaka Takeshita, Kazuyoshi Kuwano, and Takahiro Ochiya. 2013. "RNAi Therapeutic Platforms for Lung Diseases" Pharmaceuticals 6, no. 2: 223-250. https://doi.org/10.3390/ph6020223
APA StyleFujita, Y., Takeshita, F., Kuwano, K., & Ochiya, T. (2013). RNAi Therapeutic Platforms for Lung Diseases. Pharmaceuticals, 6(2), 223-250. https://doi.org/10.3390/ph6020223